

SUMMARY
Mitochondrial oxidative phosphorylation (OXPHOS) and substrate utilization critically regulate the function of hypothalamic proopiomelanocortin (POMC)-expressing neurons. Here, we demonstrate that inactivation of apoptosis-inducing factor (AIF) in POMC neurons mildly impairs mitochondrial respiration and decreases firing of POMC neurons in lean mice. In contrast, under diet-induced obese conditions, POMC-Cre-specific inactivation of AIF prevents obesity-induced silencing of POMC neurons, translating into improved glucose metabolism, improved leptin, and insulin sensitivity, as well as increased energy expenditure in AIFΔPOMC mice. On a cellular level, AIF deficiency improves mitochondrial morphology, facilitates the utilization of fatty acids for mitochondrial respiration, and increases reactive oxygen species (ROS) formation in POMC neurons from obese mice, ultimately leading to restored POMC firing upon HFD feeding. Collectively, partial impairment of mitochondrial function shifts substrate utilization of POMC neurons from glucose to fatty acid metabolism and restores their firing properties, resulting in improved systemic glucose and energy metabolism in obesity.
In Brief
Timper et al. show that mild impairment of mitochondrial OXPHOS upon deletion of AIF or MPC1 in hypothalamic POMC-expressing neurons increases their fatty acid utilization and ROS formation, prevents obesity-induced silencing of these neurons, and improves systemic glucose metabolism in obesity.
Graphical Abstract
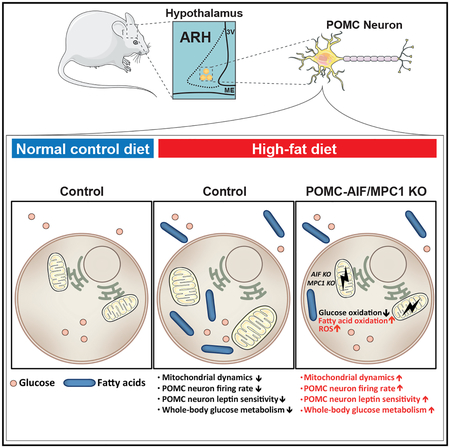
INTRODUCTION
The central regulation of glucose and energy homeostasis is controlled via the melanocortin system, comprising agouti-related peptide (AgRP)- and neuropeptide (NPY)-coexpressing neurons and their functionally antagonistic, anorexigenic proopiomelanocortin (POMC) neurons (Myers and Olson, 2012). These neurons are located in the arcuate nucleus of the hypothalamus (ARH), which exhibits facilitated access to peripheral hormonal and nutrient signals (Rodríguez et al., 2010). Thus, AgRP/NPY and POMC neurons can directly sense signals originating from the periphery of the body and integrate this information into a coordinated central feedback response to orchestrate food intake, glucose, and energy homeostasis.
Mitochondria are organelles that sense and respond to changes in the cellular energetic state in a dynamic fashion. Mitochondria adapt not only their respiratory chain activity, but also their morphology to better serve cellular energetic demands (Wai and Langer, 2016). Mitochondrial dynamics play a crucial role in the physiological adaptation of AgRP/NPY and POMC neuron activity to different energy states (Andrews et al., 2008; Diano et al., 2011; Schneeberger et al., 2013).
Upon high-fat diet (HFD) feeding, mitochondrial network complexity and dynamics are impaired in POMC neurons (Diano et al., 2011; Schneeberger et al., 2013), causing deterioration of mitochondrial Ca2+ handling and decreased excitability in these neurons (Paeger et al., 2017). The expression of mitofusin 2 (MFN2), an ubiquitously expressed mitochondrial transmembrane dynamin-like GTPase protein mediating mitochondrial fusion processes (Chen et al., 2003) and mitochondrial-ER interactions (de Brito and Scorrano, 2008), is reduced in the hypothalamus as early as 4 days after beginning of a HFD feeding, and deletion of MFN2 specifically in POMC neurons results in increased body weight gain due to enhanced food intake as well as reduced energy expenditure (EE) and BAT thermogenesis (Schneeberger et al., 2013). In addition, mitochondrial ultrastructure is disrupted in POMC neurons lacking MFN2, displaying mitochondrial enlargement and altered shape as well as smaller and more rounded cristae (Schneeberger et al., 2013). These changes are associated with impaired complex I activity and impaired oxidative phosphorylation (OXPHOS) but increased reactive oxygen species (ROS) production. Of note, ROS formation in POMC neurons plays an important role in proper POMC neuron function since ROS directly activates POMC neurons and enhances leptin sensitivity in these neurons (Diano et al., 2011).
Apoptosis-inducing factor (AIF), first described as a caspase-independent inducer of apoptosis (Susin et al., 1999), is involved in the assembly as well as the function of complex I of the mitochondrial respiratory chain (Vahsen et al., 2004). AIF deficiency leads to impaired mitochondrial OXPHOS, while cellular ROS levels remain unaltered (Pospisilik et al., 2007; Vahsen et al., 2004) or are even reduced (Urbano et al., 2005) likely due to increased respiratory chain coupling (Pospisilik et al., 2007). Interestingly, mild impairment of OXPHOS upon tissue-specific AIF deletion in liver or skeletal muscle as well as upon global reduction of AIF activity, as displayed in the harlequin mouse (Klein et al., 2002), protects against the development of diet-induced obesity (DIO) and associated alterations in glucose metabolism (Pospisilik et al., 2007). Berberine, a natural plant product, inhibits mitochondrial complex I activity (Turner et al., 2008) and improves energy and glucose metabolism in insulin-resistant, obese rodents (Lee et al., 2006) and humans (Wei et al., 2012). Similarly, metformin, a first-line treatment in type 2 diabetes mellitus, exerts its antidiabetic effects, at least in part, via inhibition of respiratory chain complex I activity (El-Mir et al., 2000). Direct application of metformin to the brain in rodents decreases food intake and improves glucose metabolism (Portela et al., 2015). However, the consequences and potential therapeutic use of a partial impairment in mitochondrial OXPHOS at the level of hypothalamic POMC neurons in the control of systemic energy and glucose homeostasis have not been investigated so far.
Here, we demonstrate that inactivation of AIF in POMC neurons results in a slightly impaired mitochondrial respiration chain complex I activity. This mild impairment in mitochondrial OXPHOS decreases POMC neuron activity in lean mice but prevents HFD-induced inhibition of POMC neurons in obese animals. AIF-deficient POMC neurons exhibit restored mitochondrial morphology and an increased ability to utilize fatty acids (FAs) for mitochondrial respiration and ROS formation, enabling their increased firing activity upon HFD feeding. Overall, AIF deficiency in POMC neurons results in improved leptin and insulin sensitivity as well as increased thermogenesis and improved glucose metabolism in obesity. Also POMC-Cre-dependent deletion of the mitochondrial pyruvate carrier (MPC)-1, which transports the critical complex I substrate pyruvate into the mitochondrial matrix, protects from HFD-induced glucose intolerance. Collectively, partial impairment of mitochondrial OXPHOS shifts substrate utilization in POMC neurons, restores their firing properties in obesity, and improves obesity-associated deteriorations in systemic glucose and energy metabolism, pointing to the therapeutic potential for mild complex I inhibition in hypothalamic neurons for the treatment of obesity and obesity-associated insulin resistance.
RESULTS
POMC-Cre-Dependent AIF Deficiency Protects from Obesity-Associated Deteriorations in Glucose Metabolism
We aimed at investigating the effect of a mild impairment of mitochondrial complex I activity in POMC neurons on the regulation of whole-body energy and glucose homeostasis. To generate POMC-specific AIF-deficient mice, we crossed female mice carrying a loxP-flanked AIFm1 allele (AIFflox/flox) (Joza et al., 2005) with male mice expressing the Cre-recombinase specifically under control of the POMC promoter (Balthasar et al., 2004). Since AIF is encoded on the X chromosome, the resulting male offspring represented either control mice (AIFflox/YPOMC-Cre−/−) or those with POMC-Cre-dependent AIF deficiency (AIFflox/YPOMC-Cre+/−, i.e., AIFΔPOMC mice). POMC neuron deletion of AIFm1 mRNA expression was investigated in control and AIFΔPOMC mice via fluorescent in situ hybridization (RNAscope) using a specific, custom-designed probe targeting exons 7–8 of the AIFm1 transcript. Although AIFm1 tissue expression is ubiquitous (Daugas et al., 2000), likely due to the small size of the probe designed to specifically detect the AIFm1 mRNA encoded by loxP-flanked region of the AIFm1 gene, AIFm1 expression was detected in only 60% of POMC-expressing neurons in control mice (Figures 1A and 1B). Nevertheless, AIFm1 mRNA expression was largely reduced in POMC neurons of AIFΔPOMC mice (Figures 1A and 1B). Tissue-specific recombination of the AIFm1 floxed-allele in AIFΔPOMC mice was further confirmed by PCR on genomic DNA isolated from ARH and the pituitary (Figure S1A), while no Cre-mediated recombination of the AIFflox-allele was detected in other brain areas or in peripheral tissues such as liver and skeletal muscle of AIFΔPOMC or control mice (Figures S1A and S1B). In line, AIF protein levels were not altered in liver and skeletal muscle in AIFΔPOMC compared to control mice (Figure S1C).
Figure 1. POMC-Cre-Dependent AIF Deletion Attenuates Obesity-Associated Deteriorations in Glucose Metabolism.

(A and B) Representative confocal images (A) and quantification (B) of in situ hybridization of mRNA of AIFm1 (red) and POMC (green) and corresponding nuclear counterstaining (DAPI, blue) in the arcuate nucleus of the hypothalamus (ARH) of AIFΔPOMC and AIFfl control mice (n = 3 per genotype group). Scale bars, 50 μm.
Data are presented as mean ± SEM of the percentage of AIFm1-positive POMC neurons.
(C and D) Glucose tolerance test (GTT) (C) and area under curve (AUC) (D) of glucose levels in lean normal chow diet (NCD)-fed (n = 18–22 per group) or high-fat diet (HFD)-fed obese (n = 24–26 per group) AIFΔPOMC and AIFfl control mice.
(E and F) Fasting plasma glucose levels (E) and fasting plasma insulin levels (F) in NCD- (n = 18–22 per group) or HFD-fed (n = 24–26 per group) AIFΔPOMC and AIFfl control mice.
(G–K) Glucose infusion rate (GIR) (G) and organ-specific glucose uptake rates in skeletal muscle (SKM) (H), perigonadal white adipose tissue (WAT) (I), brown adipose tissue (BAT) (J), and brain (K) during hyperinsulinemic-euglycemic clamp studies in HFD-fed AIFΔPOMC and AIFfl control mice (n = 9–13 per group).
Data are represented as mean ± SEM. *p ≤ 0.05, **p < 0.01, ***p < 0.001 as determined by two-tailed, unpaired Student’s t test (B and H–K) or two-way ANOVA followed by Bonferroni post hoc test (C–G).
See also Figures S1 and S2.
Of note, anatomical assessment of the ARH via POMC-Cre-mediated expression of a nuclear-localized β-galactosidase (NLS-LacZ) revealed no alteration in POMC neuron numbers upon AIF deletion in the ARH (Figures S1D and S1E). We further crossed male AIFwt/YPOMC-Cre+/−mice or AIFflox/YPOMC-Cre+/− mice with female mice allowing for Cre-dependent expression of a red fluorescent protein variant (tdTomatoflox/flox mice), resulting in mice with permanent tdTomato expression in POMC-Cre neurons throughout development. POMC and tdTomato mRNA expression in the adult ARH was investigated via fluorescent in situ hybridization (RNAscope) (Figure S1F). Our analysis revealed no difference in the number of POMC-(Figure S1G) or tdTomato-expressing neurons (Figure S1H) in adult mice with POMC-Cre-specific AIF deletion compared to control mice. While ~60% of tdTomato-expressing neurons also expressed POMC (Figure S1I), ~99% of POMC-expressing neurons also expressed tdTomato (Figure S1J). These findings are in line with previous reports by Padilla et al. revealing that POMCCre mice efficiently mark POMC neurons, but also a subpopulation of AgRP neurons, which during development temporarily express POMC (Padilla et al., 2010). Thus, since POMC-Cre-mediated recombination inherent to the use of this widely applied POMC-Cre mouse model (Ramirez et al., 2017; Santoro et al., 2017; Schneeberger et al., 2013) also occurs in a subpopulation of hypothalamic neurons, which only transiently express POMC during development, we cannot exclude an additional effect of removing AIFm1 from these cells.
To investigate the role of AIF in POMC neurons in energy and glucose homeostasis upon different dietary states, AIFΔPOMC and control mice were exposed either to a normal control diet (NCD) or to a HFD for 12–16 weeks. Whereas mice on HFD showed increased body weight (Figures S2A and S2B) and elevated leptin levels compared to mice fed a NCD (Figure S2C), POMC-Cre-dependent AIF deletion did not affect body weight (Figures S2A and S2B) or leptin levels (Figure S2C) within each diet group. Moreover, POMC-Cre-dependent AIF deficiency did not affect glucose tolerance under NCD-fed conditions (Figures 1C and 1D). However, while HFD feeding impaired glucose tolerance in control mice (Figures 1C and 1D), POMC-Cre-dependent AIF deletion partially protected the animals from HFD-induced glucose intolerance (Figures 1C and 1D). While fasting plasma levels of glucose were increased upon HFD feeding both in AIFΔPOMC and control mice (Figure 1E), fasting insulin levels were significantly elevated upon HFD feeding in control mice, but this response was attenuated in AIFΔPOMC mice (Figure 1F). On the other hand, assessment of insulin sensitivity via insulin tolerance test (ITT) revealed no differences in insulin sensitivity between mice of the different genotypes neither upon NCD nor upon HFD feeding (Figures S2E and S2F). POMC-Cre-dependent AIF deletion did not affect corticosterone plasma levels in mice on NCD or HFD (Figure S2D).
To gain deeper insights into the regulation of systemic glucose homeostasis and insulin sensitivity in obese AIFΔPOMC and litter-mate control mice, we subjected these animals to hyperinsuline-mic-euglycemic clamp studies (Figure S2G) (Kim, 2009). These experiments revealed improved insulin sensitivity in HFD-fed AIFΔPOMC compared to control mice, as reflected by an increased glucose infusion rate (GIR) during the steady state of the experiment (Figure 1G). Moreover, we assessed tissue-specific glucose uptake rates, revealing an increased glucose uptake in skeletal muscle (SKM) (Figure 1H) and white adipose tissue (WAT) (Figure 1I), but not in brown adipose tissue (BAT) (Figure 1J) or brain (Figure 1K) in HFD-fed AIFΔPOMC compared to control mice. Furthermore, insulin-dependent suppression of hepatic glucose production was improved in AIFΔPOMC compared to littermate control mice, although both groups presented with negative hepatic glucose production (HGP) rates during the clamp, thus limiting the interpretation of these results (Figure S2H). Nevertheless, our experiments indicate that POMC-Cre-dependent AIF deletion attenuates HFD-induced alterations in systemic insulin sensitivity through increased insulin action in SKM, adipose tissue, and potentially in liver.
POMC-Cre-Dependent AIF Deficiency Enhances EE and Leptin Sensitivity upon DIO
We further assessed whether POMC-Cre-dependent AIF deletion also improved energy metabolism in NCD- and HFD-fed mice. Neither body weight (Figure S2I) nor body composition (Figure S2J), food (Figure S2K), or fluid intake (Figure S2L) were altered in AIFΔPOMC compared to littermate control mice on NCD. Indirect calorimetry revealed a decrease in VO2 (Figure S2M), VCO2 (Figure S2N), and EE (Figure S2O), while locomotor activity was not changed (Figure S2P). Similarly, in AIFΔPOMC mice on HFD neither body weight (Figure S2Q) nor body composition (Figure S2R), food (Figure S2S), or fluid intake (Figure S2T) were altered. In contrast, VO2 (Figure S2U), VCO2 (Figure S2V), and EE (Figure S2W) were increased in HFD-fed AIFΔPOMC compared to littermate control mice, while locomotor activity was not enhanced (Figure S2X). Although, these minor alterations in EE did not translate into overall changes in body weight and obesity, these experiments indicated a differential and opposing role for AIF in POMC neurons in the regulation of EE depending on diet.
Leptin action in POMC neurons enhances EE and improves glucose homeostasis (Berglund et al., 2012). However, under obese conditions, POMC neurons become resistant to the regulatory actions of leptin (Enriori et al., 2007). Since POMC-Cre-dependent AIF-deficient mice on HFD display improved glucose and energy metabolism, we further assessed leptin’s ability to regulate POMC neurons upon AIF deletion. To this end, we performed leptin-sensitivity studies in lean, NCD- and obese, HFD-fed AIFΔPOMC and control mice (Figures 2A–2C and S3A–S3C). Both NCD-fed AIFΔPOMC mice and control mice responded equally to leptin administration with a reduction in food intake (Figures S3A and S3B) and body weight (Figure S3C). Consistent with previous reports (Enriori et al., 2007), control mice on HFD were resistant to the food-intake- and body-weight-reducing actions of leptin. In contrast, leptin administration induced a significant decrease in food intake (Figures 2A and 2B) and body weight (Figure 2C) in HFD-fed AIFΔPOMC mice. Moreover, while peripheral leptin administration failed to significantly induce phosphorylation of STAT3 in POMC neurons of HFD-fed control mice, leptin was capable of evoking STAT3 phosporylation in POMC neurons of HFD-fed AIFΔPOMC mice (Figures 3D and 3E).
Figure 2. AIF Deficiency in POMC Neurons Enhances Leptin Sensitivity in Obesity.

(A–C) Absolute (A) and percentage of (B) 3-day food intake and body-weight change (C) of HFD-fed obese AIFΔPOMC and AIFfl control mice after 3 days of twice daily intraperitoneal injection of leptin (2 mg/kg) versus vehicle (0.1 M PBS) (n = 7–9 per group).
(D and E) Representative confocal images (D) and quantification (E) of pSTAT3-immunoreactive POMC neurons (pSTAT3, green; POMC, red) and corresponding nuclear counterstaining (DAPI, blue) in the ARH of 16-hr-fasted HFD-fed AIFΔPOMC and AIFfl control mice 45 min after i.p. injection of leptin (2 mg/kg) versus vehicle (0.1 M PBS) (n = 3–6 per group).
Scale bars, 100 μm; 3V, third ventricle. Data are represented as mean ± SEM. *p ≤ 0.05 and **p < 0.01 as determined by two-way ANOVA followed by Bonferroni post hoc test. See also Figure S3.
Figure 3. POMC-Cre-Dependent AIF Deletion Prevents POMC Neuron Inhibition in Obesity.

(A and B) Basal action potential (AP) frequency and representative original recordings in POMC neurons from NCD-fed (n = 18 versus 27) (A and A1) and HFD-fed (n = 27 versus 17) (B and B1) AIFΔPOMC and AIFfl control mice.
(C and D) AP frequency in POMC neurons upon bath application of tolbutamide from NCD-fed (n = 5 versus 8) (C and C1) and HFD-fed (n = 7 versus 9) (D and D1) AIFΔPOMC and AIFfl control mice.
Data are presented as mean ± SEM. Mean depicted as horizontal line (red horizontal line in A and B; black horizontal line in C and D), median depicted as black horizontal line (A and B) or as + (C and D). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 as determined by Mann-Whitney test (A) and two-tailed, unpaired Student’s t test (B) or by two-tailed, paired Student’s t test (C and D). See also Figure S4.
POMC-Cre-Dependent AIF Deletion Attenuates Obesity-Induced POMC Neuron Inhibition
Since AIFΔPOMC mice were partially protected from HFD-induced leptin resistance and alterations in energy and glucose metabolism, we assessed baseline POMC neuron activity via perforated patch-clamp recordings from synaptically isolated neurons. Interestingly, AIF deficiency resulted in reduced spontaneous firing of action potentials in POMC neurons from NCD-fed mice (Figures 3A and 3A1). In line with previous reports (Diano et al., 2011; Jo et al., 2009; Paeger et al., 2017), HFD feeding strongly impaired POMC neuron firing activity in control mice (Figures 3B and 3B1), which was attenuated in HFD-fed AIFΔPOMC mice (Figures 3B and 3B1).
POMC neurons are activated in response to increases in extra-cellular glucose levels via regulation of ATP-sensitive potassium (KATP) channels (Parton et al., 2007). Therefore, we hypothesized that decreased POMC neuron activity upon AIF deletion under NCD feeding might result from impaired mitochondrial OXPHOS with a concomitant reduction in cellular ATP levels (Vahsen et al., 2004). However, bath application of tolbutamide, a specific KATP -channel blocker, resulted in a significant depolarization and a concomitant increase in action potential frequency in POMC neurons from control mice under NCD (Figures 3C and 3C1) and HFD (Figures 3D and 3D1) conditions, but not in AIF-deficient POMC neurons in either diet group (Figures 3, 3C1, 3D, and 3D1C). Thus, altered KATP-channel activation due to reduced cellular ATP content unlikely accounts for the reduced basal firing activity in AIF-deficient POMC neurons in NCD-fed mice.
Mitochondrial Morphology Is Improved in AIF-Deficient POMC Neurons from HFD-Fed Mice
Adaptations of the mitochondrial network dynamics play a pivotal role in the adaptive response of POMC neuron activity to different metabolic states (Diano et al., 2011; Santoro et al., 2017). To examine potential alterations in mitochondrial morphology in AIF-deficient POMC neurons, mitochondrial architecture and morphology were analyzed via electron microscopy in POMC neurons from AIFΔPOMC and control mice both upon NCD- and HFD-feeding conditions (Figure S4A). In line with previous reports (Diano et al., 2011; Schneeberger et al., 2013; Yi et al., 2017), mitochondrial architecture in control animals was altered upon HFD feeding with an increase in mitochondrial area (Figure S4B) and perimeter (Figure S4C), a decrease in maximal cristae width (Figure S4D), and a tendency to decreased mitochondrial-ER contacts (Figure S4F). However, these alterations were attenuated upon AIF deletion in POMC neurons in HFD-fed mice. Of note, mitochondrial area (Figure S4B), perimeter (Figure S4C), and aspect ratio (Figure S4E) were also significantly increased in AIF-deficient POMC compared to control neurons from NCD-fed mice.
AIF Deficiency in Obesity Shifts Substrate Preference of POMC Neurons from Glucose to Fatty Acid
Next, we investigated whether glucose sensitivity was altered in POMC neurons from the different diet groups upon AIF deletion. While the proportion of POMC neurons exhibiting an increase in firing frequency or depolarization of membrane potential upon rising extracellular glucose concentrations from 5 to 10 mM was slightly reduced in POMC neurons of AIFΔPOMC mice compared to controls under NCD conditions (20% versus 40%) (Figure 4A), the proportion of POMC neurons responding to increasing glucose concentrations did not differ between the genotypes, when the animals were exposed to HFD feeding (Figure 4B). Interestingly, increased extracellular glucose concentrations did not significantly increase the action potential frequency in the whole population of POMC neurons of either genotype under both dietary conditions (Figures 4A and 4B).
Figure 4. AIF Deficiency in Obesity Increases Fatty Acid Sensing of POMC Neurons.

(A and B) Action potential (AP) frequency during low (5 mM) and high (10 mM) glucose concentrations in POMC neurons from NCD-fed (n = 10 versus 10) (A) and HFD-fed (n = 13 versus 11) (B) AIFΔPOMC and AIFfl control mice.
(C and D) AP frequency in POMC neurons upon bath application of oleate (OA) from NCD-fed (n = 11 versus 11) (C) and HFD-fed (n = 11 versus 13) (D) AIFΔPOMC and AIFfl control mice. Data are presented as mean ± SEM. The mean is depicted as horizontal line, the median is depicted as +. **p ≤ 0.01 as determined by two-tailed, paired Student’s t test.
While POMC neurons are activated in the postprandial state and use glucose as their main substrate (Ibrahim et al., 2003), fatty acids represent the main fuel for AgRP neurons, which are activated in the fasting state (Andrews et al., 2008). We hypothesized that impaired mitochondrial energy generation due to reduced complex I activity in AIF-deficient POMC neurons might be compensated by a metabolic switch toward enhanced fatty acid sensing and increased mitochondrial fatty acid oxidation. Therefore, we investigated fatty acid sensing in POMC neurons applying the long-chain fatty acid (LCFA) oleate (OA) in perforated patch-clamp recordings. It has been reported that OA induces depolarization and excitation in a subpopulation (~50%) of POMC neurons via mitochondrial fatty acid oxidation (Jo et al., 2009). In line, 55% of POMC neurons of NCD control mice showed an increase in firing or depolarization of the membrane potential upon bath application of OA, while this was observed in only 37% of POMC neurons of NCD-fed AIFΔPOMC mice. Overall, OA failed to significantly enhance firing frequencies of the POMC neurons as a whole population of either genotype upon NCD feeding (Figure 4C). In contrast, in POMC neurons of HFD-fed mice, OA increased firing, or depolarized the membrane potential in a higher proportion of AIFΔPOMC mice neurons compared to controls (69% versus 55%). As a result, only in POMC neurons of HFD-fed AIFΔPOMC mice, OA was able to significantly increase the firing frequencies in the whole population of cells (Figure 4D).
AIF Deficiency Impairs Mitochondrial OXPHOS and Increases Fatty Acid Oxidation and ROS Generation in POMC-Expressing Hypothalamic Neuronal Cells
To further investigate the underlying mechanisms of the enhanced substrate preference for FAs in AIF-deficient POMC neurons from HFD-fed mice, we employed the POMC-expressing mouse hypothalamic cell line N43/5 (Choi et al., 2010). Using locked nucleic acids (LNAs), deletion of AIF was achieved with an efficiency of 80% in these cells (Figure 5A). While AIF deletion did not impact mitochondrial mass (Figure 5B), it resulted in reduced expression of complex I subunits as assessed by western blotting (Figure 5C), whereas expression of complex III and V subunits was not affected (Figure 5C). These findings are in line with previous reports on the effects of AIF deletion in a range of different cell types (Joza et al., 2005; Milasta et al., 2016; Pospisilik et al., 2007; Vahsen et al., 2004). Assessment of mitochondrial OXPHOS in hypothalamic N43/5 cells upon AIF deletion (Figure 5D) revealed reduced baseline respiration both in the absence and presence of glucose (Figure 5E), a reduced ATP-linked and maximum respiration capacity (Figure 5F) alongside with a decreased proton leak and unaltered coupling efficiency (Figure 5G). Log2-transfomed proportional values from pooled experiments confirmed the reduction in baseline (Figure 5H), ATP-linked and maximal respiration (Figure 5I), and a decreased proton leak while coupling efficiency was increased (Figure 5J) upon AIF deletion. However, while mitochondrial OXPHOS upon glucose utilization was reduced in AIF-deficient hypothalamic cells, oxidation of exogenous fatty acids was increased both at basal and maximal levels as evidenced by an enhanced ability of the CPT1 inhibitor etomoxir to inhibit oxygen consumption rate (OCR) in AIF-deficient cells compared to controls (Figures 5K and 5L). In line, mRNA expression of carnitine palmitoyltransferase (CPT)1b was increased while expression of peroxisome proliferator-activated receptor (PPAR)g, involved in lipogenesis (Zhang et al., 2006), was decreased in AIF-deficient hypothalamic cells (Figure 5M). As mitochondrial fatty acid oxidation is accompanied by generation of ROS (McFadden et al., 2014; Rosca et al., 2012), and, as increased ROS levels can promote POMC neuron activation (Diano et al., 2011; Schneeberger et al., 2013), we investigated whether increased mitochondrial fatty acid oxidation in hypothalamic cells upon AIF deletion might enhance the generation of respiration-associated ROS. Indeed, generation of mitochondrial ROS was enhanced in AIF-deficient but not in control cells upon fatty acid exposure (Figures 5N and S5).
Figure 5. AIF Deficiency Primes Increased Fatty Acid Oxidation and mitochondrial ROS Generation in N43/5 Cells.

(A) Knockdown efficiency of AIFm1 mRNA expression in hypothalamic cell line N43/5 (AIF knockout [KO]) compared to control-locked nucleic acid (LNA)-treated (Control) and untreated cells (n = 4 per group).
(B and C) Assessment of mitochondrial content in AIF-deficient (AIF KO) and control hypothalamic cells by determining the ratio of mitochondrial DNA to nuclear DNA (mtDNA/nDNA) using quantitative real-time PCR (n = 4 per group) (B) and western blots and quantification of expression of respiratory chain subunits (n = 3 per group) in AIF KO and control hypothalamic cells (C).
(D–G) Representative oxygen consumption rate (OCR) levels (D) and quantification of baseline OCR before (w/o gluc) and after glucose (with gluc) administration
(E), ATP-linked and maximum OCR (F), and proton leak and coupling efficiency (G) in AIF-deficient and control hypothalamic cells (n = 1 representative experiment).
(H–J) Log2-transfomed proportional values from pooled experiments assessing OCR in AIF-deficient and control cells of baseline OCR before (w/o gluc) and after glucose (with gluc) administration (H), ATP-linked and maximum respiration OCR (I), and proton leak and coupling efficiency (J) in AIF KO and control cells (n = 11 independent experiments).
(K and L) Representative OCR upon palmitate in the presence or absence of the CPT1 inhibitor etomoxir (n = 1 representative experiment) (K) and quantification of basal and maximum OCR levels upon PA oxidation (depicted are results for exogenous fatty acid oxidation = OCR PA – OCR control BSA; n = 13 independent experiments) (L) in AIF KO and control hypothalamic cells.
(M) mRNA expression of CPT1b and PPARg in AIF KO and control cells (n = 3 independent experiments).
(N) Mitochondria-specific superoxide (mitosox) production in AIF KO and control cells upon incubation with palmitate (PA) or BSA.
Data are presented as mean ± SEM. *p ≤ 0.05, **p < 0.01, ***p < 0.001 as determined by unpaired two-tailed Student’s t test. See also Figure S5.
Increased ROS Generation Contributes to Improved Glucose Metabolism in Obese AIFΔPOMC Mice
Next, we investigated whether ROS levels were also increased in AIF-deficient POMC neurons upon HFD feeding in vivo. To this end, 1-hr-refed HFD-fed obese control and AIFΔPOMC mice were administered intracerbroventricularly (i.c.v.) with the ROS indicator dihydroethidium (DHE) (Andrews et al., 2008; Diano et al., 2011). Comparison of fluorescence intensity in POMC neurons revealed that ROS levels were increased in POMC neurons from AIF-deficient compared to control mice upon HFD feeding (Figures 6A and 6B). Since ROS derived from mitochondrial OXPHOS can activate POMC neurons, we investigated whether improved glucose metabolism, as observed upon AIF deletion in POMC neurons in obesity, might be linked to enhanced ROS generation. To this end, obese AIFΔPOMC and control mice were centrally applied with either vehicle or the ROS scavenger honokiol (Diano et al., 2011; Ramirez et al., 2017; Schneeberger et al., 2013). While acute i.c.v. administration of honokiol did not affect glucose tolerance in obese control mice (Figures 6C and 6D), it impaired glucose tolerance in obese AIFΔPOMC mice (Figures 6E and 6F).
Figure 6. Increased ROS Generation Contributes to Improved Glucose Metabolism in Obese AIFΔPOMC Mice.

(A) Representative confocal images of the ROS-indicator dihydroethidium (DHE, red) in POMC neurons (POMC, green) and corresponding nuclear counter-staining (DAPI, blue) in the ARH of HFD-fed AIFΔPOMC and AIFfl control mice and (B) quantification DHE intensity (n = 6 per group). Scale bars: 10 μm. Data are presented as mean ± SEM of the percentage DHE intensity of AIFfl control mice.
(C–F) Glucose tolerance tests (GTTs) (C and E) and area under curve for glucose levels (D and F) HFD-fed AIFfl control mice (n = 14 per group) and HFD-fed AIFΔPOMC mice (n = 12 per group) upon i.c.v. administration of honokiol or vehicle.
Data are presented as mean ± SEM. *p ≤ 0.05, **p < 0.01, ***p < 0.001 as determined unpaired Student’s t test (B), by paired Student’s t test (D and F), or by two-way ANOVA followed by Bonferroni post hoc test (C and E).
POMC-Cre-Dependent Inactivation of MPC1 Protects from Diet-Induced Deteriorations in Glucose Metabolism
To further substantiate whether a metabolic switch toward enhanced fatty acid oxidation in POMC neurons protects from diet-induced impairments in glucose metabolism, we employed a complementary model of POMC-Cre-dependent inhibition of glucose-dependent mitochondrial OXPHOS. Ablation of MPC1 and MPC2, the genes encoding the multimeric MPC complex (Bricker et al., 2012; Herzig et al., 2012), results in a reduction in mitochondrial OXPHOS in the presence of glucose and a compensatory increase in fatty acid oxidation (Gray et al., 2015; Schell et al., 2017; Vacanti et al., 2014) through providing acetyl-CoA as an alternative substrate to fuel the tricarboxylic acid (TCA) cycle. Interestingly, liver-specific ablation of MPC1 protects from diet-induced glucose intolerance and is associated with increased liver and whole-body fat oxidation (Gray et al., 2015). We therefore generated mice, which lack MPC1 in POMC neurons and investigated their metabolic phenotype upon NCD and HFD feeding. POMC-Cre-dependent deletion of MPC1 expression was confirmed in MPC1ΔPOMC mice via fluorescent in situ hybridization (RNAscope) using a probe targeting exons 3–5 of the MPC1 transcript (Figure 7A). POMC-Cre-dependent MPC1 deletion did not affect body weight until 18 weeks of age (Figures 7B and 7C), or fasting plasma leptin (Figure 7D), insulin (Figure S6A), glucose (Figure S6B), or plasma corticosterone concentrations (Figure 7E) in the different diet groups. However, POMC-Cre-dependent MPC1 ablation partially protected from HFD-induced glucose intolerance (Figures 7F and 7G), while insulin sensitivity assessed via ITT was not significantly altered (Figures 7H and 7I). Moreover, glucose tolerance (Figures S6C and S6D) and insulin sensitivity (Figures S6E and S6F) were not different in mice with POMC-Cre-dependent MPC1 deletion compared to controls upon NCD feeding. Interestingly, evaluation of the body composition of POMC-Cre-dependent MPC1-deficient mice and littermate controls at 22 weeks of age revealed a decrease in body weight (Figure 7J) and a reduction in fat mass (Figure 7K) in HFD-fed, POMC-Cre-dependent MPC1-deficient mice compared to littermate controls. On the other hand, no differences in body weight (Figure S6G) or body composition (Figure S6H) were observed between the different genotypes upon NCD feeding.
Figure 7. POMC-Cre-Dependent MPC1 Ablation Attenuates Obesity-Associated Deterioration of Glucose Metabolism.

(A) Representative confocal images of in situ hybridization of mRNA of MPC1 (red) and POMC (green) and corresponding nuclear counterstaining (DAPI, blue) in the ARH of MPC1ΔPOMC and MPC1fl control mice (n = 3 per genotype group). Scale bars, 50 μm.
(B–D) Body weight development (B and C) until 18 weeks of age and fasting plasma leptin levels (D) of NCD-fed (n = 11–16 per group) and HFD-fed (n = 19–26) MPC1ΔPOMC and MPC1fl control mice.
(E) Random-fed plasma corticosterone levels in NCD-fed (n = 8 per group) and HFD-fed (n = 9–13) MPC1ΔPOMC and MPC1fl control mice.
(F–I) Glucose tolerance test (GTT) (F) and glucose area under curve (AUC) (G) as well as insulin tolerance test (ITT) (H) and (I) glucose AUC (I) in HFD-fed MPC1ΔPOMC and MPC1fl control mice (n = 19–26 per group).
(J and K) Body weight (J) and body composition (K) at 22 weeks of age in HFD-fed MPC1ΔPOMC and MPC1fl control mice (n = 9–13 per group).
See also Figure S6.
DISCUSSION
Mitochondrial dynamics and OXPHOS play a major role in the sensing of nutrient signals as well as in the adaptive feedback action of hypothalamic AgRP/NPY and POMC neurons in the regulation of energy and glucose homeostasis (for comprehensive reviews, see Shadel and Horvath, 2015; Wai and Langer, 2016). Here we demonstrate that deletion of AIF in POMC neurons results in a mild reduction of mitochondrial OXPHOS and respiratory chain complex I in line with previous reports investigating the effects of AIF deficiency in various different cell types (Joza et al., 2005; Milasta et al., 2016; Pospisilik et al., 2007; Vahsen et al., 2004). Furthermore, while OXPHOS is decreased upon AIF deletion, basal cellular ROS levels have been shown to remain unaltered (Milasta et al., 2016; Pospisilik et al., 2007; Vahsen et al., 2004) or even decrease (Urbano et al., 2005) most probably due to increased respiratory chain coupling (Pospisilik et al., 2007). This was also observed upon AIF deletion in our hypothalamic POMC cell line in vitro. Interestingly, POMC neurons from obese AIFΔPOMC mice and AIF-depleted POMC-expressing hypothalamic cells when challenged with LCFAs showed an increase in fatty acid utilization possibly via increased expression of CPT1b and accompanied by enhanced mitochondrial ROS generation.
ROS are known to activate POMC neurons and to promote leptin sensitivity in these neurons, leading to an overall improvement in glucose and energy homeostasis, although the mechanistic basis for ROS-dependent stimulation of neuronal activity has not yet been fully unraveled (Diano et al., 2011; Santoro et al., 2017). Consistent with this model, we found that increased fatty acid oxidation upon AIF deletion in hypothalamic POMC cells is coupled to increased ROS generation in vitro as well as in obesity in vivo, accounting for the improvement in glucose tolerance in obese AIFΔPOMC mice.
A switch toward enhanced mitochondrial fatty acid oxidation as a compensatory mechanism upon impaired mitochondrial glucose oxidation has also been described upon ablation of MPC1 (Schell et al., 2017; Vacanti et al., 2014). Interestingly, POMC-Cre-dependent ablation of MPC1 also improved glucose metabolism upon HFD feeding and even partially protected from diet-induced body weight and fat gain. The finding that impairment of glucose-dependent mitochondrial OXPHOS in POMC neurons in these complementary models protects from HFD-induced deteriorations in glucose tolerance further underscores the potential of alternative mitochondrial substrate usage in POMC neurons in control of glucose metabolism in obesity. However, inhibition of CPT1 activity, and thus associated mitochondrial fatty acid transport and fatty acid oxidation, in the hypothalamus has been shown to result in reduced food intake and improved glucose metabolism in obese mouse models (Obici et al., 2003). Although it has been hypothesized that an increase in LCFA-CoA levels at the level of the hypothalamus upon CPT1 inhibition might be responsible for the beneficial effect on food intake and glucose homeostasis regulation, the underlying molecular mechanisms how increased LCFA-CoA levels drive these improvements in energy and glucose metabolism remain elusive.
Mice lacking AIF either globally, as in the harlequin mouse (Klein et al., 2002), or upon tissue-specific deletion in liver or SKM (Pospisilik et al., 2007) are protected against HFD-induced obesity and associated impairment in glucose metabolism. Interestingly, it has also been shown that saturated LCFA protect from cell death and improve respiratory features in complex-I-deficient fibroblasts and that HFD feeding delays the progression of neurodegeneration in the harlequin mouse (Schiff et al., 2011). In line, embryonic lethality, brain morphological disruptions, and major metabolic alterations in MPC1-deficient mice can be almost completely prevented by feeding the pregnant dams a high-fat, ketogenic diet providing acetyl-CoA as an alternative substrate to the TCA cycle (Vanderperre et al., 2016). Similarly, patients suffering from mitochondrial OXPHOS defects with complex I deficiency (Theunissen et al., 2017) or from mitochondrial pyruvate dehydrogenase (PDH) deficiency (Wexler et al., 1997) benefit from high-fat, ketogenic diets, indicating that an impaired mitochondrial OXPHOS due to complex I or PDH deficiency leads to a beneficial compensatory switch in utilization of alternative substrates derived from increased fatty acid oxidation.
Of note, while impaired mitochondrial OXPHOS upon AIF deletion in POMC neurons led to improved glucose and energy homeostasis in obese mice, it was associated with a reduced basal POMC firing rate and decreased EE in the NCD setting without impacting glucose metabolism or body-weight control. However, reduced POMC neuron activity upon AIF deletion in the NCD setting was not attributable to KATP-channel opening but might have resulted from decreased ROS levels in the absence of fatty acids as indicated in tendency in the basal state in our in vitro experiments.
AIF is not only involved in mitochondrial respiration but also affects mitochondrial shape and cristae morphology. In line with a previous report (Cheung et al., 2006), we observed increased maximum cristae width equivalent to a dilated cristae ultrastructure in AIF-deficient POMC neurons. These findings are consistent with a previous study showing that cristae shape is directly linked to complex-I-dependent mitochondrial respiratory function and that cristae remodeling causes complex-I-dependent changes in mitochondrial bioenergetics (Cogliati et al., 2013). Another study investigating the role of AIF in immune cell function revealed not only disrupted cristae organization, but also grossly enlarged mitochondria in AIF-deficient T cells (Milasta et al., 2016). In line, AIF-deficient POMC neurons from NCD mice displayed enlarged, elongated mitochondria as evident from increased mitochondrial area, perimeter, and aspect ratio. Of note, in HFD-fed obese mice, mitochondrial size was increased in POMC neurons from control mice but normalized upon AIF deficiency, and enlarged mitochondria were also observed in POMC neurons upon MFN-2 deletion, which was associated with leptin resistance and disrupted energy and glucose homeostasis (Schneeberger et al., 2013).
Our findings of an overall beneficial effect resulting from mildly impaired mitochondrial OXPHOS upon either deletion of AIF or MPC1 in POMC neurons in obesity are further supported by a study investigating the role of tumor necrosis factor α (TNF-α) in POMC neurons in HFD-induced obese mice (Yi et al., 2017). This study revealed not only enhanced TNF-α expression in the mediobasal hypothalamus in DIO mice, but further demonstrated that TNF-α enhances mitochondrial OXPHOS and complex I expression in POMC neurons alongside with mitochondrial enlargement and increased expression of the mitochondrial fusion factor optic atrophy 1 (Opa1) (Yi et al., 2017). Accordingly, ablation of TNF-α signaling in the mediobasal hypothalamus (MBH) of DIO mice reversed diet-induced alterations in mitochondrial dynamics in POMC neurons and improved energy and glucose homeostasis.
POMC-expressing neurons also reside in the nucleus of the solitary tract (NTS) of the brainstem (Padilla et al., 2012; Zhan et al., 2013) and acute activation of NTS POMC neurons results in reduced food intake (Zhan et al., 2013). Therefore, we cannot exclude that AIF or MPC1 deletion in brainstem POMC neurons might have contributed, at least in part, to the phenotypes observed in the present study. However, while ablation of POMC neurons in the ARH is associated with disturbed whole-body energy homeostasis and impaired glucose tolerance, none of these phenotypes have been observed upon ablation of POMC neurons in the NTS (Zhan et al., 2013). Therefore, we conclude that the observed metabolic phenotypes upon POMC-Cre-dependent AIF or MPC1 deletion are primarily caused by alterations of ARH POMC neuron function.
In summary, our findings highlight the importance of mitochondrial respiration for proper POMC neuron activity and specifically unravel that partial impairment of mitochondrial OXPHOS shifts substrate utilization toward increased fatty acid oxidation alongside with enhanced ROS generation, which restores POMC firing in obesity and improves obesity-associated deteriorations in systemic energy and glucose metabolism. Therefore, targeting mitochondrial complex I activity in POMC neurons might represent a possible therapeutic approach to tackle obesity and associated insulin resistance in the future.
STAR⋆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jens C. Brüning (bruening@sf.mpg.de).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Care
All animal procedures were conducted in compliance with protocols approved by the local government authorities (Bezirksregierung Köln) and were in accordance with NIH guidelines. Mice were housed in groups of 3–5 at 22 C°–24 C° using a 12-h light/12-h dark cycle. Animals had ad libitum access to water and the prescribed diet at all times, and food was only withdrawn if required for an experiment. For all experiments only male mice were used, except for analysis of LacZ-positive POMC neurons, where males and female mice were used. All experiments were performed in adult mice at the age of 16–25 weeks. Unless otherwise stated, animals were fed a regular chow diet containing 57% calories from carbohydrates, 34% calories from protein, and 9% calories from fat (R/M-H; Ssniff Diet). DIO mice were fed a HFD containing 21% calories from carbohydrates, 19% calories from protein, and 60% calories from fat (D12492-(I) mod; Ssniff Diet) while corresponding chow control mice received a normal control diet (NCD) containing 67% calories from carbohydrates, 20% calories from protein, and 13% calories from fat (D12450-(B) mod; Sniff Diet). POMC-EGFP;AIFft/Y;POMC-Cre+/− (POMC-EGFP:AIFΔPOMC) and POMC-EGFP;AIFft/Y;POMC-Cre−/− (POMC-EGFP: AIFfl) litter-mate control animals were either fed a HFD containing 22.7% carbohydrates, 16.1% protein, and 61.3% fat (55.2% of calories from fat) 23% calories from carbohydrates, 16% calories from protein and 61% calories from fat (Altromin Diet #C1057) or a regular chow diet containing 58% carbohydrates, 24% protein, and 18% fat (Harlan Teklad Global Rodent#2918).
Generation of transgenic mice
AIFΔPomc mice
Pomc-Cre mice (Balthasar et al., 2004) were mated with AIF fl/fl females (Joza et al., 2005), and breeding colonies were maintained by mating AIF fl/fl females to POMC-Cre;AIF fl/Y males. Mice were kept on a 100% C57 BL/6 background throughout the experiment.
ROSA26-fl-LacZ; AIFflox/flox;POMC-Cre+/− mice
ROSA26-fl-LacZ mice (Plum et al., 2006) were mated with POMC Cre AIF fl/Y males. Further intercrossings resulted in ROSA26-fl-LacZ; AIFfl/Y;POMC-Cre+/− or ROSA26-fl-LacZ; AIFfl/fl;POMC-Cre+/− experimental and ROSA26-fl-LacZ; AIFwt/Y;POMCCre+/− or ROSA26-fl-LacZ; AIFwt/wt;POMC-Cre+/− littermate control animals.
POMC-EGFP, AIFflox/flox;POMC-Cre+/− mice
POMC-EGFP mice (Cowley et al., 2001) were mated with POMC Cre AIF fl/Y males. Further intercrossings resulted in POMC-EGFP; AIFfl/Y;POMC-Cre+/− (P-GFP:AIFΔPOMC) experimental and POMC-EGFP; AIFfl/Y;POMC-Cre−/− (P-GFP: AIFfl) littermate control animals.
MPC1ΔPomc mice:
Pomc-Cre mice (Balthasar et al., 2004) were mated with MPC1 fl/fl females (Gray et al., 2015) and breeding colonies were maintained by mating MPC1 fl/fl females to POMC-Cre;MPC1 fl/fl males. Mice were kept on a 100% C57 BL/6 background throughout the experiment.
In vitro culture of hypothalamic cell line N43/5
N43/5 cells were cultured in vitro in DMEM+GlutaMAX (#61965–026, GIBCO, ThermoFisher Scientific) supplemented with 1% penicillin/Streptomycin (#15140–122, GIBCO, ThermoFisher Scientific), 1% Sodium Pyruvate (#11360–039, GIBCO, ThermoFisher Scientific) and 10% heat-inactivated fetal-bovine serum (#P30–3302, PAN Biotech).
METHOD DETAILS
Glucose tolerance and insulin tolerance tests
Glucose tolerance tests
Glucose tolerance tests were performed in the morning after an overnight fasting period of 16 h. Glucose concentrations in blood were measured from whole venous blood using an automatic glucose monitor (Bayer Contour, Bayer, Germany). After the fasting period, each mouse received an intraperitoneal injection of 20% glucose (2 g per kg body weight; bela-pharm) and blood glucose concentrations were measured before and after 15, 30, 60 and 120 min.
Insulin tolerance tests
Insulin tolerance tests were performed in the morning in random fed mice. After determination of basal blood glucose concentrations, each mouse received an intraperitoneal injection of insulin (0.75 U per kg body weight; Insuman Rapid; Sanofi Aventis) and glucose concentrations in blood were measured after 15, 30, 60 and 120 min.
Serum analyses
Serum insulin and leptin levels as well as plasma corticosterone levels were measured by enzyme-linked immunosorbent assay according to vendor’s instructions (mouse ultra-sensitive Insulin ELISA, mouse Leptin ELISA; Crystal Chem, USA; mouse corticosterone ELISA; Enzo Life Sciences, USA).
Euglycemic-Hyperinsulinemic clamp studies in awake animals
Implantation of catheters into the jugular vein and the clamp procedure employed were performed as described before (Könner et al., 2007). In brief, mice were anesthetized using a ketamine-dexmedetomidine combination and then a saline filled catheter (Micro-Renathane, MRE 025; Braintree Scientific Inc., Braintree, Massachusetts, USA) was inserted into the right internal jugular vein and advanced to the level of the superior vena cava. The catheter was filled with heparin, tunneled, and left in a subcutaneous pocket at the back of the neck. A 3–0 silk suture was attached to the free end of the catheter and exteriorized in such a fashion as to allow retrieval of the catheter on the day of experimentation. After 5–6 days of recovery, mice that had lost less than 15% of their preoperative weight were subjected to the clamp study. On the day of experimentation, each animal was deprived of food for 4 h in the morning. For clamp studies, D-[3–3H]-glucose (PerkinElmer) was administered as a bolus (5 μCi), and then infused continuously (0.05 μCi/min). All solutions infused were prepared with 3% plasma added, obtained from 4 h fasted donor mice. Hyperinsulinemia was achieved by a bolus infusion of insulin (Insuman Rapid; Sanofi Aventis) of 20 μU/g BW, and thereafter by infusing insulin at a fixed rate of 4 μU/g/min. Blood glucose concentrations were monitored regularly according to a fixed scheme from tail vein bleedings (B-glucose analyzer, Hemocue), and maintained at around 120–140 mg/dl by administering 20% glucose (bela-pharm). An infusate of 2-deoxy-D-[1–14C]-glucose (10 μCi; American Radiolabeled Chemicals) was given approx. 120 min before steady state for analysis of tissue specific glucose uptake. Steady state was considered achieved, when a fixed glucose-infusion rate maintained the glucose concentration in blood constant for 30 min. At the end of the experiment, mice were killed by decapitation and liver, gonadal white adipose tissue, BAT, SKM and brain were dissected, snap-frozen in liquid nitrogen and stored at −80°C until further analysis. The [3–3H]glucose content in serum during basal conditions and at steady state was measured as described earlier (Könner et al., 2007). The radioactivity of 2-[1–14C]-deoxy-d- glucose in plasma was measured directly in a liquid scintillation counter. Lysates of adipose tissue, SKM and brain were processed through ion-exchange chromatography columns (AGR1-X8 formate resin, 200–400 mesh dry; Poly-Prep Prefilled Chromatography Columns; Bio Rad Laboratories) for the separation of 2-[1–14C]-deoxy-d- glucose from 2-[1–14C]-deoxy-d-glucose- 6-phosphate, 2-[1–14C] (2DG6P). The glucose-turnover rate (mg/kg/min) was calculated as described (Konner et al., 2007). Tissue specific glucose uptake in brain, WAT, BAT and SKM was calculated on the basis of the accumulation of 2-[1–14C]-deoxy-d-glucose-6-phosphate, 2-[1–14C] in the respective tissue and the disappearance rate of 2-[1–14C]-deoxy-d-glucose from plasma (Ferré et al., 1985) and expressed as nanomol per gram of tissue per minute (nmol/g/min).
Indirect Calorimetry
Indirect calorimetry was performed using an open-circuit, indirect calorimetry system (PhenoMaster, TSE systems) as previously described (Mauer et al., 2014). In brief, mice were trained for three days before data acquisition to adapt to the food/drink dispenser of the PhenoMaster system. Afterwards, mice were placed in regular type II cages with sealed lids at room temperature (22°C) and allowed to adapt to the chambers for at least 24 h. Food and water were provided ad libitum. All parameters were measured continuously and simultaneously.
Assessment of body composition
Lean and fat mass were determined via nuclear magnetic resonance (NMR Analyzer minispeq mq7.5; Bruker Optik, Ettlingen, Germany) in alive mice. Alternatively, body composition was analyzed by computed tomography (CT) in isoflurane-anesthetized mice (Dräger and Piramal Healthcare). For data acquisition on an IVIS Spectrum CT scanner (Caliper LifeScience, USA) we used IVIS LivingImage Software V4.3.1. Quantification of lean and fat mass contents were determined with a modification of the previously described Vinci software package 4.61.0 (Cízek et al., 2004) developed at our institution (available at: http://www.nf.mpg.de/vinci3/).
Leptin Sensitivity Test
Mice were singly housed and acclimatized for 1 week prior to study. Food intake and body weight was measured for 4 consecutive days. Leptin tests were conducted in a crossover fashion. Mice were i.p. injected with either 2 mg/kg body weight of mouse leptin (Sigma Aldrich) or vehicle (0.1 M phosphate-buffered saline (PBS)) twice a day (6 p.m. and at 6.00 a.m.) for 3 consecutive days.
Intracerbroventricular (ICV) cannulation and honokiol administration
ICV surgery
ICV cannulation was performed as previously described (Timper et al., 2017). In brief, mice were anesthetized using a ketamine-dexmedetomidine combination and were placed into a stereotaxic apparatus. 26 gauge cannulas were implanted to the left lateral ventricle using appropriate coordinates (bregma, −0.2 mm, +1 mm; dorsal surface, −2.1 mm). Dental acrylic (Super Bond C&B) was used to secure the cannula to the skull surface. Animals were allowed to recover for at least 1 week prior to experiment. After experiments, stereotaxic implants were anatomically verified.
ICV honokiol administration
Mice were acclimatized to ICV injections for 1 week prior to study. After an overnight fasting period of 16 h, mice were icv injected with 2 μL of either vehicle (Lipovenös® MCT 20% MCT, FRESENIUS KABI Germany) or Honokiol (37.6 mM; Sigma-Aldrich) 30 min before subjecting the mice to GTT as described above. ICV honokiol administration with subsequent GTT was conducted in a crossover fashion with a one week wash-out period in-between the tests.
PCR for detection of AIFm1 recombination events
To assess AIFm1 recombination events DNA samples from different tissues from AIFΔPOMC and AIFfl littermate control mice were subjected to conventional PCR using the following primers: P1: 5′-GAATCTGGAATATGGCACAGAGG-3′; P2: 5′-GTAGATCAGGTTGGCCAGAAACTC-3′; P3: 5′-GTAGTACATCTGGATGTAAGAGG-3′.
Analysis of gene expression and quantification of mitochondrial DNA using real-time PCR
RNA from N43/5 cells was extracted using the RNeasy® Kit (QIAGEN). RNA was reversely transcribed with High Capacity cDNA RT Kit and amplified using TaqMan® Universal PCR-Master Mix (Applied Biosystems). All samples were treated with DNase. Relative expression of target mRNAs was adjusted for total RNA content by Hprt (Mm01545399_m1). Inventoried Taqman probes were used: AIFm1 (Mm00442545_m1), CPT1b (Mm00487191_g1), PPARg (Mm00440945_m1). Calculations were performed by a comparative method (2−ΔΔCT). Mitochondrial DNA present per nuclear genome was determined using the following primer pairs: mitochondrial Nd2: fw: 5′- AGGGATCCCACTGCACATAG-3′; rev: 5′- CTCCTCATGCCCCTATGAAA-3′; mitochondrial D-Loop: fw: 5′- GGTTCT TACTTCAGGGCCATCA-3′, rev: 5′-GATTAGACCCGATACCATCGAGAT-3′; nuclear Nduv: fw: 5′- CTTCCCCACTGGCCTCAAG-3′; rev: 5′- CCAAAACCCAGTGATCCAGC-3′. Relative mitochondrial DNA content was calculated relative to nuclear DNA content based on the 2 × 2ΔCT method (Rooney et al., 2015). Quantitative PCR was performed on an AB-QuantStudio 7 Flex (Applied Biosystems).
Western blotting
Western blot analysis was conducted as previously described (Timper et al., 2017). In brief, membranes were blocked with 10% WB-blocking reagent (Roche, Switzerland) in Tris-buffered saline containing 0.1% Tween-20 (TBS-T), and incubated with primary antibodies, made up in TBS-T (0.1% Tween 20) and 5% Western Blotting-Reagent, at 4°C overnight. The following primary antibodies and dilutions were used: anti-AIF (1:1000, #ab32516, Abcam), anti-NDUFA9 (1:1000, #459100, Invitrogen), anti-NDUFB8 (1:1000, #459210, Invitrogen), anti-Complex II 70 kDa Fp Subunit (1:1000, #459200, Invitrogen), anti-UQCRC1 (1:1000, #459140, Invitrogen), anti-Complex IV (1:1000, #459110, Invitrogen), anti-ATP synthase subunit β (1:1000, #21351, Invitrogen) and, as loading control: anti-β-actin (1:2000, #A2228, Sigma Aldrich) or anti-calnexin (1:4000, #208880, Merck Milipore). Quantification of chemiluminescent signals was performed with the ImageJ (Fiji)-software package (Schneider et al., 2012).
N43/5 cell transfection with Antisense LNA GapmeRs
N43/5 cells were passaged at least twice before they were transfected at 50% confluence with custom-design Aifm1 antisense LNA: 5′-TAGCGGAGGCAATGGC-3′ (#300600, design-ID: 657181–3; Exiquon) or negative control A antisense LNA: 5′-AACACGTCTATACGC-3′ (#300610,Exiquon). Aifm1 and control A LNAs were pe- and co-incubated with Lipofectamin® RNAiMAX transfection reagent (Invitrogen) according to manufacturer’s instructions. Cells were subjected to measurements of oxygen consumption rates or directly lysed for RNA or DNA extraction a total of 24 h after LNA transfection and directly lysed for protein extraction a total of 48 h after LNA transfection.
Assessment of Oxygen Consumption Rate in N43/5 cells
Oxygen consumption rate (OCR) in AIF-deficient or control N43/5 cells was measured using the XFe96 Extracellular Flux Analyzer (Seahorse, Agilent Technologies) in XF Base Medium (Agilent) supplemented with 1 mM glutamine under basal conditions and in response to 10 mM glucose (Sigma), 1.0 μM oligomycin, 1.0 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP) and 1.0 μM Rotenone/Antimycin A (all XF Cell Mito Stress Test Kit # 101706–100, Agilent Technologies) according to manufacturer’s instructions.
For assessment of exogenous fatty acid oxidation, N43/5 cells were first starved for 20 h in substrate-limited DMEM (#11966–025, Thermo Fisher Scientific) containing 4 mM glutamine, 0.5 mM glucose (Sigma), 0.5 mM L-Carnitine hydrochloride (Sigma), 1% heat-inactivated fetal bovine serum (Invitrogen), and 1% penicillin/streptomycin (Invitrogen), OCR was then measured in FAO Assay Medium (Krebs-Ringer-Buffer (111 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 2 mM MgSO4, 1.2 mM NaH2PO4) supplemented with 2.5 mM glucose, 0.5 mM L-Carnitine hydrochloride (Sigma), and 5 mM HEPES (Sigma), adjusted to pH 7.4) in the presence or absence of 40 μM Etomoxir (Sigma, preincubation 15 min) and upon addition of 167 μM XF PA-BSA or 28 μM XF BSA Control (#102720–100, Agilent Technologies) in response to 1.0 μM oligomycin, 1.0 μM FCCP and 1.0 μM Rotenone/Antimycin A (all XF Cell Mito Stress Test Kit # 101706–100, Agilent Technologies) according to manufacturer’s instructions.
Data were analyzed and depicted relative to DNA content per well. DNA content was assessed by CyQUANT® Cell Proliferation Assay Kit (ThermoFisher Scientific) following manufacturer’s intructions.
Detection of mitochondrial superoxide production by quantitative flow cytometry
AIF-deficient or control N43/5 cells were treated as outlined above for assessment of exogenous fatty acid oxidation. Cells were incubated for 20 h in substrate-limited DMEM and then switched to FAO medium (see above) and incubated in FAO medium for 60 min before addition of 167 μM XF PA-BSA or 28 μM XF BSA Control (#102720–100, Agilent Technologies) for 90 min. Cells were then trypsinized, dissociated in FACS Buffer (0.1% low-endotoxin, fatty-acid free BSA (Sigma) in PBS) and centrifuged. Mitochondrial-specific superoxide production was measured by quantitative flow cytometry (MACSQuant VYB, Miltenyi Biotec) using MitoSox Red Mitochondrial Superoxide Indicator (5 μM, Invitrogen) and MitoTracker Green FM (100nM, Invitrogen). Cells were incubated for 10 min at 37°C in FACS Buffer to allow loading of MitoSox Red/MitoTracker Green FM and afterward washed 3 times in FACS Buffer. Cell debris as represented by distinct low forward and side scatter were gated out for analysis followed by exclusion of doublets. Dead cells were excluded using MitoTracker Green FM. 100,000 cells were acquired for each sample. Cells treated with the electron transport inhibitor Antimycin A (5 μM, Sigma) were used as positive control. Respective data analysis was performed with FlowJo (Ver. 10.0.8r1, Treestar).
Electrophysiology methods
Brain slice preparation
The animals were anesthetized with isoflurane (B506; AbbVie Deutschland GmbH & Co KG, Ludwigshafen, Germany) and subsequently decapitated. The brain was rapidly removed and a block of tissue containing the hypothalamus was immediately cut out. Coronal slices (270 – 300 μm) were cut with a vibration microtome (HM-650 V; Thermo Scientific, Walldorf, Germany) under cold (4°C), carbogenated (95% O2 and 5% CO2), glycerol-based modified artificial cerebrospinal fluid (GaCSF) to enhance the viability of neurons. GaCSF contained (in mM): 250 Glycerol, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.2 NaH2PO4, 10 HEPES, 21 NaHCO3, 5 Glucose and was adjusted to pH 7.2 with NaOH resulting in an osmolarity of ~310 mOsm. Brain slices were transferred into carbogenated artificial cerebrospinal fluid (aCSF). aCSF contained (in mM): 125 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.2 NaH2PO4, 21 NaHCO3, 10 HEPES, and 5 Glucose and was adjusted to pH 7.2 with NaOH resulting in an osmolarity of ~310 mOsm. First, they were kept for at least 20 min in a 36°C ‘recovery bath’ and then stored at room temperature (24°C) for at least 30 min prior to recording. For the recordings, slices were transferred to a Sylgard-coated (Dow Corning Corp., Midland, MI, USA) recording chamber (~3 mL volume) and, if not mentioned otherwise, continuously perfused with carbogenated aCSF at a flow rate of ~2 ml/min. To reduce GABAergic and glutamatergic synaptic input 10−4 M PTX, 5 × 10−5 M D-AP5, and 10−5 M CNQX was added to the aCFS. Recordings in the ARH were performed at 24°C using an inline solution heater (SH27B; Warner Instruments, Hamden, CT, USA) operated by a temperature controller (TC-324B; Warner Instruments).
Patch clamp recordings
Current-clamp recordings of POMC-expressing neurons of the ARH were performed during the light cycle in the perforated patch clamp configuration. Neurons were visualized with a fixed stage upright microscope (BX51WI, Olympus, Hamburg, Germany) using 40 × and 60 × water-immersion objectives (LUMplan FL/N 40 ×, 0.8 numerical aperture, 2 mm working distance; LUMplan FL/N 60 ×, 1.0 numerical aperture, 2 mm working distance, Olympus) with infrared differential interference contrast optics and fluorescence optics. POMC neurons were identified by their anatomical location in the ARH and by their GFP fluorescence that was visualized with an X-Cite 120 illumination system (EXFO Photonic Solutions, Ontario, Canada) in combination with a Chroma 41001 filter set (EX: HQ480/40×, BS: Q505LP, EM: HQ535/50m, Chroma, Rockingham, VT, USA). Electrodes with tip resistances between 5 and 6 MΩ were fashioned from borosilicate glass (0.86 mm inner diameter; 1.5 mm outer diameter; GB150–8P; Science Products) with a vertical pipette puller (PP-830; Narishige, London, UK). All recordings were performed with an EPC10 patch-clamp amplifier (HEKA, Lambrecht, Germany) controlled by the program PatchMaster (version 2.32; HEKA) running under Windows. In parallel, data were recorded using a micro1410 data acquisition interface and Spike 2 (version 7) (both from CED, Cambridge, UK). Data were sampled at 10 kHz and low-pass filtered at 2 kHz with a four-pole Bessel filter. The calculated liquid junction potential of 14.6 mV between intracellular and extracellular solution was compensated or subtracted offline (calculated with Patcher’s Power Tools plug-in from https://www.mpibpc.mpg.de/groups/neher/index.php?page=software for IGOR Pro 6 [Wavemetrics, Lake Oswego, OR, USA]).
Perforated patch clamp recordings
Perforated patch experiments were conducted using protocols modified from (Horn and Marty, 1988) and (Akaike and Harata, 1994). Recordings were performed with ATP and GTP free pipette solution containing (in mM): 128 K-gluconate, 10 KCl, 10 HEPES, 0.1 EGTA, 2 MgCl2 adjusted to pH 7.3 with KOH resulting in an osmolarity of ~300 mOsm. ATP and GTP were omitted from the intracellular solution to prevent uncontrolled permeabilization of the cell membrane (Lindau and Fernandez, 1986). The patch pipette was tip filled with internal solution and back filled with internal solution, which contained the ionophore to achieve perforated patch recordings.
Gramicidin (G5002; Sigma) was dissolved in dimethyl sulfoxide (DMSO; D8418, Sigma) following the protocols of (Rae et al., 1991) and (Kyrozis and Reichling, 1995). The used DMSO concentration (0.1 – 0.3%) had no obvious effect on the investigated neurons. The ionophore was added to the modified pipette solution shortly before use. The final concentration of gramicidin was ~10–25 μg/ml. During the perforation process access resistance (Ra) was constantly monitored and experiments were started after Ra had reached steady state (~15 – 20 min) and the action potential amplitude was stable.
Glucose and fatty acid sensing and KATP experiments
To study glucose and fatty acid sensing we used modified protocols from (Jo et al., 2009; Parton et al., 2007), respectively. Glucose concentrations were increased from 5 to 10 mM in aCSF. According to (Jo et al., 2009), a free form of oleic acid (OA, #O1008; Sigma) was dissolved in ethanol as a stock solution of 50mM and added to external solution just before the experiments to yield final concentrations of 50μM. In line with previous reports, we found POMC neuron sensitivity to glucose and OA was not homogeneous. Therefore, we used the ‘3 times standard deviation’ criterion (Dhillon et al., 2006) and considered a neuron glucose or OA responsive when the change in firing frequency between different glucose concentrations or upon OA, respectively, was 3 times larger than the standard deviation. For each neuron, the firing rate averaged from 30 s intervals was taken as one data point. To determine the mean firing rate and standard deviation under control and treatment conditions 10 data points at stable firing rates were averaged. For neurons without spontaneous generation of action potentials, the same analysis was applied to the membrane potential. In order to block KATP channels, tolbutamide (#T0891, Sigma) was dissolved in DMSO and added to the aCSF closely before the experiments to yield final concentrations of 200μM. In each experiment tolbutamide was applied for at least 10–15 min. Control firing rates where compared to peak depolarization rates within this time period.
Data analysis
Data analysis was performed with Spike2 (version 7; Cambridge Electronic Design Ltd., Cambridge, UK), Igor Pro 6 (Wavemetrics, Portland, OR, USA) and Graphpad Prism (version 5.0b; Graphpad Software Inc., La Jolla, CA, USA). If not stated otherwise, all calculated values are expressed as means ± SEM (standard error of the mean). The ‘+’ signs in the boxplots show the mean, the horizontal line the median of the data. The whiskers were calculated according to the ‘Tukey’ method. For comparisons of dependent and independent normal distributions paired and unpaired t tests were used, respectively. For comparisons of dependent and independent not normal distributions Wilcoxon matched-pairs signed-ranks tests and Mann-Whitney U-tests and were used, respectively. For multiple comparisons ANOVA with Tukey post hoc analysis was performed. A significance level of 0.05 was accepted for all tests. Significance levels were: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Histology
β-galactosidase staining and analysis of NLS-LacZ-positive POMC neurons
ROSA26-fl-LacZ; AIFfl/Y;POMC-Cre+/− experimental and ROSA26-fl-LacZ; AIFwt/Y;POMC-Cre+/− littermate control animals were deeply anesthetized and perfused transcardially with 0.9% saline followed by 4% paraformaldehyde (PFA). Brains were removed, postfixed overnight in 4% PFA and then transferred to 20% sucrose in 0.1 M PBS (pH 7.4) overnight. Perfused brains were frozen at −80°C and afterward 25 μm thick sections were cut on a cryostat. Every second section throughout the ARH was processed for β-galactosidase staining as follows. Sections were first incubated with 100% Ethanol at −20°C for 5 min and then washed twice for 10 min in PBS before they were incubated with pre-warmed (37°C) β-galactosidase staining solution (5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl 1 mg/ml (all Sigma Aldrich), 1 mg/ml β-galactosidase (PeQLab)) for 12 h at room temperature (RT). Afterward sections were quickly washed with PBS and then with tap water. Sections were counterstained with nuclear fast red for 10 min, washed again with tap water, dried at RT and then embedded in Kaisers Glyceringelatine (Millipore). Every second section throughout the ARH was taken and allocated rostral to caudal to visualize the distribution of POMC neurons throughout the ARC. Images were acquired using a Leica DM 1000 LED microscope equipped with a 20× objective. β-galactosidase positive neurons were counted and marked digitally to prevent multiple counts using ImageJ analysis software (NIH). Cell counts were performed on 5 animals per group.
Electron microscopy (EM) and analysis of mitochondrial architecture
Mice were transcardially perfused with 0.9% saline with heparine followed by fixative solution (PFA 4%, glutaraldehyde 0.1%, picric acid 15% in PBS 0.1 M, pH = 7.4). Brains were removed and fixed overnight at 4°C with the same fixative without gluteraldehyde. Brains were washed vigorously with ice-cold PBS, and sliced at 50 mm in a vibratome. Sections containing the ARH were stained for POMC (1:5000; 48 h at 4°C with gentle shaking; Phoenix Pharmaceuticals). After extensive washes, slices were incubated with secondary antibody, then with ABC and finally developed using 3,30-Diaminobenzidine. After developing, slices were put in osmium tetroxide (1%, 15 min) and then dehydrated in an ethanol gradient. Uranyl acetate (1%) was added to 70% ethanol to enhance ultrastructural contrast. Slices were then embedded in Durcupan, cut in an ultra-microtome and collected in grids for posterior analyzes. A Tecnai 12 Biotwin electron microscope was used to visualize the ultrastructure of the samples, and POMC neurons were imaged at 4800× magnification for posterior offline analyzes. For mitochondria analyzes, random sections of POMC neurons cut throughout the middle of the cell body were analyzed. Most of these sections contained the nucleus. ImageJ software was used to manually outline each individual mitochondrion in the digital images. All samples were checked twice for consistency of mitochondria labeling. We used mitochondria cross-sectional area and perimeter for the respective measurements. Only mitochondria with distinct inner architecture were chosen for maximal cristae width analyses. All measurements from biological replicates were pooled and the cumulative density function was calculated using the SciPy python package (https://www.scipy.org/). The results are presented in a plot where x axis represents the measurement and the y axis the probability of obtaining a value of xi or less. To identify significance of differences, we displayed the data in a boxplot and calculate the p value of a Whitney-Mann U test (two-sided).
pSTAT3 immunohistochemistry
For pSTAT3 immunohistochemistry, mice were starved for 16 h and ip injected with mouse leptin (2 mg/kg body weight, Sigma Aldrich) or vehicle (0.1 M PBS). 45 min thereafter mice were deeply anesthetized and perfused transcardially with 0.9% saline followed by 2% PFA, pH 7.4. Brains were removed, postfixed for 2 h in 2% PFA and transferred to 20% sucrose in 0.1 M PBS (pH 7.4) overnight. Perfused brains were frozen on dry ice and stored at −80°C. Afterward 30 μm thick sections were cut on a cryostat. Every fourth section throughout the ARH was processed for POMC immunohistochemistry and for pSTAT3 immunohistochemistry using a commercially available TSA Plus Fluorescence kit (#NEL741001KT, Perkin Elmer) as previously described (Timper et al., 2017). To ensure comparable immunostaining, sections were processed simultaneously under identical conditions. All incubations were done at room temperature unless otherwise stated. Briefly, for pSTAT3 immunohistochemistry, sections were allowed to dry before they were incubated with 2% PFA for 5 min. Thereafter, sections were washed 2 times for 10 min with PBS and then endogenous peroxidases were quenched via a 15 min incubation in 0.3% H2O2 (diluted in 0.1 M PBS, 0.25% Triton X), followed by additional rinses in PBS (2×5 min). Thereafter, sections were permeabilized by incubation with ice-cold methanol for 5 min followed by a washing step (3× 10 min 0.1 M PBS). Sections were then incubated in blocking solution (0.1 M TRIS-HCL, ph 7.5; 0.15 M NaCl; 0.5% Blocking Reagent (#FP1012; Perkin Elmer)) for 30 min. Thereafter, sections were incubated with the primary phospho-STAT3 (Tyr705) antibody (#9145, Cell Signaling; 1:100, diluted in Signal Stain, Cell Signaling) for 48 h at 4°C. After a washing step (3×5 min with 0.1 M PBS), the sections were incubated for 30 min with the secondary antibody (goat-anti-rabbit, horse raddish peroxidase, Perkin Elmer, 1:100 diluted in 0.1 M PBS, 0.25% Triton X). Slides were then again washed 5 times for 5 min with 0.1 M PBS, 0.1% Triton X and then incubated for 3 min with FITC thyramide signal amplification (TSA, Perkin Elmer, 1:100 diluted in amplification buffer, Perkin Elmer). Thereafter sections were washed once again (5×5 min in 0.1 M PBS) before they were quickly dipped in 4% PFA and the washed in 0.1 M PBS. Sections were then incubated for 60 min in blocking solution (2% donkey serum in 0.1 M PBS, 0.3% Triton X) before they were incubated with the primary POMC antibody (#H029–30, Phoenix; 1:1000, diluted in Signal Stain, Cell Signaling) for 48 h at 4°C. After a washing step (3×5 min with 0.1 M PBS, 0.1% Triton X), the sections were incubated for 60 min with the secondary antibody (donkey-anti-rabbit, Alexa 647, 1:500 diluted in 0.1 M PBS, 0.25% Triton X). After a washing step 3 times for 10 min with 0.1 M PBS, 0.1% Triton X) sections were mounted with DAPI-containing vectashield (Vector Laboratories Inc.). Images were acquired using a confocal Leica TCS SP-8-X microscope equipped with a 20× objective. To ensure similar imaging conditions for all pictures of all brains, exact same microscope set-up (including objective, zoom, laser power, gain) was used to acquire all pictures within the same experiment. All images were taken with a pinhole of 1 airy unit (AU). For representative images, maximum intensity projections were made in FIJI (NIH) and images were equally adjusted for brightness and contrast.
Quantitative analysis of pSTAT3 positive nuclei in POMC neurons
The total number of POMC neurons as well as the number of pSTAT3-positive POMC neurons was quantified unilaterally in images from every fourth section of every animal throughout the ARH using ImageJ analysis software (NIH) as previously reported (Timper et al., 2017). For quantification, only nuclei that were clearly immunoreactive were counted and only pSTAT3 nuclei that depicted a corresponding DAPI-positive staining were included in the analysis. The average percentage of pSTAT3-positive POMC neurons counted in 3 hemisections of the anterior and 3 hemisections of the middle ARH from each animal was used for statistical comparison.
Fluorescent in situ hybridization of AIFm1, MPC1 or tdTomato and POMC
Mice were deeply anesthetized and perfused transcardially with 0.9% saline followed by 4% PFA. Brains were removed, postfixed overnight in 4% PFA and then transferred to 20% sucrose in 0.1 M PBS (pH 7.4) overnight. Perfused brains were frozen and afterwards 14 μm thick sections (for detection of AIFm1 or MPC1, respectively, and POMC) or 30 μm thick sections (for detection of tdTomato and POMC) were cut on a cryostat. Fluorescent in situ hybridization for the simultaneous detection of the AIFm1, MPC1 or tdTomato, respectively, and the POMC transcripts was performed using RNAscope (ACD; Advanced Cell Diagnostics Inc., Hayward, CA). A custom designed probe was made targeting exon 7–8 of the AIFm1 transcript (target region: 901–1081, Acc. No: NM_012019.3; ACD). Accordingly, a custom designed probe was made targeting exon 3–5 of the MPC1 transcript (target region: 155–885, Acc. No: NM_018819.4; ACD. The tdTomato probe targeted the region 7 – 1382, Catalog No: 317041, ACD) and the POMC probe targeted the region 19–995 (Acc. No: NM_008895.3; ACD).
Negative and positive control probes recognizing dihydrodipicolinate reductase, DapB (a bacterial transcript) and cyclophilin and PolR2A, respectively, were processed in parallel with the target probes to ensure tissue RNA integrity and optimal assay performance. Thereby a negative control section of the hypothalamus from every animal was run in parallel. All pretreatment reagents, detection kit, and wash buffer were purchased from ACD. All incubation steps were performed at 40°C using a humidified chamber and a HybEz oven (ACD) unless indicated otherwise. On the day before the assay, sections were defrosted and briefly dipped in diethyl-pyrocarbonate (DEPC) treated Millipore water and dried at 60°C overnight. On the day of the assay, slides were first submerged in H2O2 for 10 min, then steamed at 100°C in Target Retrieval (ACD) for 20 min, followed by two brief rinses in DEPC water. The slides were then quickly dehydrated in 100% ethanol and allowed to air dry overnight. The following day, sections were incubated with Protease III (ACD) for 30 min. The subsequent steps, i.e., hybridization of the probe, the amplification steps and detection of the probe, were performed according to the online protocol for RNAscope Enhancer Fluorescent Kit v2 (#323100, ACD). In brief, the procedure included the following steps: The AIFm1 or MPC1 or tdTomato probe (channel 1) and the POMC probe (channel 2) were mixed 500:1 and hybridized to the sections for 2 h, followed by 2×2 min washes in wash buffer (ACD), incubation with Amp1 for 30 min, two washes, Amp2 for 30 min, two washes and finally Amp3 for 15 min followed by two washes. Sections were then incubated in HRP-C2 solution for 15 min followed by two washes and then incubated in channel 2-probe associated TSA® Plus Cyanine 3 (1:7500, #NEL744E001KT, Perkin Elmer) for 30 min followed by two washes and a subsequent incubation in HRP blocker solution for 15 min followed by two washes. Sections were then incubated in HRP-C1 solution for 15 min followed by two washes and then incubated in channel 1-probe associated TSA® Plus Cyanine 5 (1:750, #NEL745E001KT, Perkin Elmer) for 30 min followed by two washes and a subsequent incubation in HRP blocker solution for 15 min followed by two washes. Sections were then incubated in DAPI for 30 s and immediately coverslipped with ProLong® Gold Antifade Mountant. Images were acquired using a confocal Leica TCS SP-8 microscope equipped with a 40× objective for acquisition of AIFm1 or MPC1, respectively, and POMC staining or a 20× objective for acquisition of tdTomato and POMC staining. To ensure similar imaging conditions for all pictures of all brains, exact same microscope set-up (including objective, zoom, laser power, gain) was used to acquire all pictures within the same experiment. All pictures were taken with a pinhole of 1 airy unit (AU). For representative images maximum intensity projections were made in FIJI (NIH) and images were equally adjusted for brightness and contrast.
Quantitative analysis of fluorescent in situ hybridization
Maximum intensity projections were made in FIJI (NIH) and the raw images were imported into Halo software (Indica Labs) for quantification of cellular AIFm1 and POMC expression, which was quantified unilaterally in images depicting comparable sections of the ARH from 3 AIFΔPOMC and 3 control animals as well as in the negative control sections from every animal. The radial area surrounding each cellular nucleus defining the area where cell-associated staining was counted and the threshold for consideration of positive staining was defined upon analysis of average values of negative control sections from every animal. Labeling above average intensity of negative control section + 3 × SD was considered to be true signal. For quantification of tdTomato and POMC expression, the total number of tdTomato expressing neurons as well as the total number of POMC expressing neurons was quantified unilaterally in images from every fourth section from 3 AIFΔPOMC and 3 control animals throughout the ARH using ImageJ analysis software (NIH) as previously reported (Timper et al., 2017). The average number of tdTomato-positive or POMC-positive neurons counted in 3 hemi-sections of the anterior, 3 hemisections of the middle and 3 hemisections of the posterior ARH from each animal was used for statistical comparison and the average percentage of POMC-expressing tdTomato as well as of tdTomato-expressing POMC neurons was calculated. Analysis settings were kept the same throughout quantification of all pictures.
Administration of DHE and quantification of ROS in POMC neurons
ROS levels in POMC neurons were measured by injecting dihydroethidium (DHE, Invitrogen) icv (2 μl, DHE 1mg/ml in 20% DMSO (Koch et al., 2015)) in 16 h overnight fasted HFD-fed AIFΔPOMC and control mice. 60 min after the DHE injection, mice were deeply anesthetized and perfused transcardially with 0.9% saline followed by 4% PFA. From then on, brains were processed in the dark. Brains were removed, postfixed overnight in 4% PFA and then transferred to 20% sucrose in 0.1 M PBS (pH 7.4) overnight. Brains were then frozen on dry ice and directly 30 μm thick sections were cut on a cryostat. Every fourth section throughout the ARH was mounted on glass slides and stored at −80°C overnight. To ensure comparable immunostaining, sections were processed the following day simultaneously under identical conditions. All washing steps and incubations were performed in the dark at room temperature unless stated otherwise. Briefly, for POMC immunohistochemistry, sections were allowed to dry before they were incubated with 2% PFA for 5 min. Thereafter, sections were washed 2 times for 10 min with PBS and incubated for 10 min with 0.3% glycine, washed again for 10 min in PBS and incubated for 10 min in 0.03% SDS. Sections were then incubated for 60 min in blocking solution (2% donkey serum in 0.1 M PBS, 0.3% Triton X) before they were incubated with the primary POMC antibody (#H029–30, Phoenix; 1:1000, diluted in Signal Stain, Cell Signaling) for 48 h at 4 C. After a washing step (3×5 min with 0.1 M PBS, 0.1% Triton X), the sections were incubated for 60 min with the secondary antibody (donkey-anti-rabbit, #ab150073, Abcam Alexa 647, 1:500). After a washing step 3 times for 10 min with 0.1 M PBS, 0.1% Triton X, sections were mounted with DAPI-containing vectashield (Vector Laboratories Inc.) and kept at 4°C overnight. The next day all slides were imaged using a confocal Leica TCS SP-8-X microscope equipped with a 40× objective. To ensure similar imaging conditions for all pictures of all brains, exact same microscope set-up (including objective, zoom, laser power, gain) was used to acquire all pictures within the same experiment. All images were taken with a pinhole of 1 airy unit (AU). For representative images, maximum intensity projections were made in FIJI (NIH) and images were equally adjusted for brightness and contrast.
Quantitative analysis of DHE intensity in POMC neurons
DHE signal in POMC neurons was quantified as previously described (Koch et al., 2015). In brief, the specific region of interest in each image was defined by using the selection tool on the image from the adjacent POMC-staining unilaterally in images from every fourth section of every animal throughout the ARH using ImageJ analysis software (NIH). Mean density of the DHE signal in the respective POMC neurons was determined using ImageJ analysis software (NIH). To avoid false-positive results, the autofluorescence tissue background in every analyzed picture was subtracted from the DHE signal. The average percentage of background-corrected DHE intensity in POMC neurons counted in 2 hemisections of the anterior and 2 hemisections of the middle ARH from each animal was used for statistical comparison.
QUANTIFICATION AND STATISTICAL ANALYSIS
All values were expressed as mean ± SEM. Statistical analyses were conducted using GraphPad PRISM (version 7.0) unless indicated otherwise. Statistical details of experiments can be found in the figure legends and the respective methods section. Unless otherwise specified in the figure legends, datasets with only two independent groups were analyzed for statistical significance using unpaired, two-tailed Student’s t test. Datasets with more than two groups were analyzed using one-way ANOVA, followed by Bonferroni post hoc test. Datasets subjected to two independent factors were analyzed using two-way ANOVA, followed by Bonferroni post hoc test. All p values below or equal to 0.05 were considered significant. * p ≤ 0.05, ** p < 0.01, and *** p < 0.001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-AIF | Abcam | Cat# ab32516; RRID: AB_726995 |
| Anti-NDUFA9 | Invitrogen | Cat# 459100; RRID: AB_2532223 |
| Anti-NDUFB8 | Invitrogen | Cat# 459210; RRID:AB_2532232 |
| Anti-Complex II 70 kDa Fp Subunit | Invitrogen | Cat# 459200; RRID: AB_2532231 |
| Anti-UQCRC1 | Invitrogen | Cat# 459140; RRID: AB_2532227 |
| Anti-Complex IV | Invitrogen | Cat# 459110; RRID: AB_2532224 |
| Anti-ATP synthase subunit β | Invitrogen | Cat# 21351; RRID: AB_221512 |
| Anti-β-actin | Sigma Aldrich | Cat# A2228; RRID: AB_476697 |
| Anti-calnexin | Merck Millipore | Cat# 208880; RRID: AB_2069031 |
| Anti-phospho-STAT3 (Tyr705) | Cell Signaling | Cat# 9145; RRID: AB_2491009 |
| Anti-POMC antibody | Phoenix | Cat# H029–30; RRID: AB_2037442 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Insulin | Sanofi Aventis, Germany | Insuman rapid |
| Mouse Leptin | Sigma Aldrich | Cat# L3772–1MG |
| Honokiol | Sigma Aldrich | Cat# H4914–10MG |
| Seahorse XF Palmitate-BSA FAO Substrate | Agilent Technologies | Cat# 102720–100 |
| MitoSox Red Mitochondrial Superoxide Indicator | Invitrogen | Cat# M36008 |
| MitoTracker Green FM | Invitrogen | Cat# M7514 |
| TSA® Plus Cyanine 3 | Perkin Elmer | Cat# NEL744E001KT |
| TSA® Plus Cyanine 5 | Perkin Elmer | Cat# NEL745E001KT |
| Dihydroethidium (DHE) | Invitrogen | Cat# D1168 |
| Critical Commercial Assays | ||
| Arcturus picopure RNA isolation Kit | Thermo Fisher Scientific | Cat# KIT0204 |
| Mouse ultra-sensitivity insulin ELISA | Crystal Chem, USA | Cat# 90080 |
| Mouse Leptin ELISA | Crystal Chem, USA | Cat# 90030 |
| Mouse Corticosterone ELISA | Enzo Life Sciences, USA | Cat# ADI-900–097 |
| Dental acrylic Super Bond C&B | Sun Medical | Cat# 7100 |
| CyQUANT® Cell Proliferation Assay Kit | ThermoFisher Scientific | Cat# C7026 |
| XF Cell Mito Stress Test Kit | Agilent Technologies | Cat# 101706–100 |
| TSA Plus Fluorescence kit | Perkin Elmer, USA | Cat# NEL741001KT |
| RNAscope Enhancer Fluorescent Kit v2 | ACD | Cat# 323100 |
| Experimental Models: Cell Lines | ||
| Embryonic Mouse Hypothalamus Cell Line N43/5 | (Choi et al., 2010) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Pomc-Cre | (Balthasar et al., 2004) | N/A |
| Mouse: AIF-flox | (Joza et al., 2005) | N/A |
| Mouse: ROSA26-flox-LacZ | (Plum et al., 2006) | N/A |
| Mouse: POMC-EGFP | (Cowley et al., 2001) | N/A |
| Mouse: MPC1 | (Gray et al., 2015) | N/A |
| Oligonucleotides | ||
| Taqman probe AIFm1 | Thermo Fisher | Mm00442545_m1 |
| Taqman probe CPT1b | Thermo Fisher | Mm00487191_g1 |
| Taqman probe PPARg | Thermo Fisher | Mm00440945_m1 |
| mitochondrial Nd2: fw: 5′- AGGGATCCCACTGCACATAG-3′; | Eurogentec | https://secure.eurogentec.com/life-science.html |
| mitochondrial Nd2: rev: 5′- CTCCTCATGCCCCTATGAAA-3′ | Eurogentec | https://secure.eurogentec.com/life-science.html |
| mitochondrial D-Loop: fw: 5′- GGTTCTTACTTCAGGGCCATCA-3′ | Eurogentec | https://secure.eurogentec.com/life-science.html |
| mitochondrial D-Loop: rev: 5′-GATTAGACCCGATACCATCGAGAT-3′ | Eurogentec | https://secure.eurogentec.com/life-science.html |
| nuclear Nduv: fw: 5′- CTTCCCCACTGGCCTCAAG-3′; | Eurogentec | https://secure.eurogentec.com/life-science.html |
| nuclear Nduv: rev: 5′- CCAAAACCCAGTGATCCAGC-3′ | Eurogentec | https://secure.eurogentec.com/life-science.html |
| Aifm1 antisense LNA: 5′-TAGCGGAGGCAATGGC-3′ | Exiquon | Cat# 300600, design-ID: 657181–3 |
| Negative control A antisense LNA: 5′-AACACGTCTATACGC-3′ | Exiquon | 300610 |
| Custom-designed RNA scope probe targeting exons 7–8 of the AIFm1 transcript (target region: 901–1081) | ACD | Cat# NM_012019.3 |
| Custom-designed RNA scope probe targeting exons 3–5 of the MPC1 transcript (target region: 155–885) | ACD | Cat# NM_018819.4 |
| RNA scope tdTomato probe (target region: 7 – 1382) | ACD | Cat# 317041 |
| RNA scope POMC probe (target region 19–995) | ACD | Cat# NM_008895.3 |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji (ImageJ) Software Package (incl. Adiposoft Plugin) | (Schneider et al., 2012) | http://imagej.net/Adiposoft |
| IVIS LivingImage Software V4.3.1 | Caliper LifeScience, Perkin Elmer, USA | http://www.perkinelmer.com:80/category/in-vivo-imaging-software |
| Vinci software package 4.61.0 | (Cízek et al., 2004) | http://www.nf.mpg.de/vinci3 |
| FlowJo (Ver. 10.0.8r1) | Treestar | https://www.flowjo.com/ |
| PatchMaster (version 2.32) | HEKA, Lambrecht, Germany | https://www.heka.com/downloads/downloads_main.html |
| Patcher’s Power Tools plug-in | Wavemetrics, Lake Oswego, OR, USA | httpx://www.mpibpc.mpg.de/groups/neher/index.php?page=software |
| Spike2 (version 7) | Cambridge Electronic Design Ltd., Cambridge, UK | http://ced.co.uk/de/ |
| Other | ||
| Regular Chow Diet | sniff Spezialdiäten GmbH, Germany | R/M-H Phytoestrogenarm |
| Regular Chow Diet (Teklad) | Envigo, USA | Cat#2918 |
| High Fat Diet | sniff Spezialdiäten GmbH, Germany | Cat# D12492-(I) mod. |
| Normal Control Diet | sniff Spezialdiäten GmbH, Germany | Cat# D12450-(B) mod LS |
| High Fat Diet (Altromin) | Altromin Spezialfutter GmbH & Co. KG, Germany | Cat# C1057 |
| NMR Analyzer minispeq mq7.5 | Bruker Optik, Ettlingen, Germany | https://www.bruker.com/ |
| IVIS Spectrum CT scanner | Caliper LifeScience, Perkin Elmer, USA | Cat# 128201 |
| Indirect calorimetry system “PhenoMaster” | TSE systems, Chesterfield, USA | http://www.tse-systems.com/product-details/phenomaster/ |
| Lipovenös® MCT 20% | FRESENIUS KABI Germany | PZN:276848 |
| XFe96 Extracellular Flux Analyzer, “Seahorse” | Agilent Technologies | https://www.agilent.com/en/products/cell-analysis/seahorse-analyzers/seahorse-xfe96-analyzer |
| MACSQuant VYB | Miltenyi Biotec | https://www.miltenyibiotec.com/DE-en/ |
| Vibration microtome HM-650 V | Thermo Scientific, Walldorf, Germany | https://uk.vwr.com/store/product/13046604/vibrating-microtome-hm-650-v |
| Inline solution heater | Warner Instruments, Hamden, CT, USA | Cat# SH27B |
| Temperature controller | Warner Instruments, Hamden, CT, USA | Cat# TC-324B |
| Electrodes | Science Products | Cat# GB150–8P |
| Vertical pipette puller | Narishige, London, UK | Cat# PP-830 |
| EPC10 patch-clamp amplifier | HEKA, Lambrecht, Germany | https://www.heka.com/ |
Highlights.
AIF deletion in POMC neurons enhances fatty acid oxidation and ROS formation
POMC-specific inactivation of AIF prevents obesity-induced POMC neuron silencing
POMC-specific AIF deletion improves leptin sensitivity and glucose metabolism in obesity
POMC-specific MPC1 deletion improves glucose metabolism in obesity
ACKNOWLEDGMENTS
We acknowledge Hella Brönneke for outstanding administrative assistance, Pia Scholl, Nadine Spenrath, and Helmut Wratil for outstanding technical assistance, and Astrid Schauss and Christian Jüngst from the CECAD Imaging Facility for support with confocal imaging. J.C.B. received funding by the Deutsche Forschungsgemeinschaft (DFG) within the Excellence Initiative by German Federal and State Governments (CECAD) and the SFB 1218 TP A08. K.T. was funded by Advanced Postdoc Grants from the Swiss National Science Foundation (SNCF) and received an Early Career Research Grant from the University of Basel, Switzerland. E.B.T. was funded by the NIH (R01-DK-104998). T.L.H. was funded by the NIH (R01-DK-111178). P.K. received funding from SFB 1218 TP B07 and TR SFB 134 TP A03.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.034.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Akaike N, and Harata N (1994). Nystatin perforated patch recording and its applications to analyses of intracellular mechanisms. Jpn. J. Physiol 44, 433–473. [DOI] [PubMed] [Google Scholar]
- Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, et al. (2008). UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 454, 846–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC Jr., Elmquist JK, and Lowell BB (2004). Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42, 983–991. [DOI] [PubMed] [Google Scholar]
- Berglund ED, Vianna CR, Donato J Jr., Kim MH, Chuang JC, Lee CE, Lauzon DA, Lin P, Brule LJ, Scott MM, et al. (2012). Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J. Clin. Invest 122, 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, et al. (2012). A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, and Chan DC (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol 160, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Joza N, Steenaart NA, McClellan KA, Neuspiel M, McNamara S, MacLaurin JG, Rippstein P, Park DS, Shore GC, et al. (2006). Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J 25, 4061–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SJ, Kim F, Schwartz MW, and Wisse BE (2010). Cultured hypothalamic neurons are resistant to inflammation and insulin resistance induced by saturated fatty acids. Am. J. Physiol. Endocrinol. Metab 298, E1122–E1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cízek J, Herholz K, Vollmar S, Schrader R, Klein J, and Heiss WD (2004). Fast and robust registration of PET and MR images of human brain. Neuroimage 22, 434–442. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, and Low MJ (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480–484. [DOI] [PubMed] [Google Scholar]
- Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, and Kroemer G (2000). Apoptosis-inducing factor (AIF): A ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett 476, 118–123. [DOI] [PubMed] [Google Scholar]
- de Brito OM, and Scorrano L (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. [DOI] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, et al. (2006). Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191–203. [DOI] [PubMed] [Google Scholar]
- Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, et al. (2011). Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat. Med 17, 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, and Leverve X (2000). Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem 275, 223–228. [DOI] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, et al. (2007). Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab 5, 181–194. [DOI] [PubMed] [Google Scholar]
- Ferré P, Leturque A, Burnol AF, Penicaud L, and Girard J (1985). A method to quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the anaesthetized rat. Biochem. J 228, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray LR, Sultana MR, Rauckhorst AJ, Oonthonpan L, Tompkins SC, Sharma A, Fu X, Miao R, Pewa AD, Brown KS, et al. (2015). Hepatic mitochondrial pyruvate carrier 1 is required for efficient regulation of gluconeogenesis and whole-body glucose homeostasis. Cell Metab 22, 669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, and Martinou JC (2012). Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96. [DOI] [PubMed] [Google Scholar]
- Horn R, and Marty A (1988). Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol 92, 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Rønnekleiv OK, Low MJ, and Kelly MJ (2003). Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology 144, 1331–1340. [DOI] [PubMed] [Google Scholar]
- Jo YH, Su Y, Gutierrez-Juarez R, and Chua S Jr. (2009). Oleic acid directly regulates POMC neuron excitability in the hypothalamus. J. Neurophysiol 101, 2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Oudit GY, Brown D, Bénit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, et al. (2005). Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol. Cell. Biol 25, 10261–10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK (2009). Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods Mol. Biol 560, 221–238. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, and Ackerman SL (2002). The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419, 367–374. [DOI] [PubMed] [Google Scholar]
- Koch M, Varela L, Kim JG, Kim JD, Hernández-Nuño F, Simonds SE, Castorena CM, Vianna CR, Elmquist JK, Morozov YM, et al. (2015). Hypothalamic POMC neurons promote cannabinoid-induced feeding. Nature 519, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, et al. (2007). Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab 5, 438–449. [DOI] [PubMed] [Google Scholar]
- Kyrozis A, and Reichling DB (1995). Perforated-patch recording with gramicidin avoids artifactual changes in intracellular chloride concentration. J. Neurosci. Methods 57, 27–35. [DOI] [PubMed] [Google Scholar]
- Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, et al. (2006). Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55, 2256–2264. [DOI] [PubMed] [Google Scholar]
- Lindau M, and Fernandez JM (1986). IgE-mediated degranulation of mast cells does not require opening of ion channels. Nature 319, 150–153. [DOI] [PubMed] [Google Scholar]
- Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, Hausen AC, Schmitz J, Brönneke HS, et al. (2014). Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol 15, 423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden JW, Aja S, Li Q, Bandaru VV, Kim EK, Haughey NJ, Kuhajda FP, and Ronnett GV (2014). Increasing fatty acid oxidation remodels the hypothalamic neurometabolome to mitigate stress and inflammation. PLoS ONE 9, e115642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milasta S, Dillon CP, Sturm OE, Verbist KC, Brewer TL, Quarato G, Brown SA, Frase S, Janke LJ, Perry SS, et al. (2016). Apoptosis-Inducing-Factor-Dependent Mitochondrial Function Is Required for T Cell but Not B Cell Function. Immunity 44, 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG Jr., and Olson DP (2012). Central nervous system control of metabolism. Nature 491, 357–363. [DOI] [PubMed] [Google Scholar]
- Obici S, Feng Z, Arduini A, Conti R, and Rossetti L (2003). Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat. Med 9, 756–761. [DOI] [PubMed] [Google Scholar]
- Padilla SL, Carmody JS, and Zeltser LM (2010). Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nat. Med 16, 403–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla SL, Reef D, and Zeltser LM (2012). Defining POMC neurons using transgenic reagents: Impact of transient Pomc expression in diverse immature neuronal populations. Endocrinology 153, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paeger L, Pippow A, Hess S, Paehler M, Klein AC, Husch A, Pouzat C, Bruning JC, and Kloppenburg P (2017). Energy imbalance alters Ca(2+) handling and excitability of POMC neurons. eLife, Published online August 1, 2017. 10.7554/eLife.25641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al. (2007). Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449, 228–232. [DOI] [PubMed] [Google Scholar]
- Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Münzberg H, Shanabrough M, Burdakov D, Rother E, Janoschek R, et al. (2006). Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J. Clin. Invest 116, 1886–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela LV, Gnoatto J, Brochier AW, Haas CB, de Assis AM, de Carvalho AK, Hansel G, Zimmer ER, Oses JP, and Muller AP (2015). Intracerebroventricular metformin decreases body weight but has pro-oxidant effects and decreases survival. Neurochem. Res 40, 514–523. [DOI] [PubMed] [Google Scholar]
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, Ebersberger I, Nakashima T, Sarao R, Neely G, et al. (2007). Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131, 476–491. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, and Watsky M (1991). Low access resistance perforated patch recordings using amphotericin B. J. Neurosci. Methods 37, 15–26. [DOI] [PubMed] [Google Scholar]
- Ramirez S, Gomez-Valades AG, Schneeberger M, Varela L, Haddad-Tovolli R, Altirriba J, Noguera E, Drougard A, Flores-Martinez A, Imbernon M, Chivite I, Pozo M, Vidal-Itriago A, Garcia A, Cervantes S, Gasa R, Nogueiras R, Gama-Perez P, Garcia-Roves PM, Cano DA, Knauf C, Servitja JM, Horvath TL, Gomis R, Zorzano A, and Claret M (2017). Mitochondrial dynamics mediated by Mitofusin 1 is required for POMC neuron glucose-sensing and insulin release control. Cell Metab 25, 1390–1399. [DOI] [PubMed] [Google Scholar]
- Rodríguez EM, Blázquez JL, and Guerra M (2010). The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 31, 757–776. [DOI] [PubMed] [Google Scholar]
- Rooney JP, Ryde IT, Sanders LH, Howlett EH, Colton MD, Germ KE, Mayer GD, Greenamyre JT, and Meyer JN (2015). PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol 1241, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, and Hoppel CL (2012). Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 61, 2074–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro A, Campolo M, Liu C, Sesaki H, Meli R, Liu ZW, Kim JD, and Diano S (2017). DRP1 Suppresses Leptin and Glucose Sensing of POMC Neurons. Cell Metab 25, 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell JC, Wisidagama DR, Bensard C, Zhao H, Wei P, Tanner J, Flores A, Mohlman J, Sorensen LK, Earl CS, et al. (2017). Control of intestinal stem cell function and proliferation by mitochondrial pyruvate metabolism. Nat. Cell Biol 19, 1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiff M, Bénit P, El-Khoury R, Schlemmer D, Benoist JF, and Rustin P (2011). Mouse studies to shape clinical trials for mitochondrial diseases: High fat diet in Harlequin mice. PLoS ONE 6, e28823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, Garcia A, Esteban Y, Gonzalez-Franquesa A, Rodríguez IC, Bortolozzi A, et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155, 172–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel GS, and Horvath TL (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, et al. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446. [DOI] [PubMed] [Google Scholar]
- Theunissen TEJ, Gerards M, Hellebrekers DMEI, van Tienen FH, Kamps R, Sallevelt SCEH, Hartog ENMM, Scholte HR, Verdijk RM, Schoonderwoerd K, et al. (2017). Selection and characterization of palmitic acid responsive patients with an OXPHOS complex I defect. Front. Mol. Neurosci 10, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timper K, Denson JL, Steculorum SM, Heilinger C, Engström-Ruud L, Wunderlich CM, Rose-John S, Wunderlich FT, and Brüning JC (2017). IL-6 improves energy and glucose homeostasis in obesity via enhanced central IL-6 trans-signaling. Cell Rep 19, 267–280. [DOI] [PubMed] [Google Scholar]
- Turner N, Li JY, Gosby A, To SW, Cheng Z, Miyoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, et al. (2008). Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: A mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes 57, 1414–1418. [DOI] [PubMed] [Google Scholar]
- Urbano A, Lakshmanan U, Choo PH, Kwan JC, Ng PY, Guo K, Dhakshinamoorthy S, and Porter A (2005). AIF suppresses chemical stress-induced apoptosis and maintains the transformed state of tumor cells. EMBO J 24, 2815–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, and Metallo CM (2014). Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol. Cell 56, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, et al. (2004). AIF deficiency compromises oxidative phosphorylation. EMBO J 23, 4679–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderperre B, Herzig S, Krznar P, Hörl M, Ammar Z, Montessuit S, Pierredon S, Zamboni N, and Martinou JC (2016). Embryonic lethality of mitochondrial pyruvate carrier 1 deficient mouse can be rescued by a ketogenic diet. PLoS Genet 12, e1006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T, and Langer T (2016). Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab 27, 105–117. [DOI] [PubMed] [Google Scholar]
- Wei W, Zhao H, Wang A, Sui M, Liang K, Deng H, Ma Y, Zhang Y, Zhang H, and Guan Y (2012). A clinical study on the short-term effect of berberine in comparison to metformin on the metabolic characteristics of women with polycystic ovary syndrome. Eur. J. Endocrinol 166, 99–105. [DOI] [PubMed] [Google Scholar]
- Wexler ID, Hemalatha SG, McConnell J, Buist NR, Dahl HH, Berry SA, Cederbaum SD, Patel MS, and Kerr DS (1997). Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology 49, 1655–1661. [DOI] [PubMed] [Google Scholar]
- Yi CX, Walter M, Gao Y, Pitra S, Legutko B, Kälin S, Layritz C, García-Cáceres C, Bielohuby M, Bidlingmaier M, et al. (2017). TNFα drives mitochondrial stress in POMC neurons in obesity. Nat. Commun 8, 15143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan C, Zhou J, Feng Q, Zhang JE, Lin S, Bao J, Wu P, and Luo M (2013). Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. J. Neurosci 33, 3624–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YL, Hernandez-Ono A, Siri P, Weisberg S, Conlon D, Graham MJ, Crooke RM, Huang LS, and Ginsberg HN (2006). Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J. Biol. Chem 281, 37603–37615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.