

Abstract
Oncostain M, a member of the IL-6 family of cytokines, is produced by immune cells in response to infections and tissue injury. OSM has a broad, often context dependent effect on various cellular processes including differentiation, hematopoiesis, cell proliferation and cell survival. OSM signaling is initiated by binding to type I (LIFRβ/gp130) or type II (OSMRβ/gp130) receptor complexes and involves activation of JAK/STAT, MAPK and PI3K. High levels of OSM have been detected in many chronic inflammatory conditions characterized by fibrosis, giving a rationale to target OSM for the treatment of these diseases. Here we discuss the current knowledge on the role of OSM in various stages of the fibrotic process including inflammation, vascular dysfunction and activation of fibroblasts.
Keywords: OSM, inflammation, vascular injury, ECM, fibrosis
Introduction
Fibrosis is defined by deposition of collagen and other extracellular matrix components. During the physiological tissue repair, fibrosis occurs as a part of the healing process, however chronic tissue injury results in excessive and uncontrolled ECM deposition [1]. Pathophysiological stages of fibrosis typically involve recruitment of immune cells including T cells, dendritic cells and monocytes as well as activation of mesenchymal cells. In some fibrotic disorders such as Systemic Sclerosis (SSc, scleroderma), fibrosis is also characterized by activation and damage of endothelial cells [2,3]. Fibrosis is a major pathological feature in many chronic inflammatory diseases, including idiopathic pulmonary fibrosis (IPF), non-alcoholic fatty liver disease, cardiac fibrosis or scleroderma [4–7]. While fibrosis is increasingly recognized as a major cause of morbidity and mortality, currently there is no cure available for the fibrotic component of the disease.
The IL-6 cytokine family encompasses a group of pleiotropic cytokines produced by a variety of cells in response to inflammatory stimuli. This cytokine family shares a common signal transducer gp130 in the receptor complex. Besides IL-6, the members of this family include IL-11, ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), oncostatin M (OSM), cardiotrophin 1 (CT-1), cardiotrophin-like cytokine (CLC), and IL-27. Members of the IL-6 family of cytokines have both overlapping and distinct biologic functions and are involved in many biological processes including differentiation, hematopoiesis, cell proliferation and cell survival [8]. Increased levels of IL-6 and other family members has been reported in many pathological conditions characterized by chronic inflammation, vascular injury and fibrosis [9,10].
The IL-6 family has been comprehensively reviewed elsewhere [8,11–13]; this review focuses on Oncostatin M, a pleiotropic cytokine that has emerged as a significant player in various stages of the fibrotic process including inflammation, vascular dysfunction and activation of fibroblasts.
Oncostatin M signaling
Oncostatin M (OSM), a member of the IL-6 family, is a secreted glycoprotein that was first identified in the conditioned media of phorbol 12-myristate 13-acetate (PMA)-treated U937 monocytic cells [14]. Among the IL-6 family members, OSM displays the broadest signaling profile comprised of Janus kinase/Signal transducer and activator of transcription (JAK/STAT), Mitogen-Activated Protein Kinases (MAPK) including ERK, p38 and JNK, the phosphatidylinositol-3-kinase/AKT (PI3K/AKT), and the protein kinase C delta (PKCδ) pathways [15]. The biological activities of OSM vary in different species and depend on ligand concentration and the specific cell type [15,16]. In humans, OSM signaling is initiated by binding of OSM to its specific type I receptor complex (LIFRβ/gp130) or type II receptor complex (OSMRβ/gp130). This brings in close proximity members of the JAK family: JAK1 and JAK2. JAKs activate each other by transphosphorylation of the tyrosine residues located in their tyrosine kinase domains. Activated JAKs phosphorylate tyrosine residues located on the receptors and create a binding site for the proteins containing SH-2 domains (Src Homology 2 domains) such as STATs [17]. STATs bind to this phosphorylated site on the receptors and are subsequently phosphorylated by JAKs. Phosphorylated STATs detach from the receptors, dimerize and translocate to the nucleus where they act as transcription factors to regulate gene expression. OSMRβ/GP130 complex activates STAT1, 3, and 5, whereas LIFRβ/GP130 complex only activates STAT1 and 3 [18,19]. The MAPK signaling is activated by OSM via binding of the adapter protein GRB2 that contains the SH-2 domain. GRB2 then forms a complex with RAS and activates ERK, p38 and JNK [20]. OSM has also been reported to activate PI3K and PKCδ, however the molecular mechanism has not yet been identified [21–23]. Figure 1 displays a schematic illustration of OSM signaling in human cells. Negative regulation of Oncostatin M signaling is predominantly mediated by SOCS proteins, in particular SOCS3 [24,25]. SOCS3 inhibits JAK/STAT signaling by binding directly to tyrosine-phosphorylated sites of gp130 receptor and/or to phosphorylated JAKs and targets them for degradation [26]. OSMRβ subunit can also be utilized by IL-31 [15,27]. IL-31 is the only member of the IL-6 family that lacks gp130 subunit in the receptor complex. Instead, IL-31 binds to heterodimerized complex of IL31Rα and OSMRβ and activates downstream pathways including JAK/STAT, MAPK and PI3K/AKT [15,27]. IL-31 shares some biological activities with OSM, including regulation of cell proliferation and tissue remodeling [28–30]. Similarly to OSM, IL-31 is elevated in chronic inflammatory diseases including atopic dermatitis, liver fibrosis and Scleroderma [31–33].
Figure 1.
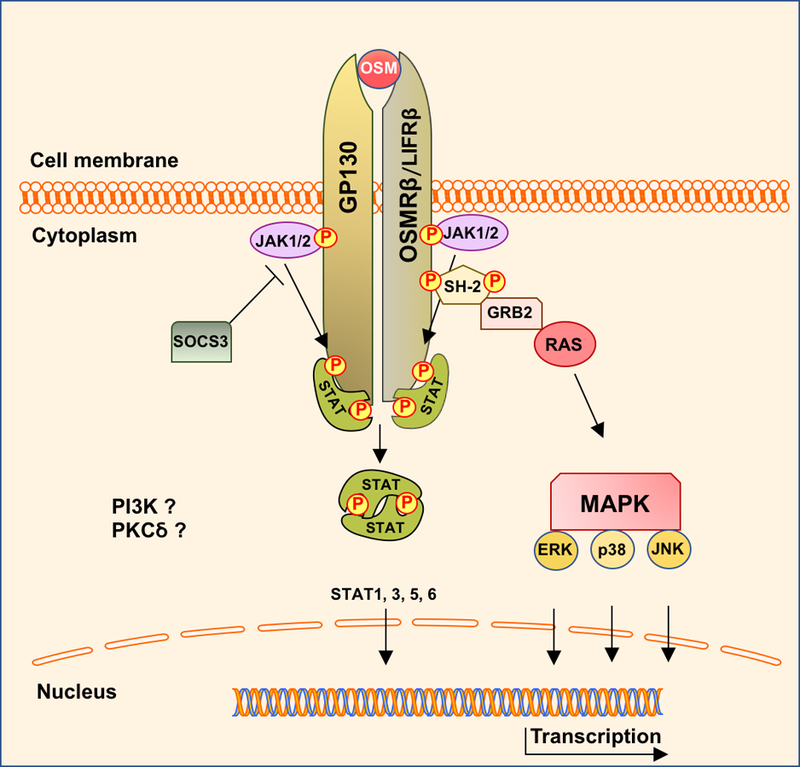
Schematic illustration of OSM signaling in human cells.
In the following sections of the review we will discuss the involvement of OSM in cellular processes that contribute to the development of tissue fibrosis, including inflammation, vascular injury, and the fibroblast activation.
Activation of the inflammatory cells
OSM is expressed in a variety of immune cells including activated T cells, monocytes, dendritic cells, neutrophils, activated mast cells and eosinophils and its expression is increased upon stimulation with pro-inflammatory mediators such as LPS [16,34–36]. Biological activities of OSM at the local sites of damage are dependent on tissue context and cellular microenvironment. Notably, OSM was shown to display both pro- and anti-inflammatory properties. These contradictory effects of OSM could be explained by activation of distinct signaling pathways downstream of OSMRβ or LIFRβ. Most of the studies looking at the role of OSM in inflammation were performed in mouse models using either human or murine OSM. However, unlike the human OSM, which activates both OSMRβ and LIFRβ, murine OSM can only signal through its specific murine OSMRβ [37–39]. Moreover, human OSM cannot bind to murine OSMRβ but can signal through murine LIFRβ [37]. In general, the anti-inflammatory effects of OSM were observed in mouse models utilizing human OSM, which suggests that these responses were dependent on the LIFRβ complex activation. For example, in a mouse model of RA, administration of human OSM attenuated the inflammatory response and helped restoring tissue homoeostasis by blocking LPS-induced expression of TNF-α in a dose dependent manner [40]. The anti-inflammatory effect of OSM was also demonstrated in a diabetic wound healing model in mice. OSM inhibited the production of IL-8, which blocked infiltration of neutrophils to the sites of local tissue damage, and at the same time increased proliferation and collagen synthesis by dermal fibroblast, resulting in accelerated wound closure [41].
On the other hand, the pro-inflammatory effects of OSM were demonstrated in mouse models utilizing murine OSM. Langdon and colleges demonstrated that administration of the murine OSM in the synovial space resulted in increased infiltration of mononuclear cells accompanied by matrix remodeling [42]. Furthermore, in a mouse model of inflammatory arthritis, murine OSM in combination with either IL-1 or TNF-α exacerbated bone destruction by increasing expression of TNF-α family member RANKL and its receptor RANK in the inflammatory cells in inflamed synovium and in articular chondrocytes [43]. Consistent with the pro-inflammatory role of OSM, OSM KO mice treated with H. hepaticus by oral gavage showed decreased chemokine production and recruitment of CD4+ T cells and granulocytes in the intestinal wall, resulting in reduced inflammation in this mouse model of colitis [44]. Furthermore, mice injected intradermally with adenovirus encoding mouse OSM (AdOSM), showed increased production of chemokines and cytokines, including CXCL3, CCL2, CCL5, and CCL20, that correlated with neutrophil and macrophage infiltration and activation of keratinocytes [45]. Interestingly, OSMRβ KO mice were protected from inflammation and epidermal thickening in response to AdOSM but not to psoriasis-like inflammation induced by imiquimod treatment [45]. The authors suggested that, since OSM failed to increase expression of IL17A/C, as compared to imiquimod, it can act independently or in parallel to IL-23/IL17 axis in inducing inflammation [45]. In bleomycin-induced lung fibrosis, increased mRNA levels of IL-6 family members, including OSM and LIF, were found in endothelial cells during the inflammatory stage [46]. Moreover, transient adenoviral-mediated overexpression of OSM or IL-6 in the course of the bleomycin-induced lung fibrosis was shown to increase the number of arginase-positive M2-like macrophages in the regions of tissue damage. Interestingly, the macrophages expressed IL6R, but lacked the expression of OSMRβ suggesting that the M2 phenotype was activated by OSM in a paracrine manner by upregulating IL-6 levels [47].
Vascular injury
Endothelial cells line the insides of blood and lymphatic vessels throughout the body and have an important role in the coagulation cascade, inflammation, maintenance of blood pressure and angiogenesis. Endothelial cell damage and dysfunction is associated with many pathological conditions, including fibrosis. Damaged ECs may contribute to perivascular ECM remodeling by increasing inflammation and vasoconstriction, and by transitioning to mesenchymal cells through a process called EndoMT (endothelial to mesenchymal transition), as well as through more direct interactions with fibroblasts [48,49]. The contribution of OSM to vascular injury and especially EC activation has not yet been fully investigated in the context of fibrosis. However, the fact that ECs express high levels of OSMR makes them one of the primary targets of OSM.
Studies have shown that endothelial cells contribute to OSM-induced inflammatory response by increasing production of proinflammatory cytokines and chemokines. For example, OSM increased expression of IL-6 and MCP1 in human cerebral endothelial cells (HCEC) [50], as well as expression of CCL21, in human dermal microvascular endothelial cells (HDMEC) and lung microvascular endothelial cells (LMEC)[51]. Proinflammatory effects of OSM were also associated with increased vessel permeability. As an example, OSM treated human brain microvascular endothelial cells showed SNAIL dependent decreased levels of tight-junction-associated MARVEL proteins (TAMPs) indicating endothelial barrier dysfunction [52]. OSM was also shown to increase the levels of adhesion molecules including P-selectin (in HUVECs) [53] and I-CAM1 (in HCECs) [50]. These data suggest that OSM could directly induce changes in ECs that can result in increased vascular permeability and perivascular infiltration of immune cells at the sites of tissue damage.
Chronic inflammatory conditions are often accompanied by a dysregulated process of angiogenesis. OSM was shown to regulate this process both in vitro and in vivo by stimulating the expression of proangiogenic factors in ECs, including VEGF, uPAR or COX-2 [54,55]. ECs treated with OSM also showed increased proliferation and migration, suggesting activation of vessel repair during inflammatory conditions [54]. Interestingly, HUVECs treated with OSM showed increased levels of angiopoietin 2, which was shown to be important in the formation of new blood vessels as well as in sensitizing ECs to TNF-α and maintaining an inflammatory response [56].
OSM has been shown to regulate plasticity of cancer cells by EMT (epithelial to mesenchymal transition), a process whereby epithelial cells lose their cell-cell attachment properties and undergo a transition to fibroblast like cells [57–60]. Similar to this process, EndoMT has been recently demonstrated in various mouse models of fibrosis as an alternative source of activated fibroblasts [61]. The effect of OSM on EC morphology was first observed in an in vitro study utilizing bovine aortic endothelial cells (BAEC). The authors demonstrated that BAECs treated with OSM became spindle-shaped and exhibited increased proliferation and migration [62]. We further confirmed these results in HDMECs. Cells treated with Oncostatin M showed increased migration, proliferation and secretion of collagen type I [63]. Moreover, we demonstrated that in HDMECs, OSM induces the mRNA expression of EndoMT genes, including TGFβ3 and SNAIL1, as well as ECM genes including TIMP1 [63]. These data suggest that OSM may play a key role in inducing EC plasticity.
Many pathophysiological responses in the vessel wall are caused by an imbalance between molecules promoting vasodilatation and vasoconstriction. Increased expression levels of vasoconstrictors such as ET- 1 and Ang II, together with decreased production of vasodilators such as nitric oxide (NO), result in increased vascular stiffness and ECM deposition [64–66]. OSM increased mRNA and protein levels of a potent vasoconstrictor, ET-1 in HUVECs [67]. Interestingly, ET-1 enhanced the TGFβ−induced EndoMT, both in vitro and in vivo [68]. OSM also increased levels of ACE (angiotensin converting enzyme) in HUVECs [69]. ACE is a main component of the renin–angiotensin system (RAS) that converts Angiotensin I to the active vasoconstrictor Ang II. Ang II has been reported to play a critical role in renal and heart fibrosis through the upregulation of inflammation and TGFβ-dependent matrix deposition, and has also been linked to the pathogenesis of skin fibrosis in SSc [70–72]. Moreover, Ang II is a potent inducer of inflammation and collagen deposition in mouse models of cardiac, renal, liver, and dermal fibrosis [70,71,73,74].
These effects of OSM on endothelial cells support the importance of this cytokine in vascular remodeling during inflammatory and fibrotic conditions.
The role of OSM in ECM regulation
High levels of OSM were also observed in inflammatory diseases characterized by fibrotic complications. For example, high levels of OSM were detected in the synovial fluid of RA [75], in the intestine of inflammatory bowel disease (IBD) [76], in the bronchoalveolar lavage (BAL) fluid of SSc associated interstitial lung disease [77], in the serum of patients with diffuse cutaneous SSc [78], and in the liver of patients with cirrhosis [79]. Biological activities displayed by OSM are consistent with activation of fibroblasts and regulation of ECM remodeling. For instance, in in vitro studies utilizing lung fibroblasts, OSM was shown to induce mRNA expression levels of collagen and glycosaminoglycans [80], as well as MAPK and STAT3 dependent proliferation, migration and decreased apoptosis [80,81]. Moreover, in dermal fibroblasts, OSM was shown to bind cis-regulatory element in the α2(I) collagen promotor and directly stimulate its expression [82,83]. Furthermore, Canady and colleges reported that cultured keratinocytes stimulated with keratinocyte growth factor (KGF) secrete high levels of OSM and stimulate fibroblasts migration as well as mRNA expression of collagen type I [84]. In addition, the authors used the OSM inhibitor S3I-201 to demonstrate that the KGF-induced profibrotic effects were OSM dependent [84]. In contrast, Sarkozi and colleges showed a potent inhibitory effect of OSM on the TGFβ-induced tubulointerstitial fibrogenesis. Specifically, OSM inhibited TGFβ induced mRNA levels of matricellular proteins including, SPARC, TSP-1, TNC, and CTGF in human kidney cells (HK-2) [85]. Moreover, the knockdown of STAT3 proximal tubular cells prevented the OSM mediated inhibition of TGFβ induced CTGF levels [86].
Interestingly, an association between OSM and CHI3L1 (YKL4) was reported by Ho and colleagues in SSc fibroblasts [87]. They showed endogenous production of CHI3L1 that was further enhanced by OSM only in a subset of fibroblasts isolated from SSc patients, but not from the healthy controls [87]. CHI3L1 is a secreted glycoprotein whose high levels are associated with increased inflammation and fibrosis in many pathological conditions including different type of cancers, RA, osteoarthritis, pneumonia, liver cirrhosis and SSc [88–95]. Moreover, CHI3L1 was identified in the serum as a marker of inflammation and tissue damage in patients with early – nonfibrotic SSc [96]. This could suggest that OSM plays an important role in the early stages of fibrosis.
Besides the effects on collagen production, proliferation and migration, OSM could influence fibrotic process by regulating the balance of metalloproteinases (MMPs) and their inhibitors in connective tissue cells. OSM was shown to induce the expression levels of TIMP1, an MMP1 inhibitor, in human lung and cardiac fibroblast, thus favoring matrix accumulation [97,98]. This was further supported by the fact that OSM can directly bind to the extracellular matrix components (including collagen, laminin, and fibronectin) and possibly promote their stabilization and increased deposition [99].
The profibrotic properties of OSM were further confirmed in in vivo experiments. For example, overexpression of OSM in mouse liver resulted in excessive fibrosis, characterized by increased expression of fibrogenic factors TGFβ and PDGF in hepatic macrophages, as well as increased levels of TIMP1 in hepatic stellate cells [100]. Interestingly, the intranasal administration of OSM in the BALB/c, RAG2KO, as well as C57BL/6J mice pretreated with the pan-anti-TGF-β 1,2,3 Ab, resulted in increased collagen deposition in the lung, suggesting that the profibrotic properties of OSM are independent of TGF-β and IL-4/IL-13 pathways [101]. In a different study Wong et al confirmed that OSM-induced ECM deposition in the lung of BALB/c, and C57Bl/6J mice, is independent of TGFβ signaling as well as Th2 cytokines [102]. Furthermore, Fritz et al demonstrated that the intratracheal administration of OSM induced Th2 phenotype via STAT6 signaling, however collagen deposition in the lung was STAT6 independent [103]. These findings provided an alternative mechanism of ECM remodeling within the lung and confirmed a direct role for OSM in activating fibroblasts. Moreover, in the mouse models of diabetic cardiomyopathy, as well as inflammatory dilated cardiomyopathy, targeting the OSMRB by using OSMRB KO mice or an antibody directed against the extracellular domain of OSMRB, resulted in decreased collagen deposition in the heart and prevented heart failure [104,105]. On the other hand, a protective role for OSM was described in other experimental models. For example, in the rat model of Dimethylnitrosamine (DMN) induced liver fibrosis, OSM gene therapy resulted in attenuation of liver damage by promoting proliferation and inhibiting apoptosis of hepatocytes [106]. Furthermore, in a bleomycin induced lung fibrosis, transplantation of OSM‐preconditioned mesenchymal stem cells (MSCs) markedly improved pulmonary respiratory functions and inhibited expression levels of inflammatory and fibrotic factors such as IL-6, collagen I and fibronectin [107].
These variable outcomes of the effects of OSM suggest that the its actions are complex and may depend on a specific cell type and tissue microenvironment; therefore, to fully dissect its profibrotic effects more detailed investigations are required. Figure 2 illustrates the input of OSM on the process of fibrosis.
Figure 2.
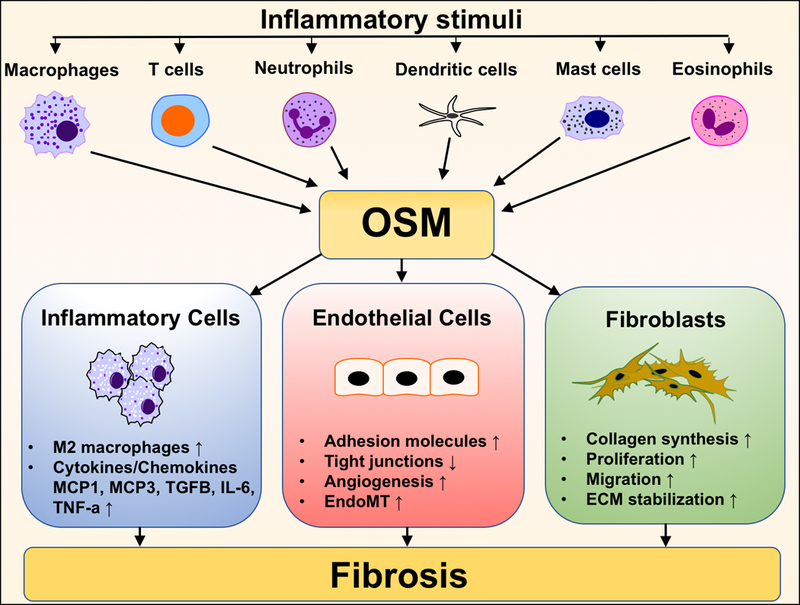
The role of OSM in pathogenesis of fibrosis.
Targeting OSM in fibrotic diseases.
In humans, targeting IL-6, the main cytokine of this family, was shown to be beneficial in many diseases including those characterize by ECM remodeling [108,109]. For example, neutralization of IL-6 activity by the IL-6R-specific monoclonal antibody tocilizumab showed beneficial effects in RA, Castleman’s disease and juvenile idiopathic arthritis [109]. In fact, tocilizumab has been approved in more than 100 countries as a treatment of autoimmune and chronic inflammatory diseases. In systemic sclerosis patients, treatment with tocilizumab was associated with beneficial effects on skin and lung fibrosis, however correlated with increased risk of infections [110]. Sirukumab, another monoclonal antibody that binds with high affinity to IL-6, reduced symptoms and improved quality of life of RA patients [111] and could potentially be beneficial in systemic sclerosis patients. Furthermore, blocking JAK1, the central signaling pathway induced by the IL-6 family members, has been shown to be beneficial in many diseases, including rheumatoid arthritis, solid tumors or other inflammatory diseases [108,112–114].
The first human study investigating the blockade of OSM using a humanized anti-OSM Immunoglobulin G1 (IgG1) monoclonal antibody (GSK315234) has verified its efficacy and acceptable safety profile for patients with RA [115]. However, results from this study suggested that a higher affinity anti-OSM antibody is needed to fully evaluate the role of OSM in RA [115]. A new generation anti-OSM antibody, GSK2330811, is now tested in a double-blinded, placebo controlled clinical trial in male and female participants with diffuse cutaneous SSc (dcSSc) [116]. Interestingly, OSMRβ was recently identified as a prognostic biomarker that corelates with progression of the skin disease in patients with dcSSc [117]. Moreover, OSMR antigen binding proteins are under development for the treatment of inflammatory disorders associated with extracellular matrix deposition or remodeling.
Conclusions
In summary, OSM is a pleiotropic cytokine whose increased expression levels were observed in a variety of inflammatory diseases, including those with fibrotic complications. Its role in the process of fibrosis is still under investigation, however it is already well documented that this cytokine has the ability to promote inflammation, vascular injury and fibroblast activation. This provides a rationale for therapeutically targeting OSM signaling in the context of fibrotic diseases.
Acknowledgments
Funding
This work was supported by the National Institutes of Health (NIAMS) grant RO1 AR42334–19 and the NIH T32 5T32AR007598–19
Footnotes
Declaration of Interest
The authors report no conflicts of interest.
Bibliography
- [1].Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med 2012;18:1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Trojanowska M Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat. Rev. Rheumatol 2010;6:453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Varga J, Trojanowska M, Kuwana M. Pathogenesis of systemic sclerosis: recent insights of molecular and cellular mechanisms and therapeutic opportunities. J. Scleroderma Relat. Disord 2017;2:137–152. [Google Scholar]
- [4].Ho YY, Lagares D, Tager AM, et al. Fibrosis--a lethal component of systemic sclerosis. Nat. Rev. Rheumatol 2014;10:390–402. [DOI] [PubMed] [Google Scholar]
- [5].Sgalla G, Iovene B, Calvello M, et al. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir. Res 2018;19:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018; [DOI] [PMC free article] [PubMed]
- [7].Kong P, Christia P, Frangogiannis NG. The Pathogenesis of Cardiac Fibrosis. Cell. Mol. Life Sci. CMLS 2014;71:549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rose-John S Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rincon M Interleukin-6: from an inflammatory marker to a target for inflammatory diseases. Trends Immunol 2012;33:571–577. [DOI] [PubMed] [Google Scholar]
- [10].Denton CP, Ong VH. Interleukin-6 and related proteins as biomarkers in systemic sclerosis. J. Scleroderma Relat. Disord 2017;2:13–19. [Google Scholar]
- [11].Dittrich A, Hessenkemper W, Schaper F. Systems biology of IL-6, IL-12 family cytokines. Cytokine Growth Factor Rev 2015;26:595–602. [DOI] [PubMed] [Google Scholar]
- [12].Komori T, Morikawa Y. Oncostatin M in the development of metabolic syndrome and its potential as a novel therapeutic target. Anat. Sci. Int 2018;93:169–176. [DOI] [PubMed] [Google Scholar]
- [13].Jones GW, Hill DG, Cardus A, et al. IL-27: a double agent in the IL-6 family. Clin. Exp. Immunol 2018; [DOI] [PMC free article] [PubMed]
- [14].Zarling JM, Shoyab M, Marquardt H, et al. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc. Natl. Acad. Sci. U. S. A 1986;83:9739–9743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hermanns HM. Oncostatin M and interleukin-31: Cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev 2015;26:545–558. [DOI] [PubMed] [Google Scholar]
- [16].Richards CD. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm 2013;2013:512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jatiani SS, Baker SJ, Silverman LR, et al. JAK/STAT Pathways in Cytokine Signaling and Myeloproliferative Disorders. Genes Cancer 2010;1:979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hemmann U, Gerhartz C, Heesel B, et al. Differential activation of acute phase response factor/Stat3 and Stat1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. II. Src homology SH2 domains define the specificity of stat factor activation. J. Biol. Chem 1996;271:12999–13007. [DOI] [PubMed] [Google Scholar]
- [19].Stahl N, Farruggella TJ, Boulton TG, et al. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science 1995;267:1349–1353. [DOI] [PubMed] [Google Scholar]
- [20].Böing I, Stross C, Radtke S, et al. Oncostatin M-induced activation of stress-activated MAP kinases depends on tyrosine 861 in the OSM receptor and requires Jak1 but not Src kinases. Cell. Signal 2006;18:50–61. [DOI] [PubMed] [Google Scholar]
- [21].Demyanets S, Kaun C, Rychli K, et al. Oncostatin M-enhanced vascular endothelial growth factor expression in human vascular smooth muscle cells involves PI3K-, p38 MAPK-, Erk1/2- and STAT1/STAT3-dependent pathways and is attenuated by interferon-γ. Basic Res. Cardiol 2011;106:217–231. [DOI] [PubMed] [Google Scholar]
- [22].Smyth DC, Kerr C, Richards CD. Oncostatin M-Induced IL-6 Expression in Murine Fibroblasts Requires the Activation of Protein Kinase Cδ. J. Immunol 2006;177:8740–8747. [DOI] [PubMed] [Google Scholar]
- [23].Smyth DC, Takenaka S, Yeung C, et al. Oncostatin M regulates osteogenic differentiation of murine adipose-derived mesenchymal progenitor cells through a PKCdelta-dependent mechanism. Cell Tissue Res 2015;360:309–319. [DOI] [PubMed] [Google Scholar]
- [24].Magrangeas F, Boisteau O, Denis S, et al. Negative regulation of onconstatin M signaling by suppressor of cytokine signaling (SOCS-3). Eur. Cytokine Netw 2001;12:309–315. [PubMed] [Google Scholar]
- [25].Liu X, Liu R, Croker BA, et al. Distinctive pro-inflammatory gene signatures induced in articular chondrocytes by oncostatin M and IL-6 are regulated by Suppressor of Cytokine Signaling-3. Osteoarthritis Cartilage 2015;23:1743–1754. [DOI] [PubMed] [Google Scholar]
- [26].Kershaw NJ, Murphy JM, Liau NPD, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat. Struct. Mol. Biol 2013;20:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ferretti E, Corcione A, Pistoia V. The IL-31/IL-31 receptor axis: general features and role in tumor microenvironment. J. Leukoc. Biol 2017;102:711–717. [DOI] [PubMed] [Google Scholar]
- [28].Zhang Q, Putheti P, Zhou Q, et al. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev 2008;19:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Singh B, Jegga AG, Shanmukhappa KS, et al. IL-31-Driven Skin Remodeling Involves Epidermal Cell Proliferation and Thickening That Lead to Impaired Skin-Barrier Function. PloS One 2016;11:e0161877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Perrigoue JG, Li J, Zaph C, et al. IL-31-IL-31R interactions negatively regulate type 2 inflammation in the lung. J. Exp. Med 2007;204:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Furue M, Yamamura K, Kido-Nakahara M, et al. Emerging role of interleukin-31 and interleukin-31 receptor in pruritus in atopic dermatitis. Allergy 2018;73:29–36. [DOI] [PubMed] [Google Scholar]
- [32].Ming D, Yu X, Guo R, et al. Elevated TGF-β1/IL-31 Pathway Is Associated with the Disease Severity of Hepatitis B Virus-Related Liver Cirrhosis. Viral Immunol 2015;28:209–216. [DOI] [PubMed] [Google Scholar]
- [33].Arumalla N, Zafar S, Rosario H, et al. OP0048 IL-31 Is An Inflammatory Pro-Fibrotic Factor Elevated in A Subset of Scleroderma Patients with Severe Pruritus. Ann. Rheum. Dis 2016;75:72–72. [Google Scholar]
- [34].Salamon P, Shoham NG, Puxeddu I, et al. Human mast cells release oncostatin M on contact with activated T cells: possible biologic relevance. J. Allergy Clin. Immunol 2008;121:448–455.e5. [DOI] [PubMed] [Google Scholar]
- [35].Hoermann G, Cerny-Reiterer S, Sadovnik I, et al. Oncostatin M is a FIP1L1/PDGFRA-dependent mediator of cytokine production in chronic eosinophilic leukemia. Allergy 2013;68:713–723. [DOI] [PubMed] [Google Scholar]
- [36].Wallace PM, MacMaster JF, Rouleau KA, et al. Regulation of inflammatory responses by oncostatin M. J. Immunol. Baltim. Md 1950 1999;162:5547–5555. [PubMed] [Google Scholar]
- [37].Lindberg RA, Juan TS, Welcher AA, et al. Cloning and characterization of a specific receptor for mouse oncostatin M. Mol. Cell. Biol 1998;18:3357–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tanaka M, Hara T, Copeland NG, et al. Reconstitution of the functional mouse oncostatin M (OSM) receptor: molecular cloning of the mouse OSM receptor beta subunit. Blood 1999;93:804–815. [PubMed] [Google Scholar]
- [39].Ichihara M, Hara T, Kim H, et al. Oncostatin M and leukemia inhibitory factor do not use the same functional receptor in mice. Blood 1997;90:165–173. [PubMed] [Google Scholar]
- [40].Wahl A, Wallace P. Oncostatin M in the anti-inflammatory response. Ann. Rheum. Dis 2001;60:iii75–iii80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shin SH, Han S-K, Jeong S-H, et al. Potential of oncostatin M to accelerate diabetic wound healing. Int. Wound J 2014;11:398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Langdon C, Kerr C, Hassen M, et al. Murine Oncostatin M Stimulates Mouse Synovial Fibroblasts in Vitro and Induces Inflammation and Destruction in Mouse Joints in Vivo. Am. J. Pathol 2000;157:1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hui W, Cawston TE, Richards CD, et al. A model of inflammatory arthritis highlights a role for oncostatin M in pro-inflammatory cytokine-induced bone destruction via RANK/RANKL. Arthritis Res Ther 2004;7:R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hegazy AN, Owens BMJ, Bullers SJ, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med 2017;23:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pohin M, Guesdon W, Mekouo AAT, et al. Oncostatin M overexpression induces skin inflammation but is not required in the mouse model of imiquimod-induced psoriasis-like inflammation. Eur. J. Immunol 2016;46:1737–1751. [DOI] [PubMed] [Google Scholar]
- [46].Leach HG, Chrobak I, Han R, et al. Endothelial cells recruit macrophages and contribute to a fibrotic milieu in bleomycin lung injury. Am. J. Respir. Cell Mol. Biol 2013;49:1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ayaub EA, Dubey A, Imani J, et al. Overexpression of OSM and IL-6 impacts the polarization of pro-fibrotic macrophages and the development of bleomycin-induced lung fibrosis. Sci. Rep 2017;7:13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr. Opin. Rheumatol 2014;26:615–620. [DOI] [PubMed] [Google Scholar]
- [49].Daehn I, Bottinger EP. Microvascular Endothelial Cells Poised to Take Center Stage in Experimental Renal Fibrosis. J. Am. Soc. Nephrol. JASN 2015;26:767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ruprecht K, Kuhlmann T, Seif F, et al. Effects of oncostatin M on human cerebral endothelial cells and expression in inflammatory brain lesions. J. Neuropathol. Exp. Neurol 2001;60:1087–1098. [DOI] [PubMed] [Google Scholar]
- [51].Sugaya M, Fang L, Cardones AR, et al. Oncostatin M Enhances CCL21 Expression by Microvascular Endothelial Cells and Increases the Efficiency of Dendritic Cell Trafficking to Lymph Nodes. J. Immunol 2006;177:7665–7672. [DOI] [PubMed] [Google Scholar]
- [52].Wertz TS, Hoettels BA, Tawara K, et al. Oncostatin M Induces Barrier Dysfunction in Human Brain Microvascular Endothelium via Snail-Dependent Repression of TAMPs. FASEB J 2017;31:679.9–679.9. [Google Scholar]
- [53].Yao L, Pan J, Setiadi H, et al. Interleukin 4 or oncostatin M induces a prolonged increase in P-selectin mRNA and protein in human endothelial cells. J. Exp. Med 1996;184:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vasse M, Pourtau J, Trochon V, et al. Oncostatin M Induces Angiogenesis In Vitro and In Vivo. Arterioscler. Thromb. Vasc. Biol 1999;19:1835–1842. [DOI] [PubMed] [Google Scholar]
- [55].Jérôme Pourtau, Farrokh Mirshahi, Hong Li, et al. Cyclooxygenase‐2 activity is necessary for the angiogenic properties of oncostatin M. FEBS Lett 1999;459:453–457. [DOI] [PubMed] [Google Scholar]
- [56].Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol 2006;27:552–558. [DOI] [PubMed] [Google Scholar]
- [57].Pollack V, Sarközi R, Banki Z, et al. Oncostatin M-induced effects on EMT in human proximal tubular cells: differential role of ERK signaling. Am. J. Physiol. Renal Physiol 2007;293:F1714–1726. [DOI] [PubMed] [Google Scholar]
- [58].Junk DJ, Bryson BL, Smigiel JM, et al. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017;36:4001–4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Smigiel JM, Parameswaran N, Jackson MW. Potent EMT and CSC Phenotypes Are Induced By Oncostatin-M in Pancreatic Cancer. Mol. Cancer Res. MCR 2017;15:478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kucia-Tran JA, Tulkki V, Smith S, et al. Overexpression of the oncostatin-M receptor in cervical squamous cell carcinoma is associated with epithelial–mesenchymal transition and poor overall survival. Br. J. Cancer 2016;115:212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to Mesenchymal Transition (EndoMT) in the Pathogenesis of Human Fibrotic Diseases. J. Clin. Med 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wijelath ES, Carlsen B, Cole T, et al. Oncostatin M induces basic fibroblast growth factor expression in endothelial cells and promotes endothelial cell proliferation, migration and spindle morphology. J. Cell Sci 1997;110 ( Pt 7):871–879. [DOI] [PubMed] [Google Scholar]
- [63].Nevin K, Stawski L, Feeney M, et al. THU0026 Osm is more effective than il-6 at inducing endomt of human dermal microvascular cells. Ann. Rheum. Dis 2017;76:209. [Google Scholar]
- [64].Schulz E, Gori T, Münzel T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. Off. J. Jpn. Soc. Hypertens 2011;34:665–673. [DOI] [PubMed] [Google Scholar]
- [65].Cotton SA, Herrick AL, Jayson MI, et al. Endothelial expression of nitric oxide synthases and nitrotyrosine in systemic sclerosis skin. J. Pathol 1999;189:273–278. [DOI] [PubMed] [Google Scholar]
- [66].Peng X, Haldar S, Deshpande S, et al. Wall stiffness suppresses Akt/eNOS and cytoprotection in pulse-perfused endothelium. Hypertens. Dallas Tex 1979 2003;41:378–381. [DOI] [PubMed] [Google Scholar]
- [67].Saijonmaa O, Nyman T, Pacek P, et al. Oncostatin M regulates endothelin-1 production in human endothelial cells. Am. J. Physiol 1998;275:H662–667. [DOI] [PubMed] [Google Scholar]
- [68].Wermuth PJ, Li Z, Mendoza FA, et al. Stimulation of Transforming Growth Factor-β1-Induced Endothelial-To-Mesenchymal Transition and Tissue Fibrosis by Endothelin-1 (ET-1): A Novel Profibrotic Effect of ET-1. PloS One 2016;11:e0161988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Saijonmaa O, Nyman T, Kosonen R, et al. Induction of angiotensin-converting enzyme by oncostatin m in human endothelial cells. Cytokine 2000;12:1253–1256. [DOI] [PubMed] [Google Scholar]
- [70].Angiotensin Brecher P. II and Fibrosis Cardiac. Trends Cardiovasc. Med 1996;6:193–198. [DOI] [PubMed] [Google Scholar]
- [71].Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertens. Dallas Tex 1979 2001;38:635–638. [DOI] [PubMed] [Google Scholar]
- [72].Kawaguchi Y, Takagi K, Hara M, et al. Angiotensin II in the lesional skin of systemic sclerosis patients contributes to tissue fibrosis via angiotensin II type 1 receptors. Arthritis Rheum 2004;50:216–226. [DOI] [PubMed] [Google Scholar]
- [73].Bataller R, Sancho-Bru P, Ginès P, et al. Liver fibrogenesis: a new role for the renin-angiotensin system. Antioxid. Redox Signal 2005;7:1346–1355. [DOI] [PubMed] [Google Scholar]
- [74].Stawski L, Han R, Bujor AM, et al. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res. Ther 2012;14:R194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Okamoto H, Yamamura M, Morita Y, et al. The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum 1997;40:1096–1105. [DOI] [PubMed] [Google Scholar]
- [76].West NR, Hegazy AN, Owens BMJ, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med 2017;23:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Luzina IG, Atamas SP, Wise R, et al. Occurrence of an activated, profibrotic pattern of gene expression in lung CD8+ T cells from scleroderma patients. Arthritis Rheum 2003;48:2262–2274. [DOI] [PubMed] [Google Scholar]
- [78].Oncostatin M As a Potential Molecular Target in Systemic Sclerosis [Internet]. ACR Meet. Abstr [cited 2018. March 22]. Available from: http://acrabstracts.org/abstract/oncostatin-m-as-a-potential-molecular-target-in-systemic-sclerosis/.
- [79].Levy MT, Trojanowska M, Reuben A. Oncostatin M: a cytokine upregulated in human cirrhosis, increases collagen production by human hepatic stellate cells. J. Hepatol 2000;32:218–226. [DOI] [PubMed] [Google Scholar]
- [80].Scaffidi AK, Mutsaers SE, Moodley YP, et al. Oncostatin M stimulates proliferation, induces collagen production and inhibits apoptosis of human lung fibroblasts. Br. J. Pharmacol 2002;136:793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nagahama KY, Togo S, Holz O, et al. Oncostatin M modulates fibroblast function via signal transducers and activators of transcription proteins-3. Am. J. Respir. Cell Mol. Biol 2013;49:582–591. [DOI] [PubMed] [Google Scholar]
- [82].Duncan MR, Hasan A, Berman B. Oncostatin M Stimulates Collagen and Glycosaminoglycan Production by Cultured Normal Dermal Fibroblasts: Insensitivity of Sclerodermal and Keloidal Fibroblasts. J. Invest. Dermatol 1995;104:128–133. [DOI] [PubMed] [Google Scholar]
- [83].Ihn H, LeRoy EC, Trojanowska M. Oncostatin M Stimulates Transcription of the Human α2(I) Collagen Gene via the Sp1/Sp3-binding Site. J. Biol. Chem 1997;272:24666–24672. [DOI] [PubMed] [Google Scholar]
- [84].Canady J, Arndt S, Karrer S, et al. Increased KGF expression promotes fibroblast activation in a double paracrine manner resulting in cutaneous fibrosis. J. Invest. Dermatol 2013;133:647–657. [DOI] [PubMed] [Google Scholar]
- [85].Sarközi R, Hauser C, Noppert S-J, et al. Oncostatin M is a novel inhibitor of TGF-β1-induced matricellular protein expression. Am. J. Physiol. Renal Physiol 2011;301:F1014–1025. [DOI] [PubMed] [Google Scholar]
- [86].Sarközi R, Flucher K, Haller V, et al. Oncostatin M inhibits TGF-β1-induced CTGF expression via STAT3 in human proximal tubular cells. Biochem. Biophys. Res. Commun 2012;424:801–806. [DOI] [PubMed] [Google Scholar]
- [87].Ho YY, Baron M, Recklies AD, et al. Cells from the skin of patients with systemic sclerosis secrete chitinase 3-like protein 1. BBA Clin 2014;1:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Johansen JS, Christensen IJ, Riisbro R, et al. High serum YKL-40 levels in patients with primary breast cancer is related to short recurrence free survival. Breast Cancer Res. Treat 2003;80:15–21. [DOI] [PubMed] [Google Scholar]
- [89].Cintin C, Johansen JS, Christensen IJ, et al. High serum YKL-40 level after surgery for colorectal carcinoma is related to short survival. Cancer 2002;95:267–274. [DOI] [PubMed] [Google Scholar]
- [90].Johansen JS, Jensen HS, Price PA. A new biochemical marker for joint injury. Analysis of YKL-40 in serum and synovial fluid. Br. J. Rheumatol 1993;32:949–955. [DOI] [PubMed] [Google Scholar]
- [91].Johansen JS, Christoffersen P, Møller S, et al. Serum YKL-40 is increased in patients with hepatic fibrosis. J. Hepatol 2000;32:911–920. [DOI] [PubMed] [Google Scholar]
- [92].La Montagna G, D’Angelo S, Valentini G. Cross-sectional evaluation of YKL-40 serum concentrations in patients with systemic sclerosis. Relationship with clinical and serological aspects of disease. J. Rheumatol 2003;30:2147–2151. [PubMed] [Google Scholar]
- [93].Nordenbaek C, Johansen JS, Halberg P, et al. High serum levels of YKL-40 in patients with systemic sclerosis are associated with pulmonary involvement. Scand. J. Rheumatol 2005;34:293–297. [DOI] [PubMed] [Google Scholar]
- [94].Kronborg G, Ostergaard C, Weis N, et al. Serum level of YKL-40 is elevated in patients with Streptococcus pneumoniae bacteremia and is associated with the outcome of the disease. Scand. J. Infect. Dis 2002;34:323–326. [DOI] [PubMed] [Google Scholar]
- [95].Wcislo-Dziadecka D, Kotulska A, Brzezińska-Wcislo L, et al. Serum human cartilage glycoprotein-39 in patients with systemic sclerosis: relationship to skin and articular manifestation. Clin. Rheumatol 2010;29:933–935. [DOI] [PubMed] [Google Scholar]
- [96].Cossu M, van Bon L, Preti C, et al. Earliest Phase of Systemic Sclerosis Typified by Increased Levels of Inflammatory Proteins in the Serum. Arthritis Rheumatol. Hoboken NJ 2017;69:2359–2369. [DOI] [PubMed] [Google Scholar]
- [97].Weiss TW, Kvakan H, Kaun C, et al. The gp130 ligand oncostatin M regulates tissue inhibitor of metalloproteinases-1 through ERK1/2 and p38 in human adult cardiac myocytes and in human adult cardiac fibroblasts: a possible role for the gp130/gp130 ligand system in the modulation of extracellular matrix degradation in the human heart. J. Mol. Cell. Cardiol 2005;39:545–551. [DOI] [PubMed] [Google Scholar]
- [98].Richards CD, Shoyab M, Brown TJ, et al. Selective regulation of metalloproteinase inhibitor (TIMP-1) by oncostatin M in fibroblasts in culture. J. Immunol. Baltim. Md 1950 1993;150:5596–5603. [PubMed] [Google Scholar]
- [99].Ryan RE, Martin B, Mellor L, et al. Oncostatin M binds to extracellular matrix in a bioactive conformation: implications for inflammation and metastasis. Cytokine 2015;72:71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Matsuda M, Tsurusaki S, Miyata N, et al. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatol. Baltim. Md 2018;67:296–312. [DOI] [PubMed] [Google Scholar]
- [101].Mozaffarian A, Brewer AW, Trueblood ES, et al. Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis. J. Immunol. Baltim. Md 1950 2008;181:7243–7253. [DOI] [PubMed] [Google Scholar]
- [102].Wong S, Botelho FM, Rodrigues RM, et al. Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 Dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab. Invest 2014;94:1003–1016. [DOI] [PubMed] [Google Scholar]
- [103].Fritz DK, Kerr C, Fattouh R, et al. A mouse model of airway disease: oncostatin M-induced pulmonary eosinophilia, goblet cell hyperplasia, and airway hyperresponsiveness are STAT6 dependent, and interstitial pulmonary fibrosis is STAT6 independent. J. Immunol. Baltim. Md 1950 2011;186:1107–1118. [DOI] [PubMed] [Google Scholar]
- [104].Zhang X, Ma S, Zhang R, et al. Oncostatin M-induced cardiomyocyte dedifferentiation regulates the progression of diabetic cardiomyopathy through B-Raf/Mek/Erk signaling pathway. Acta Biochim. Biophys. Sin 2016;48:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Pöling J, Gajawada P, Richter M, et al. Therapeutic targeting of the oncostatin M receptor-β prevents inflammatory heart failure. Basic Res. Cardiol 2014;109:396. [DOI] [PubMed] [Google Scholar]
- [106].Hamada T, Sato A, Hirano T, et al. Oncostatin M Gene Therapy Attenuates Liver Damage Induced by Dimethylnitrosamine in Rats. Am. J. Pathol 2007;171:872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Lan Y, Theng S, Huang T, et al. Oncostatin M‐Preconditioned Mesenchymal Stem Cells Alleviate Bleomycin‐Induced Pulmonary Fibrosis Through Paracrine Effects of the Hepatocyte Growth Factor. Stem Cells Transl. Med 2017;6:1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Schwartz DM, Kanno Y, Villarino A, et al. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov 2017;16:843–862. [DOI] [PubMed] [Google Scholar]
- [109].Tanaka T, Kishimoto T. Targeting Interleukin-6: All the Way to Treat Autoimmune and Inflammatory Diseases. Int. J. Biol. Sci 2012;8:1227–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Khanna D, Denton CP, Lin CJ, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann. Rheum. Dis 2017;annrheumdis-2017–211682. [DOI] [PMC free article] [PubMed]
- [111].Takeuchi T, Thorne C, Karpouzas G, et al. Sirukumab for rheumatoid arthritis: the phase III SIRROUND-D study. Ann. Rheum. Dis 2017;annrheumdis-2017–211328. [DOI] [PMC free article] [PubMed]
- [112].Welsch K, Holstein J, Laurence A, et al. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur. J. Immunol 2017;47:1096–1107. [DOI] [PubMed] [Google Scholar]
- [113].Zhang H, Watanabe R, Berry GJ, et al. Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis. Circulation 2017; [DOI] [PMC free article] [PubMed]
- [114].Thomas SJ, Snowden JA, Zeidler MP, et al. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015;113:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Choy EH, Bendit M, McAleer D, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti- oncostatin M monoclonal antibody in rheumatoid arthritis: results from phase II randomized, placebo-controlled trials. Arthritis Res. Ther 2013;15:R132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Proof of Mechanism Study of GSK2330811 in Diffuse Cutaneous Systemic Sclerosis - Full Text View - ClinicalTrials.gov [Internet]. [cited 2018. April 18]. Available from: https://clinicaltrials.gov/ct2/show/NCT03041025.
- [117].Stifano G, Sornasse T, Rice LM, et al. Skin Gene Expression Is Prognostic for the Trajectory of Skin Disease in Patients With Diffuse Cutaneous Systemic Sclerosis. Arthritis Rheumatol. Hoboken NJ 2018;70:912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]