

Abstract
Objectives:
We sought to describe the genetic complexity of 14 UM-SCC oral cavity cancer cell lines that have remained uncharacterized despite being used as model systems for decades.
Materials and Methods:
We performed exome sequencing on 14 oral cavity UM-SCC cell lines and denote the mutational profile of each line. We used a SNP array to profile the multiple copy number variations of each cell line and use immunoblotting to compare alterations to protein expression of commonly amplified genes (EGFR, PIK3CA, etc.). RNA sequencing was performed to characterize the expression of genes with copy number alterations.
Results:
The cell lines displayed a highly complex network of genetic aberrations that was consistent with alterations identified in the HNSCC TCGA project including PIK3CA amplification, CDKN2A deletion, as well as TP53 and CASP8 mutations, enabling genetic stratification of each cell line in the panel. Copy number FISH and spectral karyotyping analysis demonstrate that cell lines retain chromosomal heterogeneity.
Conclusions:
Collectively, we developed an important resource for future oral cavity HNSCC cell line studies and highlight the complexity of genomic aberrations in cell lines.
Keywords: HNSCC, Oral Cancer, UM-SCC, Cell Lines, Exome
Introduction
Head and neck squamous cell carcinomas (HNSCCs) are the sixth most common cancer worldwide and consist of malignant tumors of the oral cavity, oropharynx, hypopharynx, and larynx [1], which are thought to arise due to a variety of etiologic factors including tobacco-exposure, alcohol consumption and high risk human papilloma virus (HPV) infection. Importantly, clinical outcome and treatment course vary by anatomic site with 5-year survival rates ranging from 40-80% depending on stage, subsite, and HPV status. As such, it is important to build models representing each specific HNSCC subsite in order to model differences between subsites. This is especially true for HNSCCs of the oral cavity, which are the most common HNSCCs, have less than 60% overall survival at 5 years [2], and are not currently associated with a high rate of HPV infection. With the results of The Cancer Genome Atlas [3] and other genomic sequencing studies [4–7], the mutational landscape of primary untreated HNSCCs is beginning to be characterized [8]. However, there is still a need for follow-up in vitro studies to investigate key regulatory pathways, confirm malignant drivers, and discriminate potential therapeutic targets in genetically characterized models.
Indeed, it is clear from early precision medicine literature that the effectiveness of “matched” or “companion” therapies (e.g. those that target specific molecular lesions such as Imatinib and BCR-ABL gene fusions) can be tissue type specific, which may be due to the inherent genetic complexity or unique compensatory pathways of each cancer type [9]. In order to assess potential compensatory pathways for advancing matched therapies across different tissues, cell line models have historically have been valuable tools for investigating the role of focused genetic alterations in tumor behavior and response to therapy, especially for HNSCC [10–13]. In particular, the University of Michigan has created a repository of HNSCC cell lines (UM-SCC) [14], which have been extensively used for in vitro and in vivo modeling of HNSCCs [15]. Despite the extensive use of UM-SCCs in the literature and characterization of some lines using cytogenetics and loss of heterozygosity assessments [16–18], full genetic characterization of these cell lines has not yet been performed. Given the potential for wide phenotypic variations based on genetic mutations [19] as well as the move towards genetics based personalized medicine approaches [20–22], it is increasingly important to understand the genetic architecture of cell lines used for in vitro studies. While studies have started characterizing the genetic implications of therapeutic response in other cell line models [19, 23–25], this analysis has been limited in HNSCC.
Accordingly, whole exome characterization of UM-SCC cell lines is critical to accurately understand critical pathways and mechanistic factors that may be involved in UM-SCC phenotypes and therapeutic response to advancing precision therapies. In this study, we sought to catalog the mutational landscape of oral cavity UM-SCC cell lines. To identify genetic subsets of the disease that are well- or under-represented by our models, we then classified UM-SCCs based on disruptive genomic events and compared the mutational and copy number profiles in our panel with those of other HNSCC cell lines and primary HNSCCs. Ultimately, characterization of UM-SCCs can potentially identify tumor drivers in cell line models, and genetic biomarkers for applicability to specific targeted therapies [12] in translational models of HNSCCs.
Materials and Methods
UM-SCC models.
Cell lines were derived and characterized in the Head and Neck Oncology laboratory at the University of Michigan after consent of the patient donors [14]. The oral cavity cell lines studied in this report were selected from this panel. Cell lines were grown in DMEM with 10% FBS, 7μg/mL penicillin/streptomycin and 1% Non-essential amino acids in 5% CO2 incubator. Cell lines were maintained in exponential growth phase and whole genomic DNA was isolated using the Qiagen DNeasy kit according to manufacturer’s instructions. All cell lines were genotyped as previously described [14].
Exome Sequencing.
Exome Capture Library Construction was done using the Roche NimbleGen V2 (44.1 Mbp) Exome Enrichment Kit as described [12] or by using the Roche NimbleGen V3. Paired-end sequencing (2 × 100 bp) of the captured exons was carried out on an Illumina Genome Analyzer IIx Platform. Paired-end sequencing (2 × 150 bp) for NimbleGen V3 libraries on an Illumina HiSEQ 4000 at the University of Michigan DNA sequencing core according to standard protocol.
Variant Calling.
Read quality was assessed using FastQC [26]. Reads were aligned to hg19 reference genome using BWA v0.7.8 [27]. Mapping was followed by marking duplicates using PicardTools v1.79 (Broad Institute). INDEL realignment and base quality score recalibration was done using GATK v3.2-2 [28]. Variant calling was performed using the HaplotypeCaller and Genotype GVCFs following the GATK best practices workflow guideline [29] for jointly calling variants across all samples. To filter low quality calls, Variant Quality Score Recalibration (VQSR) was applied to the variant call set. Since the suggested sample size for applying VQSR is 30, samples from the 1000 genomes project [30] were combined along with our cell lines to reach this sample size. Varseq v1.4.0 (Golden Helix, Inc., Bozeman, MT) was used to annotate and filter the variants of interest. Filters were set to eliminate false positive variant calls due to sequencing artifacts. The variants were required to have 5 or more reads supporting the alternate allele and be found in less than 1% in a normal population according to the 1000 genomes project [30]. Additional annotations were included to annotate each alterations with COSMIC and dbSNP, which are provided in the supplement. Intronic and intergenic variants were filtered out with the exception of the variants in splice donor or accepter regions.
Sanger Sequencing Validation.
Genomic DNA was isolated following Gentra PureGene protocol (Qiagen) and PCR amplified with Platinum Taq DNA Polymerase High Fidelity (Invitrogen) according to manufacturer’s instructions. Primer sequences for CASP8 are listed in SFig2. PCR products were cloned out using pCR8 TOPO vector (Invitrogen) and submitted for Sanger sequencing at the University of Michigan DNA Sequencing Core on the 3730XL DNA Sequencer (Applied Biosystems). Sequences were aligned using the DNASTAR Lasergene software suite.
Copy Number Analysis.
The OncoScan FFPE Assay Kit (Affymetrix) was used to analyze copy number variations in our samples. Due to a lack of matched normal samples for the cell lines, a common issue for most cell lines in culture, the kit uses an internal pooled normal sample as a comparison to make copy number variation calls. CEL files generated from the kit were combined using the OncoScan Console software to generate OSCHP files. These OSCHP files were then analyzed using the TuScan algorithm of the Nexus Express for OncoScan Software. We also used keratinocyte DNA (ATCC® PCS-200-011) to generate additional OncoScan results as an additional control. We noted that in case of some homozygous deletion calls (CN=0), the B-Allele Frequency plot did not agree with the copy number estimate made by the TuScan algorithm. To provide more accurate copy number calls, we used the presence or absence of exome sequencing reads to validate complete loss of the gene. In cases that we observed a copy number call of zero but the presence of exome sequencing reads, we modified the copy number in STable 5 to one copy, noted with an asterisk.
Western blot Analysis.
Western blot analysis was performed as previously described [31]. Briefly, UM-SCC cell lines at 70-80% confluency were rinsed with PBS and lysed in buffer (150 mM NaCl, 10% Glycerol, 1% NP40, 0.1% Triton X-100, 1 mM PIPES, 1 mM MgCl, 50 mM Tris) containing protease and phosphatase inhibitors (Thermo 186129, 1861277) as described [32]. See STable 6 for primary and secondary antibodies used.
Spectral Karyotyping,
Cell lines in exponential growth phase were treated with Colcemid to capture metaphases. SKY images of UM-SCC-69 and UM-SCC-92 were then prepared and imaged by the Molecular Cytogenetic Core at Albert Einstein College of Medicine using Applied Spectral Imaging’s protocol for DNA spectral karyotyping hybridization and detection.
Fluorescent In-Situ Hybridization.
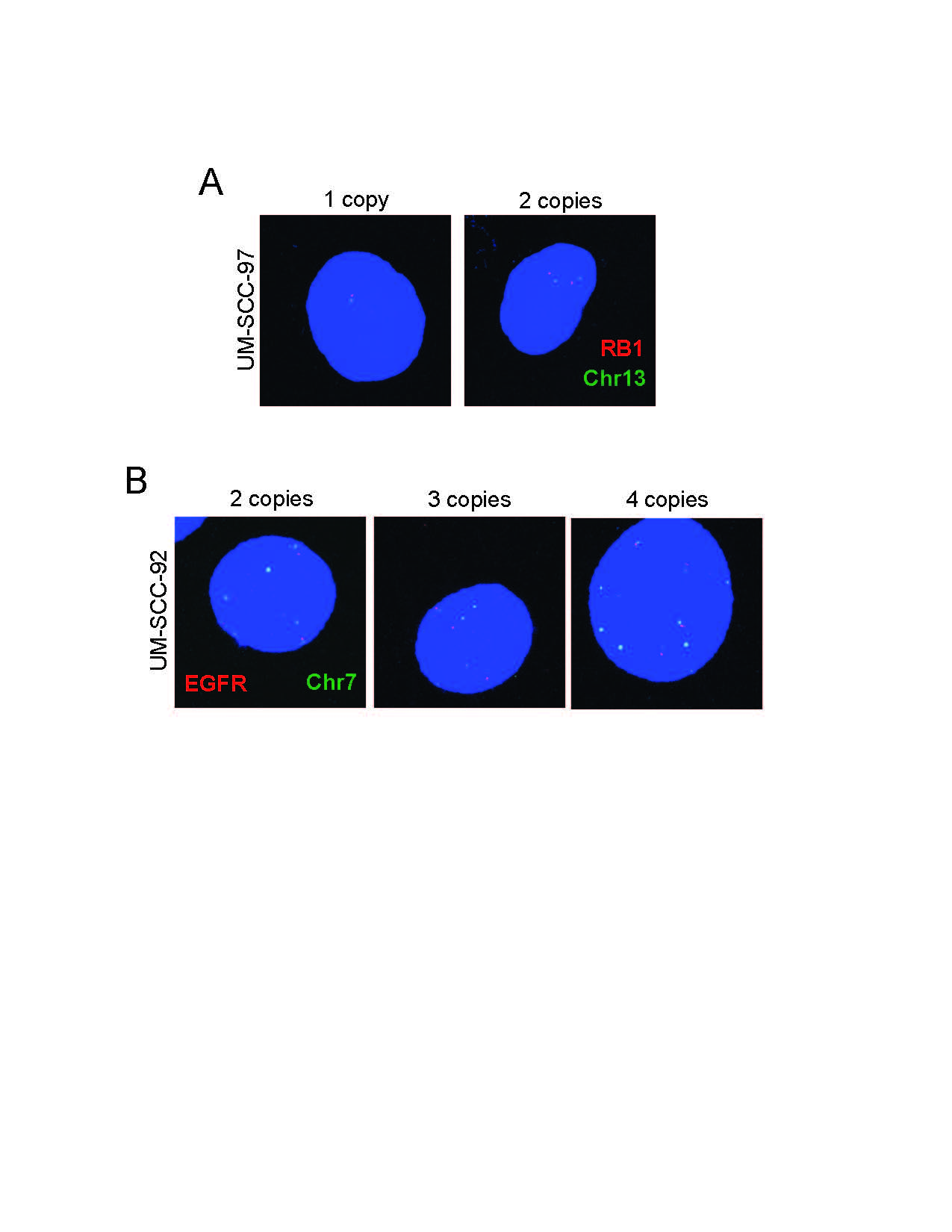
Cell lines UM-SCC-92 ad UM-SCC-97 were treated with Colcemid to arrest cells in metaphase as previously described by our group [31]. Slides were prepared and then probed for EGFR or RB1 with respective chromosome controls (Empire Genomics). Representative images were taken on Leica SP8 confocal.
RNA Sequencing and Bioinformatic Analysis.
RNA isolated with the Qiagen Allprep kit was submit to the University of Michigan DNA Sequencing core and processed using the Illumina HiSeq 4000 by paired end 75nt sequencing. Libraries were prepared according to manufacturer’s protocols with the Illumina Total RNA kit. Read quality was assessed for each cohort using FastQC (v0.11.5). No quality issues were detected in the sample set. Read alignment was performed using STAR (v2.5.3a) according to the two-step alignment protocol recommended in the user manual. Cufflinks (v2.2.1) was used to compute FPKM and values were loaded into MEV for visualization of relative expression between models.
Results
We first performed exome sequencing on 14 UM-SCC cell lines from patients with oral cavity SCC. This patient cohort consists of a mix of seven men and seven women with stage II through stage IV oral cavity cancers arising at a variety of oral cavity sites. Six patients (4 female, 2 male) were under age 40 (range 26-39yrs) and eight patients (5 male, 3 female) were 58 years of age or older (range 58-76) (Table 1). Our analysis found a large mutational load with over 1300 non-synonymous variants per cell line (SFig 1, Stable 1, Stable 2), but as with many cell line studies was limited by a lack of normal controls for each cell line model accounting for the large number of mutation calls relative to those in tumor samples from the TCGA. Nonetheless, we characterized common aberrations found in oral cavity HNSCC tumors in the data set. Similar to TCGA HNSCC tumor studies, we found high frequencies of mutations in 13/14 (93%) affecting TP53, 6/14 (43%), affecting NOTCH1, and 5/14 (36%) affecting CDKN2A (Fig 1). Mutations found in other oral cavity lines from a previous study [25] are in provided SFig 2. In our panel, we observed a range of mutations occurring in the coding regions and in splice sites as well as several frameshift alterations in common tumor suppressor genes like NOTCH1 and CASP8. We validated a set of these mutations by Sanger sequencing for CASP8 and CDKN2A (SFig 3). To then define copy number alterations in these models, we performed high density SNP arrays on all 14 oral cavity cell lines. Analysis of all 14 cell lines by summing copy number alterations at each specific SNP probe site demonstrated copy number common in oral cavity HNSCC. These include amplifications of chromosome 3q, 11q13 and 20, and loss of 3p, 8p, and 18q (Fig 2A). Genome wide analysis was performed for each cell line and demonstrated numerous differences in each cell line model (SFig 4), and held true when compared to an additional keratinocyte control (SFig 5). At the gene level, we identified frequent focal copy number variations in several canonical HNSCC genes, including amplifications of EGFR and deletions of CDKN2A. The copy number calls of our panel in relation to a list of commonly altered genes in HNSCC as identified from TCGA is shown in Fig 2B, and shows complex copy number profiles for each of our cell lines.
Table 1.
Clinical statistics of patients from which the oral cavity SCC cell lines were derived.
| Cell Line | Age | Gender | Race | Clinical TNM | Grade | Stage | Subsite | Type of Lesion | Previous Tx | Smoking | Alcohol | Postoperative Tx? | Recurrence? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UMSCC-1 | 73 | M | - | T2N0M0 | Moderately Differentiated | II | Floor of mouth | Recurrence | S,RT | Heavy | Rare | No | Yes (local) |
| UMSCC-8 | 76 | F | White | T2N1M0 | Moderately to Well Differentiated | III | Alveolar Ridge | Recurrence | RT | 50 pack-year | None | No | No |
| UMSCC-14A | 58 | F | White | T1N0M0 | Moderately to Poorly Differentiated | I | Floor of mouth | Recurrence | S,S,RT,S | 30 pack-year | Occasional | No | Yes (local) |
| UMSCC-29 | 66 | M | White | T3N2aM0 | Well Differentiated | IV | Alveolar Ridge | Persistent primary | CX | Tobacco (heavy) | Heavy | RT | Yes (metastatic) |
| UM-SCC-43 | 73 | M | - | - | - | - | Hard Palate | Primary (lymph node) | None | - | - | - | - |
| UMSCC-49 | 63 | M | Black | T3N1M0 | Moderately to Well Differentiated | III | Tongue | Primary | None | 10 pack-year | Occasional | RT | Yes (persistence) |
| UMSCC-55 | 65 | M | White | T2N0M0 | - | II | Floor of mouth | Primary | None | 90 pack-year | Heavy | Left hemimandibulectomy for ORN | Yes (Tonsillar fossa) |
| UMSCC-59 | 71 | F | White | T3N2bM0 | - | IV | Tongue | Primary | None | None | None | RT | - |
| UMSCC-69 | 35 | M | - | T4N0M0 | Moderately to Well Differentiated | IV | Hard Palate | Persistent primary | CX | 50 pack-year | Heavy | RT | - |
| UMSCC-92 | 38 | F | White | T2N0M0 | - | II | Tongue | Second Primary | None | None | None | No | No |
| UMSCC-97 | 38 | F | White | T2N0M0 | - | II | Tongue | Primary | None | None | Rare | No | Yes (local) |
| UMSCC-103 | 26 | F | White | T4N2bM0 | Well Differentiated | IV | Tongue | Primary | None | Former (6-pack years) | None | CX | Yes (local, regional) |
| UMSCC-108 | 30 | F | Asian | rT4N0M0 (T3N1) | Moderately Differentiated | IV | Tongue | Second Primary | CX,RT | None | None | Palliative chemo | Yes |
| UMSCC-110 | 39 | M | White | T3N0M0 | Moderately Differentiated | III | Tongue | Primary | None | 44 pack-year | Heavy | RT | Yes |
Figure 1. Single nucleotide variants identified in the UM-SCC oral cavity cell line panel.

(A) Mutations in oral cavity UM-SCC cell lines were annotated by color code as indicated as called from Nimblegen capture-based exome sequencing. The mutation list contains the common single nucleotide variants identified in the HNSCC TCGA project, and the percentage of cell lines with mutation in each gene is shown on the right. Schematics were created to show the distribution of mutations found in (B) CDKN2A or (C) CASP8 in the UM-SCC oral cavity cell lines (top) or in the HNSCC TCGA data set (bottom). Numbers next to individual mutations indicate the number of independent tissue samples in which each specific mutation was identified if it was recurrently mutated.
Figure 2. Genetic heterogeneity of UM-SCC oral cavity cell lines characterized by copy number alterations.

(A) Genomic DNA from low passage cell lines was analyzed with high density SNP arrays and compared to a commercially available pooled control. Copy number alterations were called using Affymetrix software and average copy number calls were annotated. This panel shows a summary of genetic alterations summed across the entire UM-SCC oral cavity cell line panel. Amplifications (blue) and deletions (red) were annotated. (B) Copy number variations of genes commonly altered in the HNSCC TCGA project are shown for each of the oral cavity cell lines using the probe medians. (C) Protein isolated from the cell line panel was used to perform Western blot analysis for several highly recurrent genetic drivers that are amplified in the cell lines including EGFR, PIK3CA and their downstream effectors as indicated. Estimated copy number values by the TuScan algorithm for EGFR and PIK3CA are shown above the Western blots. Representative blots are shown for each image. (D) Spectral Karyotyping (SKY) of UM-SCC-69 and UM-SCC-92 cells (top panel) and respective high density copy number plots from SNP array data (bottom panel). We performed SKY analysis on 10 individual cells from both cell lines and a representative image is shown for each line.
To then associate copy number outliers with protein expression in the cell line panel, we performed Western blot analysis on several proposed HNSCC oncogenic drivers with substantial copy number alterations across the panel. We observed that cell lines with the highest copy number amplification of EGFR, UM-SCC-59 and -69, also had the highest protein expression (Fig 2C). In contrast, PIK3CA copy number did not result in dramatic variance of the functional protein p110α. As PIK3CA is contained within the larger 3q amplicon, and focal PIK3CA amplifications are rare in HNSCC tumors, these data suggest that 3q amplification is not necessarily a marker for PIK3CA protein overexpression in the cell line models. Importantly, signaling downstream of these common tyrosine kinase aberrations through AKT, ERK, and MEK pathways were present in all cell lines assessed (Fig 2C). Accordingly, p53 expression is generally associated with mutations as wild type p53 is degraded by MDM2 in normal culture conditions. Our protein expression data was consistent with this postulate as the wild type cell line and those with splice site mutations did not express p53 protein. Similarly, the RNAseq data further validated our copy number calls from above as cell lines with at least one copy of CDKN2A, such as UM-SCC-43, -110 expressed CDKN2A, and cell lines with no copies of CDKN2A (UM-SCC-49, -55) did not express the gene (Figure 4).
Figure 4. Expression of TCGA related genes in the UM-SCC oral cavity cell lines.

Genes of interest are displayed across each row, with the cell lines across the top.
Surprisingly, the copy number analysis revealed that some chromosomes had uneven distributions in each cell line. For example, in UM-SCC-92, EGFR located on chromosome 7 was found an average of 2.33 times suggesting that some cells may contain 3 copies or more and others just 2 or fewer copies. Similarly, UM-SCC-69 contained 15.67 copies of EGFR. Given the apparent mixed chromosome content of some cell lines, it is likely that the cell lines contain heterogeneous populations with genetic diversity within each cell line population. We postulate that within the populations, driving genetic lesions will be found in all cells while passenger mutations would reside in only sub-populations. Thus, we analyzed the chromosomal content and fusion status of two representative cell lines from our collection, UM-SCC-69 and UM-SCC-92, by spectral karyotyping to determine the distribution on chromosome content between individual cells in each model (Fig 2D). This analysis demonstrated that UM-SCC-69 cells contained an average of 129 chromosomes, while UM-SCC-92 contained 71 chromosomes. These data were also consistent with the complexity of copy number data from the SNP arrays. For example, while most cells analyzed from the UM-SCC-92 population contained 3 copies of chromosome 1, 2/10 cells had 4 copies, 1/10 cells had 2 copies, and 2/10 cells harbored unique translocations of chromosome 1 to chromosomes 9 and 15, respectively t(1;9;15)(STable 3 and 4). We also identified a recurrent chromosome 5 to 17 translocation t(5;17) that was present in 10/10 UM-SCC-92 cells, though we did not identify any additional translocations that were present in all cells from the population in UM-SCC-69. This suggests that no initiating translocations were responsible for transformation of this model, though we did identify highly recurrent translocations in both models such as t(17,1) in 6/10 UM-SCC-69 cells and t(7,8) in 9/10 UM-SCC-92 cells. In addition, we performed FISH to evaluate the potential heterogeneity of two genes, EGFR and RB1, in two of our cells lines and found that we indeed had cells with differing copy numbers of genes, suggesting heterogeneity persisting in the cell lines (SFig 7). Collectively, these data support the concept that the UM-SCC cell lines contain heterogeneous populations of tumor cells even after several passages in long term cell culture.
With this understanding, we then summarized the overall representation of genetic events in our cell line panel as compared to the representation of events in the HNSCC TCGA data. This demonstrated that the disruptive genomic events found in our UM-SCC oral cavity collection represent a highly complex genetic distribution than is generally not found in primary untreated tumors, but could be more consistent with advanced HNSCC cases. In analyzing key pathways of oncogenesis similar to TCGA, we found that while there are some commonalities across all models (PIK3CA, E2F1, and TP63 amplifications were common) most events are a mixture of possible gain or loss of function aberration (Fig 3). For example, the tumor suppressor FAT1, an inhibitor of Wnt/β-catenin signaling, is found to be amplified, deleted, or mutated across multiple cell lines.
Figure 3. Summary of the oncogenic pathways genetically disrupted in the UM-SCC oral cavity cell line panel.

Alterations (mutations and copy number alterations) in common oncogenic pathways in the UM-SCC oral cavity cell lines were broken down by pathway classification, e.g. Cell Cycle pathway, Receptor Tyrosine Kinases, etc. as in the HNSCC TCGA project. Color shades indicate the frequency of alterations to each pathway, as either potential activating or inactivating alterations.
Discussion
The UM-SCC cell line panel was developed over the past 40 years at the University of Michigan from over 100 different donors [14, 33–37] and has available citations dating back to 1983. Here, we have characterized the molecular landscape of 14 of the most highly utilized oral cavity UM-SCC models. In the precision medicine era, comprehensive genetic sub-stratification of known driver mutations is critical in order to identify how and where to strategically plan targeted therapies [38]. In vitro experiments with cell lines are critical to identifying genetic profiles and connecting subsets to therapeutic responses. Until now, however, genetic characterization of the UM-SCC cell line panel has been limited despite their wide-ranging use as models for HNSCCs.
An important finding of this study is the limited genetic diversity observed amongst the existing cell line panel as compared to global distributions of common genetic drivers. For example, PIK3CA alterations in HNSCC range from 0-70% globally depending on cohort [39], but occur in 100% of our models. In contrast, we and others have recently described activating genetic alterations to ERBB2 (HER2) and FGFR1 that occur in both epidemiologically low risk and high risk HNSCC populations [31, 40–44]; interestingly, these genes harbored activating genetic alterations in 10/14 and 3/14 cell lines, respectively. This data suggests a need to continue deriving cell lines representative of different ethnic and genetic sub-groups to more accurately model the complexity of genetic alterations observed in oral cavity HNSCC.
Moving forward, studies of genetic heterogeneity and tumor evolution are becoming increasingly prevalent as sequencing and single cell technologies become more tenable. The data generated in this report suggest that the UM-SCC cell lines retain a high level of genetic heterogeneity which has both advantages and disadvantages for in vitro experiments. The use of CRISPR technology to knockout multiple alleles of a gene, for example, could produce clones that may not represent the whole cell line population. In short term experiments, genetic heterogeneity is unlikely to play a major role in outcomes, which may be hypothesized to relate to the primary driver mutations with which each cell line is characterized. However, in long-term culture experiments, such as selection of therapy resistant clones, genetic heterogeneity of the cell lines may play a profound effect similar to the in vivo clonal evolution of tumors following treatment. Further follow up from single cell analysis techniques [45, 46] could be very interesting in exploring this cell line heterogeneity we observed, especially over time. Nonetheless, these consequences of the genetic heterogeneity in the HNSCC remain to be explored, though previous work has shown that cell lines reflect the cytogenetic changes that are present in the tumor tissue from which they were developed [47, 48].
The data collected here suggest that many of the highly recurrent aberrations found in the HNSCC TCGA project are well represented in the UM-SCC oral cavity cell line panel. Interestingly, the distribution of mutations is distinctive. Whereas most primary untreated HNSCC tumors contain a single aberration in multiple pathways (e.g. EGFR amplification OR PIK3CA amplification plus CDKN2A deletion OR CCND1 (Cyclin D1) amplification), the majority of cell lines harbor multiple aberrations in a single pathway (e.g. EGFR amplification AND PIK3CA amplification plus CDKN2A deletion AND CCND1 (Cyclin D1) amplification). Whether this is associated with selection of successful adaptation to in vitro culture or represents the evolution of the tumor within the patient is unknown, but suggests that the cell lines represent a highly complex and genetically distinct subset of HNSCC tumors. This subset may be of particular use in representing responses in a more metastatic setting, in which the tumor may have acquired additional mutations, than of modeling the phenotypes of primary patient tumors. Sequencing patients in a metastatic setting, and understanding the genomic landscape of those tumors, will be particularly interesting in comparison. Despite this observation of mutation accumulation, a subset of cell lines such as UM-SCC-108, contain fewer established “driver” aberrations than other models and begin to add to the genetic diversity of the panel.
Collectively, this panel of UM-SCC oral cavity cell lines has immense utility for studies of HNSCC as evidenced by the vast array of publications from labs around the world over the past four decades. We report comprehensive genetic characterizations on the models that can be leveraged to validate cell line identity and just as importantly to put individual studies in the context of genetic alterations. Our study shows that UM-SCC oral cavity cell lines contain models with an array of genetic alterations that are commonly found in HNSCC, and suggests that the field may benefit from the derivation of additional models with unique genetics. As we strive towards improved personalized medicine protocols for HNSCC patients, the cell lines continue to represent important models for discovery of both HNSCC pathogenesis and therapeutic protocols that aim to improve overall survival.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
Genetic characterization of frequently used UM-SCC oral cavity cell lines
Cells had complex network of genetic aberrations like The Cancer Genome Atlas project
Copy number and spectral karyotyping showed cell lines kept chromosomal heterogeneity
Acknowledgements
The authors thank Drs. Carol R Bradford and Mark EP Prince for their support on this project and manuscript.
Grant Support: J.C.B. and T.E.C. received funding from NIH Grants U01-DE025184, P30-CA046592 and R01-CA194536. M.L.L. was funded by F31 CA20634 and J.E.M. by F31 DE027600. H.J. received funding from NIH Grant U01-DE025184 and P30-CA046592. J.C.B also received funding from the American Head and Neck Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
“The University of Michigan holds an invention disclosure on the UM-SCC cell line panel and has licensed the distribution of UM-SCC-1”
References:
- [1].Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- [2].National Cancer Institute: Surveillance E, and End Results (SEER) Program. Oral Cancer 5-Year Survival Rates by Race, Gender, and Stage of Diagnosis. In: NIDCR, editor.2006. [Google Scholar]
- [3].The Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mountzios G, Rampias T, Psyrri A. The mutational spectrum of squamous-cell carcinoma of the head and neck: targetable genetic events and clinical impact. Annals of oncology : official journal of the European Society for Medical Oncology. 2014;25:1889–900. [DOI] [PubMed] [Google Scholar]
- [7].Seiwert TY, Zuo Z, Keck MK, Khattri A, Pedamallu CS, Stricker T, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21:632–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Giefing M, Wierzbicka M, Szyfter K, Brenner JC, Braakhuis BJ, Brakenhoff RH, et al. Moving towards personalised therapy in head and neck squamous cell carcinoma through analysis of next generation sequencing data. European journal of cancer. 2016;55:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Birkeland AC, Ludwig ML, Meraj TS, Brenner JC, Prince ME. The Tip of the Iceberg: Clinical Implications of Genomic Sequencing Projects in Head and Neck Cancer. Cancers (Basel). 2015;7:2094–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Grenman R, Carey TE, McClatchey KD, Wagner JG, Pekkola-Heino K, Schwartz DR, et al. In vitro radiation resistance among cell lines established from patients with squamous cell carcinoma of the head and neck. Cancer. 1991;67:2741–7. [DOI] [PubMed] [Google Scholar]
- [11].Bradford CR, Zhu S, Ogawa H, Ogawa T, Ubell M, Narayan A, et al. P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head Neck. 2003;25:654–61. [DOI] [PubMed] [Google Scholar]
- [12].Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110:20224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Akervall J, Guo X, Qian CN, Schoumans J, Leeser B, Kort E, et al. Genetic and expression profiles of squamous cell carcinoma of the head and neck correlate with cisplatin sensitivity and resistance in cell lines and patients. Clin Cancer Res. 2004;10:8204–13. [DOI] [PubMed] [Google Scholar]
- [14].Brenner JC, Graham MP, Kumar B, Saunders LM, Kupfer R, Lyons RH, et al. Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell lines. Head Neck. 2010;32:417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wolter KG, Wang SJ, Henson BS, Wang S, Griffith KA, Kumar B, et al. (-)-gossypol inhibits growth and promotes apoptosis of human head and neck squamous cell carcinoma in vivo. Neoplasia. 2006;8:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jones JW, Raval JR, Beals TF, Worsham MJ, Van Dyke DL, Esclamado RM, et al. Frequent loss of heterozygosity on chromosome arm 18q in squamous cell carcinomas. Identification of 2 regions of loss--18q11.1-q12.3 and 18q21.1-q23. Archives of otolaryngology--head & neck surgery. 1997;123:610–4. [DOI] [PubMed] [Google Scholar]
- [17].Van Dyke DL, Worsham MJ, Benninger MS, Krause CJ, Baker SR, Wolf GT, et al. Recurrent cytogenetic abnormalities in squamous cell carcinomas of the head and neck region. Genes, chromosomes & cancer. 1994;9:192–206. [DOI] [PubMed] [Google Scholar]
- [18].Carey TE, Vandyke DL, Worsham MJ. Nonrandom Chromosome-Aberrations and Clonal Populations in Head and Neck-Cancer. Anticancer Res. 1993;13:2561–7. [PubMed] [Google Scholar]
- [19].Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Birkeland AC, Brenner JC. Personalizing Medicine in Head and Neck Squamous Cell Carcinoma: The Rationale for Combination Therapies. Medical research archives. 2015;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Birkeland AC, Ludwig ML, Spector ME, Brenner JC. The potential for tumor suppressor gene therapy in head and neck cancer. Discovery medicine. 2016;21:41–7. [PMC free article] [PubMed] [Google Scholar]
- [22].Birkeland AC, Uhlmann WR, Brenner JC, Shuman AG. Getting personal: Head and neck cancer management in the era of genomic medicine. Head Neck. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Greshock J, Bachman KE, Degenhardt YY, Jing J, Wen YH, Eastman S, et al. Molecular target class is predictive of in vitro response profile. Cancer Res. 2010;70:3677–86. [DOI] [PubMed] [Google Scholar]
- [25].Li H, Wawrose JS, Gooding WE, Garraway LA, Lui VW, Peyser ND, et al. Genomic analysis of head and neck squamous cell carcinoma cell lines and human tumors: a rational approach to preclinical model selection. Molecular cancer research : MCR. 2014;12:571–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Andrews S FastQC A Quality Control tool for High Throughput Sequence Data. http://wwwbioinformaticsbabrahamacuk/projects/fastqc/.
- [27].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].The Genomes Project C. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tillman BN, Yanik M, Birkeland AC, Liu CJ, Hovelson DH, Cani AK, et al. Fibroblast growth factor family aberrations as a putative driver of head and neck squamous cell carcinoma in an epidemiologically low-risk patient as defined by targeted sequencing. Head Neck. 2016;38 Suppl 1:E1646–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Michmerhuizen NMLE, Kulkarni A, Brenner JC Differential Compensation mechanisms Define Resistance to PI3K inhibitors in PIK3CA amplified HNSCC. Oto Head and Neck Surgery. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Owen JH, Hauff SJ, Tang AL, Graham MP, Czerwinski MJ, Kaddoura M, et al. UM-SCC-103: a unique tongue cancer cell line that recapitulates the tumorigenic stem cell population of the primary tumor. The Annals of otology, rhinology, and laryngology. 2014;123:662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Carey TE, Van Dyke DL, Worsham MJ, Bradford CR, Babu VR, Schwartz DR, et al. Characterization of human laryngeal primary and metastatic squamous cell carcinoma cell lines UM-SCC-17A and UM-SCC-17B. Cancer research. 1989;49:6098–107. [PubMed] [Google Scholar]
- [35].Grenman R, Virolainen E, Shapira A, Carey T. In vitro effects of tamoxifen on UM-SCC head and neck cancer cell lines: correlation with the estrogen and progesterone receptor content. International journal of cancer. 1987;39:77–81. [DOI] [PubMed] [Google Scholar]
- [36].Shapira A, Virolainen E, Jameson JJ, Ossakow SJ, Carey TE. Growth inhibition of laryngeal UM-SCC cell lines by tamoxifen. Comparison with effects on the MCF-7 breast cancer cell line. Archives of otolaryngology--head & neck surgery. 1986;112:1151–8. [DOI] [PubMed] [Google Scholar]
- [37].Krause CJ, Carey TE, Ott RW, Hurbis C, McClatchey KD, Regezi JA. Human squamous cell carcinoma. Establishment and characterization of new permanent cell lines. Archives of otolaryngology. 1981;107:703–10. [DOI] [PubMed] [Google Scholar]
- [38].Ludwig ML, Birkeland AC, Hoesli R, Swiecicki P, Spector ME, Brenner JC. Changing the paradigm: the potential for targeted therapy in laryngeal squamous cell carcinoma. Cancer biology & medicine. 2016;13:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Michmerhuizen NL, Birkeland AC, Bradford CR, Brenner JC. Genetic determinants in head and neck squamous cell carcinoma and their influence on global personalized medicine. Genes & cancer. 2016;7:182–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Birkeland AC, Yanik M, Tillman BN, Scott MV, Foltin SK, Mann JE, et al. Identification of Targetable ERBB2 Aberrations in Head and Neck Squamous Cell Carcinoma. JAMA otolaryngology-- head & neck surgery. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pollock NI, Grandis JR. HER2 as a Therapeutic Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Koole K, Brunen D, van Kempen PM, Noorlag R, de Bree R, Lieftink C, et al. FGFR1 Is a Potential Prognostic Biomarker and Therapeutic Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2016;22:3884–93. [DOI] [PubMed] [Google Scholar]
- [43].Goke F, Franzen A, Hinz TK, Marek LA, Yoon P, Sharma R, et al. FGFR1 Expression Levels Predict BGJ398 Sensitivity of FGFR1-Dependent Head and Neck Squamous Cell Cancers. Clin Cancer Res. 2015;21:4356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].von Massenhausen A, Franzen A, Heasley L, Perner S. FGFR1 as a novel prognostic and predictive biomarker in squamous cell cancers of the lung and the head and neck area. Annals of translational medicine. 2013;1:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017;171:1611–24.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Worsham MJ, Van Dyke DL, Grenman SE, Grenman R, Hopkins MP, Roberts JA, et al. Consistent chromosome abnormalities in squamous cell carcinoma of the vulva. Genes, chromosomes & cancer. 1991;3:420–32. [DOI] [PubMed] [Google Scholar]
- [48].Worsham MJ, Wolman SR, Carey TE, Zarbo RJ, Benninger MS, Van Dyke DL. Chromosomal aberrations identified in culture of squamous carcinomas are confirmed by fluorescence in situ hybridisation. Mol Pathol. 1999;52:42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.