

Abstract
Halogenated ligands can participate in nonbonding interactions with proteins via halogen bond (XB) or halogen-hydrogen bond donor (X-HBD) interactions. In the context of molecular dynamics (MD) simulations, the accuracy of the simulations depends strongly on the force field (FF) used. To assure good reproduction of XB and X-HBD interactions with proteins, we optimized the previously developed additive CHARMM36/CHARMM General force field (CGenFF) and Drude polarizable force field by including atom pair-specific Lennard-Jones parameters for aromatic halogen-protein interactions. The optimization targeted quantum mechanical interaction energy surfaces with the developed parameters then examined for their ability to reproduce experimental halogen-containing ligand-protein interactions in MD simulations. The calculated halogenated ligand interaction geometries were in good overall agreement with the experimental crystal data for both the polarizable and additive FFs, showing that these models can accurately treat both XB and X-HBD interactions. Analysis of the ligand-protein interactions shows significant contributions of polarizability to binding occurring in the Drude FF, with self-polarization energy making both favorable and unfavorable contributions to binding. Further analysis of the dipole moments from aqueous solution to protein indicates the polarizable FF accounts for subtle changes of the environment of the ligands that can impact binding. The present work demonstrates the utility of the updated additive CHARMM36/CGenFF and polarizable Drude FFs for the study of halogenated ligand-protein interactions in computer-aided drug design.
Graphical Abstract
Introduction
The inclusion of the halogens during ligand optimization represents an important aspect of drug design. This is due to the potential of halogens to improve selectivity and binding affinity as well as physiochemical properties related to bioavailability, thereby leading to better therapeutic agents.1,2 These properties lead to a significant number of drug candidates and launched drugs containing halogens.3 In earlier drug design campaigns the lipophilic or hydrophobic contributions of halogen were mainly considered with respect to improving biological activity.2,4 Recently, specific ligand-protein interactions involving halogens are being considered due to studies showing halogens to participate in halogen bonds (XB).1,2,5 In addition, halogens may contribute to ligand binding via halogen-hydrogen bond donor (X-HBD) interactions that are potentially more favorable than halogen bonds as indicated by quantum mechanical (QM) calculations and survey data from the Protein Data Bank (PDB).6
In drug discovery, computer-aided drug design (CADD) is able to speed up the identification of lead compounds as well as facilitate their optimization.8–11 Molecular dynamics (MD) simulation-based methods are often used to predict binding orientations and provide thermodynamic information, including the prediction of the absolute or relative binding affinity of ligands. In order to apply these approaches to halogen-containing compounds, well-developed force fields are required. Recent efforts have extended the widely used additive force fields to explicitly treat halogenated ligands, including extensions of CGenFF (CHARMM General Force Field),12–16 OPLS (Optimized Potentials for Liquid Simulations),17–20 and GAFF (Generalized AMBER Force Field).21–24 In addition, polarizable force fields that explicitly treat electronic polarizability have been introduced,25–27 including both the classical Drude28–41 and AMOEBA force fields (FFs).42–44 With the above additive and Drude polarizable FFs, improved treatment of halogens has involved attaching a charged virtual particle to the halogen to mimic the σ-hole in halogen and adequately reproduce XBs.13,20,23,24,45,46
An important feature of halogens is their asymmetric van der Waals (vdW) surface associated with the shift of the electron density resulting in a shorter electron repulsion distance along the halogen linear to the C-X bond.47 Moreover, a recent study indicates that the vdW contribution of halogens could significantly affect ligand-protein binding energies.48 Accordingly, it is important for a force field to capture the asymmetric vdW surface of the halogen. In the region linear to C-X bond, this requires the electropositive σ-hole to have an effective smaller radius when interacting with nucleophilic atoms to allow for the shorter interaction distances, while the perpendicular peripheral region of halogens should have a larger effective radius, allowing for relatively longer interaction distances with electrophilic atoms.47 However, to date only a few halogen FFs considered such features during parametrization.47,49 In our recent halogen polarizable force field49 based on the classical Drude oscillator model50 the model was parametrized to reproduce the asymmetric vdW surface of halogens to more accurately treat both XB and X-HBD interactions. This was achieved by assigning atom pair-specific Lennard-Jones (LJ) parameters51 between the halogen Drude particles and hydrogens on hydrogen bond donors (i.e. an NBFIX term in CHARMM nomenclature52–54). This term in combination with the inclusion of polarizability yielded improved modeling of halogen-water interactions as well as reproducing pure solvent properties and hydration free energies.
In this study, atom pair-specific LJ parameters on halogen Drude particles were extended to selected hydrogen bond donors and acceptors present in proteins and were optimized targeting representative small model compounds (Figure 1). The halogenated model compounds in this work focused on chlorobenzene (CHLB) and bromobenzene (BROB), as chlorine and bromine are often used during drug development. Iodobenzene was not considered as only 1.8 % of the organohalogens include iodine.3 These parameters were initially optimized targeting quantum mechanical (QM) interaction energy surfaces between halogen and protein model compounds. In addition, atom pair-specific LJ parameters were added to the additive CHARMM36/CGenFF FF to improve the agreement with the QM X-HBD interactions. The optimized atom pair-specific LJ parameters were then examined with respect to their ability to reproduce non-bonded interactions of halogen-containing ligands or a halogen-modified residue with the surrounding proteins occurring in crystallographic structures using MD simulations. With these parameters, the calculated halogenated-ligand/residue interaction geometries were in good overall agreement with the experimental data for both the polarizable and additive FFs. These results indicate that the developed polarizable and additive halogen force fields can accurately treat both XB and X-HBD interactions, thereby indicating their utility in computer-aided drug design.
Figure 1.

Abbreviations and 2D structures of the protein model compounds.
Computational methods
Quantum mechanical calculations
QM geometry optimizations were performed at the MP2 level of theory with the aug-cc-pVDZ basis set55 for chlorobenzene (CHLB) and protein model compounds (Figure 1), including N-methylacetamide (NMA), methanol (MEOH), and phenol (PHEN), acetamide (ACEM), imidazole (IMID), indole (INDO), methanethiol (MESH), methylammonium (MAMM), methylguanidinum (MGUAN), and acetate (ACET). Bromobenzene (BROB) was optimized at the MP2 level of theory with the aug-cc-pVDZ-PP basis set.56,57 All the QM geometry optimizations were performed using Gaussian03.58
Single-point interaction energies for CHLB and BROB with the protein model compounds were obtained at the RIMP2 level of theory with the cc-pVQZ basis set55 using PSI4.59 The basis set superposition error (BSSE) was corrected using the counterpoise method.60 In the interaction energy calculations, the intramolecular geometries of the two interacting molecules were kept rigid in their gas-phase QM optimized geometries. In calculating the QM interaction energies, when the halogen acts as a hydrogen bond acceptor (HBA), the hydrogen bond donor (HBD) hydrogen of each moiety was directed toward the halogen in both the perpendicular and linear orientations (Figure S2 and S4). When the halogen participates in a halogen bond interacting with a HBA, the HBA atom of each moiety was directed toward the halogen along C-X direction (linear orientation, Figure S3 and S5). The interaction energy profiles were calculated by varying the distance between the halogen and the interacting atom from 1.5 to 5.0 Å in intervals of 0.1 Å.
Molecular mechanical calculations
Additive force field calculations used the CHARMM General Force Field53,54 for the small model compounds and the CHARMM36 protein model.52 We note that the present parameters will also be compatible with the earlier C22/CMAP61,62 and more recent C36m parameters63 as well as the remainder of the C36 macromolecular force field.64–70 Polarizable calculations used the Drude-2013 protein force field71 with the parameters for the ligands built based on the published Drude polarizable parameters for small molecules32,34,38 as well as the new halogen-protein non-bond parameters optimized in the present study. In addition, dihedral linkages in the ligands not previously optimized were refined as part of the present study (see Supporting Information Table S17). Molecular mechanics calculations were performed with the programs CHARMM52–54 and NAMD72.
To calculate the interaction energies of the halogenated benzenes with the protein model compounds, the geometries in each interaction orientation were identical to those used in the QM calculations. In the additive model, the interaction energies were obtained by taking the energy difference between the CHLB/BROB-protein model compound complex and the CHLB/BROB/protein model compound monomers (e.g. equivalent to the interaction energy between the halogenated benzenes and protein model compound monomers). In the Drude model, relaxation of the Drude particles (i.e. self-consistent field (SCF) calculation) was performed via minimization by the steepest-descent (SD) and adopted-basis Newton-Raphson (ABNR) algorithms to reach a force gradient of 10−5 kcal·mol−1·Å−1 while the atomic positions were restrained with a force constant of 107 kcal/mol/Å2. This was performed for the individual monomers (Emonomer) and then for the complex (Ecomplex) with the interaction energy calculated as Einter = Ecomplex – (Emonomer1 + Emonomer2).
MD simulations were performed on eight systems including seven complexes that include halogenated ligands and one protein crystal with a halogenated lysine. Starting coordinates for all the protein-ligand complexes/modified protein were taken from the Protein Data Bank.6 Systems under study include human thrombin (PDB id: 2ZC9),73 protein kinase (PDB id: 2UW8),74 eukaryotic initiation factor 4E (PDB id: 4DT6),75 human MutT Homolog1 (MTH1) protein (PDB id: 4N1T),76 transcriptional activator protein LasR (PDB id: 3IX8),77 PDZ domain with 4-bromobenzoic acid modified lysine (LYS9) (PDB id: 4NMV),78 aminoacyl-tRNA synthetase (PDB id: 2AG6),79 and phenazine biosynthesis protein (PDB id: 3JUM).80 The protein coordinate and structure files were initially prepared in CHARMM additive formats using the PDB Reader module in the CHARMM-GUI.81 These additive coordinate and structure files were then submitted to the Drude Prepper module in the CHARMM-GUI81 to obtain files in Drude format.
For the polarizable Drude simulations the systems were solvated in a rectangular box with a 10 Å minimum distance between the edge of the box and the protein with the SWM4-NDP water82 and Na+ or Cl− ions83,84 were added to neutralize the systems as required. MD simulations were carried out using NAMD.72 The dual Langevin thermostat integrator was used with a 20 ps−1 damping coefficient for Drude oscillators and a 5 ps−1 damping coefficient for the atoms.85 SHAKE was used to fix bonds involving hydrogen atoms.86 Short-range LJ forces were switched to zero from 10–12 Å.87 Electrostatic interactions were computed with the smooth particle mesh Ewald method with a real space cutoff of 12 Å, a kappa factor of 0.34 and a 6-order spline.88,89 Each system was simulated under isothermal-isobaric (NPT) conditions at the temperature where the crystals were obtained, and the pressure was set at 1 atm using Langevin piston pressure control with a piston oscillation period of 200 fs and a relaxation time of 100 fs. A 100ps equilibration was performed with a 0.5 fs time step with all heavy atom restrained using a harmonic force constant of 1 kcal/mol·Å2 followed by a 20 ns production simulation with a 1.0 fs time step. In the production simulations weak restraints were applied to the backbone Cα atoms of all residues that did not have one or more atoms within 8 Å of the ligand in the crystal structure using a harmonic force constant of 0.5 kcal/mol·Å2. MD simulations were performed with and without the updated atom pair-specific LJ parameters (NBFIX in CHARMM nomenclature) for all the systems respectively based on the Drude model. All analyses were carried out using facilities within CHARMM,52–54 and statistical analysis was based on three independent simulations using different random seeds to assign the initial velocities.
Additive MD simulations were performed following the same protocols as those for the Drude calculations with the following exceptions. The CHARMM36 force field90 was used for the protein. Ligand topologies were generated using CGenFF15,16 and water was treated using the CHARMM TIP3P model.91 LJ forces were force switched to zero from 10–12 Å.87 Pressure control was based on a Nosé–Hoover Langevin piston algorithm.92,93 A 100ps equilibration was performed with a 1.0 fs time step with all heavy atoms restrained using a harmonic force constant of 1 kcal/mol·Å2, followed by a 20 ns production simulation with a 2.0 fs time step. As with the Drude production simulations weak restraints were applied to the backbone Cα atoms of all residues that did not have one or more atoms within 8 Å of the ligand in the crystal structure using a harmonic force constant of 0.5 kcal/mol·Å2. Similarly, MD simulations were performed with and without the updated NBFIX parameters, respectively, based on the additive model. Note that in the previously released additive halogen force field,13 NBFIX parameters were included between only aromatic chlorine and the carbonyl oxygen on the protein backbone. In the present study, additional NBFIX parameters were applied between chlorine/bromine and other protein model compounds.
Interaction energies for the additive model were calculated using the “INTER” command in CHARMM that yields the sum of the LJ and electrostatic interactions between the interacting partners.52–54 For the polarizable model, it is necessary to perform a SCF calculation on each of the individual interacting partners as well as on the full complex to allow for relaxation of the electronic degrees of freedom in analogy to the QM Born-Oppenheimer approximation. The SCF calculation in the Drude model was performed by allowing the Drude particles to relax to their local energy minima for the given fixed configuration of their parent nuclei. This was performed by constraining all real atoms (e.g. applying the “CONS FIX” command in CHARMM52–54) and relaxing the Drude particles by minimizing them through the steepest-descent algorithm with a step size of 0.01 to a force gradient of 10−2 kcal·mol−1·Å−1 followed by the adopted basis Newton-Raphson algorithm with a step size of 0.02 to a force gradient of 10−5 kcal·mol−1·Å−1. Energies of the systems were then calculated following removal of constraints on the real atoms. Interaction energies were then calculated using the following three methods (Figure 2).
Figure 2.


Schematic depictions of the interaction energy calculations based on methods a) 1, b) 2, and c) 3. Protein is colored in yellow, ligand in green, and water are labeled as “w”.
Method 1:
The relaxation of the Drude particles was performed after the water molecules were removed followed by calculation of the total energy of the complex (E1comp_relaxD_E_w/o_water), the protein alone (E1prot_relaxD_E_w/o_water) and the ligand alone (E1ligand_relaxD_E_w/o_water). The interaction energy (Einter1) was then calculated through equation 1:
| (Eq. 1) |
Method 2:
The relaxation of the Drude particles was performed for the complex, protein-alone, and ligand-alone systems in the presence of the water (comp_relaxD_w/_water, prot_relaxD_w/_water, ligand_relaxD_w/_water, respectively). Then, the water molecules were removed for calculating the total energies of the complex (E2comp_w/o_water), protein-alone (E2prot_w/o_water) and ligand-alone (E2ligand_w/o_water) systems, respectively, without additional relaxation of the Drude particles. The interaction energy (E2inter) was calculated through equation 2:
| (Eq. 2) |
Method 3:
The relaxation of the Drude particles was performed for the complex and protein-alone systems in the presence of water, respectively, to obtain total energy for the complex (E3comp_relaxD_w/_water) and protein alone (E3prot_relaxD_w/_water). Water was not included in the relaxation of Drude particles in the ligand-alone systems for calculation of the ligand energies (E3ligand_relaxD_w/o_water, which is equal to E1ligand_relaxD_w/o_water). The interaction energy (E3inter) was calculated through equation 3:
| (Eq. 3) |
To calculate the variation of dipole moments in different environments for the neutral ligands, the ligand conformations from the crystal structures were submitted to aqueous phase simulations for both the additive and Drude models in NAMD.72 Aqueous phase simulations were performed by solvating the ligands in a cubic water box with the box size determined by a 10 Å distance between the longest edge of the box and the ligand, with a harmonic force constant of 0.5 kcal/mol·Å2 applied to the non-hydrogen atoms of the ligands. The rest of the parameters are the same as those performed in ligand-protein simulations for the additive and Drude models, respectively. A 10 ns NPT simulation of pure liquid water was performed using CHARMM52–54 with a 19×19×19 Å cubic box of 216 water molecules of the SWM4-NDP water model,82 from which the mean dipole moment of water in the bulk phase was calculated based on the mean of the average dipole moment of each water molecule over the entire simulation. In addition, the root-mean-square fluctuations of the water dipole were calculated from the trajectory.
Results and Discussion
Halogens, such as chlorine, bromine and iodine, can act as hydrogen bond acceptors (HBA) to interact favorably with hydrogen bond donors (HBD) on proteins, referred to as X-HBD interactions, along with participating in halogen bonds (XB) with HBAs. XB interactions have also been referred to as X-HBA interactions.7 In the development of the halogen polarizable force field, good reproduction of both the QM X-HBD and XB water interactions was achieved by applying NBFIX parameters between the X-Drude particle and water hydrogen as well as X and water oxygen atoms.49 Based on the same strategy, NBFIX parameters are optimized targeting QM interactions with protein model compounds with the goal of better X-HBD/XB interactions with protein residues. While comparing the QM results with the additive model, the agreement with QM X-HBD interactions was found not to be satisfactory, although the NBFIX between the chlorine and the carbonyl oxygen of the protein backbone model compound (NMA) was previously added. To improve the additive model, additional NBFIX parameters were also included and optimized. Finally, both the polarizable and additive force fields with optimized NBFIX parameters were applied in the MD simulations of protein-halogenated ligand (or modified residue) complexes to examine the ability of the resulting models in reproducing the halogenated-ligand geometries. A schematic illustration of the overall parametrization procedure is shown in Figure S1.
QM interaction energies with protein model compounds
Optimization of atom pair-specific LJ parameters targeted both QM perpendicular X-HBD (X-HBD90°) and linear X-HBD/XB (X-HBD180° and XB180°) interaction energies with the protein model compounds. NMA served to model the protein backbone while other protein model compounds were used to model the side chains (Figure 1). Without the NBFIX parameters, the interaction energies with NMA displayed poor agreement with the QM target data in the Drude model (Figure 3, Init-Drude in gray). The X-HBD90° interaction energies between the halogens and NMA are too favorable with shorter interaction distances whereas the XB180° interaction energies are less favorable with longer interaction distances compared to the QM data. To overcome this disagreement, NBFIX terms between the halogen-Drude (X-Drude) and the hydrogen on amide NH in NMA as well as NBFIX between the X and carbonyl oxygen in NMA were added, resulting in better reproduction of both the X-HBD and XB (halogen bond) interactions (Figure 3, Opt-Drude in black). For the side-chain protein model compounds, the inclusion of NBFIX parameters yielded significant improvement in the balance of these interaction orientations (Figure S2–S3 for CHLB and Figure S4–S5 for BROB, Init-Drude in gray and Opt-Drude in black). Note that as the atom type of all polar hydrogens among the HBDs are the same,39 the perpendicular X-HBD90° interactions among all the protein model compounds required compromise. The NBFIX parameters for the Drude model can be found in Table S15.
Figure 3.

Interaction energies between chlorobenzene (CHLB)/bromobenzene (BROB) and NMA acting as hydrogen bond donors (HBD) and a hydrogen bond acceptor (HBA). a-b are X-HBD90° and X-HBD180° interactions, c-d are XB180° interactions. QM results are in blue, initial Drude force field without NBFIX parameters (Init-Drude) are in gray, and optimized Drude force field with NBFIX parameters (Opt-Drude) are in black. Hydrogens are white, carbons are cyan, nitrogens are blue, oxygens are red, chlorines are green, and bromines are dark red.
In the additive model, the NBFIX between chlorine and carbonyl oxygen were already parametrized based on NMA13 such that XB180° interactions with NMA are quite good from the initial CHARMM36/CGenFF model (Figure 4c). However, the X-HBD90 ° interactions displayed too favorable interaction energies with shorter interaction distance (Figure 4a, Init-CGenFF in gray). To improve these interactions, the NBFIX approach was again taken. Considering that the atom type of the hydrogen is already shared with many other compounds, the NBFIX parameters are added for the X atom with the hydrogen-bond donor or acceptor non-hydrogen atoms (e.g. nitrogen or oxygen) of the model compounds (except MESH, explained below). With NBFIX parameters between the halogen and the nitrogen on the amide in NMA, the X-HBD90 ° interactions with NMA are significantly improved (Figure 4a–b, Opt-CGenFF in black). As for the interaction energies with the protein side-chain model compounds, most of the X-HBD90°/XB180° are in good agreement with QM data using the optimized NBFIX parameters (Figure S6–S9, Opt-CGenFF). With the MEOH/PHEN interactions, no NBFIX terms were required as the initial CGenFF parameters already achieved an acceptable balance of the various interactions, though some compromise of the X-HBD90 ° and XB180 ° interactions is present. With the charged species, ACET/MAMM/MGUAN, larger well-depth parameters were required to better reproduce the QM interaction energy surfaces. This use of larger well-depth parameters has been previously applied to molecules participating in interactions with significant electrostatic contributions.94 Given that the hydrogen atom type on MESH is unique to thiol groups in the additive model, NBFIX parameters were applied between the halogen and hydrogen on MESH to improve X-HBD interactions. The NBFIX parameters for the additive model can be found in Table S16.
Figure 4.

Interaction energies between chlorobenzene (CHLB)/bromobenzene (BROB) and NMA acting as hydrogen bond donors (HBD) and a hydrogen bond acceptor (HBA). a-b are X-HBD90° and X-HBD180° interactions, c-d are XB180° interactions. QM results are in blue, initial additive CGenFF force field without NBFIX parameters (Init-CGenFF) are in gray, and optimized additive CGenFF force field with NBFIX parameters (Opt-CGenFF) are in black. Hydrogens are white, carbons are cyan, nitrogens are blue, oxygens are red, chlorines are green, and bromines are dark red.
A summary of the differences between QM and the Drude/additive minimum interaction energies and distances with the protein model compounds is shown in Table 1. The individual interaction energies and distances are shown in Table S1–S4. All the interaction energies computed using the NBFIX parameters (Opt-Drude and Opt-CGenFF) yield average difference (AVG), averaged unsigned errors (AUD), and root-mean-square differences (RMSD) generally smaller than those in the initial parameters that lacked the NBFIX parameters (Init-Drude and Init-CGenFF). The AVG of Cl-HBD interaction is somewhat larger in the Opt-Drude result compared to the Init-Drude; however, its AUD and RMS are smaller as are those for XB (X=Cl) or X-HBD/XB (X=Br) interactions. Although some of the HBD180° interactions, which are less favorable than the HBD90° or XB interactions, are still poorly treated both with and without the NBFIX parameters (Tables S1–S4), similar to those with water,49 it is evident that including the NBFIX parameters for both the optimized Drude and additive models achieved substantial improvements in the balance of gas phase X-HBD/XB interaction orientations.
Table 1.
Statistical comparison of the Drude and CGenFF computed minimum interaction energies (Emin, kcal/mol) and distances (R, Å) with QM data for chlorobenzene (CHLB) or bromobenzene (BROB) interacting with the protein model compounds. Presented are average differences and standard errors. (AVG), absolute unsigned error (AUD), and root-mean-square differences (RMSD) without or with atom pair-specific LJ terms compared with their QM target data of halogen-hydrogen bond donor (X-HBD) interactions and halogen bond (XB) interactions or ALL. Individual interaction results are shown in Table S1–S4. The errors are the standard errors.
| Mol. | Init-Drude | Opt-Drude | Init-CGenFF | Opt-CGenFF | ||||
|---|---|---|---|---|---|---|---|---|
| CHLB | Emin | R | Emin | R | Emin | R | Emin | R |
| ALL | ||||||||
| AVG | 0.30 ± 0.14 | 0.0 ± 0.1 | 0.24 ± 0.11 | 0.0 ± 0.0 | 0.92 ± 0.33 | 0.1 ± 0.1 | 0.54 ± 0.20 | 0.1 ± 0.1 |
| AUD | 0.51 | 0.3 | 0.34 | 0.1 | 1.05 | 0.4 | 0.60 | 0.3 |
| RMSD | 0.73 | 0.3 | 0.57 | 0.2 | 1.84 | 0.5 | 1.12 | 0.4 |
| Cl-HBD | ||||||||
| AVG | 0.21 ± 0.16 | −0.1 ± 0.1 | 0.33 ± 0.13 | 0.0 ± 0.0 | 0.94 ± 0.40 | 0.1 ± 0.1 | 0.65 ± 0.27 | 0.1 ± 0.1 |
| AUD | 0.48 | 0.3 | 0.40 | 0.2 | 1.10 | 0.5 | 0.73 | 0.3 |
| RMSD | 0.70 | 0.3 | 0.65 | 0.2 | 1.89 | 0.5 | 1.29 | 0.4 |
| XB, X=Cl | ||||||||
| AVG | 0.59 ± 0.27 | 0.3 ± 0.1 | −0.04 ± 0.08 | 0.0 ± 0.0 | 0.89 ± 0.64 | 0.1 ± 0.1 | 0.21 ± 0.05 | 0.0 ± 0.0 |
| AUD | 0.59 | 0.3 | 0.16 | 0.1 | 0.89 | 0.2 | 0.21 | 0.1 |
| RMSD | 0.84 | 0.3 | 0.19 | 0.1 | 1.69 | 0.2 | 0.24 | 0.1 |
| BROB | Emin | R | Emin | R | Emin | R | Emin | R |
| ALL | ||||||||
| AVG | 0.31 ± 0.08 | 0.2 ± 0.1 | 0.11 ± 0.06 | 0.2 ± 0.1 | 0.99 ± 0.35 | 0.3 ± 0.1 | 0.58 ± 0.22 | 0.3 ± 0.1 |
| AUD | 0.38 | 0.4 | 0.26 | 0.2 | 1.11 | 0.5 | 0.62 | 0.4 |
| RMSD | 0.48 | 0.6 | 0.32 | 0.3 | 1.95 | 0.7 | 1.20 | 0.5 |
| Br-HBD | ||||||||
| AVG | 0.29 ± 0.10 | 0.2 ± 0.1 | 0.21 ± 0.06 | 0.2 ± 0.1 | 0.92 ± 0.39 | 0.4 ± 0.2 | 0.69 ± 0.28 | 0.4 ± 0.1 |
| AUD | 0.39 | 0.4 | 0.26 | 0.2 | 1.07 | 0.7 | 0.71 | 0.5 |
| RMSD | 0.50 | 0.5 | 0.33 | 0.3 | 1.84 | 0.8 | 1.36 | 0.6 |
| XB, X=Br | ||||||||
| AVG | 0.36 ± 0.11 | 0.3 ± 0.1 | −0.20 ± 0.10 | 0.1 ± 0.0 | 1.23 ± 0.85 | 0.2 ± 0.0 | 0.28 ± 0.16 | 0.0 ± 0.1 |
| AUD | 0.37 | 0.3 | 0.27 | 0.1 | 1.23 | 0.2 | 0.34 | 0.1 |
| RMSD | 0.43 | 0.3 | 0.30 | 0.1 | 2.26 | 0.2 | 0.46 | 0.1 |
MD simulations of ligand-protein crystal structures
Validation of the force field was performed by examining its ability to capture the halogenated ligand (or modified residue)-protein interactions with the aim of reproducing their binding geometries from MD simulations initiated from the crystal structures. In both the Drude and additive models, NBFIX parameters directly obtained from targeting QM CHLB- and BROB-protein model compounds interactions yielded good reproduction of the ligand-protein geometries as compared to the crystal structures, as described below. In the MD simulations weak harmonic restraints were applied to all Cα atoms on residues that did not have one or more atoms within 8 Å of the halogenated ligands or residue to limit deviations of the overall protein structures thereby assuring that the results from simulations were dominated by the ligand-protein interactions.
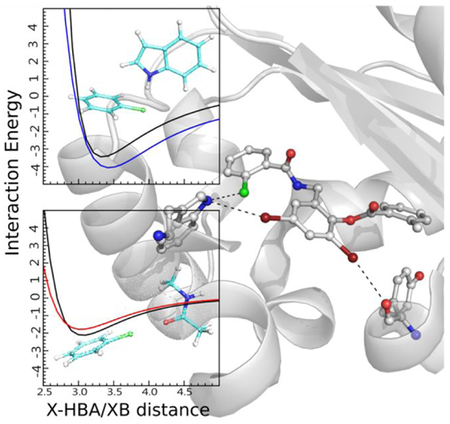
An example of one of the systems studied, the LasR ligand-protein complex,77 is shown in Figure 5. The ligand contains both Cl and Br. The Cl and one Br participate in X-HBD interactions with the indole NH moiety of Trp60 in approximately perpendicular and linear orientations, respectively. The other Br participates in an XB interaction with the backbone O of Tyr47. Analysis of the remaining complexes in Figures S10 to S17 of the supporting information shows them to encompass a range of halogen-protein interactions as required to assure a diverse range of interactions were being tested.
Figure 5.

Illustration of X-HBD and XB interactions in the LasR protein-ligand complex (PDB id: 3IX8, 1.80 Å).77 a. 2D representation of the halogenated ligand TX3. b. The ligand structure and the region of the protein in the vicinity of the ligand halogen atoms are shown. Chlorine is colored in green, bromine in dark red, nitrogen in blue, oxygen in red and carbon in white.
For quantitative analysis all the X-HBD or XB interactions from the crystal structures, the X…heavy atom distances within 4.5 Å were used as target data. These target data include the X…HBD and X…HBA distances and the C-X…HBD and C-X…HBA angles. Distances and angles from the MD trajectories were calculated to compare with those in the crystals. The deviations of the halogenated ligand (or modified residue) interaction geometries obtained from the MD simulations based on the developed force field parameters are summarized in Table 2, with detailed analysis for each crystal MD simulation presented in Tables S5–S12. With the human MutT Homolog1 (MTH1) protein (PDB id: 4N1T) the ligand geometry was poorly reproduced with both the initial Drude and CGenFF models;13 however, significantly better agreement with the crystal structure was achieved with the optimized additive and Drude models (see Table S8). Accordingly, the global analysis in Table 2 was performed both with and without those results.
Table 2.
Analysis of the angle (ϕ, deg) and distance (r, Å) deviations of the protein-ligand intermolecular simulated geometries compared to the crystal structures for all eight protein systems. Average differences (AVG) and standard error (SE), absolute unsigned error (AUD), root-mean-square differences (RMSD), and the number of each type of interaction (Count) were obtained from three independent simulations.
| All systems | All systems excluding PDB:4NIT | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ALLa | Init-Drude | Opt-Drude | Init-CGenFF | Opt-CGenFF | Init-Drude | Opt-Drude | Init-CGenFF | Opt-CGenFF | |
| AVG | ϕ | −9.59 | −3.53 | −9.13 | −5.13 | −2.19 | −2.19 | −4.43 | −4.44 |
| r | 0.57 | 0.21 | 0.63 | 0.17 | 0.33 | 0.18 | 0.20 | 0.16 | |
| SE | ϕ | 3.77 | 1.49 | 2.71 | 1.45 | 1.24 | 1.51 | 1.23 | 1.60 |
| r | 0.14 | 0.07 | 0.23 | 0.06 | 0.08 | 0.07 | 0.09 | 0.07 | |
| AUD | ϕ | 12.34 | 7.09 | 8.74 | 7.09 | 5.15 | 6.05 | 6.14 | 6.88 |
| r | 0.63 | 0.33 | 0.58 | 0.29 | 0.40 | 0.30 | 0.32 | 0.30 | |
| RMSD | ϕ | 23.06 | 9.02 | 14.62 | 9.47 | 6.80 | 8.17 | 7.78 | 9.43 |
| r | 0.95 | 0.44 | 1.21 | 0.37 | 0.52 | 0.42 | 0.51 | 0.38 | |
| Count | 32 | 28 | |||||||
| X-HBDb,c | Init-Drude | Opt-Drude | Init-CGenFF | Opt-CGenFF | Init-Drude | Opt-Drude | Init-CGenFF | Opt-CGenFF | |
| AVG | ϕ | 1.66 | 4.30 | −4.82 | −3.41 | ||||
| r | 0.40 | 0.18 | 0.23 | 0.12 | |||||
| SE | ϕ | 0.97 | 1.76 | 3.22 | 3.20 | ||||
| r | 0.15 | 0.10 | 0.07 | 0.09 | |||||
| AUD | ϕ | 2.64 | 5.37 | 8.56 | 7.00 | ||||
| r | 0.44 | 0.26 | 0.24 | 0.23 | |||||
| RMSD | ϕ | 2.90 | 6.08 | 9.25 | 8.54 | ||||
| r | 0.55 | 0.31 | 0.29 | 0.26 | |||||
| Count | 7 | ||||||||
| XBd | Init-Drude | Opt-Drude | Init-CGenFF | Opt-CGenFF | Init-Drude | Opt-Drude | Init-CgenFF | Opt-CGenFF | |
| AVG | ϕ | −23.82 | −10.54 | −13.71 | −4.14 | −9.82 | −9.93 | −0.55 | −2.55 |
| r | 0.79 | 0.30 | 0.91 | −0.08 | 0.23 | 0.17 | −0.14 | −0.16 | |
| SE | ϕ | 14.21 | 2.36 | 13.21 | 2.43 | 3.20 | 2.95 | 1.42 | 2.37 |
| r | 0.57 | 0.17 | 1.06 | 0.17 | 0.16 | 0.14 | 0.20 | 0.19 | |
| AUE | ϕ | 23.82 | 10.54 | 14.70 | 5.27 | 9.82 | 9.93 | 1.79 | 3.97 |
| r | 0.87 | 0.40 | 1.27 | 0.33 | 0.34 | 0.29 | 0.32 | 0.35 | |
| RMSD | ϕ | 37.09 | 11.55 | 29.76 | 6.38 | 11.28 | 11.17 | 2.53 | 4.83 |
| r | 1.39 | 0.46 | 2.30 | 0.35 | 0.36 | 0.29 | 0.37 | 0.37 | |
| Count | 5 | 4 | |||||||
All indicates all interactions based on all protein nitrogen and oxygen atoms within 4.5 Å of the ligand halogen atoms in the crystal structures.
X-HBD results limited to interaction geometries in Table S5–S12 where C-X..O/NHBD angles from crystals are less than 120° and belong to HBD groups (i.e. Cl…NSER in PDB id 2ZC9, Cl…NTYR and Cl…OSER in PDB id 4DT6, Br…NHIS in PDB id 3JUM, Br…OSER in PDB id 4NMV, BR…NHIS and Br…OTYR in PDB id 2AG6 of Table S5–S12).
As there are no X-HBD interaction with PDB id: 4N1T, only one set of X-HBD interactions are presented.
XB results are limited to interaction geometries in Table S5–S12 where C-X..O/NHBA angles from crystals are between 150–180° and belong to HBA groups (i.e. Cl…NGLY in PDB id 2UW8, Cl…OTRP and Cl…NPHE in PDB id 4DT6, Br…OTYR in PDB id 3IX8, Cl…OTHR in PDB id 4N1T of Table S5–S12).
When all the complexes and ligand-protein interactions are taken into account, both the optimized Drude and additive CHARMM36/CGenFF models yield substantial overall improvements in both the interaction distances and angles. Omission of the 4NIT system leads to the systematic improvement in the angles no longer being present, though the interaction distances are improved.
Analysis was also performed separately on X-HBD and XB interactions as defined by the criteria listed in the legend of Table 2. With the X-HBD interactions the distances are significantly better with the optimized Drude parameters while there is a degradation of the angles, though the standard errors of the angle differences are large. Optimization of additive CHARMM36/CGenFF leads to a systematic improvement in the treatment of the X-HBD interactions. With the XB interactions improvements occur with all interaction angles and distances with the optimized parameters when complex 4N1T is included in the analysis. When that structure is omitted, the optimized Drude model yield better agreement for the distances while the angles are similar and again show large standard errors. With the additive CHARMM36/CGenFF the optimized model performs slightly worse for the C-X…HBA angles while the distance differences are similar. The lack of differences is due, in part, to the presence of an NBFIX term in the initial CHARMM36/CGenFF model for the interaction of Cl with the peptide carbonyl O that is present in 5 of the complexes studied. Thus, the inclusion of atom pair-specific halogen-protein LJ parameters leads to improvements in the treatment of X-HBD interactions, especially with the distances in the Drude model. This trend was also present in the XB interactions for the Drude model though improvements did not occur with the additive CGenFF model. Notably, the inclusion of the optimized parameters avoided the extremely poor treatment of the 4N1T system.
To check if the deviations of the ligand-protein interactions were associated with shifts in the ligand or protein, conformational analysis was performed on the root-mean-square differences (RMSD) of the ligands and of the unrestrained, local protein backbone structure. In addition, root-mean-square fluctuation (RMSF) analysis of the local protein backbone structure was performed. Supporting information Table S13 shows only minimal conformational changes in the protein, with all RMSD values less than 1.0 Å. Larger differences are present for the ligands with the average RMSDs for both optimized FFs being below 1.5 Å. The largest RMSD values were obtained with the 4N1T system, with the significant improvement associated with the optimized parameters reflected in decreased RMSD values. Similar improvements were obtained with the 3JUM system. While within the standard errors, the Drude FF typically has larger RMSD values as compared to the additive FF. Contributing to this may be slightly larger fluctuations for the ligand with the optimized Drude model versus the additive, though the protein RMSF were similar (Table S14). These analyses indicate that more global structural or dynamical changes were not significantly impacting the Drude vs. additive results, which is expected given the use of harmonic restraints on protein Cα atoms remote from the ligands.
Ligand-Protein Interaction Energies
An accurate prediction of ligand-protein interaction energies is critical in the development of the scoring function in drug design.95 Recent studies have shown that the electronic polarization contributes to the electrostatic interaction energy based on the QM/MM studies.96–98 Accordingly, analysis was undertaken on the contribution of polarization to the interaction energy in the Drude model as such analysis, to our knowledge, has not yet been reported. Four approaches were used to calculate the protein-ligand interaction energies for the Drude model using the trajectories from the crystal MD simulations, which were compared to interaction energies obtained with the additive FF. In Table 3 the interaction energies using Drude-E1inter~Drude-E3inter methods that include the relaxation of the Drude particles are systematically more favorable than the additive values as well as Drude values without relaxation of the electronic degrees of freedom (i.e. Drude-E4inter using INTER command in the CHARMM program52–54) with the exception of 4DT6. These differences indicate that the treatment of the polarization effect contributes to the ligand-protein interaction energy as expected based on previous QM/MM studies.96–98
Table 3.
Ligand–protein interaction energies based on the optimized additive and Drude polarizable force fields.
| PDB | Opt-CGenFF | Opt-Drude | |||
|---|---|---|---|---|---|
| Metdodb | Einter | E1inter | E2inter | E3inter | E4inter |
| 2ZC9 | −59.1 ± 1.3 | −106.1 ± 1.6 | −104.5 ± 2.0 | −177.1 ± 0.4 | −66.4 ± 0.8 |
| 2UW8 | −54.8 ± 0.6 | −136.2 ± 7.22 | −135.9 ± 6.1 | −117.8 ± 2.2 | −68.0 ± 2.1 |
| 4DT6 | −119.9 ± 0.3 | −195.3 ± 6.7 | −192.6 ± 7.0 | −344.6 ± 2.8 | −85.5 ± 1.2 |
| 4N1T | −37.2 ± 0.6 | −42.6 ± 2.9 | −42.0 ± 3.1 | −57.2 ± 2.5 | −44.3 ± 3.0 |
| 31X8 | −75.6 ± 0.2 | −96.5 ± 0.1 | −96.7 ± 0.2 | −99.2 ± 0.2 | −92.6 ± 0.4 |
| 2AG6 | −53.8 ± 0.1 | −77.0 ± 0.2 | −71.6 ± 0.5 | −146.9 ± 3.2 | −74.6 ± 0.2 |
| 3JUM | −83.3 ± 6.8 | −149.8 ± 3.2 | −145.0 ± 2.4 | −345.9 ± 10.9 | −83.2 ± 4.0 |
Comparison of the interaction energies calculated with the Drude model with those from the additive model shows the correlation to be relatively high (Figure 5). The negative y-intercepts show the Drude interactions to be more favorable than the additive values while the larger slopes suggest the polarizable interaction energies that include relaxation of the Drude may be more sensitive to the nature of the interactions with the Drude model than with the additive. Such an effect suggests that polarizable models may be more sensitive to ligand modifications, though explicit studies addressing this point are necessary. Interestingly, the Drude interaction energy without electronic relaxation, E4inter, is less sensitive to the environment than the additive. This may be due to the overpolarization of the charges in the additive model designed to take into account the omission explicit treatment of electronic polarization99 while such contributions are not present in the Drude model when the electronic degrees of freedom are not allowed to relax in the different environments. The results clearly indicate the importance of allowing the electronic degrees of freedom to relax when calculating interaction energies with the Drude or any polarizable force field.
Concerning the three Drude interaction energy methods that include relaxation of the electronic degrees of freedom, method 1 and 2 give similar results with method 3 differing significantly. The absence and inclusion of water when calculating the polarization response in methods 1 and 2, respectively, while yielding similar results suggest that the overall impact of the water environment is not significant (E1inter and E2inter in Table 3). When water is included as part of the protein and the complex during the interaction energy calculation (i.e. method 3), the energies are often more favorable, though exception exists with complexes 2UW8 and 3IX8 where they are slightly less favorable or similar, respectively (E3inter in Table 3). This variation indicates the complex nature of the contribution of water to binding, another area that requires additional study. Given the similarity of the E1inter or E2inter methods and their high correlation with the additive interaction energy calculations (Figure 5), the present analyses suggest that the E1inter or E2inter methods are most appropriate for interaction energy analysis using the Drude or other polarizable force field.
An important contribution to binding comes from the electronic self-polarization energy when applying polarizable MM or QM methods. Analysis of the contribution of the electronic self-polarization energy (or self-energy) to the interaction energies varies widely (Table 4). These contributions go from large favorable to large unfavorable contributions, with those ranges of variations occurring for both between and within the individual systems. While interaction energies E1inter and E2inter are similar, the Drude self-energy contributions (E1self and E2self) are substantially anticorrelated (R=−0.72). This suggests differential impact of the aqueous environment on the protein versus ligand, though these contributions balance leading to the similar interaction energies (R=1.00). On the other hand, the self-energy contributions of E2self and E3self, in which the Drude particle relaxation includes contributions from water in both the ligand and the protein or just the protein, respectively, show good correlation (R=1.00). This similarity suggests that the impact of water is dominating the polarization of the protein over the ligand, a results that may be considered obvious given the relative size of the proteins and extent of exposure of the protein versus the ligand to the aqueous solvent.
Table 4.
Ligand–protein interaction self-energies (Eself) based on the optimized Drude polarizable force field.a Errors shown are standard errors based on the three independent simulations for each system.
| PDB | Opt-Drude-El | Opt-Drude-E2 | Opt-Drude-E3 |
|---|---|---|---|
| Metdod | E1self | E2self | E3self |
| 2ZC9 | −101.0 ± 2.7 | −5.6 ± 7.6 | 15.7 ± 6.1 |
| 2UW8 | −42.0 ± 12.0 | −93.3 ± 11.4 | −99.2 ± 11.6 |
| 4DT6 | −126.2 ± 6.9 | 39.9 ± 12.0 | 51.0 ± 13.4 |
| 4N1T | 2.2 ± 1.6 | 8.9 ± 5.5 | 13.1 ± 6.5 |
| 3IX8 | −2.5 ± 0.8 | −11.3 ± 2.5 | −6.0 ± 2.3 |
| 2AG6 | 78.6 ± 15.7 | 9.2 ± 24.9 | 24.9 ± 35.2 |
| 3JUM | −237 ± 25.3 | 292.7 ± 21.8 | 334.0 ± 17.3 |
In methods 1–3, each self-energy contribution from the complex, protein and ligand were obtained (e.g. Eself, comp, Eself, prot, and Eself, ligand). The self-energy for each component is calculated from the total bond energy minus the bond energy with the Drude particles omitted based on the geometry after Drude particles are relaxed in the SCF calculation. Eself is calculated as Eself, comp - Eself, prot - Eself, ligand. Self-energies from method 4 are zero, i.e. Eself, comp = Eself, prot + Eself, ligand, as no relaxation of Drude particles is performed.
Favorable self-energy contributions to ligand-protein interactions energies are somewhat counterintuitive. They indicate that the self-energies are larger (i.e. less favorable) in the monomers than in the complex, indicating that the effective electric field acting on the protein and ligand is smaller in the complex than in the individual monomers. Such an effect is due to the intramolecular contributions to the electric field along with condensed phase environment intermolecular contributions. These latter contributions lead to the anticorrelated nature of the E1self and E2self terms. It is interesting that despite this difference the E1inter and E2inter terms are similar. Given that the difference between the two calculations is the absence or presence of water in the SCF calculation, the similarity of the interactions energies suggest that the assumption of an isotropic polarization contribution inherent in additive models is not unreasonable.99 However, it should be emphasized that the similarity between the E1inter and E2inter terms does not indicate that water is not contributing to the overall binding affinity of ligands to proteins. Indeed the significant difference in E3inter versus E1inter and E2inter indicates a significant contribution of water itself. Additional studies are required to develop approaches to appropriately account for the contribution of water to binding in the context of both additive and polarizable force field.
Variation of the Dipole moments
To demonstrate the effect of the explicit treatment of polarization, including the response to different environments, dipole moments of the neutral ligands were obtained from a 20 ns MD simulations of the ligands in the aqueous phases and from the three 20 ns protein-ligand simulations (Table 5). In the aqueous phase simulations the ligand conformation was restrained to that present in the crystal structure to limit the analysis to the impact of the protein vs. aqueous environment on the dipole moments. Significantly larger changes in the dipole moments would also occur in most systems due to change in the ligand conformation, though such analysis requires full conformational sampling of the ligand in solution. The difference in the dipole moments of the full ligands vary from the aqueous phases to the protein with both the additive and polarizable models generally by 1 Debye or less, though with the polarizable FF there is a significant change of almost 3 Debye with 4NIT. Comparison of the RMS fluctuations shows generally higher values with the Drude model. This is anticipated as the fluctuations occurring with the additive force field are associated with changes in conformation of the ligands, as no change in electronic structure is possible, while both conformational and electronic effects contribute to the changes with the Drude model.
Table 5.
Dipole moments and RMS fluctuations (Debye) of each neutral ligand and their halogenated aromatic groups (Ar-X) in the aqueous solution and in the ligand-protein (w/Protein) simulations for the optimized Drude and additive force fields.
| PDBID | Aqueous | w/Protein | Watera (Aqueous) | Waterb (w/Protein) | |
|---|---|---|---|---|---|
| Full ligand | |||||
| Additive | |||||
| 4N1T | 5.14 ± 0.73 | 4.18 ± 1.63 | |||
| 31X8 | 6.63 ± 0.62 | 7.83 ± 0.50 | |||
| 2AG6 | 15.04 ± 0.33 | 14.91 ± 0.36 | |||
| Drude | 2.46 ± 0.16c | ||||
| 4N1T | 7.17 ± 0.99 | 4.32 ± 1.06 | 2.46 ± 0.16 | 2.42 ± 0.19 | |
| 31X8 | 9.70 ± 1.02 | 9.00 ± 0.60 | 2.48 ± 0.16 | 2.36 ± 0.22 | |
| 2AG6 | 16.10 ± 0.88 | 15.96 ± 0.74 | 2.46 ± 0.16 | 2.44 ± 0.18 | |
| Halogenated aromatic group | |||||
| Additive | |||||
| PDBid | Ar-Xd | ||||
| 4N1T | CL | 3.11 ± 0.07 | 3.11 ± 0.07 | ||
| 31X8 | CL | 2.29 ± 0.08 | 2.29 ± 0.07 | ||
| BR | 2.50 ± 0.09 | 2.52 ± 0.09 | |||
| 2AG6 | BR | 0.73 ± 0.08 | 0.72 ± 0.08 | ||
| Drude | |||||
| PDBid | Ar-Xd | ||||
| 4N1T | CL | 3.94 ± 0.46 | 3.50 ± 0.60 | ||
| 31X8 | CL | 3.46 ± 0.42 | 3.61 ± 0.29 | ||
| BR | 3.08 ± 0.45 | 3.72 ± 0.35 | |||
| 2AG6 | BR | 1.17 ± 0.50 | 0.86 ± 0.41 | ||
Dipole of waters within 3 Å of the full ligand are calculated.
Dipole of waters within 3 Å of both the protein and the full ligand are calculated.
Average and RMS fluctuations of the dipole moment of water in pure aqueous solution.
Ar-X indicates which halogen is included. The halogenated aromatic group includes all the functional groups on the same aromatic ring.
Analysis was also performed on the halogenated aromatic moieties in the ligands. With the additive FF the dipoles are approximately the same in both environments. With the Drude FF changes of the dipole moment up to 0.5 Debye from the aqueous phase to the protein binding pocket are seen. In addition, the RMS fluctuations in the dipole moments are significantly larger than in the additive FF as the halogenated aromatic groups are relatively rigid such that the fluctuations of the dipoles are dominated by the electronic response.
The dipole moments of waters in the vicinity of the protein and ligand were also analyzed for the Drude model. In aqueous solution there is little change in the average dipoles or their fluctuations. However, in the presence of the proteins some larger differences are observed in both the average and RMS fluctuations, especially with 3IX8 and 2AG6. This difference is evident in probability distributions of the water dipoles shown in Figure S18 of the supporting information. Similar variations are seen in the water molecules around the charged ligands (Figure S19). The results for the ligands and water indicate that variations in dipole moments can occur in the ligands upon binding as well as with water in the vicinity of the bound ligand, though the presence and magnitude of the effect is system dependent.
Conclusion
Presented are optimized polarizable Drude and additive CHARMM36/CGenFF force field parameters for the interaction of halogen containing aromatic compounds with proteins. While developed in the context of proteins, the improvements will be applicable to analogous functional groups on other classes of macromolecules. The improvements were implemented using atom pair-specific LJ or NBFIX parameters. These NBFIX parameters were developed to improve the halogen-hydrogen bond donor and acceptor interactions by targeting QM interaction energies between chlorobenzene or bromobenzene and a series of protein model compounds for both the additive CHARMM and polarizable Drude FFs. The optimized parameters were then evaluated in their ability to reproduce the crystallographic geometry of halogenated ligands or a modified halogenated residue with the proteins. The optimized model shows significant improvements against the target QM data for both XB and X-HBD interactions, with those results yielding a moderate level of improvement in the interactions with proteins. We note that the initial CHARMM36/CGenFF model already included NBFIX terms for the interaction of Cl with the carbonyl O of NMA, such that improvements in this class of interaction, which are present in PDB files 2ZC9, 2UW8, 4DT6, 3IX8, and 2AG6, were expected to be minimal. With system 4N1T large deviations in the ligand-protein geometry occurred with both the original additive and Drude models, an issue that was overcome with the optimized parameters in both cases. While the halogen model compounds in this study are limited to chloro- and bromobenzene, the optimized NBFIX parameters are expected to be applicable to aromatic species substituted with multiple chlorine and bromine atoms, including combinations of the two halogens. Such transferability was demonstrated in our previous study where pure solvent properties for a range of halogenated derivatives were shown to reproduce the experimental values when using halogen nonbonded parameters developed based on halogenated benzene systems.49
The results from MD simulation of the ligand-protein complexes are also useful for studying properties of the ligand in the presence of the protein with the Drude model. Methods to compute the ligand-protein interaction energies are illustrated and compared. Instead of simply calculating the energy contributed from atoms and Drude particles, a more physically correct way is to perform a self-consistent field calculation by allowing the Drude particles to relax to their local energy minima for the complex as well as the individual interacting partners. Application of this approach with all species in the absence and presence of water yield similar results, which are well correlated with interaction energy analysis performed using the additive model. Interestingly, the contribution of the electronic self-polarization energy is shown to both favor and disfavor the ligand-protein interaction energy, indicating the complex nature of the contribution of polarizability to ligand binding. Analysis of the dipole moments of ligands suggests that explicit treatment of induced polarization captures local variations in the environment of the ligands that impact their electronic structure, a phenomena that is not accounted by the mean-field approximation in the additive model.
In summary, an improved model of the interactions of halogenated aromatic ligands with proteins is presented. Improvements are made in both the additive CHARMM36/CGenFF and polarizable Drude force fields. It is anticipated that these improvements will facilitate investigations of ligand-protein interactions including efforts in the area of computer-aided drug design.
Supplementary Material
Figure 6.

Correlation of the interaction energies (kcal/mol) from the optimized additive and Drude force fields based on the data in Table 3. Drude-E1inter, Drude-E2inter, and Drude-E3inter are based on equations 1, 2 and 3, respectively. Drude-E4inter is directly calculated using INTER command in the CHARMM program.52–54
Acknowledgement
This work was supported by National Institutes of Health grants GM070855 and GM072558 and the Samuel Waxman Cancer Foundation. The University of Maryland Computer-Aided Drug Design Center and XSEDE are acknowledged for their generous allocations of computer time.
Footnotes
Competing financial interests: ADM is co-founder and CSO of SilcsBio LLC.
Associated Content: The Supporting Information is available free of charge on the ACS Publications website. Tables are included showing minimum interaction energies and distances between halogenated and protein model compounds, geometric parameters for ligands, and root-mean-square deviations as well as average root mean square fluctuations of the unrestrained protein backbone and ligand non-hydrogen atoms of each simulation system. Figures are included showing the overall parametrization procedure, interaction energies between the halogenated and protein model compounds, halogenated ligand structures and the region of the proteins in the vicinity of the ligands, and normalized distributions of the water dipole moments from each simulation system.
References
- (1).Auffinger P; Hays FA; Westhof E; Ho PS Halogen Bonds in Biological Molecules. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Scholfield MR; Zanden CMV; Carter M; Ho PS Halogen Bonding (X-Bonding): A Biological Perspective. Protein Sci. Publ. Protein Soc 2013, 22, 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Xu Z; Yang Z; Liu Y; Lu Y; Chen K; Zhu W Halogen Bond: Its Role beyond Drug–Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Model 2014, 54, 69–78. [DOI] [PubMed] [Google Scholar]
- (4).Naumann K Influence of Chlorine Substituents on Biological Activity of Chemicals: A Review. Pest Manag. Sci 2000, 56, 3–21. [Google Scholar]
- (5).Cavallo G; Metrangolo P; Milani R; Pilati T; Priimagi A; Resnati G; Terraneo G The Halogen Bond. Chem. Rev 2016, 116, 2478–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lin F-Y; MacKerell AD Jr. Do Halogen–Hydrogen Bond Donor Interactions Dominate the Favorable Contribution of Halogens to Ligand–Protein Binding? J. Phys. Chem. B 2017, 121, 6813–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ganesan A; Coote ML; Barakat K Molecular Dynamics-Driven Drug Discovery: Leaping Forward with Confidence. Drug Discov. Today 2017, 22, 249–269. [DOI] [PubMed] [Google Scholar]
- (9).Koebel MR; Schmadeke G; Posner RG; Sirimulla S AutoDock VinaXB: Implementation of XBSF, New Empirical Halogen Bond Scoring Function, into AutoDock Vina. J. Cheminformatics 2016, 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yang Z; Liu Y; Chen Z; Xu Z; Shi J; Chen K; Zhu W A Quantum Mechanics-Based Halogen Bonding Scoring Function for Protein-Ligand Interactions. J. Mol. Model 2015, 21, 138. [DOI] [PubMed] [Google Scholar]
- (11).Liu Y; Xu Z; Yang Z; Chen K; Zhu W A Knowledge-Based Halogen Bonding Scoring Function for Predicting Protein-Ligand Interactions. J. Mol. Model 2013, 19, 5015–5030. [DOI] [PubMed] [Google Scholar]
- (12).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; MacKerell AD Jr. CHARMM General Force Field (CGenFF): A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Soteras Gutiérrez I; Lin F-Y; Vanommeslaeghe K; Lemkul JA; Armacost KA; Brooks CL III; MacKerell AD Jr. Parametrization of Halogen Bonds in the CHARMM General Force Field: Improved Treatment of Ligand–Protein Interactions. Bioorg. Med. Chem 2016, 24, 4812–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yu W; He X; Vanommeslaeghe K; MacKerell AD Jr. Extension of the CHARMM General Force Field to Sulfonyl-Containing Compounds and Its Utility in Biomolecular Simulations. J. Comput. Chem 2012, 33, 2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vanommeslaeghe K; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model 2012, 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Vanommeslaeghe K; Raman EP; MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Jorgensen WL; Tirado-Rives J The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- (18).Jorgensen WL; Maxwell DS; Tirado-Rives J Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- (19).Jorgensen WL; Schyman P Treatment of Halogen Bonding in the OPLS-AA Force Field: Application to Potent Anti-HIV Agents. J. Chem. Theory Comput 2012, 8, 3895–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (21).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (22).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (23).Ibrahim MAA Molecular Mechanical Study of Halogen Bonding in Drug Discovery. J. Comput. Chem 2011, 32, 2564–2574. [DOI] [PubMed] [Google Scholar]
- (24).Ibrahim MAA Molecular Mechanical Perspective on Halogen Bonding. J. Mol. Model 2012, 18, 4625–4638. [DOI] [PubMed] [Google Scholar]
- (25).Kaminski GA; Stern HA; Berne BJ; Friesner RA Development of an Accurate and Robust Polarizable Molecular Mechanics Force Field from Ab Initio Quantum Chemistry. J. Phys. Chem. A 2004, 108, 621–627. [Google Scholar]
- (26).Patel S; Brooks CL III. CHARMM Fluctuating Charge Force Field for Proteins: I Parameterization and Application to Bulk Organic Liquid Simulations. J. Comput. Chem 2004, 25, 1–15. [DOI] [PubMed] [Google Scholar]
- (27).Patel S; MacKerell AD Jr.; Brooks CL III. CHARMM Fluctuating Charge Force Field for Proteins: II Protein/Solvent Properties from Molecular Dynamics Simulations Using a Nonadditive Electrostatic Model. J. Comput. Chem 2004, 25, 1504–1514. [DOI] [PubMed] [Google Scholar]
- (28).Harder E; Anisimov VM; Whitfield T; MacKerell AD Jr.; Roux B Understanding the Dielectric Properties of Liquid Amides from a Polarizable Force Field. J. Phys. Chem. B 2008, 112, 3509–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Vorobyov IV; Anisimov VM; MacKerell AD Jr. Polarizable Empirical Force Field for Alkanes Based on the Classical Drude Oscillator Model. J. Phys. Chem. B 2005, 109, 18988–18999. [DOI] [PubMed] [Google Scholar]
- (30).Noskov SY; Lamoureux G; Roux B Molecular Dynamics Study of Hydration in Ethanol−Water Mixtures Using a Polarizable Force Field. J. Phys. Chem. B 2005, 109, 6705–6713. [DOI] [PubMed] [Google Scholar]
- (31).Anisimov VM; Lamoureux G; Vorobyov IV; Huang N; Roux B; MacKerell AD Jr. Determination of Electrostatic Parameters for a Polarizable Force Field Based on the Classical Drude Oscillator. J. Chem. Theory Comput 2005, 1, 153–168. [DOI] [PubMed] [Google Scholar]
- (32).Lopes PEM; Lamoureux G; Roux B; MacKerell AD Jr. Polarizable Empirical Force Field for Aromatic Compounds Based on the Classical Drude Oscillator. J. Phys. Chem. B 2007, 111, 2873–2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Anisimov VM; Vorobyov IV; Roux B; MacKerell AD Jr. Polarizable Empirical Force Field for the Primary and Secondary Alcohol Series Based on the Classical Drude Model. J. Chem. Theory Comput 2007, 3, 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Vorobyov I; Anisimov VM; Greene S; Venable RM; Moser A; Pastor RW; MacKerell AD Jr. Additive and Classical Drude Polarizable Force Fields for Linear and Cyclic Ethers. J. Chem. Theory Comput 2007, 3, 1120–1133. [DOI] [PubMed] [Google Scholar]
- (35).Lopes PEM; Lamoureux G; MacKerell AD Jr. Polarizable Empirical Force Field for Nitrogen-Containing Heteroaromatic Compounds Based on the Classical Drude Oscillator. J. Comput. Chem 2009, 30, 1821–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Baker CM; MacKerell AD Jr. Polarizability Rescaling and Atom-Based Thole Scaling in the CHARMM Drude Polarizable Force Field for Ethers. J. Mol. Model 2010, 16, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Zhu X; MacKerell AD Jr. Polarizable Empirical Force Field for Sulfur-Containing Compounds Based on the Classical Drude Oscillator Model. J. Comput. Chem 2010, 31, 2330–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lin B; Lopes PEM; Roux B; MacKerell AD Jr. Kirkwood-Buff Analysis of Aqueous N-Methylacetamide and Acetamide Solutions Modeled by the CHARMM Additive and Drude Polarizable Force Fields. J. Chem. Phys 2013, 139, 084509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lopes PEM; Huang J; Shim J; Luo Y; Li H; Roux B; MacKerell AD Jr. Force Field for Peptides and Proteins Based on the Classical Drude Oscillator. J. Chem. Theory Comput 2013, 9, 5430–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Small MC; Aytenfisu AH; Lin F-Y; He X; MacKerell AD Jr. Drude Polarizable Force Field for Aliphatic Ketones and Aldehydes, and Their Associated Acyclic Carbohydrates. J. Comput. Aided Mol. Des 2017, 31, 349–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lin F-Y; Lopes PEM; Harder E; Roux B; MacKerell AD Jr. Polarizable Force Field for Molecular Ions Based on the Classical Drude Oscillator. J. Chem. Inf. Model 2018, 58, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ren P; Ponder JW Polarizable Atomic Multipole Water Model for Molecular Mechanics Simulation. J. Phys. Chem. B 2003, 107, 5933–5947. [Google Scholar]
- (43).Ponder JW; Wu C; Ren P; Pande VS; Chodera JD; Schnieders MJ; Haque I; Mobley DL; Lambrecht DS; DiStasio RA; Head-Gordon M; Clark GNI; Johnson ME; Head-Gordon T Current Status of the AMOEBA Polarizable Force Field. J. Phys. Chem. B 2010, 114, 2549–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shi Y; Xia Z; Zhang J; Best R; Wu C; Ponder JW; Ren P Polarizable Atomic Multipole-Based AMOEBA Force Field for Proteins. J. Chem. Theory Comput 2013, 9, 4046–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Adluri ANS; Murphy JN; Tozer T; Rowley CN Polarizable Force Field with a σ-Hole for Liquid and Aqueous Bromomethane. J. Phys. Chem. B 2015, 119, 13422–13432. [DOI] [PubMed] [Google Scholar]
- (46).Mu X; Wang Q; Wang L-P; Fried SD; Piquemal J-P; Dalby KN; Ren P Modeling Organochlorine Compounds and the σ-Hole Effect Using a Polarizable Multipole Force Field. J. Phys. Chem. B 2014, 118, 6456–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Carter M; Rappé AK; Ho PS Scalable Anisotropic Shape and Electrostatic Models for Biological Bromine Halogen Bonds. J. Chem. Theory Comput 2012, 8, 2461–2473. [DOI] [PubMed] [Google Scholar]
- (48).Ibrahim MAA; Hasb AAM; Mekhemer GAH Role and Nature of Halogen Bonding in Inhibitor⋅⋅⋅receptor Complexes for Drug Discovery: Casein Kinase-2 (CK2) Inhibition as a Case Study. Theor. Chem. Acc 2018, 137, 38. [Google Scholar]
- (49).Lin F-Y; MacKerell AD Jr. Polarizable Empirical Force Field for Halogen-Containing Compounds Based on the Classical Drude Oscillator. J. Chem. Theory Comput 2018, 14, 1083–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lamoureux G; Roux B Modeling Induced Polarization with Classical Drude Oscillators: Theory and Molecular Dynamics Simulation Algorithm. J. Chem. Phys 2003, 119, 3025–3039. [Google Scholar]
- (51).Baker CM; Lopes PEM; Zhu X; Roux B; MacKerell AD Jr. Accurate Calculation of Hydration Free Energies Using Pair-Specific Lennard-Jones Parameters in the CHARMM Drude Polarizable Force Field. J. Chem. Theory Comput 2010, 6, 1181–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Brooks BR; Brooks CL III; MacKerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: The Biomolecular Simulation Program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Brooks BR; Bruccoleri RE; Olafson DJ; States DJ; Swaminathan S; Karplus M CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- (54).MacKerell AD Jr.; Brooks CL III; Nilsson L; Roux B; Won Y; Karplus M CHARMM: The Energy Function and Its Parameterization with an Overview of the Program; Schleyer P. v. R., Allinger NL, Clark T, Gasteiger J, Kollman PA, Schaefer HF III, Schreiner PR, Eds.; The Encyclopedia of Computational Chemistry; John Wiley & Sons: Chichester, 1998; Vol. 1, pp 271–277. [Google Scholar]
- (55).Woon DE; Dunning TH Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The Atoms Aluminum through Argon. J. Chem. Phys 1993, 98, 1358–1371. [Google Scholar]
- (56).Peterson KA; Figgen D; Goll E; Stoll H; Dolg M Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post-d Group 16–18 Elements. J. Chem. Phys 2003, 119, 11113–11123. [Google Scholar]
- (57).Peterson KA; Shepler BC; Figgen D; Stoll H On the Spectroscopic and Thermochemical Properties of ClO, BrO, IO, and Their Anions. J. Phys. Chem. A 2006, 110, 13877–13883. [DOI] [PubMed] [Google Scholar]
- (58).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 03, Revision D.01. Gaussian, Inc., Wallingford CT, 2004. [Google Scholar]
- (59).Turney JM; Simmonett AC; Parrish RM; Hohenstein EG; Evangelista FA; Fermann JT; Mintz BJ; Burns LA; Wilke JJ; Abrams ML; Russ NJ; Leininger ML; Janssen CL; Seidl ET; Allen WD; Schaefer HF; King RA; Valeev EF; Sherrill CD; Crawford TD Psi4: An Open-Source Ab Initio Electronic Structure Program. Wiley Interdiscip. Rev. Comput. Mol. Sci 2012, 2, 556–565. [Google Scholar]
- (60).Boys S; Bernardi F The Calculation of Small Molecular Interactions by the Differences of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys 1970, 19, 553–566. [Google Scholar]
- (61).MacKerell AD Jr.; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiórkiewicz-Kuczera J; Yin D; Karplus M All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (62).MacKerell AD Jr.; Feig M; Brooks CL III. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- (63).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Hatcher E; Guvench O; MacKerell AD Jr. CHARMM Additive All-Atom Force Field for Acyclic Polyalcohols, Acyclic Carbohydrates and Inositol. J. Chem. Theory Comput 2009, 5, 1315–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hatcher E; Guvench O; MacKerell AD Jr. CHARMM Additive All-Atom Force Field for Aldopentofuranoses, Methyl-Aldopentofuranosides, and Fructofuranose. J. Phys. Chem. B 2009, 113, 12466–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD Jr.; Pastor RW Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Hart K; Foloppe N; Baker CM; Denning EJ; Nilsson L; MacKerell AD Jr. Optimization of the CHARMM Additive Force Field for DNA: Improved Treatment of the BI/BII Conformational Equilibrium. J. Chem. Theory Comput 2012, 8, 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Denning EJ; Priyakumar UD; Nilsson L; MacKerell AD Jr. Impact of 2’-Hydroxyl Sampling on the Conformational Properties of RNA: Update of the CHARMM All-Atom Additive Force Field for RNA. J. Comput. Chem 2011, 32, 1929–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Foloppe N; MacKerell AD Jr. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem 2000, 21, 86–104. [Google Scholar]
- (70).MacKerell AD Jr.; Banavali NK All-Atom Empirical Force Field for Nucleic Acids: II. Application to Molecular Dynamics Simulations of DNA and RNA in Solution. J. Comput. Chem 2000, 21, 105–120. [Google Scholar]
- (71).Lopes PEM; Huang J; Shim J; Luo Y; Li H; Roux B; MacKerell AD Jr. Polarizable Force Field for Peptides and Proteins Based on the Classical Drude Oscillator. J. Chem. Theory Comput 2013, 9, 5430–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Phillips JC Scalable Molecular Dynamics with NAMD. J Comput Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Baum B; Muley L; Heine A; Smolinski M; Hangauer D; Klebe G Think Twice: Understanding the High Potency of Bis(Phenyl)Methane Inhibitors of Thrombin. J. Mol. Biol 2009, 391, 552–564. [DOI] [PubMed] [Google Scholar]
- (74).Saxty G; Woodhead SJ; Berdini V; Davies TG; Verdonk ML; Wyatt PG; Boyle RG; Barford D; Downham R; Garrett MD; Carr RA Identification of Inhibitors of Protein Kinase B Using Fragment-Based Lead Discovery. J. Med. Chem 2007, 50, 2293–2296. [DOI] [PubMed] [Google Scholar]
- (75).Chen X; Kopecky DJ; Mihalic J; Jeffries S; Min X; Heath J; Deignan J; Lai S; Fu Z; Guimaraes C; Shen S; Li S; Johnstone S; Thibault S; Xu H; Cardozo M; Shen W; Walker N; Kayser F; Wang Z Structure-Guided Design, Synthesis, and Evaluation of Guanine-Derived Inhibitors of the EIF4E MRNA–Cap Interaction. J. Med. Chem 2012, 55, 3837–3851. [DOI] [PubMed] [Google Scholar]
- (76).Gad H; Koolmeister T; Jemth A-S; Eshtad S; Jacques SA; Ström CE; Svensson LM; Schultz N; Lundbäck T; Einarsdottir BO; Saleh A; Göktürk C; Baranczewski P; Svensson R; Berntsson RP-A; Gustafsson R; Strömberg K; Sanjiv K; Jacques-Cordonnier M-C; Desroses M; Gustavsson A-L; Olofsson R; Johansson F; Homan EJ; Loseva O; Bräutigam L; Johansson L; Höglund A; Hagenkort A; Pham T; Altun M; Gaugaz FZ; Vikingsson S; Evers B; Henriksson M; Vallin KSA; Wallner OA; Hammarström LGJ; Wiita E; Almlöf I; Kalderén C; Axelsson H; Djureinovic T; Puigvert JC; Häggblad M; Jeppsson F; Martens U; Lundin C; Lundgren B; Granelli I; Jensen AJ; Artursson P; Nilsson JA; Stenmark P; Scobie M; Berglund UW; Helleday T MTH1 Inhibition Eradicates Cancer by Preventing Sanitation of the DNTP Pool. Nature 2014, 508, 215–221. [DOI] [PubMed] [Google Scholar]
- (77).Zou Y; Nair SK Molecular Basis for the Recognition of Structurally Distinct Autoinducer Mimics by the Pseudomonas Aeruginosa LasR Quorum-Sensing Signaling Receptor. Chem. Biol 2009, 16, 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Amacher JF; Zhao R; Spaller MR; Madden DR Chemically Modified Peptide Scaffolds Target the CFTR-Associated Ligand PDZ Domain. PLOS ONE 2014, 9, e103650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Turner JM; Graziano J; Spraggon G; Schultz PG Structural Plasticity of an Aminoacyl-TRNA Synthetase Active Site. Proc. Natl. Acad. Sci 2006, 103, 6483–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Mentel M; Blankenfeldt W; Breinbauer R The Active Site of an Enzyme Can Host Both Enantiomers of a Racemic Ligand Simultaneously. Angew. Chem. Int. Ed 2009, 48, 9084–9087. [DOI] [PubMed] [Google Scholar]
- (81).Jo S; Kim T; Iyer VG; Im W CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- (82).Lamoureux G; Harder E; Vorobyov IV; Roux B; MacKerell AD Jr. A Polarizable Model of Water for Molecular Dynamics Simulations of Biomolecules. Chem. Phys. Lett 2006, 418, 245–249. [Google Scholar]
- (83).Yu H; Whitfield TW; Harder E; Lamoureux G; Vorobyov I; Anisimov VM; MacKerell AD Jr.; Roux B Simulating Monovalent and Divalent Ions in Aqueous Solution Using a Drude Polarizable Force Field. J. Chem. Theory Comput 2010, 6, 774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Luo Y; Jiang W; Yu H; MacKerell AD Jr.; Roux B Simulation Study of Ion Pairing in Concentrated Aqueous Salt Solutions with a Polarizable Force Field. Faraday Discuss. 2013, 160, 135–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Jiang W; Hardy DJ; Phillips JC; MacKerell AD Jr; Schulten K; Roux B High-Performance Scalable Molecular Dynamics Simulations of a Polarizable Force Field Based on Classical Drude Oscillators in NAMD. J. Phys. Chem. Lett 2011, 2, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Ryckaert J-P; Ciccotti G; Berendsen HJC Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- (87).Steinbach PJ; Brooks BR New Spherical-Cutoff Methods for Long-Range Forces in Macromolecular Simulation. J. Comput. Chem 1994, 15, 667–683. [Google Scholar]
- (88).Darden T; York D; Pedersen L Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (89).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A Smooth Particle Mesh Ewald Method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (90).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain X1 and X2 Dihedral Angles. J. Chem. Theory Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (92).Martyna GJ; Tobias DJ; Klein ML Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys 1994, 101, 4177–4189. [Google Scholar]
- (93).Feller SE; Zhang Y; Pastor RW; Brooks BR Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
- (94).Vidal-Vidal Á; Silva López C; Faza ON Lennard-Jones Potentials for the Interaction of CO2 with Five-Membered Aromatic Heterocycles. J. Phys. Chem. A 2017, 121, 9518–9530. [DOI] [PubMed] [Google Scholar]
- (95).Kroemer RT Structure-Based Drug Design: Docking and Scoring. Curr. Protein Pept. Sci 2007, 8, 312–328. [DOI] [PubMed] [Google Scholar]
- (96).Hensen C; Hermann JC; Nam K; Ma S; Gao J; Höltje H-D A Combined QM/MM Approach to Protein−Ligand Interactions: Polarization Effects of the HIV-1 Protease on Selected High Affinity Inhibitors. J. Med. Chem 2004, 47, 6673–6680. [DOI] [PubMed] [Google Scholar]
- (97).Garcia-Viloca M; Truhlar DG; Gao J Importance of Substrate and Cofactor Polarization in the Active Site of Dihydrofolate Reductase. J. Mol. Biol 2003, 327, 549–560. [DOI] [PubMed] [Google Scholar]
- (98).Ryde U; Söderhjelm P Ligand-Binding Affinity Estimates Supported by Quantum-Mechanical Methods. Chem. Rev 2016, 116, 5520–5566. [DOI] [PubMed] [Google Scholar]
- (99).MacKerell AD Jr. Empirical Force Fields for Biological Macromolecules: Overview and Issues. J. Comput. Chem 2004, 25, 1584–1604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.