

Abstract
We evaluated the influence of aerobic exercise on cardiac remodelling during the transition from compensated left ventricular (LV) hypertrophy to clinical heart failure in aortic stenosis (AS) rats. Eighteen weeks after AS induction, rats were assigned into sedentary (AS) and exercised (AS‐Ex) groups. Results were compared to Sham rats. Exercise was performed on treadmill for 8 weeks. Exercise improved functional capacity. Echocardiogram showed no differences between AS‐Ex and AS groups. After exercise, fractional shortening and ejection fraction were lower in AS‐Ex than Sham. Myocyte diameter and interstitial collagen fraction were higher in AS and AS‐Ex than Sham; however, myocyte diameter was higher in AS‐Ex than AS. Myocardial oxidative stress, evaluated by lipid hydroperoxide concentration, was higher in AS than Sham and was normalized by exercise. Gene expression of the NADPH oxidase subunits NOX2 and NOX4, which participate in ROS generation, did not differ between groups. Activity of the antioxidant enzyme superoxide dismutase was lower in AS and AS‐Ex than Sham and glutathione peroxidase was lower in AS‐Ex than Sham. Total and reduced myocardial glutathione, which is involved in cellular defence against oxidative stress, was lower in AS than Sham and total glutathione was higher in AS‐Ex than AS. The MAPK JNK was higher in AS‐Ex than Sham and AS groups. Phosphorylated P38 was lower in AS‐Ex than AS. Despite improving functional capacity, aerobic exercise does not change LV function in AS rats. Exercise restores myocardial glutathione, reduces oxidative stress, impairs JNK signalling and further induces myocyte hypertrophy.
Keywords: cardiac remodelling, echocardiogram, MAPK, oxidative stress, physical training, rat
1. INTRODUCTION
Pathological cardiac hypertrophy induced by chronic pressure overload conditions, such as aortic stenosis and arterial systemic hypertension, is often associated with impaired prognosis and increased mortality.1 Long‐term pressure overload is a major cause of cardiac remodelling evolving progressively from compensated left ventricular (LV) hypertrophy to diastolic and systolic dysfunction and clinical heart failure.2
Despite advances in the pharmacologic treatment of heart failure, morbidity and mortality levels remain elevated.1 Several authors and current guidelines recommend physical exercise for patients with stable heart failure to prevent and/or attenuate cardiac remodelling and skeletal muscle changes and improve functional capacity and quality of life.3, 4, 5, 6, 7 Physical exercise in experimental heart failure attenuated ventricular dilation, cardiac dysfunction, increased passive stiffness, myocyte hypertrophy, myocardial fibrosis, myocyte calcium handling changes and mitochondrial dysfunction.8, 9, 10, 11, 12, 13 However, as most benefits of exercise have been described in experimental models of myocardial infarction‐induced heart failure,14, 15, 16, 17 the effects of exercise during sustained LV pressure‐overload remains poorly understood. In fact, studies performed in this experimental model have returned controversial results. Aortic stenosis mice subjected to an 8‐week‐period of voluntary rotating wheel exercise showed no improvement in LV function and presented a trend towards impaired ventricular dysfunction in severe cases.18, 19 As mice perform very well in voluntary wheel running,20 excessive exercise may have contributed to these results. We have previously shown that aerobic exercise improves functional capacity and attenuates systolic dysfunction in rats with uncontrolled arterial hypertension21 or aortic stenosis.22
Ascending AS in rats has often been used to study chronic pressure overload. In this model, 3‐ to 4‐week‐old rats are subjected to thoracotomy and clip placement around the ascending aorta. Immediately after clip positioning, aorta diameter is preserved and stenosis progressively occurs as rats age. LV hypertrophy develops quickly, occurring within 1 month.23 However, ventricular dysfunction and cardiac failure develop slowly, similar to what is seen in human chronic pressure overload.23 This study evaluated the influence of aerobic exercise on cardiac remodelling in rats with AS performed during the period of transition from compensated LV hypertrophy to overt heart failure. An important effect of exercise is a reduction in oxidative stress24 associated with an attenuation in cardiac remodelling. We therefore analysed markers of systemic and myocardial oxidative stress, and myocardial antioxidant enzyme activity. The NADPH oxidase (NOX) family is a source of reactive oxygen species in various tissues including the myocardium,25 we therefore analysed subunits NOX2 and NOX4 gene expression. Since mitogen‐activated protein kinases (MAPK) may be involved in myocardial response to oxidative stress,26 we also evaluated their proteins expression in myocardium. Results for myocardial morphometric evaluation, oxidative stress analyses and protein expression from Sham and AS rats have been previously published.27
2. MATERIALS AND METHODS
2.1. Experimental animals and study protocol
The experimental protocol employed in this study was approved by the Animal Experimentation Ethics Committee of Botucatu Medical School, UNESP, SP, Brazil, and has previously been described in detail.22 Male Wistar rats (90‐100 g) were purchased from the Central Animal House, Botucatu Medical School, UNESP, and housed in a temperature controlled room at 23°C and kept on a 12‐hour light/dark cycle. Food and water were supplied ad libitum.
In short, Wistar rats (90‐100 g) were anaesthetized with a mixture of ketamine hydrochloride (50 mg/kg, i.m.) and xylazine hydrochloride (10 mg/kg, i.m.). Aortic stenosis was induced by placing a 0.6 mm stainless steel clip on the ascending aorta via a thoracic incision.22 Sham operated rats were used as controls. Eighteen weeks after surgery, rats were assigned to three groups: Sham (n = 23), sedentary AS (AS, n = 28) and exercised AS (AS‐Ex, n = 31) for 8 weeks. During euthanasia, we assessed the presence or absence of clinical and pathologic heart failure features. The clinical finding suggestive of heart failure was tachypnoea/laboured respiration. Pathologic assessment of heart failure included pleuropericardial effusion, left atrial thrombi, hepatic congestion, pulmonary congestion (lung weight/bodyweight ratio more than two standard deviations above Sham group mean) and right ventricular hypertrophy (right ventricle weight/body weight ratio more than 0.8 mg/g).28 Functional capacity was evaluated and transthoracic echocardiogram was performed before and after the exercise protocol.
2.2. Exercise testing
Rats underwent 10 min/day test environment adaption for 1 week before evaluations. Each animal was tested individually. The test consisted of an initial 5‐min warm up at 5 m/min on treadmill. The rats were then subjected to exercise at 8 m/min followed by increments of 3 m/min every 3 min until exhaustion. Exhaustion was determined when the animal refused to run even after electric stimulation or was unable to coordinate steps.21 Maximum running speed was recorded and total distance was calculated.
2.3. Exercise training protocol
Exercise was performed on a treadmill five times a week for 8 weeks.21, 29 There was an adaptation period, with a gradual increase in speed and exercise duration. Speed from the 1st to the 3rd week was 5, 7.5 and 10 m/min, and then remained constant until the end of the protocol. Exercise duration from the 1st to the 6th week was 10, 14, 18, 22, 26 and 30 min, and then remained constant until the end of the experiment.
2.4. Echocardiography
Cardiac structures and LV function were evaluated by transthoracic echocardiogram and tissue Doppler imaging using a commercially available echocardiograph (General Electric Medical Systems, Vivid S6 model, Tirat Carmel, Israel) equipped with a 5‐11.5 MHz multifrequency transducer.30, 31, 32, 33 The rats were anaesthetized with a mixture of ketamine hydrochloride (50 mg/kg) and xylazine hydrochloride (1 mg/kg) intramuscularly. A two‐dimensional parasternal short‐axis view of the left ventricle was obtained at the level of the papillary muscles. M‐mode tracings were obtained from short‐axis views of the LV at or just below the tip of the mitral‐valve leaflets, and at the level of the aortic valve and left atrium. M‐mode images of the LV were printed on a black and white thermal printer (Sony UP‐890MD, Montvale, NJ, USA) at a sweep speed of 100 mm/s. All LV structures were manually measured by the same observer (KO). Values obtained were the mean of at least five cardiac cycles on M‐mode tracings. The following structural variables were measured: left atrium diameter (LA), LV diastolic and systolic diameters (LVDD and LVSD respectively), LV diastolic (D) and systolic (S) posterior wall thickness (PWT) and septal wall thickness (SWT) and aortic diameter. LV mass (LVM) was calculated using the formula [(LVDD + DPWT + DSWT)3 − LVDD3] × 1.04. LV relative wall thickness (RWT) was calculated by the formula 2 × DPWT/LVDD. LV function was assessed by the following parameters: endocardial fractional shortening (EFS), midwall fractional shortening (MFS), ejection fraction (EF), posterior wall shortening velocity (PWSV), early and late diastolic mitral inflow velocities (E and A waves), E/A ratio, E wave deceleration time (EDT) and isovolumetric relaxation time (IVRT). A joint assessment of diastolic and systolic LV function was performed with the myocardial performance index (Tei index). The study was complemented using tissue Doppler imaging (TDI) to evaluate systolic (S′), early diastolic (E′) and late diastolic (A′) velocity of the mitral annulus (arithmetic average of the lateral and septal walls), and E/E′ ratio.
2.5. Collection of tissues for analysis
One day after final echocardiogram, the rats were weighed, anaesthetized with intraperitoneal sodium pentobarbital (50 mg/kg), and killed. After blood collecting, hearts were removed by thoracotomy. Atria and ventricles were dissected and weighed separately. LV was frozen in liquid nitrogen and stored at −80°C. The liver was macroscopically examined to determine the presence or absence of liver congestion. Lung weight was used to assess the degree of pulmonary congestion.34
2.6. Morphologic study
Left ventricular serial transverse 8 μm thick sections were cut in a cryostat, cooled to −20°C and stained with haematoxylin and eosin. At least 50 cardiomyocyte diameters were measured from each LV as the shortest distance between borders drawn across the nucleus.21 Other slides were stained with Sirius red F3BA and used to quantify interstitial collagen fraction. On average, 20 microscopic fields were analysed with a 40 X lens. Perivascular collagen was excluded from this analysis. Measurements were performed with a microscope (Leica DM LS; Nussloch, Germany) attached to a computerized imaging analysis system (Media Cybernetics, Silver Spring, MD, USA).
2.7. Oxidative stress evaluation
2.7.1. Myocardial glutathione concentration
Reduced glutathione was determined using a kinetic method in media consisting of 100 mmol L−1 phosphate buffer pH 7.4 containing 5 mmol L−1 EDTA, 2 mmol L−1 5.5′‐dithiobis‐(2‐nitrobenzoic) acid (DTNB), 0.2 mmol L−1 NADPH2 and 2 U of glutathione reductase.35 Total glutathione was measured in the presence of 0.1 mol L−1 Tris‐HCl buffer, pH 8.0 with 0.5 mmol L−1 EDTA, 0.6 mmol L−1 DTNB and 0.1 U glutathione reductase.35
2.7.2. Malondialdehyde serum concentration
Systemic lipid peroxidation was assessed by measuring malondialdehyde (MDA) by high performance liquid chromatography (HPLC). One hundred microlitre of serum was treated with 700 μL of 1% orthophosphoric acid and vortex‐mixed for 10 seconds for protein precipitation. Then, 200 μL of thiobarbituric acid (TBA) were added. The mixture was heated to 100°C for 60 minutes and cooled to −20°C for 10 minutes. Then, 200 μL of this reaction was added to a solution containing 200 μL of NaOH: methanol (1:12). The tubes were centrifuged for 3 minutes at 13 000 g; 200 μL of the supernatant was taken for injection into the equipment. Analysis was performed on a Shimadzu HPLC using a C18 5 μm Gemini Phenomenex column, and an Rf‐535 fluorescence detector, which was set to Ex 525 nm and EM 551 nm. The mobile phase consisted of a 60:40 (v/v) mixture of 10 mmol potassium dihydrogen phosphate (pH 6.8): methanol. MDA was quantified by a calibration curve, which was constructed every day for analysis.
2.7.3. Antioxidant enzymes activity and lipid hydroperoxide concentration
Left ventricular samples (~200 mg) were homogenized in 5 mL of cold 0.1 mol L−1 phosphate buffer, pH 7.0. Tissue homogenates were prepared in a motor‐driven Teflon glass Potter‐Elvehjem tissue homogenizer. The homogenate was centrifuged at 10 000 g, for 15 minutes at 4°C, and the supernatant was assayed for total protein, lipid hydroperoxide,36 and glutathione peroxidase (GSH‐Px, E.C.1.11.1.9), catalase (E.C.1.11.1.6.) and superoxide dismutase (SOD, E.C.1.15.1.1.) activities by spectrophotometry.37 Enzyme activities were analysed at 25°C using a microplate reader (μQuant‐MQX 200) with KCjunior software for computer system control (Bio‐Tech Instruments, Winooski, VT, USA). Spectrophotometric determinations were performed in a Pharmacia Biotech spectrophotometer with temperature controlled cuvette chamber (UV/visible Ultrospec 5000 with Swift II applications software for computer system control, Cambridge, UK). All reagents were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
2.8. Real‐time quantitative reverse transcription‐polymerase chain reaction (RT‐PCR)
Gene expression of NADPH oxidase subunits (NOX2, NOX4, p22 phox and p47 phox) and reference genes cyclophilin and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was analysed by RT‐PCR according to a previously described method.38 Total RNA was extracted from LV myocardium with TRIzol Reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) and treated with DNase I (Invitrogen Life Technologies). One microgram of RNA was reverse transcribed using High Capacity cDNA Reverse Transcription Kit, according to standard methods (Applied Biosystems, Foster City, CA, USA). Aliquots of cDNA were then submitted to real‐time PCR using a customized assay containing sense and antisense primers, and Taqman (Applied Biosystems) probes specific to each gene: NOX2 (Rn00576710m1), NOX4 (Rn00585380m1), p22 phox (Rn00577357m1) and p47 phox (Rn00586945m1). Amplification and analysis were performed with Step One Plus™ Real‐Time PCR System (Applied Biosystems). Expression data were normalized to reference gene expressions: cyclophilin (Rn00690933m1) and GAPDH (Rn01775763g1). Reactions were performed in triplicate and expression levels were calculated using the CT comparative method (2−ΔΔCT).
2.9. Western blotting
Protein levels were analysed by Western blotting using specific antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA): total JNK1/2 (sc‐137019), p‐JNK (sc‐6254), total p38‐MAPK (sc‐7972), p‐p38‐MAPK (sc‐17852), total ERK 1 (sc‐93) and p‐ERK1/2 (sc‐16982).39 Protein levels were normalized to GAPDH (6C5 sc‐32233). Myocardial protein was extracted using RIPA buffer (containing proteases and phosphatases inhibitors); supernatant protein content was quantified by the Bradford method. Samples were separated on a polyacrylamide gel and then transferred to a nitrocellulose membrane. After blockade, membrane was incubated with the primary antibodies. The membrane was then washed with TBS and Tween 20 and incubated with secondary peroxidase‐conjugated antibodies. Super Signal® West Pico Chemiluminescent Substrate (Pierce Protein Research Products, Rockford, IL, USA) was used to detect bound antibodies.
2.10. Statistical analysis
Data are expressed as mean ± standard deviation or median and percentile. Comparisons between groups were performed by one‐way ANOVA and Tukey test or Kruskal‐Wallis and Dunn test. Mortality was assessed by log‐rank test (Kaplan‐Meier). Frequency of heart failure features was assessed by the Goodman test. Significance level was set at 5%.
3. RESULTS
Survival rate did not differ between AS and AS‐Ex groups. Heart failure features were evaluated at euthanize. The frequency of heart failure features did not differ between AS and AS‐Ex groups. Anatomical data are presented in Table 1. Both AS groups had left and right ventricular hypertrophy, and higher atria and lung weights compared to Sham. There were no differences between AS‐Ex and AS groups.
Table 1.
Anatomical data
| Sham (n = 23) | AS (n = 19) | AS‐Ex (n = 22) | |
|---|---|---|---|
| BW (g) | 510 ± 50.7 | 482 ± 41.6 | 464 ± 57.5 |
| LVW (g) | 0.94 (0.88‐0.98) | 1.84 (1.56‐2.09)a | 1.48 (1.13‐1.95)a |
| LVW/BW (mg/g) | 1.86 (1.69‐2.05) | 3.80 (2.98‐4.33)a | 3.19 (2.23‐5.05)a |
| RVW (g) | 0.24 (0.21‐0.25) | 0.46 (0.40‐0.55)a | 0.43(0.36‐0.46)a |
| RVW/BW (mg/g) | 0.48 (0.45‐0.50) | 0.99 (0.83‐1.11)a | 0.89 (0.66‐1.05)a |
| Atria weight (g) | 0.10 (0.09‐0.11) | 0.36 (0.21‐0.41)a | 0.34 (0.31‐0.41)a |
| Atria/BW (mg/g) | 0.20 (0.19‐0.23) | 0.75 (0.66‐0.82)a | 0.71 (0.46‐0.93)a |
| Lung weight (g) | 1.80 (1.66‐1.99) | 2.89 (2.11‐3.29)a | 3.96 (2.31‐4.37)a |
| Lung/BW (mg/g) | 3.80 (3.12‐4.11) | 6.30 (4.29‐7.05)a | 8.41 (4.71‐9.74)a |
One‐way ANOVA and Tukey or Kruskal‐Wallis and Dunn test. Data are mean ± SD or median and percentile.
AS: aortic stenosis; AS‐Ex: exercised aortic stenosis; BW: body weight; LVW: left ventricle weight; RVW: right ventricle weight.
P < 0.05 vs Sham.
Maximal exercise test is shown in Figure 1. Before exercise, running time was lower in both AS and AS‐Ex than Sham. After exercise, running time and distance were lower in AS than Sham and AS‐Ex.
Figure 1.
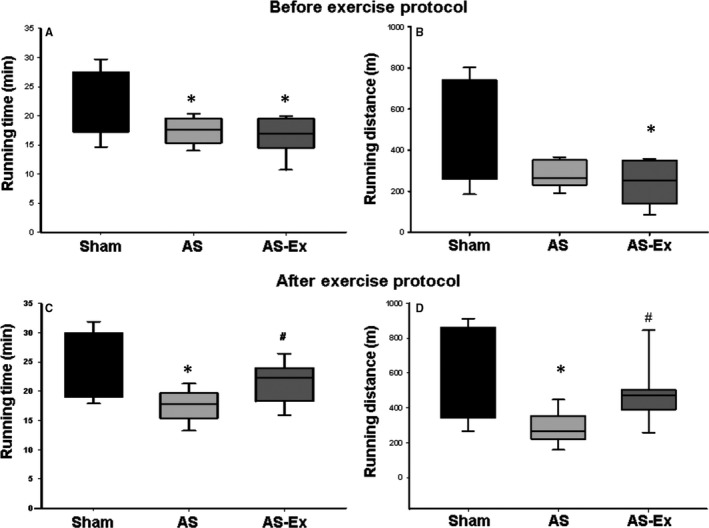
Maximal exercise test. Running time (A) and running distance (B) before exercise protocol; running time (C) and running distance (D) after exercise protocol. AS: aortic stenosis; AS‐Ex: exercised aortic stenosis. Data are median and percentile; Kruskal‐Wallis and Dunn test. *P < 0.05 vs Sham; # P < 0.05 vs AS
Before exercise (data not shown), AS and AS‐Ex presented left atrium dilation and concentric LV hypertrophy, characterized by increased LV wall thickness, LV mass and relative wall thickness. Both AS‐Ex and AS had LV systolic and diastolic dysfunction as posterior wall shortening velocity, isovolumetric relaxation time and E wave deceleration time were lower, and E wave and E‐to‐tissue Doppler imaging (TDI) of early diastolic velocity of mitral annulus ratio were higher than Sham. TDI S′ was lower in AS than Sham. There were no differences between AS‐Ex and AS.
Echocardiographic evaluation after the exercise protocol showed the same cardiac remodelling pattern as before exercise. Additionally, both stenosis groups presented increased LV systolic and diastolic diameters and E/A ratio, and decreased TDI S′ and A wave. Endocardial and midwall fractional shortening and ejection fraction were lower in AS‐Ex than Sham. No differences were observed between AS‐Ex and AS (Tables 2 and 3).
Table 2.
Echocardiographic structural data after exercise protocol
| Sham (n = 23) | AS (n = 19) | AS‐Ex (n = 22) | |
|---|---|---|---|
| BW (g) | 504 ± 58 | 478 ± 50 | 461 ± 50a |
| LVDD (mm) | 8.29 ± 0.54 | 9.16 ± 0.86a | 9.11 ± 0.84a |
| LVDD/BW (mm/kg) | 16.7 ± 2.04 | 19.3 ± 2.62a | 20.0 ± 3.14a |
| LVSD (mm) | 3.99 (3.58‐4.50) | 5.04 (4.06‐5.34)a | 5.12 (4.67‐5.99)a |
| DPWT (mm) | 1.42 (1.38‐1.46) | 2.05 (1.85‐2.11)a | 2.02 (1.84‐2.17)a |
| SPWT (mm) | 3.01 (2.75‐3.14) | 3.48 (2.87‐3.79)a | 3.49 (2.96‐3.74)a |
| DSWT (mm) | 1.43 (1.39‐1.46) | 2.11 (1.85‐2.16)a | 2.02 (1.84‐2.17)a |
| SSWT (mm) | 2.51 (2.38‐2.64) | 2.82 (2.65‐3.16)a | 3.00 (2.68‐3.25)a |
| RWT | 0.34 (0.33‐0.36) | 0.42 (0.40‐0.47)a | 0.44 (0.38‐0.49)a |
| AO (mm) | 4.02 (3.83‐4.16) | 3.94 (3.84‐4.16) | 3.97± 0.20 |
| LA (mm) | 5.29 (4.93‐5.64) | 8.54 (7.72‐8.71)a | 8.25 (7.15‐8.98)a |
| LA/AO | 1.34 (1.26‐1.38) | 2.17 (1.86‐2.29)a | 2.12 (1.75‐2.25)a |
| LA/BW (mm/kg) | 10.4 (9.53‐12.1) | 17.4 (14.8‐19.5)a | 17.6 (15.7‐19.5)a |
| LVM (g) | 0.82 (0.76‐0.89) | 1.41 (1.33‐1.96)a | 1.61 (1.18‐1.87)a |
| LVMI (g/kg) | 1.70 (1.46‐1.87) | 2.97 (2.70‐3.90)a | 3.40 (2.66‐4.16)a |
One‐way ANOVA and Tukey or Kruskal‐Wallis and Dunn test. Data are mean ± SD or median and percentile.
AS: aortic stenosis; AS‐Ex: exercised aortic stenosis; BW: body weight; LVDD and LVSD: left ventricular (LV) diastolic and systolic diameters respectively; DPWT and SPWT: LV diastolic and systolic posterior wall thickness respectively; DSWT and SSWT: LV diastolic and systolic septal wall thickness respectively; RWT: relative wall thickness; AO: aorta diameter; LA: left atrial diameter; LVM: LV mass; LVMI: LVM index.
P < 0.05 vs Sham.
Table 3.
Echocardiographic data of left ventricular function after exercise protocol
| Sham (n = 23) | AS (n = 19) | AS‐Ex (n = 22) | |
|---|---|---|---|
| HR (bpm) | 291 ± 40 | 295 ± 41 | 319 ± 41 |
| EFS (%) | 51.0 (46.4‐55.3) | 45.6 (38.7‐55.3) | 43.8 (38.3‐48.2)a |
| MFS (%) | 29.5 (27.2‐32.4) | 26.8 (21.1‐33.5) | 24.5 (21.6‐28.6)a |
| PWSV (mm/s) | 40.6 ± 5.55 | 30.8 ± 6.96a | 29.2 ± 7.15a |
| Tei index | 0.44 ± 0.08 | 0.42 ± 0.08 | 0.42 ± 0.11 |
| EF | 0.88 (0.85‐0.91) | 0.84 (0.77‐0.91) | 0.82 (0.77‐0.86)a |
| TDI S′ (average, cm/s) | 3.76 ± 0.70 | 2.95 ± 0.67a | 2.92 ± 0.49a |
| Mitral E (cm/s) | 79.0 (72.3‐83.5) | 142 (85.0‐158)a | 142 (98‐164)a |
| Mitral A (cm/s) | 58.0 (51.5‐67.0) | 25.5 (22.0‐47.0)a | 20.0 (16.0‐58.5)a |
| E/A | 1.39 (1.23‐1.52) | 5.38 (1.74‐6.82)a | 8.00 (1.49‐9.14)a |
| IVRT (ms) | 26.0 (22.0‐26.0) | 18.0 (15.0‐22.0)a | 16.0 (15.0‐22.0)a |
| IVRTn | 53.0 ± 7.08 | 42.8 ± 11.0a | 39.6 ± 10.3a |
| EDT (ms) | 46.2 ± 6.87 | 31.1 ± 8.67a | 30.1 ± 8.33a |
| TDI E′ (average, cm/s) | 4.51 ± 0.77 | 4.00 ± 1.26 | 4.23 ± 0.99 |
| TDI A′ (average, cm/s) | 4.49 ± 1.34 | 3.91 ± 1.39 | 4.07 ± 1.05 |
| E/TDI E′ (average) | 17.6 (14.7‐19.8) | 33.5 (26.5‐41.0)a | 34.3 (25.1‐40.4)a |
One‐way ANOVA and Tukey or Kruskal‐Wallis and Dunn test. Data are mean ± SD or median and percentile.
AS: aortic stenosis; AS‐Ex: exercised aortic stenosis; HR: heart rate; EFS: endocardial fractional shortening; MFS: midwall fractional shortening; PWSV: posterior wall shortening velocity; Tei index: myocardial performance index; EF: ejection fraction; TDI S′: tissue Doppler imaging (TDI) of systolic velocity of the mitral annulus; E/A: ratio between early (E)‐to‐late (A) diastolic mitral inflow; IVRT: isovolumetric relaxation time; IVRTn: IVRT normalized to heart rate; EDT: E wave deceleration time; TDI E′ and A′: TDI of early (E′) and late (A′) diastolic velocity of mitral annulus.
P < 0.05 vs Sham.
Myocyte diameter and interstitial collagen fraction were higher in AS and AS‐Ex than Sham. Myocyte diameter was higher in AS‐Ex than AS (Table 4).
Table 4.
Myocardial morphometric parameters
| Sham (n = 10) | AS (n = 12) | AS‐Ex (n = 8) | |
|---|---|---|---|
| Myocyte diameter (μm) | 13.4 ± 1.17 | 15.3 ± 0.97* | 16.7 ± 1.75* , # |
| ICF (%) | 4.30 ± 1.20 | 9.86 ± 1.69* | 8.46 ± 1.61* |
One‐way ANOVA and Tukey test. Data are mean ± SD.
AS: aortic stenosis; AS‐Ex: exercised aortic stenosis; ICF: interstitial collagen fraction.
*P < 0.05 vs Sham; # P < 0.05 vs AS.
Total and reduced myocardial concentration of glutathione was lower in AS than Sham and total glutathione concentration was higher in AS‐Ex than AS (Figure 2A and B). Malondialdehyde serum concentration did not differ between groups (Figure 2C). Lipid hydroperoxide myocardial concentration was higher in AS than Sham and AS‐Ex (Figure 2D). Myocardial antioxidant enzyme activities are shown in Figure 2 panels E, F and G. Superoxide dismutase was lower in AS and AS‐Ex than Sham and glutathione peroxidase was lower in AS‐Ex than Sham.
Figure 2.
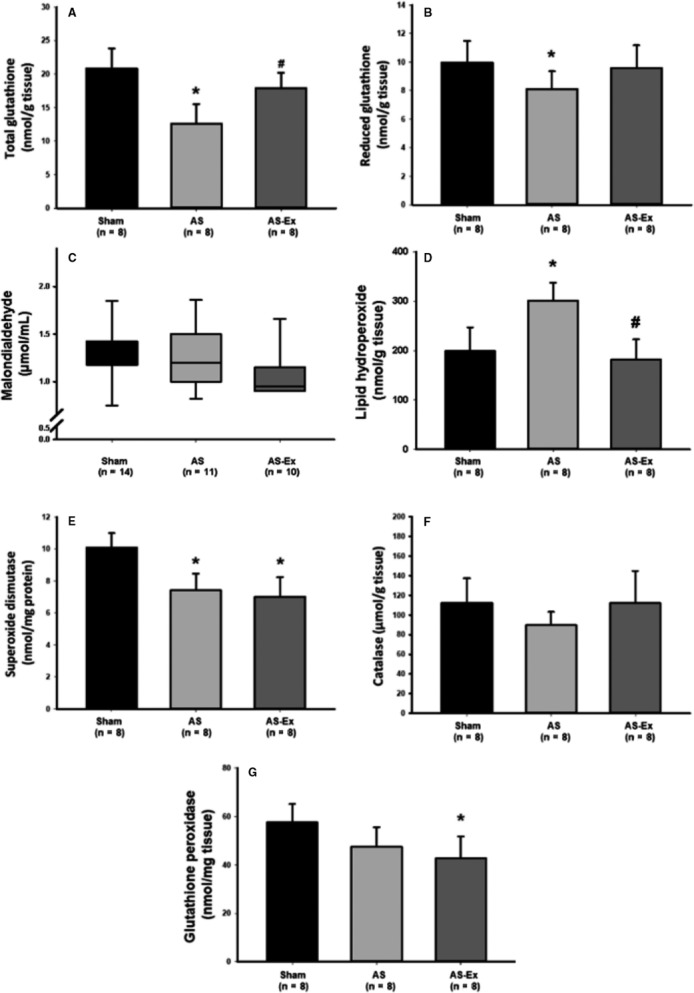
Myocardial concentration of total (A) and reduced glutathione (B); malondialdehyde serum concentration (C); lipid hydroperoxide myocardial concentration (D); and myocardial antioxidant enzyme activity: superoxide dismutase (E), catalase (F) and glutathione peroxidase (G). AS: aortic stenosis; AS‐Ex: exercised aortic stenosis; n: number of animals. Data are mean ± SD or median and percentile; ANOVA and Tukey or Kruskal‐Wallis and Dunn test; *P < 0.05 vs Sham; # P < 0.05 vs AS
Myocardial gene expression of NADPH oxidase subunits did not change between groups (Table 5). Myocardial expression of MAPK proteins is shown in Table 5. Phosphorylated JNK was higher in AS‐Ex than Sham and AS groups and total JNK was higher in AS‐Ex than Sham. Phosphorylated P38 was lower in AS‐Ex than AS.
Table 5.
Myocardial gene expression of NADPH oxidase subunits and protein expression of MAPKs
| Sham (n = 8) | AS (n = 7) | AS‐Ex (n = 8) | |
|---|---|---|---|
| p22 phox | 1.00 ± 0.51 | 1.26 ± 0.98 | 1.33 ± 0.71 |
| p47 phox | 1.00 ± 0.48 | 0.89 ± 0.61 | 0.74 ± 0.43 |
| NOX 2 | 0.74 (0.55‐1.63) | 1.64 (0.29‐4.07) | 1.12 (0.50‐2.51) |
| NOX 4 | 1.01 (0.85‐1.06) | 3.47 (0.19‐3.67) | 1.39 (1.22‐1.82) |
| p‐ERK/ERK | 1.00 ± 0.22 | 1.46 ± 0.34a | 1.33 ± 0.20 |
| p‐ERK/GAPDHH | 1.00 ± 0.16 | 1.30 ± 0.85 | 1.74 ± 0.77 |
| ERK/GAPDH | 1.00 ± 0.21 | 0.98 ± 0.41 | 1.23 ± 0.58 |
| p‐JNK/JNK | 1.01 (0.86‐1.07) | 0.78 (0.68‐1.27) | 1.03 (0.94‐1.72) |
| p‐JNK/GAPDH | 1.07 (0.95‐1.18) | 1.15 (0.92‐1.26) | 1.88 (1.46‐2.22)a , b |
| JNK/GAPDH | 1.00 ± 0.23 | 1.11 ± 0.19 | 1.48 (1.29‐1.73)a |
| p‐P38/P38 | 0.74 (0.50‐1.68) | 1.29 (1.23‐1.81) | 0.40 (0.26‐0.70)b |
| p‐P38/GAPDH | 1.00 ± 0.54 | 2.01 ± 1.30 | 0.69 ± 0.41b |
| P38/GAPDH | 1.05 (0.81‐1.13) | 1.00 (0.70‐2.58) | 1.37 (0.92‐1.92) |
One‐way ANOVA and Tukey or Kruskal‐Wallis and Dunn. Data are mean ± SD or median and percentiles.
AS: aortic stenosis; AS?Ex: exercised aortic stenosis.
P < 0.05 vs Sham;
P < 0.05 vs AS.
4. DISCUSSION
In this study, we showed that aerobic exercise improves functional capacity in AS rats during transition from LV compensated hypertrophy to clinical heart failure. However, the increased physical capacity was not related to an improvement in cardiac structures or function. We also performed the first evaluation on the influence of physical exercise on myocardial oxidative stress and MAPK signalling in AS rats with heart failure.
Aortic stenosis rats have been often used to evaluate compensated LV hypertrophy and its transition to clinical heart failure. LV hypertrophy can be observed 1 month after stenosis induction.23 Rats then remain compensated for 20‐28 weeks when they start to present heart failure features and evolve to death in 2‐4 weeks.40 In this study, the exercise protocol was initiated 18 weeks after AS surgery; at this time, no rat had tachypnoea/laboured respiration. We used a low intensity exercise protocol, adapted from published studies on aged untreated spontaneously hypertensive rats.21, 41 All rats tolerated the treadmill exercise intensity of 10 m/min. The protocol was efficient as physical capacity was improved in AS‐Ex compared with AS group.
Exercise did not change heart rate or the frequency of heart failure features and survival rate. Heart rate was assessed during echocardiographic evaluation with rats under a light anaesthesia, which may have interfered with the measurement preventing heart rate reduction after exercise protocol. Before treatment, AS rats had LV concentric hypertrophy with diastolic dysfunction and mild systolic dysfunction. The same pattern of concentric hypertrophy was observed after the exercise protocol. Additionally, both AS‐Ex and AS had LV dilation and a severe degree of diastolic dysfunction. There were no differences between AS‐Ex and AS groups before and after the exercise protocol. However, AS‐Ex presented depressed LV indexes of systolic function such as endocardial and midwall fractional shortening and ejection fraction compared to the Sham group. Despite the impaired systolic function, functional capacity was increased in AS‐Ex compared with AS. Therefore, the better functional capacity in AS‐Ex was not related to an improvement in echocardiographic parameters. Studies on heart failure have suggested that exercise‐induced improvement in skeletal muscle metabolism and performance are involved in the increase of functional capacity.22, 42
Myocardial oxidative stress, evaluated by lipid hydroperoxide concentration, was higher in AS than Sham and was normalized by exercise. Exercise has been long shown to decrease oxidative stress in different experimental models.24, 29
Oxidative stress is characterized by an increase in the levels of reactive oxygen species and/or reactive nitrogen species. Both increased generation in reactive oxygen species and decreased antioxidant capacity can lead to oxidative stress.43 Impaired antioxidant capacity can result from low concentrations of antioxidants and decreased antioxidant enzymes activity.43 At physiological concentrations, reactive oxygen species play important roles in redox signalling and cell survival by modulating the activity of different enzymes such as mitogen‐activated protein kinase (MAPK), phosphatases and gene‐dependent cascades.43 However, high levels of reactive oxygen species cause changes or injury to DNA, proteins and lipids, and can stimulate apoptotic cell death.24, 44
Myocytes consume high quantities of oxygen during muscle cell contraction and can generate a large quantity of reactive oxygen species. Reactive oxygen species are mainly generated in mitochondria.43, 44 They can also be produced in cytosol and membranes in response to different stimuli including growth factors and inflammatory cytokines.44 Several enzymes including NADPH oxidase participate in ROS generation.45 In this study, we showed for the first time that myocardial gene expression of the NADPH oxidase subunits NOX 2 and NOX 4 is not increased in AS rats with heart failure and is not modulated by physical exercise.
Oxidative stress is neutralized by the antioxidant system, which includes endogenous and exogenous molecules. The main enzymatic defences are superoxide dismutase, catalase and glutathione peroxidase, which can be modified by exercise.24, 44 In this study, superoxide dismutase was reduced in AS and AS‐Ex compared to Sham and glutathione peroxidase was lower in AS‐Ex than Sham. Exercise preserved both total and reduced myocardial glutathione. Glutathione is an endogenous tripeptide that plays a central role in cellular defence against oxidative stress.46 It is synthesized and maintained at high concentrations in most cells and is the main intracellular oxidant scavenger protecting membranes, proteins and DNA from oxidative stress.47 In heart failure, glutathione redox status is changed and its concentration is decreased in myocardium.48, 49 Considering the restored levels of glutathione and normalization of lipid hydroperoxide, it was surprising to observe a reduced glutathione peroxidase activity in AS‐Ex compared to the Sham group. We have not identified studies evaluating physical exercise and myocardial oxidative stress in AS rats. Therefore, additional studies are necessary to clarify the interplay between exercise and oxidative stress during transition from compensated LV hypertrophy to clinical heart failure.
Phosphorylated p38 levels in AS‐Ex and AS were similar to those from the Sham group. However, phosphorylated p38 was lower in AS‐Ex than AS. Myocardial MAPK expression in rodents subjected to long‐term pressure overload and chronic exercise has been poorly addressed in literature. Miyachi et al50 observed that increase in p38 phosphorylation in sedentary hypertensive rats was prevented by swimming training. In accordance, it is possible that the chronic exercise has contributed to prevent an increase in myocardial phosphorylated p38 in the AS‐Ex group. As p38 activation is related to pathological hypertrophy development,50 its decrease in AS‐Ex group suggests a beneficial effect of exercise on the pressure‐overloaded hearts. Exercise increased myocardial protein expression of total and phosphorylated JNK. The fact that JNK activation is related to myocyte hypertrophy51 may explain the larger myocyte diameter in AS‐Ex than Sham and AS. However, the role of isolated JNK increase on cardiac hypertrophy is not completely obvious. JNK activity may be required for IGF‐I‐mediated cardiomyocyte growth.52 On the other hand, myocyte stretching stimulates JNK and inhibits IGF‐I signalling and hypertrophy.53 The disparate results are probably dependent on the different experimental models. Finally, myocardial fibrosis was evident in AS rats and was not modulated by physical exercise.
A large body of research spanning several decades has shown the safety and efficacy of regular physical activity in improving outcome in heart failure patients regardless of age, sex or ethnicity.5, 11, 54 Most clinical and experimental studies, however, have evaluated the remodelling of hearts with ischaemic cardiomyopathy.55, 56 Nonetheless, the majority of experimental studies in genetic models of systemic hypertension21, 41, 50, 57 as well as some clinical studies58 point towards regular exercise having a beneficial effect on cardiac remodelling. In fact, it was recently observed that exercise‐induced improvement in functional capacity is independent of heart failure aetiology.59 A few studies have reported negative effects of rodents subjected to long‐term exercise.18, 19, 58 In these studies, animals exercised by voluntary rotating wheel, which may be associated with excessive exercise.20 To the best of our knowledge, this is the first study to show that controlled aerobic exercise is associated with impaired cardiac remodelling in AS rats.
One important component of exercise‐induced improvement in LV function during cardiac remodelling is mediated via a reduction in peripheral impedance and cardiac afterload. However, as LV afterload is fixed in aortic constriction, this beneficial effect of exercise is not seen in aortic stenosis.18 The results of this study show that the beneficial effect of exercise in reducing oxidative stress and restoring glutathione was preserved in a myocardium subjected to chronic and persistent pressure overload. However, in the face of increased afterload and during the transition from compensated hypertrophy to heart failure, the further overload caused by exercise resulted in impaired MAPK pathway signalling, additional myocyte hypertrophy and a trend towards worse systolic function compared to sedentary AS rats. It is probable that the previously observed beneficial effects of exercise in chronic and persistent pressure overload40, 60 occurred early in the remodelling process and not during the transition to clinical heart failure, when rats present advanced degrees of LV hypertrophy and dysfunction, thus preventing a reverse remodelling process.
In conclusion, aerobic exercise improves functional capacity in AS rats during the transition from LV compensated hypertrophy to clinical heart failure independent of changes in LV function. Physical exercise reduces myocardial oxidative stress and glutathione peroxidase activity, and restores myocardial concentrations of total and reduced glutathione. Despite the beneficial effects on oxidative stress, exercise impairs JNK signalling and further induces myocyte hypertrophy.
ACKNOWLEDGEMENTS
We are grateful to Jose Carlos Georgette for his technical assistance and Colin Edward Knaggs for English editing. Financial support was provided by CNPq (Proc. n. 306770/2015‐6 and 308674/2015‐4); FAPESP (Proc. n. 2014/21972‐3 e 2012/22485‐3); PROPe, UNESP; and PAEDEX/AUIP Program.
CONFLICTS OF INTEREST
The authors confirm that there are no conflicts of interest.
Reyes DRA, Gomes MJ, Rosa CM, et al. Exercise during transition from compensated left ventricular hypertrophy to heart failure in aortic stenosis rats. J Cell Mol Med. 2019;23:1235–1245. 10.1111/jcmm.14025
REFERENCES
- 1. Benjamin EJ, Virani SS, Callaway CW, et al. American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics‐2018 update: a report from the American Heart Association. Circulation. 2018;137:e67‐e492. [DOI] [PubMed] [Google Scholar]
- 2. Drazner MH. The progression of hypertensive heart disease. Circulation. 2011;123:327‐334. [DOI] [PubMed] [Google Scholar]
- 3. Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. The task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129‐2200. [DOI] [PubMed] [Google Scholar]
- 4. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;128:1810‐1852. [DOI] [PubMed] [Google Scholar]
- 5. Ellingsen O, Halle M, Conraads V, et al. High‐intensity interval training in patients with heart failure with reduced ejection fraction. Circulation. 2017;135:839‐849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Snoek JA, Eijsvogels TMH, van ‘tHof AWJ, et al. Impact of a graded exercise program on VO2peak and survival in heart failure patients. Med Sci Sports Exerc. 2018;50:2185‐2191. 10.1249/MSS.0000000000001688. [DOI] [PubMed] [Google Scholar]
- 7. Palmer K, Bowles KA, Paton M, et al. Chronic heart failure and exercise rehabilitation: a systematic review and meta‐analysis. Arch Phys Med Rehabil. 2018; In press. 10.1016/j.apmr.2018.03.015. [DOI] [PubMed] [Google Scholar]
- 8. Garciarena CD, Pinilla OA, Nolly MB, et al. Endurance training in the spontaneously hypertensive rat. Conversion of pathological into physiological cardiac hypertrophy. Hypertension. 2009;53:708‐714. [DOI] [PubMed] [Google Scholar]
- 9. Kraljevic J, Marinovic J, Pravdic D, et al. Aerobic interval training attenuates remodelling and mitochondrial dysfunction in the post‐infarction failing rat heart. Cardiovasc Res. 2013;99:55‐64. [DOI] [PubMed] [Google Scholar]
- 10. Llewellyn TL, Sharma NM, Zheng H, Patel KP. Effects of exercise training on sfo‐mediated sympathoexcitation during chronic heart failure. Am J Physiol Heart Circ Physiol. 2014;306:H121‐H131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Slater RE, Strom JG, Granzier H. Effect of exercise on passive myocardial stiffness in mice with diastolic dysfunction. J Mol Cell Cardiol. 2017;108:24‐33. [DOI] [PubMed] [Google Scholar]
- 12. Grassi B, Majerczak J, Bardi E, et al. Exercise training in Tgαq*44 mice during the progression of chronic heart failure: cardiac vs. Peripheral (soleus muscle) impairments to oxidative metabolism. J Appl Physiol. 2017;123:326‐336. [DOI] [PubMed] [Google Scholar]
- 13. Medeiros A, Rolim NP, Oliveira RS, et al. Exercise training delays cardiac dysfunction and prevents calcium handling abnormalities in sympathetic hyperactivity‐induced heart failure mice. J Appl Physiol. 2008;104:103‐109. [DOI] [PubMed] [Google Scholar]
- 14. Guizoni DM, Oliveira‐Junior SA, Noor SL, et al. Effects of late exercise on cardiac remodeling and myocardial calcium handling proteins in rats with moderate and large size myocardial infarction. Int J Cardiol. 2016;221:406‐412. [DOI] [PubMed] [Google Scholar]
- 15. Abela M. Exercise training in heart failure. Postgrad Med J. 2018;94:392‐397. [DOI] [PubMed] [Google Scholar]
- 16. Chen Z, Yan W, Mao Y, et al. Effect of aerobic exercise on Treg and Th17 of rats with ischemic cardiomyopathy. J Cardiovasc Transl Res. 2018;11:230‐235. [DOI] [PubMed] [Google Scholar]
- 17. Blumberg Y, Ertracht O, Gershon I, et al. High‐intensity training improves global and segmental strains in severe congestive heart failure. J Card Fail. 2017;23:392‐402. [DOI] [PubMed] [Google Scholar]
- 18. van Deel ED, Boer M, Kuster DW, et al. Exercise training does not improve cardiac function in compensated or decompensated left ventricular hypertrophy induced by aortic stenosis. J Mol Cell Cardiol. 2011;50:1017‐1025. [DOI] [PubMed] [Google Scholar]
- 19. van Deel ED, Octavia Y, de Waard MC, et al. Exercise training has contrasting effects in myocardial infarction and pressure overload due to divergent endothelial nitric oxide synthase regulation. Int J Mol Sci. 2018;19:E1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lerman I, Harrison BC, Freeman K, et al. Genetic variability in forced and voluntary endurance exercise performance in seven inbred mouse strains. J Appl Physiol. 2002;92:2245‐2255. [DOI] [PubMed] [Google Scholar]
- 21. Pagan LU, Damatto RL, Cezar MD, et al. Long‐term low intensity physical exercise attenuates heart failure development in aging spontaneously hypertensive rats. Cell Physiol Biochem. 2015;36:61‐74. [DOI] [PubMed] [Google Scholar]
- 22. Gomes MJ, Martinez PF, Campos DHS, et al. Beneficial effects of physical exercise on functional capacity and skeletal muscle oxidative stress in rats with aortic stenosis‐induced heart failure. Oxid Med Cell Longev. 2016;2016:8695716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ribeiro HB, Okoshi K, Cicogna AC, et al. Estudo evolutivo da morfologia e função cardíaca em ratos submetidos a estenose aórtica supravalvar. Arq Bras Cardiol. 2003;81:562‐568. [Google Scholar]
- 24. Gomes MJ, Martinez PF, Pagan LU, et al. Skeletal muscle aging: influence of oxidative stress and physical exercise. Oncotarget. 2017;8:20428‐20440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245‐313. [DOI] [PubMed] [Google Scholar]
- 26. Martinez PF, Bonomo C, Guizoni DM, et al. Modulation of MAPK and NF‐kappaB signaling pathways by antioxidant therapy in skeletal muscle of heart failure rats. Cell Physiol Biochem. 2016;39:371‐384. [DOI] [PubMed] [Google Scholar]
- 27. Reyes DRA, Gomes MJ, Rosa CM, et al. N‐acetylcysteine influence on oxidative stress and cardiac remodeling in rats during transition from compensated left ventricular hypertrophy to heart failure. Cell Physiol Biochem. 2017;44:2310‐2321. [DOI] [PubMed] [Google Scholar]
- 28. Martinez PF, Okoshi K, Zornoff LA, et al. Echocardiographic detection of congestive heart failure in postinfarction rats. J Appl Physiol. 2011;111:543‐551. [DOI] [PubMed] [Google Scholar]
- 29. Gimenes C, Gimenes R, Rosa CM, et al. Low intensity physical exercise attenuates cardiac remodeling and myocardial oxidative stress and dysfunction in diabetic rats. J Diabetes Res. 2015;2015:457848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cezar MD, Damatto RL, Pagan LU, et al. Early spironolactone treatment attenuates heart failure development by improving myocardial function and reducing fibrosis in spontaneously hypertensive rats. Cell Physiol Biochem. 2015;36:1453‐1466. [DOI] [PubMed] [Google Scholar]
- 31. Damatto RL, Martinez PF, Lima AR, et al. Heart failure‐induced skeletal myopathy in spontaneously hypertensive rats. Int J Cardiol. 2013;167:698‐703. [DOI] [PubMed] [Google Scholar]
- 32. Okoshi K, Fioretto JR, Okoshi MP, et al. Food restriction induces in vivo ventricular dysfunction in spontaneously hypertensive rats without impairment of in vitro myocardial contractility. Braz J Med Biol Res. 2004;37:607‐613. [DOI] [PubMed] [Google Scholar]
- 33. Gimenes R, Gimenes C, Rosa CM, et al. Influence of apocynin on cardiac remodeling in rats with streptozotocin‐induced diabetes mellitus. Cardiovasc Diabetol. 2018;17:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Damatto RL, Lima AR, Martinez PF, et al. Myocardial myostatin in spontaneously hypertensive rats with heart failure. Int J Cardiol. 2016;215:384‐387. [DOI] [PubMed] [Google Scholar]
- 35. Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502‐522. [DOI] [PubMed] [Google Scholar]
- 36. Rosa CM, Gimenes R, Campos DH, et al. Apocynin influence on oxidative stress and cardiac remodeling of spontaneously hypertensive rats with diabetes mellitus. Cardiovasc Diabetol. 2016;15:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martinez PF, Bonomo C, Guizoni DM, et al. Influence of N‐acetylcysteine on oxidative stress in slow‐twitch soleus muscle of heart failure rats. Cell Physiol Biochem. 2015;35:148‐159. [DOI] [PubMed] [Google Scholar]
- 38. Cezar MD, Damatto RL, Martinez PF, et al. Aldosterone blockade reduces mortality without changing cardiac remodeling in spontaneously hypertensive rats. Cell Physiol Biochem. 2013;32:1275‐1287. [DOI] [PubMed] [Google Scholar]
- 39. Lima AR, Martinez PF, Damatto RL, et al. Heart failure‐induced diaphragm myopathy. Cell Physiol Biochem. 2014;34:333‐345. [DOI] [PubMed] [Google Scholar]
- 40. Souza RW, Piedade WP, Soares LC, et al. Aerobic exercise training prevents heart failure‐induced skeletal muscle atrophy by anti‐catabolic, but not anabolic actions. PLoS ONE. 2014;9:e110020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chicco AJ, McCune SA, Emter CA, et al. Low‐intensity exercise training delays heart failure and improves survival in female hypertensive heart failure rats. Hypertension. 2008;51:1096‐1102. [DOI] [PubMed] [Google Scholar]
- 42. Okoshi MP, Capalbo RV, Romeiro FG, Okoshi K. Cardiac cachexia: perspectives for prevention and treatment. Arq Bras Cardiol. 2017;108:74‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Altenhofer S, Radermacher KA, Kleikers PW, et al. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal. 2015;23:406‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bouzid MA, Filaire E, McCall A, Fabre C. Radical oxygen species, exercise and aging: an update. Sports Med. 2015;45:1245‐1261. [DOI] [PubMed] [Google Scholar]
- 45. Ferreira LF, Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med. 2016;98:18‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fratelli M, Goodwin LO, Orom UA, et al. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc Natl Acad Sci USA. 2005;102:13998‐14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rushworth GF, Megson IL. Existing and potential therapeutic uses for N‐acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. 2014;141:150‐159. [DOI] [PubMed] [Google Scholar]
- 48. Adamy C, Mulder P, Khouzami L, et al. Neutral sphingomyelinase inhibition participates to the benefits of N‐acetylcysteine treatment in post‐myocardial infarction failing heart rats. J Mol Cell Cardiol. 2007;43:344‐353. [DOI] [PubMed] [Google Scholar]
- 49. Lombardi R, Rodriguez G, Chen SN, et al. Resolution of established cardiac hypertrophy and fibrosis and prevention of systolic dysfunction in a transgenic rabbit model of human cardiomyopathy through thiol‐sensitive mechanisms. Circulation. 2009;119:1398‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Miyachi M, Yazawa H, Furukawa M, et al. Exercise training alters left ventricular geometry and attenuates heart failure in Dahl salt‐sensitive hypertensive rats. Hypertension. 2009;53:701‐707. [DOI] [PubMed] [Google Scholar]
- 51. Okoshi MP, Yan X, Okoshi K, et al. Aldosterone directly stimulates cardiac myocyte hypertrophy. J Card Fail. 2004;10:511‐518. [DOI] [PubMed] [Google Scholar]
- 52. Wadosky KM, Rodriguez JE, Hite RL, et al. Muscle RING finger‐1 attenuates IGF‐I‐dependent cardiomyocyte hypertrophy by inhibiting JNK signaling. Am J Physiol Endocrinol Metab. 2014;306:E723‐E739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taniike M, Yamaguchi O, Tsujimoto I, et al. Apoptosis signal‐regulating kinase 1/p38 signaling pathway negatively regulates physiological hypertrophy. Circulation. 2008;117:545‐552. [DOI] [PubMed] [Google Scholar]
- 54. Coats AJS, Forman DE, Haykowsky M, et al. Physical function and exercise training in older patients with heart failure. Nat Rev Cardiol. 2017;14:550‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Haykowsky MJ, Liang Y, Pechter D, et al. A meta‐analysis of the effect of exercise training on left ventricular remodeling in heart failure patients: the benefit depends on the type of training performed. J Am Coll Cardiol. 2007;49:2329‐2336. [DOI] [PubMed] [Google Scholar]
- 56. Crimi E, Ignarro LJ, Cacciatore F, Napoli C. Mechanisms by which exercise training benefits patients with heart failure. Nat Rev Cardiol. 2009;6:292‐300. [DOI] [PubMed] [Google Scholar]
- 57. MacDonnell SM, Kubo H, Crabbe DL, et al. Improved myocardial beta‐adrenergic responsiveness and signaling with exercise training in hypertension. Circulation. 2005;111:3420‐3428. [DOI] [PubMed] [Google Scholar]
- 58. Palatini P, Visentin P, Dorigatti F, et al. Regular physical activity prevents development of left ventricular hypertrophy in hypertension. Eur Heart J. 2009;30:225‐232. [DOI] [PubMed] [Google Scholar]
- 59. Antunes‐Correa LM, Ueno‐Pardi LM, Trevizan PF, et al. The influence of aetiology on the benefits of exercise training in patients with heart failure. Eur J Prev Cardiol. 2017;24:365‐372. [DOI] [PubMed] [Google Scholar]
- 60. Wang B, Xu M, Li W, et al. Aerobic exercise protects against pressure overload‐induced cardiac dysfunction and hypertrophy via β3‐AR‐nNOS‐NO activation. PLoS ONE. 2017;12:e0179648. [DOI] [PMC free article] [PubMed] [Google Scholar]