

Abstract
Purpose of review
The hematopoietic stem cell (HSC) compartment is the cornerstone of a lifelong blood cell production but also contributes to the ability of the hematopoietic system to dynamically respond to environmental challenges. This review summarizes our knowledge about the interaction between HSCs and its inflammatory environment during life and questions how its disruption could affect the health of the hematopoietic system.
Recent findings
The latest research demonstrates the direct role of inflammatory signals in promoting the emergence of the HSCs during development and in setting their steady-state activity in adults. They indicate that inflammatory patho-physiological conditions or immunological history could shape the structure and biology of the HSC compartment, therefore altering its overall fitness.
Summary
Through instructive and/or selective mechanisms, the inflammatory environment seems to provide a key homeostatic signal for HSCs. Although the mechanistic basis of this complex interplay remains to be fully understood, its dysregulation has broad consequences on HSC physiology and the development of hematological diseases. As such, developing experimental models that fully recapitulate a normal basal inflammatory state could be essential to fully assess HSC biology in native conditions.
Keywords: Hematopoietic stem cell, Inflammation, aging, obesity, immunological history, hematological disease
INTRODUCTION
Inflammation is defined as a protective immune mechanism and can be activated in response to acute events associated with infection/tissue injury or during ordinary physical/physiological disturbances that affect tissue homeostasis [1]. As inflammatory responses are triggered by a broad range of internal and environmental insults, they are extremely variable in quality, intensity, kinetics, and duration. These processes are ubiquitous, affecting all stages of life and all body tissues while interplaying with many biological processes. Although the innate and adaptive immune cells are the prime inducers and responders of inflammatory signals, inflammation also directly affects the bone marrow (BM) hematopoietic tissue that underlies the production of all blood cells and particularly the immune cells. Hematopoiesis originates within a small pool of self-renewing and multipotent hematopoietic stem cells (HSCs), which undergo a stepwise differentiation process to produce a series of intermediary progenitors. While these progenitors are proliferating to contribute to the daily blood cell production, HSCs remain mostly quiescent and are responsible for sustaining the hematopoietic activity during the entire life of the organism. Blood production is highly dynamic and is constantly adapted to accommodate environmental challenges and respond to the needs of the organism. Inflammation is viewed as a key interface between the hematopoietic system and the broad array of internal stresses and environmental disturbances. There is now a new understanding that this interplay involves the entire hematopoietic hierarchy including the quiescent and long-lived HSC compartment. In recent years, great progress has been made in the understanding of how inflammatory signals regulate HSC fate and modulates blood production. Here, we will review new studies that describe the interaction of the HSC compartment with its inflammatory environment during the different stages of life. We will focus on recent reports that suggest the basal inflammatory state in healthy individuals contributes to shaping the HSC compartment and toning its steady-state activity. Herein, we highlight how chronic inflammatory patho-physiological conditions or diverse immunological history could impact the overall fitness of the HSC compartment. Finally, we will discuss the potential mechanisms underlying the regulation of HSC activity by inflammatory signals and their implications for our understanding of hematological diseases.
Inflammation signals as a driver of early hematopoietic development
During embryonic development, hematopoiesis occurs in discrete waves [2]. The first wave occurs in the yolk sac in vertebrates and only produces a limited set of primitive erythrocytes and macrophages. The second wave, which is the matrix of lifelong definitive hematopoiesis, is fueled by the activity of HSCs that emerged from the hemogenic endothelium lining the floor of the dorsal aorta. HSC emergence during this narrow developmental window represents a critical step for the establishment of a functional hematopoietic system. Several classical developmental pathways including Notch, Wnt and Bone morphogenetic protein (BMPs) have been shown to contribute to HSC specification [3]. Recent studies in zebrafish and mouse embryos indicate that inflammatory signals, released by primitive macrophage and neutrophils are essential cofactors of these pathways [4•]. In this context, pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ) and Interleukin-1β (IL-1β), in addition to toll-like receptors (TLR) signaling are key determinants of HSC specification [5–8]. These results were surprising because the womb is traditionally considered as a sterile environment protected from the microbial environment. As such, the mechanisms triggering inflammatory signals in such a sterile environment remain to be defined. This inflammation could be linked to the release of damage-associated molecular patterns (DAMPs) by fast-growing embryonic tissues, which undergo cell remodeling and sustain oxidative stress. Alternatively, recent reports seem to revisit the sterile womb paradigm indicating some level of maternal microbial transmission before birth, therefore suggesting an early encounter between the hematopoietic system and the microbial environment [9]. Independently of the mechanisms, these reports indicate a co-development between the emerging hematopoietic system and its inflammatory environment. This co-development continues at birth when neonates are exposed to a microbial-rich environment and develop a gut flora. During this transitional period, newborns are highly susceptible to infections as their immune system progressively matures in contact with this new environment [10]. This neonatal period also coincides with profound changes in the HSC compartment that progressively develop its adult characteristics [11]. Notably, this includes the extinction of particular fetal HSC subsets responsible for the production of primitive innate-like lymphocytes [12] and a switch in the cycling activity of the HSCs that acquire their adult quiescent status [13]. Little is known about the environmental factors able to influence this critical fetal-adult transition but the inflammatory environment is certainly one of the potential candidates. Overall these reports highlight how inflammatory signals could contribute to shaping the nascent hematopoietic tissue, a feature consistent with the evolutionary need to adapt the activity of this complex tissue to its environment.
Inflammatory basal state and adult HSC homeostasis
Adult animals are in constant interaction with microbial organisms. These interactions include daily encounters with bacteria, viruses, and fungi found in food or in the environment, which can lead to sub-clinical immune responses. These interactions also include the complex system of microbes, often referred to as the microbiota, that exist on many body surfaces, including the skin, mouth, nose, vagina, and digestive tract [14, 15]. Most of these host-microbe relationships are not pathogenic and are actually part of the normal physiological functions. A majority of these microorganisms are commensal bacteria that reside in the colon which contribute to nutrient absorption, and various metabolic functions but also regulate the innate and adaptive immune system [16••]. As such, the microbiota has been shown to play a protective role against infectious pathogens. Conversely, its perturbation (or dysbiosis) has been linked to various metabolic, inflammatory and autoimmune diseases including diabetes, inflammatory bowel disease, lupus and allergy [17].
Recent studies have highlighted the impact of the inflammatory basal state on the hematopoietic system including the HSC compartment [18]. The first lines of evidence come from the study of germ-free (GF) mice. GF mice (also named axenic mice) are produced by hysterectomy re-derivation and maintained in strict sterile condition. They are by definition free of all microorganisms, including those typically found in the gut. Compared to the common specific-pathogen-free (SPF) laboratory mice that interact with non-pathogenic microbes and have a complete gut microbiota, GF mice display an immature adaptive immune system and an increased susceptibility to infection [19]. Reports also indicate that GF conditions are associated with reduced mature myeloid compartments and reduced BM myeloid potential [20]. Notably, the number of granulo-macrophage progenitors (GMPs) in BM appears to be directly proportional to the microbiota complexity, suggesting its contribution to the dynamic production of these short-lived populations [21••]. This study also shows that the absence of microbiota reduces the size of the immature hematopoietic compartments (i.e. Lin-c-Kit+Sca-1+, LSK population) that encompasses HSCs and multipotent progenitors. Interestingly, partial reconstitution of the microbiota fails to fully restore the size of LSK compartment suggesting the existence of specific qualitative and/or quantitative requirements for the microbiota to modulate the most immature hematopoietic fractions. These results were confirmed in more refined HSC compartment phenotypically defined as LSK Flk2-CD34- or LSK CD48-CD150+CD34- Notably studies in GF mice and in mice treated with a broad-spectrum antibiotic cocktail show that disruption of the microbiota results in a two-to-three fold reduction in the number of HSCs [22••, 23•]. Complementation experiments through microbiota transfer in GF mice or interruption of the antibiotic treatment seems to confirm that the microbiota provides a dynamic and reversible regulation of HSC number. However, the impact of the microbiota on HSC functions remains to be fully explored. One study shows that HSCs isolated from GF mice show improved reconstitution activity in transplantation assay [24] but this effect was not reproduced with antibiotics-treated mice [23•, 24]. This discrepancy could indicate that chronic but not acute alterations of the microbiota are able to modulate long-term HSC functions. Alternatively, this could highlight fundamental differences between the two models. Notably, it is unclear whether constitutive GF condition affects the initial maturation of HSC compartment in addition to its adult steady state function. In this context, development of experimental schemes based on, (i) the use of auxotrophic bacterial strain to promote transient microbiota colonization [25] or (ii) the modulation of the timing/duration of the antibiotics treatment could prove informative. In addition, it is important to note that microbiota alterations in these models also affect nutrient absorption and therefore have important metabolic consequences that could disrupt the HSC compartment independently to inflammation. Despite their limitations, these recent studies suggest that signals derived from the microbiota provide, at steady state, a tonic stimulation not only to the mature immune cells and their immediate progenitors but also to the long-lived HSC compartment. However, more specific assays remains to be performed to validate this idea. Notably, it would be important to determine how the microbiota affects the continuum of dormant and active states in the HSC compartment during homeostasis [26••–28]. The impact of basal inflammatory states on the long-term HSC fitness remains to be studied through stringent serial transplantation assays and in aging condition. Finally, a full functional and molecular characterization of HSC pre- and post-microbiota complementation remain to be described.
Basal inflammatory signals controlling HSC fate
Several mechanisms have been proposed to mediate the effect of the basal inflammatory environment on the HSC compartment. It is well established that host interactions with pathogenic and commensal microbiota lead to the presence of immunogenic microbial compounds in the systemic circulation. Often referred as pathogen-associated molecular patterns (PAMPs), these molecules, which include lipopolysaccharide (LPS), RNA, and peptidoglycans, are present at low concentrations in healthy individuals [29, 22••]. As a normal part of the immune response, host cells recognize these molecules through numerous specialized pathogen-recognition receptors (PRRs) that include toll-like receptors (TLR), C-type lectin receptors, nucleotide-binding oligomerization domain-containing protein (NOD)-like receptors (NLR) and other RNA sensing-receptors. In the BM, these receptors are present on non-hematopoietic cells and on resident immune cells but also on the HSCs themselves. As such, PAMPs could alter the HSC function directly via intracellular signaling, or indirectly via alterations of the BM niche and/or production of pro-inflammatory cytokines/chemokines (Fig. 1a) [30, 31]. Determining the ability of PRRs to sense the low PAMP concentrations present in non-infectious condition and evaluating their contribution to the HSC homeostasis at steady state remains challenging, due to potential redundancy between these different signals and their potential complex effects on the various BM cell compartments. In addition, differences in the HSC definition has led to some degree of discrepancy in the published literature about the impact of these signals on the HSC compartment.
Fig. 1: Molecular network controlling the interplay between HSCs and its inflammatory environment.

(a) Direct and indirect HSC response to inflammation. Low concentration of circulating pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) can directly affect the function of the HSCs, which express corresponding pattern recognition receptors (PRRs). PAMPs and DAMPs also alter the activity of immune cells and non-hematopoietic cells that form the bone marrow niche, leading to indirect modulation to the HSC compartment. (b) Consequences of inflammatory signaling on the HSC compartment. Direct and indirect inflammatory signals impact the HSC long-term fitness by instructing particular cell fate decision or promoting the selection of specific HSC subsets.
PAMP receptors:
Seminal work by the Kincade group has shown that HSCs express TLR2 and TLR4 and respond to their respective agonists (Pam3CSK and LPS) [32]. HSCs also harbor low level of other TLRs but their impacts on HSCs have yet to be fully established [22••, 24]. Reports indicate that genetic disruptions of these signals by targeting individual TLR (including TLR2 or TLR4) or key components of their transduction pathways (such as the downstream adaptors proteins Myd88 and Trif) have no major consequences on HSCs at steady state [33, 34]. However discordant studies have described an effect of these mutations on the HSC phenotype and functions ([35–37, 24]. These studies suggest that disruption of these pathways could be associated with increased HSC quiescence and an overall competitive advantage upon transplantation. While such discrepancies remain to be explained, it is tempting to speculate that they could reflect some subtle environmental differences in research animal facilities and therefore might testify to the importance of these pathways in adapting the HSC compartment to changes in microbiota and/or hygienic conditions. In addition to TLR signaling, a recent study describes a key role of NOD-like receptor Nod1 on the HSC homeostasis [22••]. The authors found that HSCs express Nod1 and that the Nod1-deficient mice displayed a reduction in the size of the hematopoietic primitive compartment that mimics the phenotype observed in GF condition. At steady state, providing synthetic NOD1 ligand (NOD1L) to GF mice reverses their hematopoietic defect and particularly their reduced HSC number. Mechanistically, this study describes that NOD1L does not directly affect the hematopoietic compartments but provides a tonic signal necessary for the supportive function of the BM microenvironment. Nod1 signaling is particularly shown to stimulate the production of key hematopoietic cytokines (such SCF, TPO, Flt3L) by mesenchymal stem cells (MSCs). However, it is interesting to note again some discordance in the literature as one study indicates that loss of the adaptor protein RIPK2 (receptor-interacting serine-threonine kinase 2) that is required for signal transduction downstream of the NOD-like receptors does not induce a major HSC phenotype at steady state [33].
Secondary inflammatory signals:
One the consequences of the presence of PAMPs in the circulation is a basal production of various secondary inflammatory cytokines by diverse immune and hematopoietic cells. Targeted disruption of some of these pathways at steady state show limited effects on the HSC compartment. As such, receptors for tumor necrosis factor (TNF) or Interleukin-1 (IL-1) have been shown to be dispensable for maintaining HSC homeostasis [38, 39]. Although the observed effects are modest, HSCs seems to be susceptible to the basal concentration of both type I and type II interferons (IFN). As such IFN-α/β receptor (Ifnar)- or IFN-γ receptor (Ifngr1)-deficient mice showed reduced HSC numbers and lower proliferation rates [40, 41]. Consistent with these results, loss of Statl, which represents a common node for IFN pathways induces similar HSC defects and mimic some of the phenotype observed in antibiotic-treated animals [23•]. Finally, disruption of critical negative feedback mechanisms for the IFN signals in Irgm1, Irf2, and Adar1 mutant mice is associated with severe alteration of HSC compartment at steady state [42–44]. Altogether these studies suggest that baseline IFN signaling is tightly regulated in HSCs and could be critical to the tonic stimulation necessary to maintain HSC homeostasis. The mechanisms driven by IFN in HSCs at steady state remain unknown. It would be particularly interesting to determine the impact of baseline IFN signaling on the HSC gene expression program and its role in tuning signal transduction pathways associated with other key hematopoietic cytokines [45].
Intracellular molecular integrators:
TLR, NLR, and IFN signaling converge on the intracellular activation of NF-κΒ. Early studies have demonstrated the role of the different NF-κΒ subunits as integrators of inflammatory signals in the hematopoietic system [46]. However, due to developmental defects in the mutant mice, most of these results were obtained using fetal liver cells and did not specifically address the role of these molecules in adult steady state. Recent publications have extended these results, demonstrating that both the canonical and the non-canonical NF-κΒ pathways are required to maintain adult HSCs [47–50]. Indeed, genetic disruption of these pathways by targeting of NF-κΒ subunits (e.g. Re1A or Re1B/NF-κB2) or their regulators (e.g. Ikkb or NIK) leads to complex alterations of the size and, in some cases, functions of the HSC compartment at steady state. Altogether these studies suggest that a complex and subtle equilibrium of the different NF-κΒ subunits/pathways is required both intrinsically and in the BM environment to maintain HSC homeostasis. In the HSCs, these studies show that quantitative and qualitative dosage between NF-κΒ subunits could affect multiple HSC gene programs and control HSC fate. Although the NF-κΒ requirements for the HSCs are not completely understood, NF-κΒ appears to be one key regulator of the basal inflammatory tone in HSCs.
Patho-physiological disruption of the basal inflammatory state
The steady-state hematopoietic activity of an individual could be affected by multiple chronic patho-physiological conditions. Natural aging or metabolic diseases such as obesity display similar hematological characteristics. These patho-physiological conditions are associated with functional declines of the immune system, which leads to increased susceptibility to infections, low response to vaccinations, and increased vulnerability to the development of autoimmunity and hematologic malignancies [51, 52]. Both conditions show hematopoietic skewing toward the myeloid lineage and reduction of the fitness of the HSC compartment [53, 54]. Along with common metabolic and hormonal characteristics, these conditions also share a common inflammatory component. Aged-associated inflammation (also termed inflamm-aging) and metabolic inflammation (also termed meta-inflammation) are chronic states associated with an increased basal level of pro-inflammatory cytokines such as IL-6, TNF-α, IL-IRα, even in the absence of infection [55, 56]. The low-grade inflammatory state of these conditions seems to be multifactorial and has been linked with the expansion of myeloid cells and adipocytes, which are key producers of inflammatory cytokines. Aging and obesity are also associated with increased gut permeability along with changes in the composition and the diversity of the gut microbiota [57, 58]. As such, both conditions show a higher concentration of microbial products in the blood circulation [59, 60]. Due to the complexity of phenotype associated with these conditions, the exact contribution of inflammation on the HSC phenotypes observed in aging and obesity remains to be fully explored. Importantly, both inflamm-aging and meta-inflammation arise through slow kinetics. The slow development of these specific basal inflammatory states raise the intriguing possibility of the concomitant development of yet to be defined adaptive or selective mechanisms aimed at preserving the HSC functions [53].
Infectious Inflammatory response
Infectious episodes represent another type of normal recurrent alternations of the inflammatory basal state. Infections are associated with profound hematopoietic changes that contribute to the replacement of diminished immune effectors cells. Recent studies indicate that HSCs respond to inflammatory signals in case of acute and chronic infection and directly participate in the primary immune response [30]. In vivo injection of inflammatory mediators (including IFN-α/β, IFN-γ, IL-1, TNF-α or PAMPs) [40, 61, 41, 38, 39] and more physiological models mimicking bacterial and viral infections [40, 33, 62•] are invariably associated with HSC loss of quiescence, differentiation and mobilization toward the periphery. Recent reviews provide excellent summaries of these studies [63•–65]. Overall, the type, intensity and duration of the infection seem to dictate HSC fate in these models. Short-term inflammatory signals induce rapid HSC proliferation and differentiation that correlate with the increased hematopoietic production and promotes host defenses. This activation is transient allowing the HSC compartment to recover its quiescence after blood homeostasis has been re-established [28]. In contrast, sustained inflammatory signals, associated with chronic infection or autoimmune disease, alter HSC self-renewal activity and ultimately their long-term potential [40, 61, 41, 38]. Several mechanisms have been proposed to explain this negative impact of sustained inflammation (Fig. 1b). Reports have described how inflammation could disrupt key hematopoietic and non-hematopoietic elements of the BM niche that normally support HSC self-renewal, leading to HSC mobilization to the periphery and promoting their differentiation [33]. In parallel, inflammation provides instructive signals that directly control HSC proliferation, differentiation and apoptosis, all mechanisms that may contribute to the attrition of the HSC pool [32, 66, 38]. Finally, as the heterogeneity of the HSC compartment in term of levels of quiescence, self-renewal capability and potential of differentiation is increasingly appreciated [28, 67], recent studies suggest that inflammatory signals selectively affect distinct HSC subsets. To some extent, this selectivity could contribute to the adaptation of hematopoietic lineage output. For instance, IFN-γ has been proposed to preferentially activate myeloid-biased HSCs, contributing to the refurnishing of the pool of innate immune cells [68]. Similarly type-1 IFN and TNF-α have been shown to promote a megakaryocytic program in one particular HSC subset, potentially contributing to a rapid platelet recovery upon infection [69••]. The mechanisms responsible for such selectivity are yet-to-be-defined. HSC selection could be linked to the cell-intrinsic HSC ability to respond to inflammatory signals, affected by the differential expression of cytokines, chemokines, and pathogen-recognition receptors, or dependent on the presence of negative feedback responses able to modulate the transduction signals and protect some HSC subsets [42–44]. Alternatively it could be associated with a localization of the HSC subsets in more or less immunologically sensitive BM niches. Independently of the underlying mechanisms, this selectivity could promote the exhaustion of particular HSC subsets and therefore affect the overall composition of the HSC compartment. This could explain how immunological status but also the accumulation immunological events could have long-term consequences on the fitness of HSC compartment and may be linked to alteration of the blood function and development of hematopoietic malignancies.
CONCLUSION
Point 1: Interplay between HSC compartment and basal inflammatory state
Overall, the studies presented in this review highlight the ubiquitous nature of inflammation and its function in modulating the HSC activity at different stages of life (Fig. 2), and suggest a co-development of the HSC compartment with its inflammatory environment. During embryonic development, inflammatory signals collaborate with developmental pathways to stimulate the emergence of HSCs, which serve as the cornerstone of definitive hematopoiesis. At birth, encounter with the microbial environment and development of the gut microbiota contribute to shaping the immune system and may affect the maturation of the HSC compartment. In adults, several reports suggest that basal inflammatory signals are able to provide a tonic stimulation to the peripheral immune system and hematopoietic progenitors but also to the HSCs. Many questions remain about the mechanisms modulating the HSC activity in this context. A crosstalk between inflammation signals and key hematopoietic signaling pathways has been proposed [45]. We can also postulate that basal inflammation is necessary to maintain a threshold of activation by priming the signal transduction machinery but also its negative modulators [70]. Importantly, studies suggest that the in vivo inflammatory threshold required for activation is higher in HSCs than in downstream progenitors [32, 21••]. This could be linked to the intrinsic quiescent properties of the HSCs but could also be dependent on the location of HSCs receiving the inflammatory stimuli. In some instance, the BM niche has been shown to be essential in transducing the inflammatory information to the HSC compartment [33, 22••]. Alternatively, inflammatory sensing could occur during the daily HSC trafficking through the blood and peripheral tissues [71]. This immune-surveillance could be important for the PAMP sensing, as TLR activation in HSCs is at least partially dependent on the presence of the soluble CD14 co-receptor [32]. Further investigations will have to define the relative contributions of these different mechanisms and their overall impact on the health of the hematopoietic and immune systems.
Fig. 2: Multiple types of inflammation modulate the HSC compartment during life.
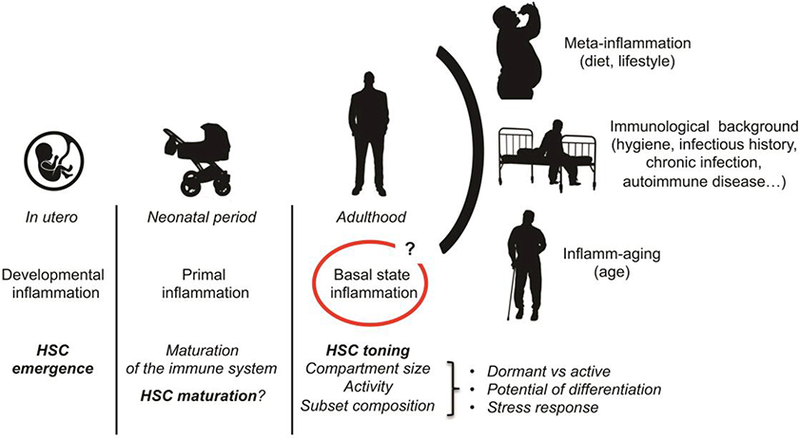
Inflammatory signals are key to the emergence of the HSCs in the embryo and may also contribute to their maturation after birth, underlying the tight relationship between the HSC compartment and its inflammatory environment. In adult, studies suggest that the presence of the microbiota and other physiological stresses maintain a basal inflammatory state that contributes to the toning of HSC activity. Chronic conditions associated with lifestyle choices, hygiene or age modify this steady state inflammatory landscape with consequences on the long-term health of the HSC compartment.
Point 2: Dysregulated inflammatory state and hematopoietic pathologies
Metabolic status, age, hygiene conditions and immunological history are some of the multiple parameters that alter the basal inflammatory state and influence HSC homeostasis. Interestingly these factors have been associated with altered immunological functions but also with hematological pathologies including myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPNs) and acute myeloid leukemia (AML) [72–76]. Whether the lone effect of inflammation on HSCs is able to promote hematological diseases remains to be formally established. However, it is clear that inflammatory cytokines and inflammatory signaling molecules can contribute to some important features of hematological disease initiation and development [77]. For example, inflammation is linked to genomic instability via DNA damage linked to the excessive production of reactive oxygen species (ROS) [78•] or via replicative stress linked to the aberrant activation of dormant HSCs [62•, 79] Similarly, hyperactive inflammatory signaling has been shown to alter gene expression and RNA processing, resulting in impaired hematopoiesis and bone marrow failure [80, 81]. In addition to cell-autonomous alterations, the inflammatory environment could modulate HSC composition by balancing the representation of functionally distinct subsets. As such, inflammation could provide a selective landscape promoting the expansion of some HSC clones at the expense of others. Restriction of the clonal hematopoietic diversity, a phenomenon known as clonal hematopoiesis of indeterminate potential (CHIP) is a frequent feature of aging that may precede the emergence of hematological malignancies [82–85]. Associated with somatic malignancy-linked mutations (such as DNMT3A, TET2, ASXL1), CHIP is considered a pre-leukemic state even if it is not clinically predictive, with only a small fraction of patients with CHIP actually developing hematological disease [86, 87]. The mechanisms contributing to CHIP development are unknown. Notably, it is unclear if inflammation is a cause or a consequence of the CHIP phenomenon. On one hand, CHIP could act as a source of inflammation as CHIP-associated mutations in genes such as TET2 and DNMT3A could promote the development of an inflammatory state [88, 89]. In this context, recent studies show an association between clonal hematopoiesis and cardiovascular diseases and provide a new perspective on the broad consequences of the alteration of HSC fitness on the health of non-hematopoietic tissues [90••, 91••]. On the other hand, it has been proposed that inflammation could, through repeated cycles of infection, systemic inflammation, or other environmental exposures, confer a competitive advantage to mutant cells [92]. Whether and how inflammation differentially impacts fitness of normal HSCs and HSCs harboring CHIP mutation are crucial open questions [93]. Similarly, whether other inflammation-associated lifestyle factors such as smoking, alcohol consumption, or diet influence such clonal expansion could constitute a clinically relevant line of investigation [84].
Point 3: Integrating the inflammatory state in experiment models
This review focuses on the broad influence of inflammation in the HSC compartment and its function. However, it is important to note that most of our experimental results are obtained in laboratory mice living in aberrant hygienic conditions. SPF mice develop an aberrant microbiota and therefore present an abnormal basal inflammatory state [94••]. In addition, these mice have a reduced immunological history that is normally associated with the occurrence of infectious episodes in “free-living” animals [95••]. As consequences, SPF mice present an immature immune system more related to human newborn than to adult [94••]. These conditions could impact the early maturation of the HSC compartment at birth and its activity in adults. As microbiota and basal inflammation appears to directly affect the size and composition of the HSC compartment but also tone its basal activity, these models may provide a distorted view of the HSC activity at steady state. We can envision that the overall inflammatory environment affects HSC state of dormancy and modulate HSC activity at steady state. This may play out in the controversy questioning the extent to the HSC contribution to the steady state hematopoietic output [96–99]. Basal inflammation could also tone the HSC response to stress. The absence of normal stimulation may limit the number of HSCs in a recently proposed primed “GAlert” phase of quiescence, a phenotype accompanied by a more robust functional response to stress [100, 101]. Similarly, recent reports indicate that sporadic inflammatory events could lead to long-lasting modifications of the HSCs and their downstream progenitors, suggesting that these hematopoietic compartments could maintain a memory of the past inflammatory challenges [102, 103]. Finally, inflammation and immune signaling are now viewed as an inherent part of the pathogenesis of the hematopoietic malignancies. As such, we can postulate that the specificity of the basal inflammatory state and immunological history of SPF mice could have a direct impact on the modeling of hematopoietic malignancies [104••]. In this context, analysis of “dirty” mice presenting a normal inflammatory environment along with GF and SPF models could help to better define the influence of basal inflammatory signals on the HSC compartment at steady state. Similarly, exploring the impact of the inflammatory environment on the HSC compartment in diverse patho-physiological conditions could improve our understanding of the long and complex path from health to disease in the hematopoietic system (Fig. 3).
Fig. 3: Modeling the impact of the inflammatory landscape on the HSC compartment and its potential long-term contribution to hematological diseases.
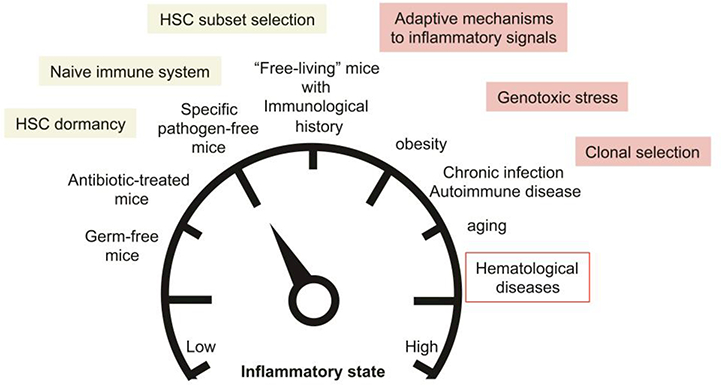
Use of animal models with low inflammatory background such as germ-free mice, antibiotic-treated mice or specific pathogen-free mice could affect key feature of the HSC compartment at steady state and alter their response to stress. Normalization of the inflammatory state to mimic the situation occurring in “free-living” animals could provide a more accurate picture of the HSC biology in physiological conditions. Finally modeling the alterations of the inflammatory landscape in common patho-physiological conditions such as obesity, chronic infection and aging could provide crucial information on the long-term contribution of inflammatory signals in dysregulating HSC molecular programs, promoting genomic instability and selecting pre-leukemic clones.
ACKNOWLEDGMENTS
The authors apologize to their colleagues whose original work could not be cited due to space limitations. The authors thank Drs. Jose Cancelas, Daniel Starczynowski and Gang Huang for critical reading of this review. This work was supported by a National Institutes of Health grant (R01HL141418) and aDODPRCRP award (DOD#W81XWH-15-1-0344).
COMPLIANCES WITH ETHICAL STANDARDS
Conflict of Interest
Vinothini Govindarajah and Damien Reynaud declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
REFERENCES
Recent papers of particular interest have been highlighted as:
• Of importance
•• Of major importance
- 1.Medzhitov R Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 2.Kauts ML, Vink CS, Dzierzak E. Hematopoietic (stem) cell development - how divergent are the roads taken? FEBS Letters. 2016;590(22):3975–86. doi: 10.1002/1873-3468.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clements WK, Traver D. Signalling pathways that control vertebrate haematopoietic stem cell specification. Nature Reviews Immunology. 2013;13(5):336–48. doi: 10.1038/nri3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.•.Espin-Palazon R, Weijts B, Mulero V, Traver D. Proinflammatory Signals as Fuel for the Fire of Hematopoietic Stem Cell Emergence. Trends in Cell Biology. 2018;28(l):58–66. doi: 10.1016/j.tcb.2017.08.003 Detailed review of the inflammatory mechanisms contributing to HSC emergence in the embryo. [DOI] [PubMed] [Google Scholar]
- 5.Espin-Palazon R, Stachura DL, Campbell CA, Garcia-Moreno D, Del Cid N, Kim AD et al. Proinflammatory signaling regulates hematopoietic stem cell emergence. Cell. 2014; 159(5): 1070–85. doi: 10.1016/j.cell.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Q, Zhang C, Wang L, Zhang P, Ma D, Lv J et al. Inflammatory signaling regulates hematopoietic stem and progenitor cell emergence in vertebrates. Blood. 2015; 125(7): 1098–106. doi: 10.1182/blood-2014-09-601542. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Esain V, Teng L, Xu J, Kwan W, Frost IM et al. Inflammatory signaling regulates embryonic hematopoietic stem and progenitor cell production. Genes Dev. 2014;28(23):2597–612. doi: 10.1101/gad.253302.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawamiphak S, Kontarakis Z, Stainier DY. Interferon gamma signaling positively regulates hematopoietic stem cell emergence. Developmental Cell. 2014;31(5):640–53. doi: 10.1016/j.devcel.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Funkhouser LJ, Bordenstein SR. Mom knows best: the universality of maternal microbial transmission. PLoS Biology. 2013; 11(8):e1001631. doi: 10.1371/journal.pbio.1001631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Basha S, Surendran N, Pichichero M. Immune responses in neonates. Expert Review of Clinical Immunology. 2014; 10(9): 1171–84. doi: 10.1586/1744666x.2014.942288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copley MR, Eaves CJ. Developmental changes in hematopoietic stem cell properties. Experimental & Molecular Medicine. 2013;45:e55. doi: 10.1038/emm.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beaudin AE, Boyer SW, Perez-Cunningham J, Hernandez GE, Derderian SC, Jujjavarapu C et al. A Transient Developmental Hematopoietic Stem Cell Gives Rise to Innate-like B and T Cells. Cell Stem Cell. 2016;19(6):768–83. doi: 10.1016/j.stem.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowie MB, McKnight KD, Kent DG, McCaffrey L, Hoodless PA, Eaves CJ. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. The Journal of Clinical Investigation. 2006;116(10):2808–16. doi: 10.1172/jci28310.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bashan A, Gibson TE, Friedman J, Carey VJ, Weiss ST, Hohmann EL et al. Universality of human microbial dynamics. Nature. 2016;534(7606):259–62. doi: 10.1038/nature18301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–10. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.••.Kundu P, Blacher E, Elinav E, Pettersson S. Our Gut Microbiome: The Evolving Inner Self. Cell. 2017;171(7):1481–93. doi: 10.1016/j.cell.2017.11.024 Comprehensive review of the evolving relationship of the gut microbiota with its host across human lifespan. [DOI] [PubMed] [Google Scholar]
- 17.Chow J, Lee SM, Shen Y, Khosravi A, Mazmanian SK. Host-bacterial symbiosis in health and disease. Advances in Immunology. 2010;107:243–74. doi: 10.1016/b978-0-12-381300-8.00008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manzo VE, Bhatt AS. The human microbiome in hematopoiesis and hematologic disorders. Blood. 2015; 126(3):311–8. doi: 10.1182/blood-2015-04-574392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Seminars in Immunology. 2007;19(2):59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 20.Khosravi A, Yanez A, Price JG, Chow A, Merad M, Goodridge HS et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host & Microbe. 2014; 15(3):374–81. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.••.Balmer ML, Schurch CM, Saito Y, Geuking MB, Li H, Cuenca M et al. Microbiota-derived compounds drive steady-state granulopoiesis via MyD88/TICAM signaling. Journal of Immunology (Baltimore, Md : 1950). 2014;193(10):5273–83. doi: 10.4049/jimmunol.1400762 Uses germ-free and genotobiotic mice to establish, at steady state, the contribution of the microbiota in providing tonic stimulation to bone marrow stem and progenitor cells. [DOI] [PubMed] [Google Scholar]
- 22.••.Iwamura C, Bouladoux N, Belkaid Y, Sher A, Jankovic D. Sensing of the microbiota by NOD1 in mesenchymal stromal cells regulates murine hematopoiesis. Blood. 2017;129(2):171–6. doi: 10.1182/blood-2016-06-723742 Provides evidence of the role of the microbiota in regulating steady state hematopoiesis and establishes the contribution of Nodi innate recognition pathway in this context. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.•.Josefsdottir KS, Baldridge MT, Kadmon CS, King KY. Antibiotics impair murine hematopoiesis by depleting the intestinal microbiota. Blood. 2017;129(6):729–39. doi: 10.1182/blood-2016-03-708594 Described the broad effect of a stringent antibiotic treatment on the hematopoietic compartment, suggesting a role of the microbiota on steady state hematopoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuettpelz LG, Borgerding JN, Christopher MJ, Gopalan PK, Romine MP, Herman AC et al. G-CSF regulates hematopoietic stem cell activity, in part, through activation of Toll-like receptor signaling. Leukemia. 2014;28(9):1851–60. doi: 10.1038/leu.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gomez de Aguero M, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H et al. The maternal microbiota drives early postnatal innate immune development. Science (New York, NY). 2016;351 (6279): 1296–302. doi: 10.1126/science.aad2571. [DOI] [PubMed] [Google Scholar]
- 26.••.Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell. 2017;169(5):807–23.el9. doi: 10.1016/j.cell.2017.04.018 Uses single cell RNA analyses to establish the heterogeneity of the quiescent HSC compartment and uncover a continuum of intermediate states from dormant HSCs to quiescent HSCs prone to activation. Proposes that this heterogeneity may reflect latent or past inflammatory events. [DOI] [PubMed] [Google Scholar]
- 27.Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15(4):507–22. doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 28.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–29. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 29.Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nature Medicine. 2010;16(2):228–31. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.King KY, Goodell MA. Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nature Reviews Immunology. 2011;11(10):685–92. doi: 10.1038/nri3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukaida N, Tanabe Y, Baba T. Chemokines as a Conductor of Bone Marrow Microenvironment in Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2017;18(8). doi: 10.3390/ijms18081824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagai Y, Garrett KP, Ohta S, Bahrun U, Kouro T, Akira S et al. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24(6):801–12. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burberry A, Zeng MY, Ding L, Wicks I, Inohara N, Morrison SJ et al. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host & Microbe. 2014;15(6):779–91. doi: 10.1016/j.chom.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Rodriguez S, Wang L, Wang S, Serezani H, Kapur R et al. Sepsis Induces Hematopoietic Stem Cell Exhaustion and Myelosuppression through Distinct Contributions of TRIF and MYD88. Stem Cell Reports. 2016;6(6):940–56. doi: 10.1016/j.stemcr.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiedler K, Kokai E, Bresch S, Brunner C. MyD88 is involved in myeloid as well as lymphoid hematopoiesis independent of the presence of a pathogen. American Journal of Blood Research. 2013;3(2):124–40. [PMC free article] [PubMed] [Google Scholar]
- 36.Ichii M, Shimazu T, Welner RS, Garrett KP, Zhang Q, Esplin BL et al. Functional diversity of stem and progenitor cells with B-lymphopoietic potential. Immunological Reviews. 2010;237(1):10–21. doi: 10.1111/j.1600-065X.2010.00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu A, Wang Y, Ding Y, Baez I, Payne KJ, Borghesi L. Cutting Edge: Hematopoietic Stem Cell Expansion and Common Lymphoid Progenitor Depletion Require Hematopoietic-Derived, Cell-Autonomous TLR4 in a Model of Chronic Endotoxin. Journal of Immunology (Baltimore, Md : 1950). 2015;195(6):2524–8.doi: 10.4049/jimmunol.1501231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nature Cell Biology. 2016;18(6):607–18. doi: 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pronk CJ, Veiby OP, Bryder D, Jacobsen SE. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. The Journal of Experimental Medicine. 2011;208(8):1563–70. doi: 10.1084/jem.20110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465(7299):793–7. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458(7240):904–8. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 42.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nature Immunology. 2009;10(1):109–15. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King KY, Baldridge MT, Weksberg DC, Chambers SM, Lukov GL, Wu S et al. Irgm1 protects hematopoietic stem cells by negative regulation of IFN signaling. Blood. 2011;118(6):1525–33. doi: 10.1182/blood-2011-01-328682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato T, Onai N, Yoshihara H, Arai F, Suda T, Ohteki T. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nature Medicine. 2009;15(6):696–700. doi: 10.1038/nm.1973. [DOI] [PubMed] [Google Scholar]
- 45.de Bruin AM, Voermans C, Nolte MA. Impact of interferon-gamma on hematopoiesis. Blood. 2014;124(16):2479–86. doi: 10.1182/blood-2014-04-568451. [DOI] [PubMed] [Google Scholar]
- 46.Bottero V, Withoff S, Verma IM. NF-kappaB and the regulation of hematopoiesis. Cell death and differentiation. 2006;13(5):785–97. doi: 10.1038/sj.cdd.4401888. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez-Murillo A, Fernandez L, Baena S, Melen GJ, Sanchez R, Sanchez-Valdepenas C et al. The NFKB Inducing Kinase Modulates Hematopoiesis During Stress. Stem Cells (Dayton, Ohio). 2015;33(9):2825–37. doi: 10.1002/stem.2066. [DOI] [PubMed] [Google Scholar]
- 48.Stein SJ, Baldwin AS. Deletion of the NF-kappaB subunit p65/RelA in the hematopoietic compartment leads to defects in hematopoietic stem cell function. Blood. 2013;121(25):5015–24. doi: 10.1182/blood-2013-02-486142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Li L, Baldwin AS Jr, Friedman AD, Paz-Priel I Loss of IKKbeta but Not NF- kappaB p65 Skews Differentiation towards Myeloid over Erythroid Commitment and Increases Myeloid Progenitor Self-Renewal and Functional Long-Term Hematopoietic Stem Cells. PloS One. 2015;10(6):e0130441. doi: 10.1371/journal.pone.0130441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao C, Xiu Y, Ashton J, Xing L, Morita Y, Jordan CT et al. Noncanonical NF-kappaB signaling regulates hematopoietic stem cell self-renewal and microenvironment interactions. Stem Cells (Dayton, Ohio). 2012;30(4):709–18. doi: 10.1002/stem.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adler BJ, Kaushansky K, Rubin CT. Obesity-driven disruption of haematopoiesis and the bone marrow niche. Nature Reviews Endocrinology. 2014;10(12):737–48. doi: 10.1038/nrendo.2014.169. [DOI] [PubMed] [Google Scholar]
- 52.Akunuru S, Geiger H. Aging, Clonality, and Rejuvenation of Hematopoietic Stem Cells. Trends in molecular medicine. 2016;22(8):701–12. doi: 10.1016/j.molmed.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee JM, Govindarajah V, Goddard B, Hinge A, Muench DE, Filippi MD et al. Obesity alters the long-term fitness of the hematopoietic stem cell compartment through modulation of Gfi1 expression. The Journal of Experimental Medicine. 2017. doi: 10.1084/jem.20170690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(26):9194–9. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–45. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 56.Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Frontiers in Immunology. 2016;7:502. doi: 10.3389/fimmu.2016.00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proceedings of the National Academy of Sciences of the United States of America. 2011. ; 108 Suppl 1:4586–91. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host & Microbe. 2008;3(4):213–23. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–72. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 60.Stehle JR Jr, Leng X, Kitzman DW, Nicklas BJ, Kritchevsky SB, High KP. Lipopolysaccharide-binding protein, a surrogate marker of microbial translocation, is associated with physical function in healthy older adults. The Journals of Gerontology Series A, Biological sciences and medical sciences. 2012;67(11): 1212–8. doi: 10.1093/gerona/gls178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esplin BL, Shimazu T, Welner RS, Garrett KP, Nie L, Zhang Q et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. Journal of Immunology (Baltimore, Md : 1950). 2011;186(9):5367–75. doi: 10.4049/jimmunol.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.•.Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell. 2017;21(2):225–40.e5. doi: 10.1016/j.stem.2017.06.013 Establishes in vivo the molecular mechanisms by which TLR signaling directly impacts of the fitness of the HSC compartment. [DOI] [PubMed] [Google Scholar]
- 63.•.Kobayashi H, Suda T, Takubo K. How hematopoietic stem/progenitors and their niche sense and respond to infectious stress. Experimental Hematology. 2016;44(2):92–100. doi: 10.1016/j.exphem.2015.11.008 Comprehensive review of the impact of various infectious conditions on the hematopoietic stem and progenitor compartment. [DOI] [PubMed] [Google Scholar]
- 64. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 2017; 130(15): 1693–8. doi: 10.1182/blood-2017-06-780882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. 2012;119(13):2991–3002. doi: 10.1182/blood-2011-12-380113. [DOI] [PubMed] [Google Scholar]
- 66.Pietras EM, Lakshminarasimhan R, Techner JM, Fong S, Flach J, Binnewies M et al. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. The Journal of Experimental Medicine. 2014;211(2):245–62. doi: 10.1084/jem.20131043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL et al. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154(5): 1112–26. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 68.Matatall KA, Shen CC, Challen GA, King KY. Type II interferon promotes differentiation of myeloid-biased hematopoietic stem cells. Stem Cells (Dayton, Ohio). 2014;32(11):3023–30. doi: 10.1002/stem.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.••.Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell. 2015;17(4):422–34. doi: 10.1016/j.stem.2015.07.007 Describes the specific activation of an HSC-like compartment promoting the rapid and efficient platelet recovery after inflammation-induced thrombocytopenia. [DOI] [PubMed] [Google Scholar]
- 70.Taniguchi T, Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nature Reviews Molecular Cell Biology. 2001;2(5):378–86. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 71.Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131(5):994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kristinsson SY, Bjorkholm M, Hultcrantz M, Derolf AR, Landgren O, Goldin LR. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. Journal of Clinical Oncology : official journal of the American Society of Clinical Oncology. 2011;29(21):2897–903. doi: 10.1200/jco.2011.34.8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kristinsson SY, Landgren O, Samuelsson J, Bjorkholm M, Goldin LR. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica. 2010;95(7):1216–20. doi: 10.3324/haematol.2009.020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Larsson SC, Wolk A. Overweight and obesity and incidence of leukemia: a meta-analysis of cohort studies. International Journal of Cancer. 2008;122(6):1418–21. doi: 10.1002/ijc.23176. [DOI] [PubMed] [Google Scholar]
- 75.Lichtman MA, Rowe JM. The relationship of patient age to the pathobiology of the clonal myeloid diseases. Seminars in Oncology. 2004;31(2): 185–97. [DOI] [PubMed] [Google Scholar]
- 76.Williamson BT, Foltz L, Leitch HA. Autoimmune Syndromes Presenting as a Paraneoplastic Manifestation of Myelodysplastic Syndromes: Clinical Features, Course, Treatment and Outcome. Hematology Reports. 2016;8(2):6480. doi: 10.4081/hr.2016.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hemmati S, Haque T, Gritsman K. Inflammatory Signaling Pathways in Preleukemic and Leukemic Stem Cells. Frontiers in Oncology. 2017;7:265. doi: 10.3389/fonc.2017.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.•.Zambetti NA, Ping Z, Chen S, Kenswil KJ, Mylona MA, Sanders MA et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell. 2016;19(5):613–27. doi: 10.1016/j.stem.2016.08.021 Shows how mutations in the bone marrow niche could lead to the development of an inflammatory environment that promotes HSC genotoxic stress and increases the risk of leukemic transformation. [DOI] [PubMed] [Google Scholar]
- 79.Walter D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015;520(7548):549–52. doi: 10.1038/naturel4131. [DOI] [PubMed] [Google Scholar]
- 80.Fang J, Bolanos LC, Choi K, Liu X, Christie S, Akunuru S et al. Ubiquitination of hnRNPA1 by TRAF6 links chronic innate immune signaling with myelodysplasia. Nature Immunology. 2017;18(2):236–45. doi: 10.1038/ni.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM et al. ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110(3): 1041–6. doi: 10.1073/pnas.1213021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nature Genetics. 2012;44(11):1179–81. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England Journal of Medicine. 2014;371(26):2477–87. doi: 10.1056/NEJMoal409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG et al. Age-related clonal hematopoiesis associated with adverse outcomes. The New England Journal of Medicine. 2014;371(26):2488–98. doi: 10.1056/NEJMoal408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jan M, Ebert BL, Jaiswal S. Clonal hematopoiesis. Seminars in Hematology. 2017;54(l):43–50. doi: 10.1053/j.seminhematol.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cooper JN, Young NS. Clonality in context: hematopoietic clones in their marrow environment. Blood. 2017;130(22):2363–72. doi: 10.1182/blood-2017-07-794362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015; 126(1):9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Experimental Hematology. 2017;55:56–70.el3. doi: 10.1016/j.exphem.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 89.Leoni C, Montagner S, Rinaldi A, Bertoni F, Polletti S, Balestrieri C et al. Dnmt3a restrains mast cell inflammatory responses. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(8):E1490–e9. doi: 10.1073/pnas.1616420114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.••.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (New York, NY). 2017;355(6327):842–7. doi: 10.1126/science.aagl381 Demonstrates in mouse model that clonal hematopoiesis associated with TET2 mutation leads to inflammation and contributes to exacerbated atherosclerosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.••.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. The New England Journal of Medicine. 2017;377(2): 111–21. doi: 10.1056/NEJMoal701719 Complementary to Fuster et al. Indicates that somatic mutations in hematopoietic cells contribute to the development of human atherosclerosis through the activation of specific inflammatory pathways. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Link DC, Walter MJ. ‘CHIP’ping away at clonal hematopoiesis. Leukemia. 2016;30(8):1633–5. doi: 10.1038/leu.2016.130. [DOI] [PubMed] [Google Scholar]
- 93.Abegunde SO, Buckstein R, Wells RA, Rauh MJ. An inflammatory environment containing TNFalpha favors Tet2-mutant clonal hematopoiesis. Experimental Hematology. 2017. doi: 10.1016/j.exphem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 94.••.Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532(7600):512–6. doi: 10.1038/naturel7655 Highlights the caveats associated with the use of experimental mouse model in aberrant hygienic conditions and the interest of the restoring normal environmental exposure for the modeling of immunological events. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.••.Reese TA, Bi K, Kambal A, Filali-Mouhim A, Beura LK, Burger MC et al. Sequential Infection with Common Pathogens Promotes Human-like Immune Gene Expression and Altered Vaccine Response. Cell Host & Microbe. 2016;19(5):713–9. doi: 10.1016/j.chom.2016.04.003 As in Beura et al, highlights the importance of providing natural immunological history to laboratory animals to better model human immunological system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518(7540):542–6. doi: 10.1038/nature14242.. [DOI] [PubMed] [Google Scholar]
- 97.Sawai CM, Babovic S, Upadhaya S, Knapp D, Lavin Y, Lau CM et al. Hematopoietic Stem Cells Are the Major Source of Multilineage Hematopoiesis in Adult Animals. Immunity. 2016;45(3):597–609. doi: 10.1016/j.immuni.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schoedel KB, Morcos MNF, Zerjatke T, Roeder I, Grinenko T, Voehringer D et al. The bulk of the hematopoietic stem cell population is dispensable for murine steady-state and stress hematopoiesis. Blood. 2016;128(19):2285–96. doi: 10.1182/blood-2016-03-706010. [DOI] [PubMed] [Google Scholar]
- 99.Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514(7522):322–7. doi: 10.1038/naturel3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Malam Z, Cohn RD. Stem cells on alert: priming quiescent stem cells after remote injury. Cell Stem Cell. 2014; 15(l):7–8. doi: 10.1016/j.stem.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 101.Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature. 2014;510(7505):393–6. doi: 10.1038/naturel3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell. 2018; 172(1–2): 176–90.e19. doi: 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 103.Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell. 2018; 172( l-2):147–61.el2. doi: 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.••.Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell. 2017; 171(5): 1015–28.el3. doi: 10.1016/j.cell.2017.09.016 Demonstrates that the gut microbiota of laboratory mice markedly differs from wild populations and highlights the impact of this difference on the outcome of infectious diseases and cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]