

Abstract
Background
Chrysin (5,7-dihydroxyflavone) is a widely distributed natural flavonoid found in many plant extracts, honey and propolis. Several studies revealed that chrysin possesses multiple biological activities including anti-cancer effects. It has been established that activation of apoptosis is the key molecular mechanism responsible for the cytotoxic potential of chrysin. The objective of this study was to design and synthesize potent chrysin analogues as potential cytotoxic agents.
Methods
A series of chrysin derivatives (3a-m) bearing N′-alkylidene/arylideneacetohydrazide moiety were designed, synthesized, and evaluated for their antiproliferative activity against two human breast cancer cell lines, MDA-MB-231 and MCF-7 by applying the MTT colorimetric assay. Selected compounds were tested for their ability to induce apoptosis through caspase 3/7 activation in MDA-MB-231 cells only since MCF-7 cells lack procaspase 3.
Results
Compounds (3a-m) were obtained as geometrical isomers (E/Z isomers) in good yields upon treatment of hydrazide 5 with different aliphatic and aromatic aldehydes. Most of the synthesized compounds demonstrated moderate-to-good activity against both cell lines. The cytotoxicity results revealed the importance of lipophilic moieties at C-4 position of ring D in imparting the cytotoxic activities to the compounds. Compound 3e with 4-benzyloxy substituent was found to be the most active among the synthesized compounds with IC50 3.3 µM against MDA-MB-231 and 4.2 µM against MCF-7 cell lines. The cytotoxic potential of compound 3e is comparable to that of the well-known anti-cancer agent doxorubicin. In addition, compounds substituted with fluoro (3b), nitro (3h), and dimethylamino (3j) exhibited good cytotoxicity with IC50 <6.5 µM against MDA-MB-231 and <12 µM against MCF-7. Selected compounds were able to induce apoptosis in MDA-MB-231 cells as indicated by caspase-3 and/or -7 activation.
Conclusion
Our results show that the newly designed chrysin derivatives exert anticancer activity in human breast cancer cell lines, MDA-MB-231 and MCF-7. Therefore, they can be considered as leads for further development of more potent and selective cytotoxic agents.
Keywords: apoptosis, chrysin, design, synthesis, cytotoxicity, caspase-3/7, antiproliferative agents
Introduction
Cancer is a worldwide epidemic that has been a primary concern in the field of medicinal chemistry research. Despite the significant work and the huge effort in finding cancer chemotherapeutic agents, many obstacles were faced including toxicity and lack of sensitivity.1,2 Therefore, there is a growing need to develop new and effective anticancer agents that can overcome the problems with the currently available agents.
Apoptosis or programmed cell death is a regulatory mechanism in multicellular organisms that plays a massive role in maintaining tissue homeostasis through balancing cell proliferation as well as eliminating unwanted and damaged cells.3–5 It has been established that the main mechanism of action for several anticancer agents involves induction of apoptosis.6 This leads to the fact that designing effective apoptosis inducers is considered a potential strategy to develop efficient anticancer agents. Caspases are well known to play a vital role in the process of apoptosis. Caspases 3 and 7 have been identified as key effector enzymes that are normally presented as inactive zymogens inside cells. Activating theses zymogens will result in irreversible apoptotic cell death.7 Accordingly, compounds that have the ability to activate caspases are of potential interest as new therapeutic anticancer agents.
Flavonoids are natural polyphenolic compounds that are abundant in plants and various common foods. Chrysin (5,7-dihydroxyflavone, compound 1 in Figure 1) is a widely distributed natural flavonoid found in many plant extracts, honey, and propolis. Chrysin exhibits a wide range of biologic activities such as antioxidant,8 antiallergic,9 antibacterial,10 anti-inflammatory,11 antidiabetic,12 anxiolytic,13 and anticancer effects.14 Several studies on a wide variety of cancer cell lines revealed that activation of apoptosis is the key molecular mechanism responsible for the anticancer activity of chrysin.14 In some types of cells, activation of apoptosis is associated with caspase-3 activation.15 To improve the pharmaceutical activity of chrysin, many recent studies have been done utilizing the synthesis of chrysin derivatives.16–20
Figure 1.

Chemical structures of chrysin (1), PAC-1 (2a), De-allyl PAC-1 (2b), and hybrid structure of 1 and 2 (3a-m).
On the other hand, it was reported that the proapototic small molecules PAC-1 and De-allyl PAC-1 (compounds 2a-b in Figure 1) induce apoptosis in different types of cancerous cells through activation of caspase-3/7.21,22 Structurally, compounds 2a-b possess an acetohydrazide moiety linked directly to the nitrogen atom of the benzylpiperazine moiety. It has been demonstrated that structural alterations on the acetohydrazide moiety of PAC-1 diminish the cytotoxic activity of the compound, whereas structural modifications on the corresponding piperazine ring retained the activity. These findings highlight the importance of the acetohydrazide moiety as a chemical feature required for the cytotoxic activity in contrast to the piperazine moiety that was found not essential for the activity. Based on these results and for obtaining potent chrysin analogs as cytotoxic agents, hybrid structures of chrysin (1) and compounds 2a-b were synthesized in which the chrysin scaffold was linked to the acetohydrazide moiety. The cytotoxic activities of the synthesized compounds were evaluated against two human breast cancer cell lines, MDA-MB-231 and MCF-7. Selected compounds were tested for their ability to induce apoptosis through caspase-3/7 activation in MDA-MB-231 cells only since MCF-7 cells lack procaspase 3.23,24 The general structure of the hybrid compounds (3a-m) is shown in Figure 1. The chemical modifications of the hybrid compounds were chosen to include a variety of aliphatic and para-substituted aromatic moieties.
Materials and methods
Chemistry
General experimental procedures Reagents and solvents were purchased from Sigma Aldrich, Acros Organics, and Cayman Chemicals and used without further purification. Reaction’s progress was qualitatively analyzed by thin-layer chromatography performed on precoated silica gel plates (Merk, Kieselgel 60 F-254, 0.2 mm). Compound spots were visualized by UV light (254 nm). Compounds were characterized by 1H-NMR and 13C-NMR using a 400 MHz Bruker Avance Ultrashield Spectrometer (Switzerland); the spectra were obtained in ppm using automatic calibration to the residual proton peak of the solvent, DMSO-d6. The 1H NMR data are presented as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet, m = multiplet), coupling constants (Hz), and integration. The 13C NMR analyses were reported in terms of chemical shift. 1H and 13C NMR spectra for all compounds are included in the Supplementary material. HRMS data were acquired using a Thermo QExactive Plus mass spectrometer equipped with an electrospray ionization source (Thermo Fisher Scientific).
Synthesis of methyl 2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetate (4)
Chrysin 1 (1.0 g, 3.93 mmol) and potassium carbonate (1.63 g, 11.8 mmol) were dissolved in 20 mL anhydrous DMF and allowed to cool down to 0°C in ice/water bath. Methyl 2-bromoacetate (0.37 mL, 3.93 mmol) was then added dropwise and the reaction mixture left stirring at the same temperature for 3 hours. After completion, the mixture was diluted with ethyl acetate (35 mL) and washed with water (35 mL × 2). The aqueous layers were combined and extracted with ethyl acetate (35 mL × 2). The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (5% methanol in chloroform) that afforded the desired product as pale yellow powder (0.77 g, 60%).
Alternative workup procedure: after reaction completion the reaction mixture was slowly poured into well-stirred ice/ water mixture. The formed precipitate was filtered, washed with water, and dried on air to give the title compound as pure yellow powder. Yield (1.2 g, 94%). 1H NMR (DMSO-d6): δ 12.80 (s, 1H), 8.11 (d, J=7.6 Hz, 2H), 7.62–7.56 (m, 3H), 7.04 (s, 1H), 6.84 (s, 1H), 6.43 (s, 1H), 4.96 (s, 2H), 3.73 (s, 3H).
Synthesis of 2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetohydrazide (5)
Method A
A suspension of ester 4 (1.0 g, 3.0 mmol) in absolute ethanol (30 mL) was cooled down to 0°C in ice/water bath. To that was added dropwise 80% hydrazine hydrate (0.58 mL, 12 mmol). The resulting mixture was allowed to stir at same temperature for 1 hour and then at room temperature for few days. After completion, the reaction mixture was poured into ice/water mixture. The formed precipitate was filtered, washed with water, and dried on air overnight to give the desired pure product as off-white powder.
Method B
To an ice-cold suspension of ester 4 (1.0 g, 3.0 mmol) in anhydrous methanol (30 mL) was added dropwise 80% hydrazine hydrate (0.58 mL, 12 mmol) and few drops of hydrochloric acid. After 1 hour, the reaction was completed. The mixture was poured into ice/water mixture and the formed precipitate was filtered, washed with water, and dried on air overnight.
Yield (0.98 g, 98%). 1H NMR (DMSO-d6): δ 12.81 (s, 1H), 9.43 (s, 1H), 8.09 (d, J=7.2 Hz, 2H), 7.65–7.57 (m, 3H), 7.05 (s, 1H), 6.82 (d, J=1.6 Hz, 1H), 6.45 (d, J=1.68 Hz, 1H), 4.64 (s, 2H), 4.38 (s, 2H). 13C NMR (DMSO-d6): δ 182.11, 165.85, 163.78, 163.62, 161.10, 157.21, 132.21, 130.57, 129.19, 126.48, 105.45, 105.27, 98.83, 93.57, 66.58. HRMS (ESI, m/z): calculated for C17H14N2O5 [M+H]+ 327.0975; found 327.0965.
General procedure for the synthesis of N′-alkylidene/arylidene-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetohydrazide (3a-m)
Method C
To a solution of hydrazide 5 (1.0 g, 3.0 mmol) in anhydrous DMF (60 mL) was added the appropriate aldehyde (3.0 mmol) and allowed to stir at room temperature for few days. Upon reaction completion, the mixture was poured into ice-cold water and the formed precipitate was filtered, washed with water exhaustively, and dried on air overnight to give pure desired compounds in good yields.
Method D
To a suspension of hydrazide 5 (1.0 g, 3.0 mmol) in anhydrous methanol (60 mL) was added the appropriate aldehyde (3.0 mmol) and few drops of hydrochloric acid. After 1 hour, the reaction was completed and the formed precipitate was separated by filtration, washed with methanol, and dried on air to give pure desired compounds in good yields.
(E, Z)-N′-benzylidene-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetohydrazide (3a)
The product was obtained as white powder. Yield (82%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.84–12.81 (m, 1.5H), 11.69–11.63 (m, 1.5H), 8.34 (s, 0.5H), 8.12–8.04 (m, 4H), 7.76–7.45 (m, 12H), 7.08–7.06 (m, 1.5H), 6.89–6.87 (m, 1.5H), 6.50–6.43 (m, 1.5H), 5.34 (s, 2H), 4.85 (s, 1H). 13C NMR (DMSO-d6): δ 182.03, 168.22, 164.40, 163.71, 163.58, 163.43, 163.41, 161.14, 161.05, 157.20, 148.07, 144.04, 134.01, 133.91, 132.10, 130.53, 130.20, 129.98, 129.08, 128.76, 127.14, 126.98, 126.44, 105.42, 105.30, 105.07, 98.70, 93.60, 93.45, 66.63, 65.32. HRMS (ESI, m/z): calculated for C24H18N2O5 [M+H]+ 415.1301; found 415.1284.
(E, Z)-N′-(4-fluorobenzylidene)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetohydrazide (3b)
The product was obtained as off-white powder. Yield (62%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.84–12.81 (m, 1.5H), 11.70–11.66 (m, 1.5H), 8.33 (s, 0.5H), 8.11–8.03 (m, 4H), 7.83–7.75 (m, 3H), 7.64–7.55 (m, 4.5H), 7.32– 7.27 (m, 3H), 7.06–7.05 (m, 1.5H), 6.88–6.85 (m, 1.5H), 6.49–6.41 (m, 1.5H), 5.33 (s, 2H), 4.84 (s, 1H). 13C NMR (DMSO-d6): δ 182.03, 168.45, 166.87, 164.37, 163.67, 163.60, 163.48, 161.15, 161.06, 157.22, 146.88, 142.95, 138.03, 137.92, 132.10, 131.87, 131.67, 130.55, 129.68, 129.08, 127.16, 127.01, 126.45, 105.43, 105.32, 105.09, 98.73, 93.61, 93.48, 66.63, 65.34. HRMS (ESI, m/z): calculated for C24H17FN2O5 [M+H]+ 433.1194; found 433.1196.
(E, Z)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)-N′-(4-hydroxybenzylidene)acetohydrazide (3c)
The product was obtained as white powder. Yield (90%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.83–12.80 (m, 1.5H), 11.48–11.42 (m, 1.5H), 9.96–9.93 (m, 1.5H), 8.21–8.09 (m, 3.5H), 7.93 (s, 1H), 7.62–7.53 (m, 7.5H), 7.06–7.05 (m, 1.5H), 6.87–6.82 (m, 4.5H), 6.49–6.41 (m, 1.5H), 5.28 (s, 2H), 4.80 (s, 1H). 13C NMR (DMSO-d6): δ 182.12, 182.07, 167.90, 164.48, 163.79, 163.68, 163.55, 163.07, 161.18, 161.09, 159.59, 159.37, 157.27, 157.24, 148.51, 144.50, 132.22, 132.16, 130.58, 129.20, 129.15, 129.01, 128.79, 126.50, 124.99, 115.75, 115.72, 105.46, 105.37, 105.34, 105.11, 98.80, 98.75, 93.66, 93.48, 66.72, 65.36. HRMS (ESI, m/z): calculated for C24H18N2O6 [M+H]+ 431.1237; found 431.1238.
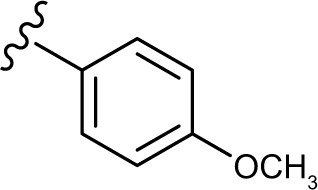
(E, Z)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)-N′-(4-methoxybenzylidene)acetohydrazide (3d)
The product was obtained as yellow powder. Yield (46%). 1H NMR (DMSO-d6) (E:Z=1:0.6): δ 12.84–12.80 (m, 1.6H), 11.56–11.49 (m, 1.6H), 8.27 (s, 0.6H), 8.11–8.09 (d, J=8 Hz, 3H), 7.97 (s, 1H), 7.71–7.55 (m, 8H), 7.06–7.00 (m, 5H), 6.87–6.84 (m, 1.6H), 6.50–6.41 (m, 1.6H), 5.30 (s, 2H), 4.82 (s, 1.2H), 3.80 (s, 4.8H). 13C NMR (DMSO-d6): δ 182.03, 167.95, 164.43, 163.74, 163.59, 163.46, 163.10, 161.13, 161.04, 160.94, 160.74, 157.21, 147.97, 143.92, 132.10, 130.54, 129.08, 128.76, 128.57, 126.45, 114.25, 105.42, 105.30, 105.05, 98.69, 93.60, 93.45, 66.66, 65.31, 55.28. HRMS (ESI, m/z): calculated for C25H20N2O6 [M+H]+ 445.1394; found 445.1395.
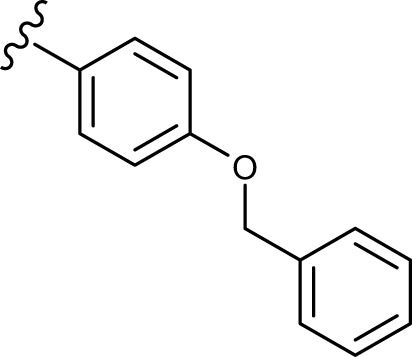
(E, Z)-N′-(4-(benzyloxy)benzylidene)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy) acetohydrazide (3e)
The product was obtained as pale yellow powder. Yield (64%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.84–12.81 (m, 1.5H), 11.56–11.50 (m, 1.5H), 8.27 (s, 0.5H), 8.11–7.97 (m, 4H), 7.70–7.32 (m, 15H), 7.10–7.05 (m, 4.5H), 6.87–6.85 (m, 1.5H), 6.49–6.42 (m, 1.5H), 5.30 (s, 2H), 5.16 (s, 3H), 4.82 (s, 1H). 13C NMR (DMSO-d6): δ 182.07, 182.02, 167.97, 164.42, 163.73, 163.56, 163.44, 163.13, 161.13, 161.04, 160.02, 159.80, 157.20, 147.90, 143.84, 136.71, 132.15, 132.09, 130.54, 129.08, 128.76, 128.57, 128.43, 127.90, 127.73, 126.72, 126.44, 115.10, 105.41, 105.29, 105.05, 98.70, 93.59, 93.44, 69.33, 66.65, 65.32. HRMS (ESI, m/z): calculated for C31H24N2O6 [M+H]+ 521.1707; found 521.1707.
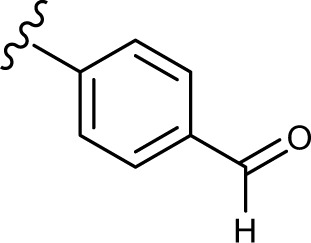
(E, Z)-N′-(4-formylbenzylidene)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetohydrazide (3f)
The product was obtained as yellow powder. Yield (57%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.84–12.81 (m, 1.5H), 11.90–11.86 (m, 1.5H), 10.04 (s, 1.5H), 8.41 (s, 0.5H), 8.11–7.97 (m, 10H), 7.62–7.57 (m, 4.5H), 7.07–7.06 (m, 1.5H), 6.88 (s, 1.5H), 6.50–6.45 (m, 1.5H), 5.38 (s, 2H), 4.88 (s, 1H). 13C NMR (DMSO-d6): δ 192.66, 182.05, 168.56, 164.38, 163.77, 163.70, 163.64, 163.51, 161.15, 161.06, 157.23, 146.65, 142.70, 139.51, 139.41, 136.88, 136.72, 132.13, 130.55, 129.87, 129.11, 127.66, 127.53, 126.47, 105.45, 105.33, 105.10, 98.74, 93.64, 93.52, 66.62, 65.38. HRMS (ESI, m/z): calculated for C25H18N2O6 [M+H]+ 443.1237; found 443.1238.
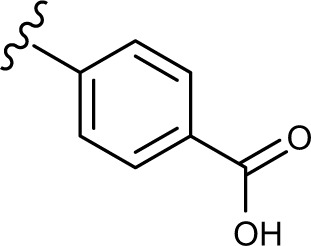
(E, Z)-4-((2-(2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetyl)hydrazono)methyl)benzoic acid (3g)
The product was obtained as white powder. Yield (65%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.84–12.81 (m, 1.5H), 11.83–11.79 (m, 1.5H), 8.39 (s, 0.5H), 8.11–8.09 (m, 4H), 8.01–7.99 (m, 3H), 7.87–7.81 (m, 3H), 7.63–7.54 (m, 4.5H), 7.06–7.05 (m, 1.5H), 6.87–6.86 (m, 1.5H), 6.50–6.43 (m, 1.5H), 5.36 (s, 2H), 4.87 (s, 1H). 13C NMR (DMSO-d6): δ 182.03, 168.46, 166.88, 164.37, 163.68, 163.60, 163.48, 161.16, 161.06, 157.22, 146.87, 142.95, 138.04, 137.92, 132.10, 131.88, 131.67, 130.55, 129.69, 129.08, 127.17, 127.01, 126.45, 105.43, 105.31, 105.10, 98.73, 93.61, 93.48, 66.63, 65.34. HRMS (ESI, m/z): calculated for C25H18N2O7 [M+H]+ 459.1186; found 459.1183.
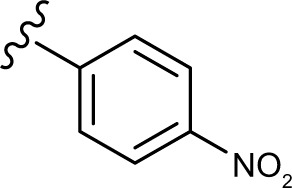
(E, Z)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)-N′-(4-nitrobenzylidene)acetohydrazide (3h)
The product was obtained as yellow powder. Yield (89%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.84–12.81 (m, 1.5H), 11.97–11.94 (m, 1.5H), 8.44 (s, 0.5H), 8.30–7.97 (m, 10H), 7.61–7.57 (m, 4.5H), 7.06 (s, 1.5H), 6.88 (s, 1.5H), 6.50–6.45 (m, 1.5H), 5.39 (s, 2H), 4.89 (s, 1H).). 13C NMR (DMSO-d6): δ 182.06, 168.68, 164.36, 163.92, 163.68, 163.63, 163.55, 161.16, 161.09, 160.06, 157.23, 147.82, 145.62, 141.69, 140.22, 132.14, 130.56, 129.55, 129.12, 128.10, 127.96, 126.47, 124.09, 123.96, 105.34, 105.11, 98.72, 98.57, 93.63, 93.55, 66.61, 65.37. HRMS (ESI, m/z): calculated for C24H17N3O7 [M+H]+ 460.1139; found 460.1136.
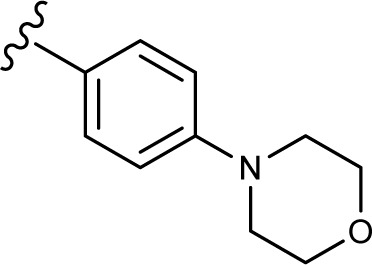
(E, Z)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)-N′-(4-morpholinobenzylidene)acetohydrazide (3i)
The product was obtained as white powder. Yield (92%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.83–12.80 (m, 1.5H), 11.48–11.41 (m, 1.5H), 8.20 (s, 0.5H), 8.10–8.08 (d, J=8 Hz, 3H), 7.92 (s, 1H), 7.60-7.55 (m, 7.5H), 7.05–6.97 (m, 4.5H), 6.86–6.83 (m, 1.5H), 6.49–6.40 (m, 1.5H), 5.28 (s, 2H), 4.80 (s, 1H), 3.73 (m, 6H), 3.19 (m, 6H). 13C NMR (DMSO-d6): δ 182.07, 182.03, 167.76, 164.45, 163.76, 163.59, 163.47, 162.89, 161.13, 161.03, 157.21, 152.27, 152.12, 148.37, 144.39, 132.15, 132.10, 130.55, 129.10, 128.35, 128.13, 126.46, 124.18, 124.13, 114.30, 105.42, 105.30, 105.05, 98.70, 93.61, 93.44, 66.70, 65.92, 65.33, 47.47, 47.41. HRMS (ESI, m/z): calculated for C28H25N3O6 [M+H]+ 500.1816; found 500.1812.
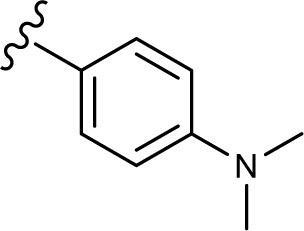
(E, Z)-N′-(4-(dimethylamino)benzylidene)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy) acetohydrazide (3j)
The product was obtained as orange powder. Yield (45%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.84–12.80 (m, 1.5H), 11.41–11.32 (m, 1.5H), 8.17–8.09 (m, 3.5H), 7.89 (s, 1H), 7.63–7.50 (m, 7.5H), 7.07–7.05 (m, 1.5H), 6.87–6.73 (m, 4.5H), 6.49–6.41 (m, 1.5H), 5.27 (s, 2H), 4.79 (s, 1H), 2.96 (s, 9H). 13C NMR (DMSO-d6): δ 182.07, 182.02, 167.55, 164.47, 163.78, 163.59, 163.45, 162.67, 161.12, 161.02, 157.21, 151.57, 151.40, 148.88, 144.89, 132.15, 132.09, 130.54, 129.08, 128.48, 128.25, 126.45, 121.23, 121.19, 111.72, 105.42, 105.29, 105.03, 98.71, 93.61, 93.42, 66.72, 65.31. HRMS (ESI, m/z): calculated for C26H23N3O5 [M+H]+ 458.1710; found 458.1711.
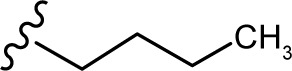
(E, Z)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)-N′-pentylideneacetohydrazide (3k)
The product was obtained as white powder. Yield (96%). 1H NMR (DMSO-d6) (E:Z=1:0.65): δ 12.82–12.79 (m, 1.65H), 11.24–11.19 (m, 1.65H), 8.10–8.09 (m, 3.3H), 7.60–7.59 (m, 5.6H), 7.35 (s, 1H), 7.04 (m, 1.65H), 6.84–6.77 (m, 1.65H), 6.46–6.35 (m, 1.65H), 5.12 (s, 2H), 4.73 (s, 1.3H), 2.22 (m, 3.3H), 1.45–1.32 (m, 6.6H), 0.89 (m, 4.95H). 13C NMR (DMSO-d6): δ 182.03, 181.98, 167.42, 164.36, 163.71, 163.57, 163.46, 162.76, 161.09, 161.00, 157.16, 152.66, 148.67, 132.07, 130.53, 129.06, 126.42, 105.40, 105.29, 105.01, 98.67, 98.57, 93.57, 93.36, 66.53, 65.15, 31.49, 31.37, 27.97, 27.86, 21.68, 13.67. HRMS (ESI, m/z): calculated for C22H22N2O5 [M+H]+ 395.1601; found 395.1599.
(E, Z)-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)-N′-(2-methylpropylidene)acetohydrazide (3l)
The product was obtained as yellow powder. Yield (99%). 1H NMR (DMSO-d6) (E:Z=1:0.6): δ 12.83–12.80 (m, 1.6H), 11.25–11.16 (m, 1.6H), 8.11 (m, 3.2H), 7.6–7.59 (m, 5.4H), 7.30 (s, 1H), 7.05 (s, 1.6H), 6.85–6.78 (m, 1.6H), 6.46–6.36 (m, 1.6H), 5.13 (s, 2H), 4.74 (s, 1.2H), 1.07 (m, 9.6H). 13C NMR (DMSO-d6): δ 182.07, 182.01, 167.67, 164.40, 163.74, 163.59, 163.50, 162.89, 161.11, 161.02, 157.19, 157.02, 152.96, 132.13, 130.56, 129.11, 126.46, 105.42, 105.31, 105.03, 98.70, 98.59, 93.59, 93.39, 66.53, 65.14, 30.95, 30.74, 19.49, 19.41. HRMS (ESI, m/z): calculated for C21H20N2O5 [M+H]+ 381.1444; found 381.1442.
(E, Z)-N′-ethylidene-2-((5-hydroxy-4-oxo-2-phenyl-4H-chromen-7-yl)oxy)acetohydrazide (3m)
The product was obtained as yellow powder. Yield (55%). 1H NMR (DMSO-d6) (E:Z=1:0.5): δ 12.82–12.78 (m, 1.5H), 11.26–11.22 (m, 1.5H), 8.12–8.10 (m, 3H), 7.62–7.58 (m, 5H), 7.37–7.36 (m, 1H), 7.04 (m, 1.5H), 6.82–6.77 (m, 1.5H), 6.44–6.35 (m, 1.5H), 5.13 (s, 2H), 4.73 (s, 1H), 1.90 (m, 4.5H). 13C NMR (DMSO-d6): δ 182.08, 182.04, 167.35, 164.39, 163.75, 163.61, 163.50, 162.76, 161.13, 161.08, 161.01, 157.21, 148.90, 145.04, 132.16, 132.14, 130.56, 129.12, 126.47, 105.43, 105.30, 105.04, 98.80, 98.68, 98.63, 93.59, 93.39, 66.56, 66.51, 65.18, 18.21. HRMS (ESI, m/z): calculated for C19H16N2O5 [M+H]+ 353.1131; found 353.1131.
Biologic assays
Cell viability assay
The human breast cancer cell lines, MDA-MB-231 and MCF7, were purchased from the American Type Culture Collection. Cell viability was assessed using MTT colorimetric assay. Four thousand cells/well were plated in a 96-well plate in triplicate for each condition. After 24 hours of incubation, treatment compounds were added to each well as indicated, and the cells were further allowed to grow for 48 hours. Cell viability was assessed by adding 10 mL of filter-sterilized MTT solution at 0.5 mg/mL to each well. After 4 hours of incubation at 37°C, the medium was removed, and 100 µL of DMSO was added to each well to dissolve the blue formazan crystals. Absorbance was measured at a wavelength of 490 nm by using a Biotek ELX800 microplate reader (Biotek, Winooski, VT, USA). Cell viability was determined by dividing the absorbance values of the treated cells to that of the controlled cells and expressed as a percentage.
Detection of caspase-3/7 activation
Apoptosis was assessed using the Apo-one homogeneous caspase-3/7 assay (Promega). A total of 4,000 cells/well were plated in 100 µL culture medium in 96 black well plates in triplicate for each condition. After 24-hour incubation, treatment compounds were added to each well and the cells were further incubated for 48 hours. Then, caspase-3/7 substrate rhodamine 110 (Z-DEVD-R110) and homogeneous caspase-3/7 buffer were mixed in a 1:1,000 ratio to prepare homogeneous caspase-3/7 reagent. The reagent was added directly to each well to maintain a 1:1 ratio with sample volume and the cells were incubated for 3 hours at room temperature. Homogeneous caspase-3/7 buffer lyses cultured cell and supports optimal caspase-3/7 enzyme activity, while the caspase-3/7 substrate rhodamine 110 (Z-DEVD-R110), which exists as a profluorescent substrate upon interaction with the caspase-3/7 enzymes and excitation at 499 nm, cleaves the nonfluorescent substrate (Z-DEVD-R110) and creates the fluorescent rhodamine 110. Enzyme activity was measured after 3 hours at an excitation wavelength of 499 nm and an emission of 521 nm on synergy 2 multimode microplate reader (Biotek).
Results and discussion
Chemistry
The synthetic routes adopted for the synthesis of chrysin derivatives (3a-m) are outlined in Scheme 1. The commercially available chrysin (5,7-dihydroxy-2-phenyl-4H-chromen-4-one) 1 was treated with K2CO3 in anhydrous DMF to facilitate the formation of the chelation-controlled alkylation product of the 7-hydroxy group. Treatment of the resulting solution with methyl 2-bromoacetate at 0°C led to the formation of ester 4 in good yield.25 Attempts to convert ester 4 into hydrazide 5 were successful using the two procedures described in the Materials and methods section (methods A and B). Briefly, in method A, ester 4 was treated with 80% hydrazine hydrate in absolute ethanol at 0°C and allowed to stir at room temperature for few days. Although the desired compound was obtained in good yield and high purity, the reaction was time-consuming.
Scheme 1.

Reagents and conditions: (A) methyl 2-bromoacetate, K2CO3, DMF, 0°C; (B) hydrazine hydrate 80%, EtOH, 0°C; (C) different aldehydes, CH3OH, HCl (cat.).
To overcome this problem, method B was used in which the ester 4 was treated with 80% hydrazine hydrate and few drops of hydrochloric acid in methanol at 0°C. After 1 hour, the reaction was completed and the resulting solid was filtered and washed with cold methanol. 1H and 13C NMR confirmed the formation of hydrazide 5 as a pure single product. For example, 1H NMR spectrum of hydrazide 5 displayed a singlet at δ 12.8 ppm indicating the presence of the chelated phenolic proton at C5 of the 4H-chromen-4-one ring (ie, OH on ring A) integrating for one proton. Proton atoms present on the phenyl ring (ring B) resonated as multiplet signals in the range δ 7.5–8.0 ppm. The three proton atoms that belong to the 4H-chromen-4-one ring (rings A and C) are resonated as doublet, doublet, and singlet signals at δ 6.4, δ 6.8, and 7.0 ppm, respectively. The –CH2–O– protons appeared as a singlet at δ 4.6 ppm, whereas hydrazide group (CONHNH2) appears as two singlets: one at δ 9.4 ppm integrating for one proton and one at δ 4.3 ppm integrating for two protons. The 13C NMR spectrum and HRMS data further confirmed the formation of the desired compound. This result reflects the stability of the α,β-unsaturated ketone functionality of 4H-chromen-4-one scaffold in the presence of the nucleophilic hydrazine hydrate reagent.
With hydrazide 5 in hand, several chrysin derivatives (3a-m) were obtained utilizing two facile procedures (methods C and D, see Materials and methods section). In both methods hydrazide 5 was treated with the appropriate aldehyde in a suitable solvent. The main difference between the two methods is the use of an acid as a catalyst (method D) that produced the desired products in short time (1–3 hours). It is important to mention that, when using method D, washing the precipitated products with water should be avoided because it leads to a reversible reaction producing hydrazide 5 as a byproduct.
All chrysin analogs (3a-m) gave adequate analytical and spectroscopic data, which were in full accordance with their depicted structures. 1H and 13C NMR spectra showed that compounds 3a-m were obtained as geometrical isomers (E/Z isomers). The E:Z ratio for each compound was determined from 1H NMR spectra utilizing the integration of the neat methylene group peaks that appeared as two separated singlets for the two isomers between δ 4.6 and 5.4 ppm. The 1H NMR spectra for compound 3a, as an example, showed two characteristic overlapped singlets for the two isomers at δ 12.8 ppm representing the chelated phenolic C5–OH of the 4H-chromen-4-one ring. The CONH protons of the two isomers resonate as two overlapped singlets at δ 11.6 ppm. The N=CH protons of the two isomers appeared as two singlets at δ 8.3 and 8.0 ppm, which can be utilized to confirm the formation of E/Z isomers and their corresponding ratios. Proton atoms present on the phenyl rings (rings B and D) of the two isomers resonated as multiplet signals in the range δ 7.4–8.1 ppm, whereas the three proton atoms that belong to the 4H-chromen-4-one ring (rings A and C) are resonated as three multiplet signals at δ 6.46, δ 6.88, and 7.07 ppm. The methylene protons (CH2O) for the two isomers appeared as two singlets at δ 5.3 and 4.8 ppm. The 13C NMR spectrum and HRMS data further confirmed the formation of the desired compound.
Biologic assays
Effect of compounds on the cytotoxicity in human cancer cells
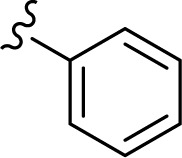
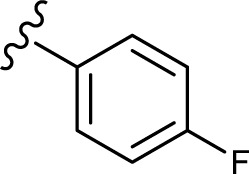
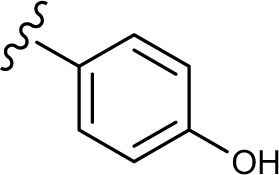
The in vitro antiproliferative effect of novel chrysin analogs 3a-m was assayed against two human breast cancer cell lines, MDA-MB-231 and MCF-7, by applying the MTT colorimetric assay. As summarized in Table 1, the results were expressed in terms of IC50 values (the minimum concentration in µM that causes 50% of cell death with respect to the control culture), where the well-known anticancer agent doxorubicin was used as a positive control.26 The results revealed that most of the synthesized compounds exhibit significant cytotoxic effect. These compounds were designed to test the importance of the phenyl ring (ring D) along with the corresponding ring substitution at the para position. The structure–activity relationship study revealed that the phenyl ring as well as the corresponding ring substitutions play important role in imparting the cytotoxic activities to the compounds. The parent compound 3a possessing an unsubstituted phenyl ring (ring D) was found to exhibit good and selective cytotoxic activity against MDA-MB-231 cell line with IC50 10.2 µM. Introducing a fluoro group at the para position (compound 3b) enhanced the cytotoxic activity toward both cell lines, significantly toward MCF-7. Replacement of the fluoro group with hydroxyl moiety produced compound 3c with significant reduced activity against both cell lines. Methylation of the hydroxyl group in 3c led to the compound 3d with considerable improved activity against both cell lines, when compared to compound 3c. Increasing the size of 4-methoxy group by replacing it with 4-benzyloxy group led to compound 3e with enhanced cytotoxic activity. Compound 3e was found to show the most potent activity against MDA-MB-231 with IC50 3.3 µM and MCF-7 with IC50 4.2 µM. The above results indicate that lipophilic groups at C-4 of ring D play a role in enhancing cytotoxic activities of the synthesized compounds. Accordingly, several derivatives (3f-3j) possessing different degrees of polarity were synthesized. Formyl (3f) and carboxy (3g) moieties were found to be detrimental for the cytotoxic activity. Nitro substituent (3h) demonstrated good cytotoxic activity against both cell lines with IC50 <9 µM. Compound 3i with morpholinyl substituent demonstrated a moderate cytotoxic activity against MDA-MB-231 and no activity against MCF-7 cell lines. Replacing the morpholinyl substituent with dimethylamino moiety led to compound 3j with drastically enhanced cytotoxicity against MDA-MB-231 and MCF-7 with IC50 2.6 and 8.4 µM, respectively. Next, we investigated the importance of the phenyl ring (ring D) on cytotoxic activity of the synthesized compounds. For that, compounds 3k-m were synthesized in which the phenyl ring was replaced with aliphatic moieties including butyl (3k), isopropyl (3l), and methyl (3m) groups. It was observed that the above aliphatic modifications provided weaker activity against MDA-MB-231 and enhanced activity against MCF-7 cell lines, when compared to compound 3a.
Table 1.
Cytotoxic activities of compounds 3a-m and doxorubicin against two cancerous cell lines; MDA-MB-231 and MCF-7
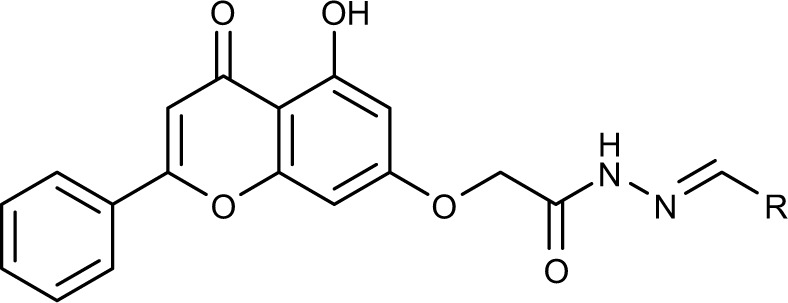
| |||
|---|---|---|---|
| No. | R | IC50 (µM)a
|
|
| MDA-MB-231 | MCF-7 | ||
|
| |||
| 3a |
|
10.2±2.2 | <60 |
| 3b |
|
5.5±2.2 | 11.7±1.8 |
| 3c |
|
54.8±5.7 | <60 |
| 3d |
|
11.9±3.5 | 35.7±7.1 |
| 3e |
|
3.3±0.8 | 4.2±2.1 |
| 3f |
|
<60 | <60 |
| 3g |
|
<60 | <60 |
| 3h |
|
6.1±1.1 | 8.6±1.4 |
| 3i |
|
21.7±2.3 | <60 |
| 3j |
|
2.6±0.6 | 8.4±5.7 |
| 3k |
|
22.2±0.5 | 18.7±5.6 |
| 3l |
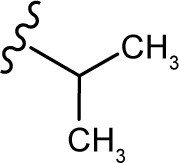
|
37.8±4.2 | 29.7±1.1 |
| 3m |
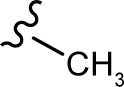
|
25.2±7.5 | 45±8.1 |
| Doxorubicin | NA | 2.3±0.3 | 2.9±0.1 |
Notes:
Result of MTT assay after 48-hour drug treatment. Compounds tested in triplicate, data expressed as mean values of independent experiments ± standard error of the mean.
Abbreviation: NA, not available.
Collectively, the results proved that the compounds with lipophilic moieties at C-4 position of ring D exhibit higher cytotoxic activity than the derivatives possessing highly or moderately polar moieties. Moreover, the data showed that aliphatic moieties are not good replacement for ring D when compared to derivatives bearing hydrophobic aromatic moieties. Compounds 3b, 3e, 3h, and 3j revealed to be the most active of the synthesized compounds with cytotoxic potential similar to that of the well-known anticancer agent doxorubicin.
Determination of the involvement of caspase-3/7 activation in MDA-MB-231 cells
As apoptosis is mediated by caspases, we investigated the ability of selected compounds (3a, 3b, 3d, 3e, and 3h) to activate the key effector caspases: caspase-3 and/or -7 in MDA-MB-231 cells. The cells were treated with different concentrations of the compounds (IC50 and 2 × IC50) for 48 hours, then analyzed for the activity of caspase-3/7 utilizing caspase-Glo assay. As shown in Figure 2, the five tested compounds caused activation of caspase-3/7 in which the best activation was provided by compound 3e that exhibited twofold increase of activity. Therefore, it can be deduced from the above results that the cytotoxic activities of the synthesized compounds are attributed to induction of apoptosis, which is mediated by caspase-3/7 activation.
Figure 2.

Activation of caspase-3/7 in MDA-MB-231 cells.
Note: The activity of caspase is expressed as percentage compared to untreated cells.
Conclusion
In summary, we have designed and synthesized a novel series of chrysin derivatives that have exhibited variable extent of cytotoxic activity against two human breast cancer cell lines, MDA-MB-231 and MCF-7. The cytotoxicity results of 3a-m established the importance of the phenyl ring (ring D) substituted with hydrophobic moieties at the para position. The data revealed that compounds substituted with benzyloxy (3e), dimethylamino (3j), nitro (3h), and fluoro (3b) are found to be potent cytotoxic agents that exhibited IC50 <6.5 µM against MDA-MB-231 and <12 µM against MCF-7. The cytotoxic potential of the above compounds was comparable to that of the well-known anticancer agent doxorubicin. Mechanistic studies utilizing caspase-Glo assay revealed that selected compounds, at their IC50 and 2 × IC50 values, were able to induce apoptosis in MDA-MB-231 cells as indicated by caspase-3 and/or -7 activation. The results provided valuable information to design further modifications for obtaining more potent and selective cytotoxic agents.
Supplementary Materials
Acknowledgments
The authors acknowledge the financial support given by the Deanship of Research at the Jordan University of Science and Technology (grant 88/2015). We also thank Dr Nizar A. Al-Shar’i for his helpful comments on this manuscript. The high-resolution mass spectrometry data were acquired in the Triad Mass Spectrometry Laboratory at the University of North Carolina at Greensboro.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Haskell CM. In: Principles of Cancer Chemotherapy Cancer Treatment. 5th ed. Haskell CM, editor. Philadelphia: WB Saunders; 2001. pp. 62–86. [Google Scholar]
- 2.Lawen A, Alfons L. Apoptosis-an introduction. Bioessays. 2003;25(9):888–896. doi: 10.1002/bies.10329. [DOI] [PubMed] [Google Scholar]
- 3.Parchment RE. The implications of a unified theory of programmed cell death, polyamines, oxyradicals and histogenesis in the embryo. Int J Dev Biol. 1993;37(1):75–83. [PubMed] [Google Scholar]
- 4.Reed JC, Tomaselli KJ. Drug discovery opportunities from apoptosis research. Curr Opin Biotechnol. 2000;11(6):586–592. doi: 10.1016/s0958-1669(00)00148-8. [DOI] [PubMed] [Google Scholar]
- 5.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21(3):485–495. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 6.Vial JP, Belloc F, Dumain P, et al. Study of the apoptosis induced in vitro by antitumoral drugs on leukaemic cells. Leuk Res. 1997;21(2):163–172. doi: 10.1016/s0145-2126(96)00102-6. [DOI] [PubMed] [Google Scholar]
- 7.Thornberry NA. Caspases: key mediators of apoptosis. Chem Biol. 1998;5(5):R97–R103. doi: 10.1016/s1074-5521(98)90615-9. [DOI] [PubMed] [Google Scholar]
- 8.Fonseca SF, Lima DB, Alves D, et al. Synthesis, characterization and antioxidant activity of organoselenium and organotellurium compound derivatives of chrysin. New J Chem. 2015;39(4):3043–3050. [Google Scholar]
- 9.Shin HD, Lee HJ, Sikder MA, et al. Effect of chrysin on gene expression and production of MUC5AC mucin from cultured airway epithelial cells. Tuberc Respir Dis (Seoul) 2012;73(4):204–209. doi: 10.4046/trd.2012.73.4.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Qiu J, Dong J, et al. Chrysin protects mice from Staphylococcus aureus pneumonia. J Appl Microbiol. 2011;111(6):1551–1558. doi: 10.1111/j.1365-2672.2011.05170.x. [DOI] [PubMed] [Google Scholar]
- 11.Shin EK, Kwon HS, Kim YH, Shin HK, Kim JK. Chrysin, a natural flavone, improves murine inflammatory bowel diseases. Biochem Biophys Res Commun. 2009;381(4):502–507. doi: 10.1016/j.bbrc.2009.02.071. [DOI] [PubMed] [Google Scholar]
- 12.Torres-Piedra M, Ortiz-Andrade R, Villalobos-Molina R, et al. A comparative study of flavonoid analogues on streptozotocin-nicotinamide induced diabetic rats: quercetin as a potential antidiabetic agent acting via 11beta-hydroxysteroid dehydrogenase type 1 inhibition. Eur J Med Chem. 2010;45(6):2606–2612. doi: 10.1016/j.ejmech.2010.02.049. [DOI] [PubMed] [Google Scholar]
- 13.Brown E, Hurd NS, McCall S, Ceremuga TE. Evaluation of the anxiolytic effects of chrysin, a Passiflora incarnata extract, in the laboratory rat. AANA J. 2007;75(5):333–337. [PubMed] [Google Scholar]
- 14.Khoo BY, Chua SL, Balaram P. Apoptotic effects of chrysin in human cancer cell lines. Int J Mol Sci. 2010;11(5):2188–2199. doi: 10.3390/ijms11052188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo KJ, Jeong YJ, Park JW, Kwon TK. Chrysin-induced apoptosis is mediated through caspase activation and Akt inactivation in U937 leukemia cells. Biochem Biophys Res Commun. 2004;325(4):1215–1222. doi: 10.1016/j.bbrc.2004.09.225. [DOI] [PubMed] [Google Scholar]
- 16.Zhu ZY, Wang WX, Wang ZQ, et al. Synthesis and antitumor activity evaluation of chrysin derivatives. Eur J Med Chem. 2014;75:297–300. doi: 10.1016/j.ejmech.2013.12.044. [DOI] [PubMed] [Google Scholar]
- 17.Liu Y, Song X, He J, Zheng X, Wu H. Synthetic derivatives of chrysin and their biological activities. Med Chem Res. 2014;23(2):555–563. [Google Scholar]
- 18.Patel RV, Mistry B, Syed R, et al. Chrysin-piperazine conjugates as antioxidant and anticancer agents. Eur J Pharm Sci. 2016;88:166–177. doi: 10.1016/j.ejps.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Xuan HZ, Zhang JH, Wang YH, Fu CL, Zhang W. Anti-tumor activity evaluation of novel chrysin-organotin compound in MCF-7 cells. Bioorg Med Chem Lett. 2016;26(2):570–574. doi: 10.1016/j.bmcl.2015.11.072. [DOI] [PubMed] [Google Scholar]
- 20.Valdez-Calderón A, González-Montiel S, Martínez-Otero D, et al. Synthesis, structural study and biological activity of new derivatives of chrysin containing a 2-mercaptopyridyl or 5-(trifluoromethyl)-2-mercaptopyridyl fragments. J Mol Struct. 2016;1110:196–207. [Google Scholar]
- 21.Putt KS, Chen GW, Pearson JM, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2(10):543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- 22.Peterson QP, Hsu DC, Goode DR, Novotny CJ, Totten RK, Hergenrother PJ. Procaspase-3 activation as an anti-cancer strategy: structure-activity relationship of procaspase-activating compound 1 (PAC-1) and its cellular co-localization with caspase-3. J Med Chem. 2009;52(18):5721–5731. doi: 10.1021/jm900722z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svingen PA, Loegering D, Rodriquez J, et al. Components of the cell death machine and drug sensitivity of the National Cancer Institute Cell Line Panel. Clin Cancer Res. 2004;10(20):6807–6820. doi: 10.1158/1078-0432.CCR-0778-02. [DOI] [PubMed] [Google Scholar]
- 24.Putt KS, Chen GW, Pearson JM, et al. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2(10):543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- 25.Choe H, Kim J, Hong S. Structure-based design of flavone-based inhibitors of wild-type and T315I mutant of ABL. Bioorg Med Chem Lett. 2013;23(15):4324–4327. doi: 10.1016/j.bmcl.2013.05.095. [DOI] [PubMed] [Google Scholar]
- 26.Pilco-Ferreto N, Calaf GM, Gmjijoo C. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int J Oncol. 2016;49(2):753–762. doi: 10.3892/ijo.2016.3558. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.