

Abstract
OBJECTIVE:
To investigate mechanisms of in utero death in normally formed fetuses by measuring amniotic fluid (AF) biomarkers for hypoxia [erythropoietin (EPO)], myocardial damage [cardiac troponin I (cTnI)], and brain injury [glial fibrillary acidic protein (GFAP)], correlated with risk factors for fetal death and placental histopathology.
MATERIAL AND METHODS:
This retrospective, observational cohort study included intrauterine deaths with transabdominal amniocentesis prior to induction of labor. Women with a normal pregnancy and an indicated amniocentesis at term were randomly selected as controls. AF was assayed for EPO, cTnI and GFAP using commercial immunoassays. Placental histopathology was reviewed, and CD15-immunohistochemistry was used. Analyte concentrations >90th centile for controls were considered ‘raised’. Raised AF-EPO, AF-cTnI, and AF-GFAP concentrations were considered evidence of hypoxia, myocardial and brain injury, respectively.
RESULTS:
There were sixty cases and sixty controls. Hypoxia was present in 88% (53/60), myocardial damage in 70% (42/60), and brain injury in 45% (27/60) of fetal deaths. Hypoxic fetuses had evidence of myocardial injury, brain injury, or both in 77% (41/53), 49% (26/53), and 13% (7/53) cases, respectively. Histopathological evidence for placental dysfunction was found in 74% (43/58) of these cases.
CONCLUSION:
Hypoxia, secondary to placental dysfunction, was found to be the mechanism of death in the majority of fetal deaths of structurally normal fetuses. Ninety-one percent of hypoxic fetal deaths sustained brain, myocardial or both brain and myocardial injuries in utero. Hypoxic myocardial injury was an attributable mechanism of death in 70% of the cases. Non-hypoxic cases may be caused by cardiac arrhythmia secondary to a cardiac conduction defect.
Keywords: amniotic fluid, brain injury, cardiac troponin I, CD15, delayed villous maturation, erythropoietin, fetal hypoxia, glial fibrillary acidic protein, myocardial damage, placental dysfunction
INTRODUCTION
The rate of fetal death in the United States is 5.96 per 1,000 live births (1), and it is twice as common in African-American than in Caucasian women (1, 2). Several maternal and fetal disorders, such as maternal hypertension (3–9), diabetes mellitus (3, 10, 11), intrauterine infection (12–14) or inflammation (15–17), placental abruption (3, 7, 18–22), placental disorders associated with maternal vascular lesions of malperfusion (23–25), massive perivillous fibrin deposition (26, 27), and small-for-gestational-age (SGA) fetuses (3, 28–31) have been associated with fetal death. However, 25%−62% of all fetal deaths are not attributed to known maternal, placental, or fetal risk factors (32–38). These ‘unexplained’ fetal deaths comprise a progressively larger proportion of all fetal deaths as pregnancy advances (39), given that fetal deaths associated with congenital anomalies occur more frequently before the third trimester (32, 34, 35).
The term ‘unexplained fetal death’ is an unfortunate designation applied to cases of fetal death that occur in the absence of known risk factors: it implies that the mechanism of death in cases associated with maternal, placental, and fetal risk factors is known. Although our understanding as to why fetuses may die in utero has improved over time, this knowledge has allowed only educated guesses about possible causes (40–43).
Hypoxia, secondary to placental dysfunction, is believed to play an important role in most fetal deaths, yet evidence is indirect (25, 44–59). The more proximal factors that lead to fetal cardiac arrest are largely conjectural. Understanding of the causes of hypoxia, and the intermediary steps between hypoxia and fetal death, may allow identification of biomarkers that could be used to predict and prevent fetal death in women at risk for this complication (23, 60–62).
There is evidence that erythropoietin (EPO), cardiac troponin I (cTnI), and glial fibrillary acidic protein (GFAP) detected in maternal serum or plasma are specific biomarkers for hypoxia (44, 63–65), myocardial damage (66–68), and brain injury (69–71), respectively, and that prenatal myocardial and brain damage occur in 20% to 66% of stillborn fetuses (72–81). Furthermore, studies have shown that EPO is of both fetal (82–85) and placental (86, 87) origin and remains elevated in amniotic fluid after the fetus dies (63); that cTnI is specific to myocardial damage (66, 88, 89), and its concentration is elevated in hypoxic neonates (90–92); and that serum concentrations of GFAP are elevated in newborns with hypoxic-ischemic encephalopathy (HIE), which correlate with the severity of HIE (70, 71, 81). Although intrauterine brain injury is not generally as a mechanism of death, abnormalities of the cardiovascular center in the brainstem, and of the cardiac conduction system, have been reported in 92% of a series of unexplained fetal or infant deaths (93).
With these considerations in mind, we measured the concentrations of EPO, cTnI, and GFAP in samples of amniotic fluid collected from 60 structurally normal fetuses that died in utero to determine if, and to what extent, hypoxic myocardial and brain injury were implicated in these fetal deaths. We also correlated the findings with the histopathology of the placenta and known risk factors for intrauterine death.
MATERIALS and METHODS
This was a retrospective observational study of fetal deaths that occurred among pregnant women recruited into cohort studies conducted between January 2004 and January 2016 at Hutzel Women’s Hospital, Detroit, Michigan, USA. All study participants provided written informed consent and were followed until delivery. The use of clinical data and biological specimens obtained from these women for research purposes was approved by the Institutional Review Boards of Wayne State University and the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, U.S. Department of Health and Human Services (NICHD/NIH/DHHS).
The study group of cases comprised all fetal deaths for which an amniocentesis had been performed prior to induction of labor, and for those patients who had sufficient amniotic fluid available for analysis. Women who had a normal pregnancy and an amniocentesis at term to assess fetal lung maturity prior to a scheduled cesarean section were selected as controls, provided they also had sufficient amniotic fluid available for analysis. We selected elective cesarean deliveries because labor is known to increase EPO concentrations in umbilical cord blood among live births (94). Some of the women had been subjects in prior studies.
Clinical Definitions
The following definitions were used:
Gestational age was determined by the last menstrual period and confirmed by ultrasound examination or by ultrasound examination alone if the sonographic determination of gestational age was inconsistent with menstrual dating by more than one week in the first trimester (95);
Fetal death or stillbirth: death of the fetus diagnosed after 20 weeks of gestation confirmed by ultrasound examination prior to delivery (96);
- Hypertensive disorders of pregnancy included four categories: preeclampsia, chronic hypertension, chronic hypertension with superimposed preeclampsia, and gestational hypertension, defined as follows:Preeclampsia: presence of new-onset systolic blood pressure ≥140 mm Hg and/or diastolic blood pressure ≥90 mm Hg on at least two occasions, 4 hours to 1 week apart, after 20 weeks of gestation, and of proteinuria ≥300 mg in a 24-hour urine collection or one dipstick with ≥1+ for protein (97);Chronic hypertension: maternal hypertension that predates pregnancy (98);Chronic hypertension with superimposed preeclampsia: presence of chronic hypertension in association with preeclampsia (98);Gestational hypertension: new-onset systolic blood pressure ≥140 mm Hg and/or diastolic blood pressure ≥90 mm Hg on at least two occasions, 4 hours to 1 week apart, after 20 weeks of gestation in the absence of systemic maternal organ damage (proteinuria, thrombocytopenia, elevated liver enzymes, elevated creatinine, pulmonary edema, and/or cerebral or visual disturbances) (98);
A small-for-gestational-age (SGA) neonate was defined as having a birthweight <10th percentile for gestational age at delivery according to a U.S. reference population (99);
An appropriate-for-gestational-age (AGA) neonate was defined as having a birthweight between the 10th and 90th percentiles for gestational age at delivery according to a U.S. reference population (99);
- Diabetes mellitus in pregnancy included two categories: pre-gestational and gestational, defined as follows (100):Pre-gestational diabetes mellitus: diabetes mellitus diagnosed prior to pregnancy based on a fasting blood glucose >126 mg/dL or A1C >6.5%;Gestational diabetes mellitus diagnosed in a pregnant woman without a prior history of diabetes and with at least two values ≥ the following on a 3-hour, 100 g oral glucose tolerance test performed between 24 and 28 weeks of gestation: fasting ≥ 95 mg/dL, 1-hour ≥180 mg/dL, 2-hour ≥ 155 mg/dL, and 3-hour ≥ 140 mg/dL;
Placental abruption was diagnosed clinically by the presence of vaginal bleeding, abdominal pain, and a retroplacental blood clot not associated with vasa previa, placenta previa, or uterine rupture (101);
Placenta previa was defined as a placenta that overlies or is proximate to the internal cervical os based on transvaginal ultrasound examination (102);
Intra-amniotic inflammation was diagnosed when the interleukin (IL)-6 concentration in the amniotic fluid was ≥2.6 ng/mL (103–112);
Intra-uterine infection was defined by the presence of intra-amniotic infection and Cytomegalovirus (CMV) placentitis. Intra-amniotic infection was defined as a positive amniotic fluid culture for microorganisms using cultivation techniques (113–116). CMV placentitis was defined by the presence of focal segmental chronic lymphoplasmacytic villitis, hemosiderin deposition, and an occasional presence of owl-like-CMV inclusions (117). The latter was subsequently confirmed by immuno-histochemistry for CMV (118);
Control group comprised women who had no medical, obstetrical, or surgical complications, who were not in labor, and who had an elective cesarean delivery of a structurally normal singleton fetus at 37–42 weeks of gestation, whose birthweight was between the 10th and 90th percentiles for gestational age (99).
Amniotic fluid samples
Amniotic fluid was collected by transabdominal amniocentesis for medical indications, such as karyotype studies or fetal lung maturity, or to rule out intra-amniotic infection, using standard cultivation techniques for aerobic and anaerobic microorganisms, genital mycoplasmas, and fungi. Samples of amniotic fluid not required for clinical purposes were centrifuged to remove cellular and particulate matter. Aliquots of amniotic fluid were stored at −70ºC until analysis.
Analyte assays
Assay for erythropoietin
Concentrations of EPO in amniotic fluid were determined with a commercially available specific immunoassay [American Laboratory Products Company (ALPCO), Salem, New Hampshire, USA]. The sensitivity of this assay was 1.8 mIU/mL, and the coefficients of variation for intra-and inter-assays were 6.1% and 9.2 %, respectively. All samples were assayed in duplicate.
Assay for cardiac troponin I
Concentrations of cTnI in amniotic fluid were determined with a commercially available specific immunoassay (LifeSpan BioSciences, Seattle, Washington, USA). The sensitivity of this assay was 26.89 pg/mL, and the coefficients of variation for intra-and inter-assays were 6.1% and 8.1 %, respectively. All samples were assayed in duplicate.
Assay for glial fibrillary acid protein
Concentrations of GFAP in amniotic fluid were determined using an enzyme-linked immunosorbent assay (Cloud Clone, Katy, Texas, USA). The sensitivity of this assay was 0.19 ng/mL, and the coefficients of variation for intra-and inter-assays were 8.2% and 15.1 %, respectively. All samples were assayed in duplicate.
Placental Histopathological Analysis
The placental disc of each patient was randomly sampled using a random sequence generator and software designed for this purpose as previously described (119). Slides stained with hematoxylin and eosin as well as cytokeratin-7 from cases and controls were reviewed by a placental pathologist (SJ) blinded to the pregnancy outcome and any previous histopathological diagnoses. The nomenclature adopted by the Amsterdam Placental Workshop Group was used to describe the vaso-occlusive lesions and villous maturational defects in these cases (120). Slides were also stained for the marker CD15; “immature” CD15-positive endothelium is a diagnostic marker of persisting villous immaturity and chronic placental dysfunction (121). The level of CD15 expression was considered as pathological placental villous immaturity in both macrovasculature (chorionic plate and stem vessels) and microvasculature (terminal villi) with ≥50% villi exhibiting positive expression.
Accelerated villous maturation was defined as premature maturation of terminal chorionic villi (resembling term villi at more than 38 weeks of gestation) with conspicuous syncytial knotting in preterm placentas (122). Delayed villous maturation is a developmental placental abnormality defined by a monotonous villous population (at least 10 such villi) with centrally placed capillaries and decreased vasculosyncytial membranes, recapitulating the histology in early pregnancy (120, 123).The presence of stromal villous-vascular karyorrhexis, avascular villi, and stem villous vessel obliteration was noted but considered as post-mortem changes secondary to cessation of the fetal circulation, and used only to estimate the time of death prior to delivery, as previously described (124).
In cases for which light microscopy and hematoxylin and eosin-stained sections displayed focal segmental chronic lymphoplasmacytic villitis, hemosiderin deposition, and the occasional presence of owl-like inclusions in the stromal cells suggestive of CMV placentitis (117), the diagnosis of CMV placentitis was subsequently confirmed by immunohistochemistry (118). Cases of chronic villitis that did not exhibit lymphoplasmacytes in the villi nor had any evident inclusions or hemosiderin deposition were not considered as CMV villitis. Immunohistochemistry was performed in cases with lymphoplasmacytic villitis by the streptavidin–biotin complex method using a monoclonal anti-CMV antibody (Leica BOND, Buffalo Grove, Illinois, USA).
Statistical Analysis
We analyzed the results as multivariate discrete data. The analyte concentrations below the limit of detection were replaced by 99% of the minimum observed value for the analyte. The analytes were then categorized at the 90th centile for the controls, and values above the 90th centile were considered ‘raised.’ Fetal deaths were dichotomized based on the presence of known risk factors (i.e., placental abruption, chronic hypertension, preeclampsia, gestational hypertension, gestational diabetes mellitus, pre-gestational diabetes mellitus, SGA neonate, intra-amniotic infection, and placenta previa) to determine whether analyte concentrations differed between what have traditionally been labeled ‘explained’ and ‘unexplained’ deaths. Because the proportion of ‘unexplained’ fetal deaths has been found to increase with gestational age (32, 34, 35, 125, 126), we also examined the effect of gestational age on analyte concentrations and dichotomized gestational age arbitrarily at 28 weeks of gestation so that this analysis could be as close as possible to equal numbers in the two gestational-age categories.
We used the statistical package R to fit hierarchically nested log linear models to multi-way contingency tables of the data to analyze associations, partial and conditional, between the three analytes, presence of risk factors, and gestational age. The relationship between the variables was first examined by fitting a three-way contingency table to the three analytes dichotomized at the 90th centile for the controls. We next examined the effects of risk factors and gestational age individually in a model containing informative analytes and retained significant terms to arrive at a final model.
A two-tailed Fisher’s exact test was used to test 2×2 marginal tables, and the Mann-Whitney U test was used to compare median analyte concentrations between the different causes of death and placental perfusion categories. Values for statistics below the 5% significance level were considered statistically significant.
RESULTS
Of 279 fetal deaths in the database, 45 had congenital anomalies. The proportion of fetal deaths associated with congenital anomalies was significantly higher before, rather than after, 28 weeks of gestation [20% (27/134) vs. 11.6% (18/155), p=0.03]. Cases with congenital anomalies were excluded from further analysis because amniotic fluid was available for only two fetal deaths associated with congenital malformations.
Of the 234 cases of structurally normal singleton fetuses that died in utero, 119 had undergone amniocentesis, and amniotic fluid was available for analysis in 60 cases. Among these, clinical abruption occurred in 10 (16.7%) cases, diabetes mellitus in 11 (18.3%; pre-gestational diabetes in 5, gestational diabetes in 6 cases), hypertensive disorders of pregnancy in 16 (26.7%; gestational hypertension in 6, chronic hypertension in 4, preeclampsia in 4, and chronic hypertension with preeclampsia in 2 cases), SGA fetuses in 18 (31%, 18/58), intra-uterine infection in 6 (10%; intra-amniotic infection in 4 and CMV-chronic villitis in 2 cases), and one case with placenta previa (1.7%). Fifteen (25%) patients had more than one of these risk factors, and 15 (25%) had no risk factors (Figure 1). Birthweight was registered in 58 cases and autopsy reports were available for 37 cases. None of the fetal deaths was hydropic.
Figure 1. Frequency of risk factors in cases of fetal death.
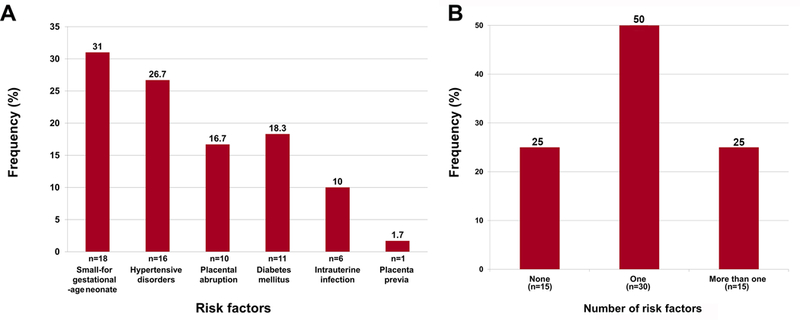
A: Frequency of different risk factors. B: Frequency of none, one, or more than one risk factor.
Sixty controls were randomly selected from 535 patients who had an amniocentesis at term when not in labor. There were no significant differences in median maternal age, body mass index, frequency of smoking status, or self-reported ethnicity (African-American) among the 60 cases and 60 controls. Cases had significant lower median gestational ages at amniocentesis, at delivery, as well as lower birthweights, and a higher frequency of nulliparity than the controls (Table 1). The estimated time interval between death and delivery in cases estimated from placental histopathology is shown in Table 2. The percentile distribution of birthweight between cases and controls is shown in Figure 2A; the difference between the groups was not statistically significant (p=0.05). The distribution of birthweight centiles by gestational age for cases is shown in Figure 2B; there was a significant correlation between birthweight centile and gestational age (Spearman’s rho =0.3, p=0.034): earlier fetal deaths had lower birthweight centiles than later fetal deaths. There was a significant difference between cases and controls in the birthweight-to-placenta weight ratio (3.94 vs. 5.72, respectively, p<0.001; Table 1).
Table 1.
Demographic and clinical characteristics of the study group
| Characteristics | Cases of fetal death (n=60) |
Controls (n=60) |
P valuea |
|---|---|---|---|
| Maternal age (years) | 25.0 [16.0–42.0] |
26.0 [17.0–42.0] |
0.77 |
| Body mass index (kg/m2)b | 28.3 [15.5–47.9] |
30.1 [15.5–55.8] |
0.38 |
| Smoking status | 19 (31.7) | 13 (21.7) | 0.30 |
| Nulliparity | 22 (35.5) | 0 | <0.001 |
| African-American ethnicity | 50 (83.3) | 50 (83.3) | 1.0 |
| Gestational age at amniocentesis (weeks) |
29.9 [20.1–40.9] |
39.1 [37.0 −41.4 ] |
<0.001 |
| Gestational age at delivery (weeks) |
30.0 [20.3–41.3] |
39.1 [37.0–41.4] |
<0.001 |
| Birthweight (grams) |
1265.0 [91.0–3885.0] |
3017.5 [2670.0–3970.0] |
<0.001 |
| Birthweight-to-placenta-weight ratio |
3.94 [0.6–9.66] | 5.72 [3.4–7.6] | <0.001 |
Continuous variables are expressed as median [range], and categorical variables are expressed as number (percentage).
Kruskal-Wallis test for continuous variables and a Fisher’s exact test for categorical variables.
Body mass index (BMI) was calculated as follows: BMI = (maternal pre-pregnancy weight [kg] / maternal height [m]2).
Table 2.
Pattern of raised amniotic fluid concentrations of cardiac troponin I (CTnI) and glial fibrillary acidic protein (GFAP) according to the estimated death-to-delivery time interval in cases of fetal death
| Death-to-delivery time interval |
Not raised cTnI or GFAP (n = 10) |
Raised cTnI only (n = 21) |
Raised GFAP only (n = 8) |
Raised cTnI and GFAP (n = 19) |
|---|---|---|---|---|
| ≤ 6 hours | 0 | 0 | 0 | 1 |
| 6–24 hours | 3 | 1 | 1 | 2 |
| ≥ 48 hours | 7 | 9 | 4 | 12 |
| 2–7 days | 0 | 6 | 3 | 3 |
| 7–14 days | 0 | 5 | 0 | 1 |
Figure 2. Percentile distribution of birthweight in cases of fetal death and the controls.
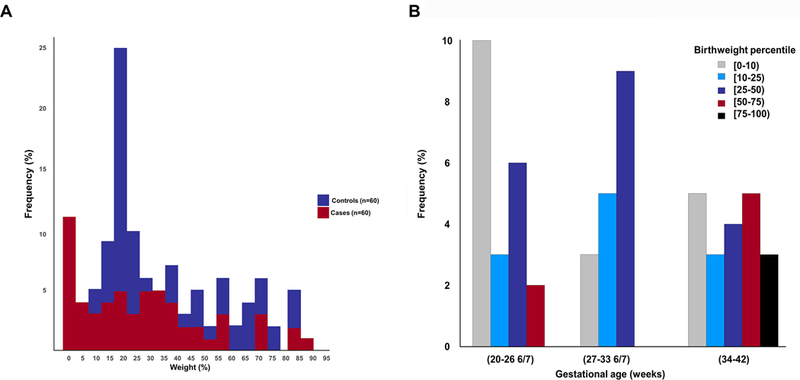
A: There was no significant difference in the percentile distribution of birthweight between cases of fetal death and the controls (p=0.05). B: Distribution of birthweight centiles by gestational age for cases of fetal deaths. There was a significant correlation between birthweight centile and gestational age (Spearman’s rho = 0.3, p = 0.03): earlier fetal deaths had lower birthweight centiles than later fetal deaths.
Analyte Concentrations
Distribution of the analyte concentrations in the cases and controls is shown for each analyte in Figures 3A–C. Amniotic fluid EPO was below the level of detection in 60% (36/60) of controls and in 1 (1.7%) case; amniotic fluid cTnI was below the level of detection in 25% (15/60) of controls and 2 (3.3%) cases; and amniotic fluid GFAP was below the level of detection in 1 (1.7%) control and 4 (6.7%) cases.
Figure 3. Amniotic fluid concentration of erythropoietin (EPO), cardiac troponin I (cTnI), and glial fibrillary acidic protein (GFAP) in controls and cases of fetal death.
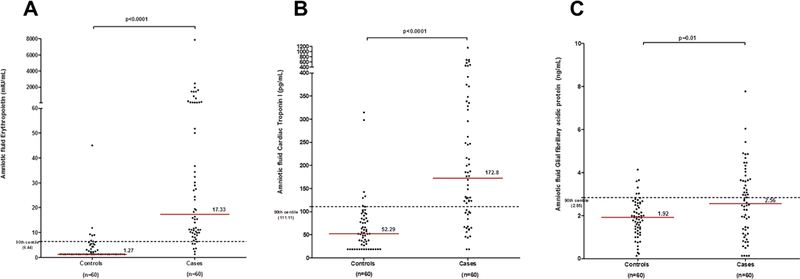
A: The median amniotic fluid concentration of EPO in fetal death was significantly higher compared to controls [median (interquartile range): 17.33 mIU/mL (9.89–77.08) versus 1.27 mIU/mL (1.27–4.03), p<0.0001]. The concentration at the 90th centile in amniotic fluid EPO for controls was 6.44 mIU/ml (dashed line). B: The median amniotic fluid concentration of cTnI in fetal death was significantly higher compared to controls [median (interquartile range): 172.8 pg/mL (97.51–326.30) versus 52.29 pg/mL (21.07–85.60), p<0.0001)]. The concentration at the 90th centile in amniotic fluid of cTnI for controls was 111.11 pg/mL (dashed line). C: The median amniotic fluid concentration of cTnI in fetal death was significantly higher compared to controls [median (interquartile range): 2.56 ng/mL (1.21–3.65) versus 1.92 ng/mL (1.32–2.56), p=0.01)]. The concentration at the 90th centile in amniotic fluid of GFAP for controls was 2.84 ng/ml (dashed line).
Median amniotic fluid concentrations of EPO, cTnI, and GFAP were significantly higher for cases than controls (EPO: 17.33 mIU/mL vs. 1.27 mIU/mL, p <0.001; cTnI: 172.8 pg/mL vs. 52.29 pg/mL, p<0.001; GFAP: 2.56 ng/mL vs. 1.92 ng/mL, p=0.01) (Figures 3A–C). Median amniotic fluid concentrations of EPO were significantly higher among cases with risk factors than cases without risk factors (cases with risk factors: 23.19 mIU/mL vs. cases without risk factors 10.89 mIU/mL, p =0.03), but median amniotic fluid cTnI and GFAP concentrations were not significantly different between cases that had risk factors and those that did not (AF cTnI: cases with risk factors: 177.08 pg/mL vs. cases without risk factors 124.48 pg/mL, p= 0.29; AF GFAP: cases with risk factors: 2.71 ng/mL vs. cases without risk factors: 2.48 ng/mL, p=0.53). There was no significant association between the presence of risk factors and gestational age (dichotomized ≥28 weeks of gestation at amniocentesis).
The percentile distribution of the analytes in amniotic fluid is shown in Table 3. The 90th centile concentration of amniotic fluid EPO, cTnI, and GFAP in controls was 6.44 mIU/mL, 111.11 pg/mL, and 2.85 ng/mL, respectively. Amniotic fluid EPO, cTnI, and GFAP concentrations were ‘raised,’ i.e., above the 90th centile for the controls, in 88% (53/60), 70% (42/60), and 45% (27/60) of fetal deaths, respectively (Table 3). The proportion of cases in which amniotic fluid EPO, cTnI, or GFAP was raised was not significantly different between cases that had or did not have risk factors for fetal death.
Table 3.
Percentile distribution of amniotic fluid concentrations of erythropoietin, cardiac troponin I and glial fibrillary acidic protein in cases of fetal death
|
Percentile |
Erythropoietin (n = 60) |
Cardiac troponin I (n = 60) |
Glial fibrillary acidic protein (n = 60) |
|---|---|---|---|
| > 95th | 48 | 33 | 19 |
| 90th–95th | 5 | 9 | 8 |
| 80th–90th | 3 | 4 | 2 |
| 70th–80th | 2 | 2 | 5 |
| 60th–70th | 1 | 5 | 3 |
| 50th–60th | 0 | 2 | 3 |
| ≤ 50th | 1 | 5 | 20 |
The relationship among the three analytes, dichotomized as above or equal or below the 90th centiles for the controls, is shown in Table 4. No amniotic fluid analyte was ‘raised’ in five (8.3%) cases. Only amniotic fluid EPO was ‘raised’ in five (8.3%) cases; only amniotic fluid cTnI or only amniotic fluid GFAP was ‘raised’ in one case each (1.7%). It should be noted that in these two latter cases, amniotic fluid EPO was above the 80th and 70th centiles, respectively. Therefore, these two cases were considered as cases with hypoxic cardiac and brain injury occurring at slightly lower concentrations of EPO than the criteria used in this study.
Table 4.
Distribution of amniotic fluid concentrations of erythropoietin (EPO), cardiac troponin I (cTnI), and glial fibrillary acidic protein (GFAP) according to the presence or absence of EPO raised (>90th percentile for controls) in cases of fetal death
| EPO > 90th percentile | EPO ≤ 90th percentile | ||||||
|---|---|---|---|---|---|---|---|
| cTnI | GFAP | Total (n=53) |
cTnI | GFAP | Total (n=7) |
||
| >90th percentile (n=26) |
≤ 90th percentile (n=27) |
>90th percentile (n=1) |
≤ 90th percentile (n=6) |
||||
| >90th percentile | 19 | 22 | 41 | >90th percentile | 0 | 1 | 1 |
| ≤90th percentile | 7 | 5 | 12 | ≤90th percentile | 1 | 5 | 6 |
Thus, when including these two cases (‘raised’ cTnI and EPO >80th and ‘raised’ GFAP and EPO >70th centiles), the concentration of GFAP in hypoxic fetal deaths was ‘raised’ in the same proportion of cases regardless of whether the concentration of cTnI was or was not raised [45.2% (19/42) vs. 44.4% (8/13), p=0.36]. In addition, there was no significant difference in median GFAP concentration between these two groups of fetal deaths (2.65 ng/mL vs. 2.98 ng/mL, p=0.95).
There was no association between cases with ‘raised’ amniotic fluid cTnI and those with ‘raised’ amniotic fluid GFAP concentrations; amniotic fluid GFAP was ‘raised’ in 45.2% (19/42) of fetal deaths with ‘raised’ amniotic fluid cTnI, and in 44.4% (8/18) of those that did not have ‘raised’ amniotic fluid cTnI (p=1). The difference between the proportion of SGA and AGA neonates with fetal death that had ‘raised’ amniotic fluid cTnI concentrations did not reach statistical significance (15/40 (37.5%) vs. 3/18 (16.7%), p=0.14; data not shown).
The mean amniotic fluid concentrations of EPO, cTnI, and GFAP, and the proportion of fetal deaths with birthweights below the 50th centile for gestational age, and of SGA neonates, as well as the birthweight-to-placenta-weight ratio for each ‘raised’ amniotic fluid EPO combination are shown in Table 5. The mean amniotic fluid cTnI concentration was significantly higher among cases with ‘raised’ concentration for all analytes (amniotic fluid EPO, cTnI and GFAP) than among cases that had amniotic fluid with either ‘raised’ cTnI and GFAP or ‘raised’ EPO alone (p<0.001). Furthermore, the mean amniotic fluid cTnI concentration in cases with ‘raised’ amniotic fluid EPO, cTnI and GFAP was higher than in those for which amniotic fluid EPO and cTnI were ‘raised’ (380.38 pg/ml vs. 232.48 pg/ml, p=0.03).
Table 5.
Birthweight below the 50th percentile, small-for-gestational-age neonate, birthweight-to-placenta-weight and mean amniotic fluid concentrations of erythropoietin (EPO), cardiac troponin I (cTnI), and glial fibrillary acidic protein (GFAP) according to the pattern of raised amniotic fluid concentration of EPO in cases of fetal death
| Raised EPO only (n=5) |
Raised EPO and cTnI (n=23) |
Raised EPO and GFAP (n=8) |
Raised EPO, cTnI and GFAP (n=19) |
P-valuea | |
|---|---|---|---|---|---|
| Birthweight <50th centile | 4 | 18/21 | 6 | 15 | 0.87 |
| Small-for-gestational-age neonate | 1 | 10/21 | 2 | 5 | 0.47 |
| Birthweight-to-placenta-weight ratio | 4.95 | 3.75 | 4.35 | 3.68 | 0.40 |
| Mean amniotic fluid concentration | |||||
| EPO (mIU/mL) | 27.24 | 226.93 | 261.62 | 724.30 | 0.41 |
| cTnI (pg/mL) | 67.27 | 232.48 | 70.93 | 380.38 | <0.001 |
| GFAP (ng/mL) | 1.1 | 1.65 | 3.66 | 4.24 | <0.001 |
Kruskal-Wallis test for continuous variables and exact tests for categorical variables.
Model Selection
The three amniotic fluid analytes were not mutually independent (Pearson’s Χ2= 18.124, df=4, p=0.001). EPO was conditionally dependent on cTnI (p=0.001)—i.e., the interaction among the biomarkers was statistically significant—but the interactions between GFAP and cTnI, and between GFAP and EPO, were not significant (p=0.96, and p=0.07, respectively). When entered into the model containing the main effects of the three analytes and the interaction between EPO and cTnI, the effects of gestational age dichotomized at 28 weeks of gestation and the presence of prognostic factors were not statistically significant.
Histopathology
Placental histology was available for 58 of 60 cases and all 60 controls. The frequency of placental lesions consistent with maternal and fetal vascular malperfusion and of placental inflammatory lesions was significantly higher in cases compared to controls (Table 6).
Table 6.
Placental histopathological findings of the study group
| Histologic lesions | Cases of fetal death (n=58) |
Controls (n=60) |
q-valuea |
|---|---|---|---|
| Acute inflammatory lesions | 11 (19%) | 2 (3.3%) | 0.01 |
| Chronic inflammatory lesions | 36 (62.1%) | 10 (16.7%) | <0.001 |
| Maternal vascular malperfusion | 41 (70.7%) | 21/ (35%) | <0.001 |
| Vascular | 35 (60.3%) | 17(28.3%) | <0.01 |
| Villous lesions | 19 (32.8%) | 7 (11.7%) | 0.01 |
| Retroplacental hemorrhage | 13 (22.4%) | 0 (0%) | <0.001 |
| Infarction | 10 (17.2%) | 1 (1.7%) | <0.01 |
| Accelerated villous maturation | 9 (15.5%) | 0 (0%) | <0.01 |
| Distal villous hypoplasia | 2 (3.4%) | 0 (0%) | 0.24 |
| Decidual arteriopathy | 23 (39.7%) | 13 (21.7%) | <0.05 |
| Fetal vascular malperfusion | 48 (82.8%) | 20 (33.3%) | <0.001 |
| Vascular | 40 (69%) | 9 (15%) | <0.001 |
| Villous | 36 (62.1%) | 12 (20%) | <0.001 |
| Delayed villous maturation | 12 (20.7%) | 3 (5%) | <0.02 |
q-values are P values adjusted for multiple comparisons using the Benjamini-Hochberg procedure.
Two cases with missing pathology slides had raised amniotic fluid EPO and cTnI concentrations: one patient had late-onset preeclampsia with an SGA neonate and delivered at 37.1 weeks; the other patient had placental abruption and delivered at 22.4 weeks.
Sections from normal term placentas may display immature villi, and this physiological immaturity occurs in less than 10% of cases (127). Therefore, to establish the pathogenicity of these immature villi, immunohistochemistry with CD 15 was performed in any placental section that showed monotonous villous population (defined as at least 10 such villi) with centrally placed capillaries and decreased vasculo-syncytial membranes, recapitulating the histology in early pregnancy. CD15 immunohistochemistry was used in 53 samples, including 40 controls and 13 cases. In the control group, 3/40 of the samples examined showed immunohistochemistery consistent with pathological delayed villous maturation, while 12/13 of the cases examined displayed this pathological pattern.
Of 53 hypoxic fetal deaths (EPO > 90th percentile), placental histology was available for 51 cases. Among these 51 hypoxic fetal deaths (EPO > 90th percentile), vaso-occlusive lesions, accelerated or delayed maturational villous defects, retroplacental hemorrhage or fetal vascular lesions were present in 38 (74.5%) cases. There were twelve cases of fetal deaths with delayed villous maturation: eleven (92%) had raised EPO and one case (8%) was associated with placental abruption. Four of the five fetal deaths in which only the amniotic fluid EPO concentration was raised had delayed or accelerated villous maturation in the placenta, and the fifth had hypervascular villi, a characteristic response to hypoxia. Three of the five fetal deaths not associated with any raised analytes in amniotic fluid had histological evidence of abruption consisting of retroplacental and/or intravillous hemorrhage, one had a massive subchorial hematoma (a Breus’ mole), and one displayed delayed villous maturation. Therefore, a histopathological explanation for placental insufficiency was found in 74% (43/58) of cases.
Eleven fetal deaths showed evidence of acute histologic chorioamnionitis: four had intermediate (stage 2) disease with intra-amniotic inflammation diagnosed in two, and seven cases were mild (stage 1), and six of them had intra-amniotic inflammation (128). Amniotic fluid cultures were negative for nine cases of histological chorioamnionitis, except for one case with candida placentitis, which was culture positive for Candida spp. and had intra-amniotic inflammation, and another with coagulase-negative Staphylococcus species without intra-amniotic inflammation.
Two 22-week-old fetal deaths displayed chronic villitis due to CMV infection with raised amniotic fluid EPO and cTnI concentrations as well as intra-amniotic inflammation.
The proportion of cases that had no significant placental pathology was not significantly different between the various amniotic fluid analyte groups, nor was there a difference between the groups in the type of pathological lesions present.
DISCUSSION
Principal findings of the study
The study indicated that 88% (53/60) of fetal deaths were hypoxic; that 91% (48/53) of hypoxic fetal deaths had sustained brain, myocardial or both brain and myocardial injuries in utero; and that circulatory failure and cardiac arrest, secondary to hypoxic myocardial damage, was the mechanisms of death in 70% (42/60) of cases.
Since we considered the two isolated cases, in which only the concentration of cTnI or GFAP was raised, as instances of hypoxic cardiac and brain injury occurring at slightly lower concentrations of EPO than the criteria used to conduct this study (>80th centile and >70th centile, respectively); we may conclude that: 1) 92% (55/60) of fetal deaths were hypoxic; 2) 91% (50/55) of hypoxic fetal deaths sustained myocardial, brain, or both myocardial and brain injuries in utero; 3) myocardial injuries alone occurred significantly more frequently than brain injury alone (23/50 vs. 8/50, p< 0.01), but myocardial and brain injuries occurred independently of each other; and 4) vaso-occlusive lesions, villous maturational defects, histologic evidence of abruption or fetal vascular lesions were present in 74.5% (38/51) of hypoxic fetal deaths.
Difficulty in determining the cause of fetal death
The use of objective data to assess the cause of fetal death, including clinical information, placental examination, and fetal autopsy, allows for the identification of potential etiologies of fetal death in 40% of cases (42). Given that the risk factors for stillbirth are also present in live born neonates, one cannot attribute the presence of these risk factors as direct causes of fetal demise (40). Instead, potential etiologies of fetal death should be classified as a probable cause, a possible cause, or a present condition based on the best available scientific evidence (41). The Stillbirth Collaborative Research Network found that 31% (161/512) of fetal deaths in 59 tertiary care and community hospitals from 2006 to 2008 had more than one probable or possible cause of death (59). In the current study, we found that 25% (15/60) of cases with fetal death had more than one risk factor. The result of these two studies supports the notion that the cause of death is multifactorial in at least one-quarter of cases of fetal death. Because multiple conditions may contribute to a stillbirth, the practice of recording the chain of events that led to death, rather than the single most probable cause of death, is recommended (40, 129, 130).
In the current study, 75% (45/60) of fetal deaths had a known risk factor, and these fetuses had significantly increased median concentrations of EPO than fetuses without a risk factor. The clinical implication of this finding is that pregnancies with risk factors for fetal death are more likely to present placental dysfunction due to fetal hypoxia in the index pregnancy. Previous studies reported that pregnancies with risk factors for fetal death, such as diabetes mellitus (131), hypertensive disorder of pregnancies (64), and fetal growth restriction (132) have increased amniotic fluid/umbilical cord concentration of EPO as compared to normal pregnancies without these risk factors. Furthermore, Lamont et al. (133) reported that, compared to women who had a live birth in their first pregnancy, those who experienced a stillbirth were almost five times more likely to experience a fetal death in their second pregnancy. In addition, a recent systematic review and meta-analysis has demonstrated that neonates of mothers who had a previous preterm birth or an SGA neonate are more likely to be stillborn; the risk of stillbirth in the following pregnancy is doubled if the previous neonate was preterm and SGA, and neonates of mothers who had a previous fetal death were more likely to be preterm or SGA (134). Collectively, these findings are consistent with the concept that the outcome of previous pregnancies influences the risk of a poor outcome in the subsequent pregnancy (133, 134).
Birth occurring before 28 weeks of gestation appears to be strongly associated with an intra-amniotic infection, whereas late preterm births are less likely to have an associated intrauterine infection (135, 136). Indeed, our study showed that 66.7% (4/6) of cases with intrauterine infection occurred before 28 weeks of gestation. In addition, we found that CMV infection accounted for 33.3% of intrauterine infection in cases with fetal death and was present in 3% of cases with fetal death. An Australian study showed that 9% of blood samples taken from fetal deaths by cardiac puncture were PCR-positive for CMV (137). A study from Greece, using PCR, showed significantly increased levels of CMV (16%) in the placentas of fetal deaths compared to controls (3%) (138). Since CMV is the most common cause of congenital infection associated with fetal growth restriction and central nervous system damage, and as a recent meta-analysis has demonstrated that maternal CMV infection increases the risk of spontaneous abortion and fetal death (139), we suggest that every placenta and fetus submitted for autopsy should be examined for CMV infection.
The usefulness of placental histopathology in evaluating fetal death
Normal placental maturation involves transition of immature intermediate villi in the first and second trimesters of pregnancy to mature intermediate and terminal villi, defined by small-caliber (40–100 μm), minimal stroma and abundant syncytio-capillary membrane formation in the third trimester (140). This transition results from an increasing number of peripherally placed, thin-walled vascular sinusoids and formation of syncytio-capillary membranes in villi, thereby shortening the distance between the maternal and fetal circulations and increasing the efficiency of gas exchange across the placenta (123, 141). In contrast, delayed villous maturation involves a higher percentage of centrally placed villous vessels, a reduced number of syncytio-capillary membranes, and an increase in the distance between the maternal and fetal circulations (121, 142, 143). A reduced number of syncytio-capillary membranes prevents maternal supply from meeting fetal demand for oxygen and nutrients, increasing the risk of antenatal fetal hypoxia (121, 142), unexplained fetal death (121, 142), and increased neonatal mortality (121, 142).
Although an assessment of the presence of delayed placental villous maturation is subjective, and the concordance for this lesion is poor among pathologists and subject to much inter-observer variability (144), the subjectivity and variability can be enhanced by the use of CD15 immunohistochemistry, a diagnostic marker of persisting villous immaturity and chronic placental dysfunction. The level of CD15 expression in the macro-and microvasculature reflects the degree of pathological placental villous immaturity (121). The usefulness of placental pathology for identification of potential causes of fetal death in the current study was enhanced by the use of an immunohistochemistry marker of CD15 to diagnose delayed villous maturation as reported by Seidmann et al. (121, 145–147). Indeed, the contribution of placental pathology for the identification of potential causes of fetal death in the current study was higher (74%) compared to that reported by Page et al. (148) who also reported that the most useful test in the diagnostic evaluation of fetal death is placental pathology (65% of the cases). Therefore, we propose that the evaluation of late fetal death should include placental pathology with the use of a CD15 immunohistochemistry marker to identify delayed villous immaturity as a marker of fetal hypoxia. Indeed, all twelve cases of fetal deaths with CD15-confirmed delayed villous maturation were hypoxic: eleven (92%) had ‘raised’ AF EPO concentration, and one case (8%), associated with placental abruption, did not have ‘raised’ AF EPO concentration since fetal death from an acute event prevents the elevation of AF EPO concentration (63). Raised amniotic fluid EPO concentration indicates chronic fetal hypoxia (65).
Placental dysfunction
Decreased placental function, due to either abnormal placental development, placental damage, or both, leads to decreased blood flood, oxygen, and nutrient transfer to the fetus (149). The results of the current study establish that placental dysfunction was the underlying cause of death in more than 90% of cases of fetal deaths. Consistent with this conclusion, birthweight was below the 50th centile in 81% (47/58) of the cases for which it was recorded, and that 31% (18/58) of fetal deaths were SGA neonates. Although the apparent birthweight centile may have been affected by the death-to-delivery time interval (150), the fact that the birthweight-to-placenta-weight ratio for the cases was significantly lower than that for the controls (3.94 vs. 5.72, p< 0.001) makes it unlikely that this was a significant factor affecting the birthweight centile. Placental pathology ‘explained’ 74% (43/58) of these fetal deaths, and the proportion may have been higher, but for the inability to differentiate post-mortem from ante-mortem histologic changes of fetal vascular malperfusion in the chorionic plate and in the stem villous and terminal villous vessels that, therefore, were excluded.
Fetal response to acute and chronic hypoxia
The likely mechanisms of death can be inferred from the fetal physiological responses to acute and chronic hypoxia, which have been investigated extensively in vivo in chronically instrumented, unanesthetized fetal lambs. Because fetal arterial partial pressure of oxygen (PO2) is only about 25% of maternal arterial PO2, comparable to the PO2 on Mount Everest (151), and fetal oxygen supply is limited by uterine and umbilical blood flow, the fetus is vulnerable to sudden interruptions of its oxygen supply, and has thus evolved cardiovascular and neuro-endocrinal mechanisms to maintain oxygen supply to its vital organs during episodes of acute hypoxic episodes and to survive in a chronically hypoxic intrauterine environment. The net effects of these compensatory mechanisms are to redistribute the fetal cardiac output away from peripheral organs and the carcass to the brain, myocardium, and adrenal glands (‘centralization of the circulation’), and to reduce oxygen consumption (152, 153).
Mechanisms for fetal blood redistribution
Redistribution of blood to vital organs is mediated partly by vasoconstriction and increased peripheral vascular resistance, and partly by increased perfusion of vital organs, which is at first passive and pressure dependent, but is maintained by active vasodilation and increased conductance in the cerebral, myocardial, adrenal, and umbilical circulations (154, 155). These and other adaptations allow the fetus to withstand moderate reductions in PO2 and to maintain cerebral and myocardial oxygen consumption at normal values and centralization of its circulation more or less indefinitely. However, this comes at the cost of reducing fetal growth (156–163), and reprograming tissue development (164), which predisposes the fetus to develop a variety of diseases in adult life (164–166).
The increase in peripheral vascular resistance in response to acute hypoxia involves cardiovascular, neuro-endocrine, and local vascular mechanisms. The cardiovascular response consists of transient fetal bradycardia, systemic hypertension, and peripheral vasoconstriction triggered by carotid sinus chemoreceptors that transmit afferent impulses to the cardiovascular center in the medulla, which then sends parasympathetic and α-and β-adrenergic efferent discharges to the heart, peripheral vasculature, and adrenal glands (154). Blood flow to the brain, heart, and adrenal glands increase, as do plasma concentrations of catecholamines, adrenocorticotropic hormone (ACTH), and cortisol, and the cortisol response to ACTH(154, 167, 168), but cardiac output and umbilical blood flow are unaltered unless there is acidosis, in which case both umbilical blood flow and cardiac output are reduced (152, 169).
If hypoxia continues, redistribution of blood flow to vital organs is maintained by continued peripheral vasoconstriction mediated by the circulating vasoconstrictors norepinephrine, arginine vasopressin, neuropeptide Y, and angiotensin II (155), and also by the local balance between vasoconstriction and vasodilation in peripheral vascular beds (170). Hypoxia increases both nitric oxide synthesis by vascular endothelium by a mechanism involving calcitonin gene-related peptide (cGRP) (170, 171) and the production of oxygen-free radicals that quench nitrogen oxide (172, 173). The ratio of oxygen-free radicals to nitric oxide determines the balance between vasoconstriction and vasodilation. In the peripheral vascular beds, this balance usually favors vasoconstriction, but this balance can change in fetuses compromised by chronic hypoxia (170–173).
Nitric Oxide
Chronic hypoxia increases nitric oxide production and attenuates the cardiovascular response to acute hypoxia by diminishing the vasoconstrictor response (174). Baseline cerebral and femoral blood flow is higher, but cerebral blood flow does not increase, and femoral blood flow increases less during acute hypoxia (175). These findings have been interpreted to indicate a shift in the compensatory mechanism to chronic hypoxia from increased peripheral vasoconstriction to enhanced central vasodilation (175). This effect is seen even if the ‘chronic’ hypoxia is transient, as when induced by reduction of umbilical flow by 30% for three days, and the effect lasts up to one week (176).
The carotid chemoreceptor
The carotid chemoreceptor response that initiates the response to acute hypoxia and the humoral response that maintains it are actually enhanced by chronic intrauterine conditions, including chronic hypoxia (177, 178). However, the vasoconstrictor effect of these responses is offset by increased nitric oxide production; the net vascular response, i.e., whether there is vasodilation or vasoconstriction, will be determined by the balance between the catecholaminergic and nitrergic activities, which may be different in different vascular beds (174). In the femoral circulation, the increased nitric oxide activity in chronically hypoxic fetuses overcomes the vasoconstrictor influences, and these fetuses have a markedly diminished capacity to increase peripheral vascular resistance in response to an episode of acute hypoxia (174–176).
Centralization of the fetal circulation
Centralization of the circulation occurs if uterine blood flow is reduced experimentally to 50% of the baseline for 15 minutes, but fails if uterine blood flow is reduced to 25% of the baseline. Respiratory and metabolic acidosis then develop, and there is generalized vasoconstriction, reduction of blood flow in all vascular beds, followed by hypotension, bradycardia, and decreased cardiac output prior to fetal death (179). Centralization of the circulation is maintained for about five minutes following complete occlusion of the umbilical cord, after which loss of vasoconstriction, increase in femoral blood flow, further decrease in fetal heart rate, and progressive hypotension develop as peripheral vasoconstriction is lost, cardiac glycogen stores are depleted, and acidosis impairs cardiac contractility (180).
Failure of centralization of fetal blood as a mechanism of fetal death
In 91% (50/55) of hypoxic fetal deaths in this study, centralization of the circulation failed or could not be maintained. In 35% (19/55) of cases, there was global failure of centralization resulting in both myocardial and brain injuries; in 56% (31/55) of cases, blood flow was insufficient to meet the metabolic requirements of the heart or brain. Myocardial damage occurred in 70% (42/60) of cases, and brain damage occurred in 45% (27/60) of cases.
The mean EPO concentration in fetal deaths that had only myocardial injury was not significantly different from those that had only brain injury (Table 4), but significantly more fetal deaths had only myocardial injury than only brain injury (23/50 vs. 8/50, p< 0.01), which implies either that the fetal heart is more vulnerable to hypoxia than the fetal brain, or that the oxygen-free radical-to-nitric-oxide ratio in the cerebral and myocardial circulations was such as to cause greater blood flow and oxygen delivery to the brain than to the heart.
Poudel et al. (181) measured blood flow to the brain, heart, and adrenal glands in fetal lambs rendered chronically hypoxic and growth-restricted by carunculectomy prior to pregnancy (a procedure that restricts placental size in a subsequent pregnancy), and found that blood flow, as well as oxygen and glucose delivery, increased only in the adrenal glands; blood flow and oxygen delivery to the whole brain did not increase; and oxygen and glucose delivery to the heart actually decreased compared to controls. A similar distribution of blood flow would explain why the frequency of myocardial injury alone was almost three times higher than brain injury alone among these fetal deaths (23/50 vs. 8/50, p< 0.01).
Myocardial injury is an insufficient cause of brain injury in fetal death
There was no association between the frequency of brain and myocardial injury, as the concentration of GFAP was raised in the same proportion of hypoxic fetal deaths in which the concentration of cTnI was raised as in those in which cTnI was not raised; nor was there a significant difference in median GFAP concentration between these two groups of fetal deaths. Therefore, myocardial injury does not appear to have been the direct cause of the brain injury where both myocardial and brain injury were present.
A role for fetal hypotension
However, the mean cTnI concentration in fetal deaths that had both myocardial and brain injuries was significantly higher than the mean concentration in fetal deaths that had only myocardial injury (380.38 pg/mL vs. 232.48 pg/mL, p= 0.03), implying that there was an indirect relationship between myocardial and brain injuries. Hypotension likely links these two types of injuries because the amount of brain damage during acute hypoxia correlates strongly with the depth and duration of hypotension (165), and hypotension is likely related to the extent of the myocardial injury, which, in turn, is reflected in the cTnI concentration.
When redistribution of the cardiac output fails to sustain myocardial oxygenation, there is cardiac glycogen depletion, anaerobic respiration, metabolic acidosis, myocardial depression, circulatory failure, and cardiac arrest (176, 179). Whether there is also brain injury will depend on the duration of hypotension prior to cardiac arrest (165). When there is no evidence of intrauterine myocardial injury, the mechanism of brain injury and death is less clear.
If hypotension is a pre-requisite for brain injury, as appears to be the case (165), it suggests that the cause of hypotension in fetal deaths that have brain injury without myocardial injury is not cardiac failure, but rather inadequate peripheral vasoconstriction, although it is far from clear how hypotension might cause brain injury and death without cardiac damage. However, we speculate that increased nitric oxide production in response to chronic hypoxemia could simultaneously decrease systemic vascular resistance and cerebral blood flow while maintaining adequate myocardial perfusion long enough for brain injury to occur before cardiac glycogen stores are depleted, anaerobic respiration has run its course, and the cycle of cardiac decompensation leading to circulatory failure and cardiac arrest commences.
Nitric oxide attenuates the sensitivity of the fetal baroreceptor reflex (182), which means that a drop in blood pressure caused by nitric oxide-mediated reduction in resistance in peripheral vascular beds would not elicit a sufficient increase in heart rate to restore blood pressure. Cerebral blood flow can be expected to drop in parallel with the decrease in systemic arterial blood pressure as autoregulation of the cerebral vasculature operates within a narrow range in the fetus (165). However, coronary blood flow could be maintained by nitric oxide-mediated coronary vasodilation sufficiently long enough for brain injury to occur before cardiac decompensation and death supervene, as the fetus has a significant coronary vasodilator reserve, 3.5-fold greater than in adults (183).
Fetal cardiac arrhythmia as a potential cause of fetal death in cases without evidence of myocardial and neuronal injuries
The mechanism of death in hypoxic fetal deaths that had no evidence of myocardial or brain injury cannot be determined from the data in this study. Each autopsy report showed some evidence of brain damage in all three of the five cases for which one was available: diffuse white matter and periventricular damage with calcifications in one case; bilateral ventriculomegaly with focal germinal matrix hemorrhage, and scattered petechial hemorrhages most prominent in white matter, in another; and evidence of brain stem gliosis in the third case. Brain injury, however, does not explain intrauterine death, and this was caused either by the previously described mechanisms without being reflected in an increase of cTnI and/or GFAP in amniotic fluid, or, we suggest, more likely by a fetal cardiac arrhythmia. A fetal arrythmia could have been caused by: 1) a cardiac conduction defect, which has been described in 39% of fetal deaths (93); 2) autosomal recessive inheritance of a gene that predisposes to atrial fibrillation (184); 3) mutation of the potassium voltage-gated channel subfamily H member 2 (KCNH2) gene that involves the pore region of the potassium channel (185); or 4) mutation in the sodium channel alpha-subunit gene (SCN5A gene) that interferes with sodium transport in cardiac sodium channels that predisposes to long QT intervals and ventricular arrhythmias (186, 187).
Placental abruption
In the remaining five cases for which none of the analytes was raised, there was histological evidence of a placental abruption in three of them, delayed villous maturation in one case, and a massive subchorial hematoma (Breus’ mole) in the fifth. Autopsy of the brain and heart showed only autolytic changes. Amniotic fluid EPO concentrations can increase exponentially with severe hypoxia—by as much as 22–25 mIU/mL daily among high-risk pregnancies (64, 131). Nevertheless, these fetal deaths were most likely hypoxic, given the histological findings, but death from cardiac arrest or a cardiac arrhythmia occurred too rapidly for the EPO or the markers for heart damage to rise in amniotic fluid (63).
Limitations
The main limitation on the conclusions of this study is the lack of knowledge of the pharmacokinetics of cTnI and GFAP in amniotic fluid given that the length of time it takes for cTnI and GFAP to become detectable in amniotic fluid after brain or myocardial injury is not known; how frequently these analytes are detectable after brain or myocardial injury, i.e., their sensitivity in amniotic fluid, is not known; and the rate at which the concentrations of these analytes can increase in amniotic fluid, as well as their half-lives, is also unknown. The time of death prior to delivery was also not known in any of these cases, although it could be estimated based on the placental histological findings (124). From these estimates, one can be reasonably certain that the analyte patterns observed in these fetal deaths were not materially affected by clearance of the analytes from amniotic fluid.
Conclusion
Hypoxia, secondary to placental dysfunction, is the mechanism of death in the majority of structurally normal fetuses after 20 weeks of gestation. Ninety-one percent of hypoxic fetal deaths sustained brain, myocardial, or both brain and myocardial injuries in utero. An attributable mechanism of death in 70% of the cases is circulatory failure and cardiac arrest secondary to hypoxic myocardial injury. A histopathological explanation for placental dysfunction was found in 74% of these cases.
Acknowledgments
Funding: This research was supported, in part, by the Perinatology Research Branch, Division of Obstetrics and Maternal-Fetal Medicine, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, U.S. Department of Health and Human Services (NICHD/NIH/DHHS); and, in part, with Federal funds from NICHD/NIH/DHHS under Contract No. HHSN275201300006C.
Footnotes
Disclosure: The authors report no conflicts of interest.
REFERENCES
- 1.MacDorman MF, Gregory EC. Fetal and Perinatal Mortality: United States, 2013. Natl Vital Stat Rep 2015;64(8):1–24. Epub 2015/07/30. [PubMed] [Google Scholar]
- 2.Willinger M, Ko CW, Reddy UM. Racial disparities in stillbirth risk across gestation in the United States. American journal of obstetrics and gynecology 2009;201(5):469 e1–8. Epub 2009/09/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smulian JC, Ananth CV, Vintzileos AM, Scorza WE, Knuppel RA. Fetal deaths in the United States. Influence of high-risk conditions and implications for management. Obstetrics and gynecology 2002;100(6):1183–9. Epub 2002/12/07. [DOI] [PubMed] [Google Scholar]
- 4.Allen VM, Joseph K, Murphy KE, Magee LA, Ohlsson A. The effect of hypertensive disorders in pregnancy on small for gestational age and stillbirth: a population based study. BMC pregnancy and childbirth 2004;4(1):17 Epub 2004/08/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aagaard-Tillery KM, Holmgren C, Lacoursiere DY, Houssain S, Bloebaum L, Satterfield R, et al. Factors associated with nonanomalous stillbirths: the Utah Stillbirth Database 1992–2002. American journal of obstetrics and gynecology 2006;194(3):849–54. Epub 2006/03/09. [DOI] [PubMed] [Google Scholar]
- 6.Ahmad AS, Samuelsen SO. Hypertensive disorders in pregnancy and fetal death at different gestational lengths: a population study of 2 121 371 pregnancies. BJOG : an international journal of obstetrics and gynaecology 2012;119(12):1521–8. Epub 2012/08/29. [DOI] [PubMed] [Google Scholar]
- 7.Stormdal Bring H, Hulthen Varli IA, Kublickas M, Papadogiannakis N, Pettersson K. Causes of stillbirth at different gestational ages in singleton pregnancies. Acta obstetricia et gynecologica Scandinavica 2014;93(1):86–92. Epub 2013/10/15. [DOI] [PubMed] [Google Scholar]
- 8.Harmon QE, Huang L, Umbach DM, Klungsoyr K, Engel SM, Magnus P, et al. Risk of fetal death with preeclampsia. Obstetrics and gynecology 2015;125(3):628–35. Epub 2015/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panaitescu AM, Syngelaki A, Prodan N, Akolekar R, Nicolaides KH. Chronic hypertension and adverse pregnancy outcome: a cohort study. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology 2017;50(2):228–35. Epub 2017/04/25. [DOI] [PubMed] [Google Scholar]
- 10.Rosenstein MG, Cheng YW, Snowden JM, Nicholson JM, Doss AE, Caughey AB. The risk of stillbirth and infant death stratified by gestational age in women with gestational diabetes. American journal of obstetrics and gynecology 2012;206(4):309 e1–7. Epub 2012/04/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Starikov R, Dudley D, Reddy UM. Stillbirth in the pregnancy complicated by diabetes. Curr Diab Rep 2015;15(3):11 Epub 2015/02/11. [DOI] [PubMed] [Google Scholar]
- 12.McClure EM, Goldenberg RL. Infection and stillbirth. Semin Fetal Neonatal Med 2009;14(4):182–9. Epub 2009/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldenberg RL, McClure EM, Saleem S, Reddy UM. Infection-related stillbirths. Lancet 2010;375(9724):1482–90. Epub 2010/03/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wijs LA, de Graaff EC, Leemaqz S, Dekker G. Causes of stillbirth in a socioeconomically disadvantaged urban Australian population -a comprehensive analysis. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2017;30(23):2851–7. Epub 2016/11/29. [DOI] [PubMed] [Google Scholar]
- 15.Blackwell S, Romero R, Chaiworapongsa T, Refuerzo J, Gervasi MT, Yoshimatsu J, et al. Unexplained fetal death is associated with changes in the adaptive limb of the maternal immune response consistent with prior antigenic exposure. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2003;14(4):241–6. Epub 2004/01/24. [DOI] [PubMed] [Google Scholar]
- 16.Blackwell S, Romero R, Chaiworapongsa T, Kim YM, Bujold E, Espinoza J, et al. Maternal and fetal inflammatory responses in unexplained fetal death. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2003;14(3):151–7. Epub 2003/12/26. [DOI] [PubMed] [Google Scholar]
- 17.Lee J, Romero R, Dong Z, Xu Y, Qureshi F, Jacques S, et al. Unexplained fetal death has a biological signature of maternal anti-fetal rejection: chronic chorioamnionitis and alloimmune anti-human leucocyte antigen antibodies. Histopathology 2011;59(5):928–38. Epub 2011/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ananth CV, Berkowitz GS, Savitz DA, Lapinski RH. Placental abruption and adverse perinatal outcomes. Jama 1999;282(17):1646–51. Epub 1999/11/30. [DOI] [PubMed] [Google Scholar]
- 19.Ananth CV, Wilcox AJ. Placental abruption and perinatal mortality in the United States. Am J Epidemiol 2001;153(4):332–7. Epub 2001/02/24. [DOI] [PubMed] [Google Scholar]
- 20.Raisanen S, Gissler M, Nielsen HS, Kramer MR, Williams MA, Heinonen S. Social disparity affects the incidence of placental abruption among multiparous but not nulliparous women: a register-based analysis of 1,162,126 singleton births. European journal of obstetrics, gynecology, and reproductive biology 2013;171(2):246–51. Epub 2013/10/08. [DOI] [PubMed] [Google Scholar]
- 21.Stanek J. Placental examination in nonmacerated stillbirth versus neonatal mortality. Journal of perinatal medicine. 2017 doi: 10.1515/jpm-2017-0198. Epub 2017/09/16. [DOI] [PubMed] [Google Scholar]
- 22.Nkwabong E, Tiomela Goula G. Placenta abruption surface and perinatal outcome. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2017;30(12):1456–9. Epub 2016/08/04. [DOI] [PubMed] [Google Scholar]
- 23.Lannaman K, Romero R, Chaiworapongsa T, Kim YM, Korzeniewski SJ, Maymon E, et al. Fetal death: an extreme manifestation of maternal anti-fetal rejection. Journal of perinatal medicine 2017;45(7):851–68. Epub 2017/09/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YM, Chaemsaithong P, Romero R, Shaman M, Kim CJ, Kim JS, et al. Placental lesions associated with acute atherosis. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2015;28(13):1554–62. Epub 2014/09/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Man J, Hutchinson JC, Heazell AE, Ashworth M, Jeffrey I, Sebire NJ. Stillbirth and intrauterine fetal death: role of routine histopathological placental findings to determine cause of death. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology 2016;48(5):579–84. Epub 2016/10/27. [DOI] [PubMed] [Google Scholar]
- 26.Whitten AE, Romero R, Korzeniewski SJ, Tarca AL, Schwartz AG, Yeo L, et al. Evidence of an imbalance of angiogenic/antiangiogenic factors in massive perivillous fibrin deposition (maternal floor infarction): a placental lesion associated with recurrent miscarriage and fetal death. American journal of obstetrics and gynecology 2013;208(4):310.e1-.e11. Epub 2013/01/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romero R, Whitten A, Korzeniewski SJ, Than NG, Chaemsaithong P, Miranda J, et al. Maternal floor infarction/massive perivillous fibrin deposition: a manifestation of maternal antifetal rejection? American journal of reproductive immunology 2013;70(4):285–98. Epub 2013/08/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gardosi J, Mul T, Mongelli M, Fagan D. Analysis of birthweight and gestational age in antepartum stillbirths. Br J Obstet Gynaecol 1998;105(5):524–30. Epub 1998/06/24. [DOI] [PubMed] [Google Scholar]
- 29.Froen JF, Gardosi JO, Thurmann A, Francis A, Stray-Pedersen B. Restricted fetal growth in sudden intrauterine unexplained death. Acta obstetricia et gynecologica Scandinavica 2004;83(9):801–7. Epub 2004/08/19. [DOI] [PubMed] [Google Scholar]
- 30.Premru-Srsen T, Verdenik I, Ponikvar BM, Steblovnik L, Gersak K, Cerar LK. Infant mortality and causes of death by birth weight for gestational age in non-malformed singleton infants: a 2002–2012 population-based study. Journal of perinatal medicine 2018; 46:547–553. Epub 2017/06/11. [DOI] [PubMed] [Google Scholar]
- 31.Pilliod RA, Page JM, Sparks TN, Caughey AB. The growth-restricted fetus: risk of mortality by each additional week of expectant management. J Matern Fetal Neonatal Med 2017. October 3:1–6. [Epub ahead of print]. Epub 2017/10/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yudkin PL, Wood L, Redman CW. Risk of unexplained stillbirth at different gestational ages. Lancet 1987;1(8543):1192–4. Epub 1987/05/23. [DOI] [PubMed] [Google Scholar]
- 33.Cnattingius S, Haglund B, Kramer MS. Differences in late fetal death rates in association with determinants of small for gestational age fetuses: population based cohort study. Bmj 1998;316(7143):1483–7. Epub 1998/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang DY, Usher RH, Kramer MS, Yang H, Morin L, Fretts RC. Determinants of unexplained antepartum fetal deaths. Obstetrics and gynecology 2000;95(2):215–21. Epub 2000/02/16. [DOI] [PubMed] [Google Scholar]
- 35.Froen JF, Arnestad M, Frey K, Vege A, Saugstad OD, Stray-Pedersen B. Risk factors for sudden intrauterine unexplained death: epidemiologic characteristics of singleton cases in Oslo, Norway, 1986–1995. American journal of obstetrics and gynecology 2001;184(4):694–702. Epub 2001/03/23. [DOI] [PubMed] [Google Scholar]
- 36.Nappi L, Trezza F, Bufo P, Riezzo I, Turillazzi E, Borghi C, et al. Classification of stillbirths is an ongoing dilemma. Journal of perinatal medicine 2016;44(7):837–43. Epub 2016/02/26. [DOI] [PubMed] [Google Scholar]
- 37.Kunjachen Maducolil M, Abid H, Lobo RM, Chughtai AQ, Afzal AM, Saleh HAH, et al. Risk factors and classification of stillbirth in a Middle Eastern population: a retrospective study. J Perinat Med 2017. December 21 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 38.Basu MN, Johnsen IBG, Wehberg S, Sorensen RG, Barington T, Norgard BM. Causes of death among full term stillbirths and early neonatal deaths in the Region of Southern Denmark. Journal of perinatal medicine 2018;46(2):197–202. Epub 2017/07/29. [DOI] [PubMed] [Google Scholar]
- 39.Fretts RC, Boyd ME, Usher RH, Usher HA. The changing pattern of fetal death, 1961–1988. Obstet Gynecol 1992;79(1):35–9. Epub 1992/01/01. [PubMed] [Google Scholar]
- 40.Silver RM, Varner MW, Reddy U, Goldenberg R, Pinar H, Conway D, et al. Work-up of stillbirth: a review of the evidence. American journal of obstetrics and gynecology 2007;196(5):433–44. Epub 2007/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dudley DJ, Goldenberg R, Conway D, Silver RM, Saade GR, Varner MW, et al. A new system for determining the causes of stillbirth. Obstetrics and gynecology 2010;116(2 Pt 1):254–60. Epub 2010/07/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Man J, Hutchinson JC, Ashworth M, Judge-Kronis L, Levine S, Sebire NJ. Stillbirth and intrauterine fetal death: role of routine histological organ sampling to determine cause of death. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology 2016;48(5):596–601. Epub 2016/10/27. [DOI] [PubMed] [Google Scholar]
- 43.International Stillbirth Alliance Collaborative for Improving Classification of Perinatal D, Flenady V, Wojcieszek AM, Ellwood D, Leisher SH, Erwich J, et al. Classification of causes and associated conditions for stillbirths and neonatal deaths. Semin Fetal Neonatal Med 2017;22(3):176–85. Epub 2017/03/14. [DOI] [PubMed] [Google Scholar]
- 44.Teramo KA. Obstetric problems in diabetic pregnancy -The role of fetal hypoxia. Best Pract Res Clin Endocrinol Metab 2010;24(4):663–71. Epub 2010/09/14. [DOI] [PubMed] [Google Scholar]
- 45.Hovatta O, Lipasti A, Rapola J, Karjalainen O. Causes of stillbirth: a clinicopathological study of 243 patients. Br J Obstet Gynaecol 1983;90(8):691–6. Epub 1983/08/01. [DOI] [PubMed] [Google Scholar]
- 46.Kidron D, Bernheim J, Aviram R. Placental findings contributing to fetal death, a study of 120 stillbirths between 23 and 40 weeks gestation. Placenta 2009;30(8):700–4. Epub 2009/06/19. [DOI] [PubMed] [Google Scholar]
- 47.Korteweg FJ, Erwich JJ, Holm JP, Ravise JM, van der Meer J, Veeger NJ, et al. Diverse placental pathologies as the main causes of fetal death. Obstetrics and gynecology 2009;114(4):809–17. Epub 2009/11/06. [DOI] [PubMed] [Google Scholar]
- 48.Ptacek I, Sebire NJ, Man JA, Brownbill P, Heazell AEP. Systematic review of placental pathology reported in association with stillbirth. Placenta 2014;35(8):552–62. [DOI] [PubMed] [Google Scholar]
- 49.Varli IH, Petersson K, Bottinga R, Bremme K, Hofsjo A, Holm M, et al. The Stockholm classification of stillbirth. Acta obstetricia et gynecologica Scandinavica 2008;87(11):1202–12. Epub 2008/10/28. [DOI] [PubMed] [Google Scholar]
- 50.Labarrere CA, DiCarlo HL, Bammerlin E, Hardin JW, Kim YM, Chaemsaithong P, et al. Failure of physiologic transformation of spiral arteries, endothelial and trophoblast cell activation, and acute atherosis in the basal plate of the placenta. American journal of obstetrics and gynecology 2017;216(3):287.e1-.e16. Epub 2016/12/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amir H, Weintraub A, Aricha-Tamir B, Apel-Sarid L, Holcberg G, Sheiner E. A piece in the puzzle of intrauterine fetal death: pathological findings in placentas from term and preterm intrauterine fetal death pregnancies. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2009;22(9):759–64. [DOI] [PubMed] [Google Scholar]
- 52.Tellefsen CH, Vogt C. How important is placental examination in cases of perinatal deaths? Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 2011;14(2):99–104. Epub 2010/08/20. [DOI] [PubMed] [Google Scholar]
- 53.Miller ES, Minturn L, Linn R, Weese-Mayer DE, Ernst LM. Stillbirth evaluation: a stepwise assessment of placental pathology and autopsy. American journal of obstetrics and gynecology 2016;214(1):115.e1–6. Epub 2015/09/01. [DOI] [PubMed] [Google Scholar]
- 54.Heazell AE, Martindale EA. Can post-mortem examination of the placenta help determine the cause of stillbirth? J Obstet Gynaecol 2009;29(3):225–8. Epub 2009/04/10. [DOI] [PubMed] [Google Scholar]
- 55.Helgadottir LB, Turowski G, Skjeldestad FE, Jacobsen AF, Sandset PM, Roald B, et al. Classification of stillbirths and risk factors by cause of death--a case-control study. Acta obstetricia et gynecologica Scandinavica 2013;92(3):325–33. Epub 2012/11/20. [DOI] [PubMed] [Google Scholar]
- 56.Pinar H, Goldenberg RL, Koch MA, Heim-Hall J, Hawkins HK, Shehata B, et al. Placental findings in singleton stillbirths. Obstetrics and gynecology 2014;123(2 Pt 1):325–36. Epub 2014/01/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Horn LC, Langner A, Stiehl P, Wittekind C, Faber R. Identification of the causes of intrauterine death during 310 consecutive autopsies. European journal of obstetrics, gynecology, and reproductive biology 2004;113(2):134–8. Epub 2004/04/06. [DOI] [PubMed] [Google Scholar]
- 58.Rahman A, Cahill LS, Zhou YQ, Hoggarth J, Rennie MY, Seed M, et al. A mouse model of antepartum stillbirth. American journal of obstetrics and gynecology 2017;217(4):443 e1-e11. Epub 2017/06/18. [DOI] [PubMed] [Google Scholar]
- 59.Stillbirth Collaborative Research Network Writing G. Causes of death among stillbirths. Jama 2011;306(22):2459–68. Epub 2011/12/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaiworapongsa T, Romero R, Korzeniewski SJ, Kusanovic JP, Soto E, Lam J, Dong Z, Than NG, Yeo L, Hernandez-Andrade E, Conde-Agudelo A, Hassan SS. Maternal plasma concentrations of angiogenic/antiangiogenic factors in the third trimester of pregnancy to identify the patient at risk for stillbirth at or near term and severe late preeclampsia. Am J Obstet Gynecol 2013. April;208(4):287.e1–287.e15. Epub 2013 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaiworapongsa T, Romero R, Erez O, Tarca AL, Conde-Agudelo A, Chaemsaithong P, et al. The prediction of fetal death with a simple maternal blood test at 24–28 weeks: a role for angiogenic index-1 (PlGF/sVEGFR-1 ratio). American journal of obstetrics and gynecology 2017;217(6):682 e1-e13. Epub 2017/10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Korzeniewski SJ, Romero R, Chaiworapongsa T, Chaemsaithong P, Kim CJ, Kim YM, et al. Maternal plasma angiogenic index-1 (placental growth factor/soluble vascular endothelial growth factor receptor-1) is a biomarker for the burden of placental lesions consistent with uteroplacental underperfusion: a longitudinal case-cohort study. American journal of obstetrics and gynecology 2016;214(5):629.e1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teramo KA, Schwartz R, Clemons GK, Widness JA. Amniotic fluid erythropoietin concentrations differentiate between acute and chronic causes of fetal death. Acta obstetricia et gynecologica Scandinavica 2002;81(3):245–51. [DOI] [PubMed] [Google Scholar]
- 64.Teramo KA, Hiilesmaa VK, Schwartz R, Clemons GK, Widness JA. Amniotic fluid and cord plasma erythropoietin levels in pregnancies complicated by preeclampsia, pregnancy-induced hypertension and chronic hypertension. Journal of perinatal medicine 2004;32(3):240–7. [DOI] [PubMed] [Google Scholar]
- 65.Teramo KA, Widness JA. Increased Fetal Plasma and Amniotic Fluid Erythropoietin Concentrations: Markers of Intrauterine Hypoxia. Neonatology 2009;95(2):105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hunkeler NM, Kullman J, Murphy AM. Troponin I isoform expression in human heart. Circ Res 1991;69(5):1409–14. Epub 1991/11/01. [DOI] [PubMed] [Google Scholar]
- 67.Larue C, Calzolari C, Bertinchant JP, Leclercq F, Grolleau R, Pau B. Cardiac-specific immunoenzymometric assay of troponin I in the early phase of acute myocardial infarction. Clinical chemistry 1993;39(6):972–9. Epub 1993/06/01. [PubMed] [Google Scholar]
- 68.Madrid AH, del Rey JM, Rubi J, Ortega J, Gonzalez Rebollo JM, Seara JG, et al. Biochemical markers and cardiac troponin I release after radiofrequency catheter ablation: approach to size of necrosis. Am Heart J 1998;136(6):948–55. Epub 1998/12/08. [DOI] [PubMed] [Google Scholar]
- 69.Pelinka LE, Kroepfl A, Leixnering M, Buchinger W, Raabe A, Redl H. GFAP versus S100B in serum after traumatic brain injury: relationship to brain damage and outcome. Journal of neurotrauma 2004;21(11):1553–61. Epub 2005/02/03. [DOI] [PubMed] [Google Scholar]
- 70.Ennen CS, Huisman TA, Savage WJ, Northington FJ, Jennings JM, Everett AD, et al. Glial fibrillary acidic protein as a biomarker for neonatal hypoxic-ischemic encephalopathy treated with whole-body cooling. American journal of obstetrics and gynecology 2011;205(3):251 e1–7. Epub 2011/07/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chalak LF, Sanchez PJ, Adams-Huet B, Laptook AR, Heyne RJ, Rosenfeld CR. Biomarkers for severity of neonatal hypoxic-ischemic encephalopathy and outcomes in newborns receiving hypothermia therapy. J Pediatr 2014;164(3):468–74 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Sa DJ. Coronary arterial lesions and myocardial necrosis in stillbirths and infants. Arch Dis Child 1979;54(12):918–30. Epub 1979/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sims ME, Turkel SB, Halterman G, Paul RH. Brain injury and intrauterine death. American journal of obstetrics and gynecology 1985;151(6):721–3. Epub 1985/03/15. [DOI] [PubMed] [Google Scholar]
- 74.Squier M, Keeling JW. The incidence of prenatal brain injury. Neuropathol Appl Neurobiol 1991;17(1):29–38. Epub 1991/02/01. [DOI] [PubMed] [Google Scholar]
- 75.Gaffney G, Squier MV, Johnson A, Flavell V, Sellers S. Clinical associations of prenatal ischaemic white matter injury. Arch Dis Child Fetal Neonatal Ed 1994;70(2):F101–6. Epub 1994/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grafe MR. The correlation of prenatal brain damage with placental pathology. J Neuropathol Exp Neurol 1994;53(4):407–15. Epub 1994/07/01. [DOI] [PubMed] [Google Scholar]
- 77.Grafe MR, Kinney HC. Neuropathology associated with stillbirth. Seminars in perinatology 2002;26(1):83–8. Epub 2002/03/06. [DOI] [PubMed] [Google Scholar]
- 78.Becher JC, Bell JE, Keeling JW, Liston WA, McIntosh N, Wyatt B. The Scottish Perinatal Neuropathology Study--clinicopathological correlation in stillbirths. BJOG : an international journal of obstetrics and gynaecology 2006;113(3):310–7. Epub 2006/02/21. [DOI] [PubMed] [Google Scholar]
- 79.Jacques SM, Kupsky WJ, Giorgadze T, Qureshi F. Fetal Central Nervous System Injury in Third Trimester Stillbirth: A Clinicopathologic Study of 63 Cases. Pediatric and Developmental Pathology 2012;15(5):375–84. [DOI] [PubMed] [Google Scholar]
- 80.Bamber AR, Pryce J, Cook A, Ashworth M, Sebire NJ. Myocardial necrosis and infarction in newborns and infants. Forensic Sci Med Pathol 2013;9(4):521–7. Epub 2013/07/13. [DOI] [PubMed] [Google Scholar]
- 81.Gazzolo D, Pluchinotta F, Bashir M, Aboulgar H, Said HM, Iman I, et al. Neurological abnormalities in full-term asphyxiated newborns and salivary S100B testing: the “Cooperative Multitask against Brain Injury of Neonates” (CoMBINe) international study. PloS one 2015;10(1):e0115194 Epub 2015/01/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zanjani ED, Pixley JS, Slotnick N, MacKintosh FR, Ekhterae D, Clemons G. Erythropoietin does not cross the placenta into the fetus. Pathobiology 1993;61(3–4):211–5. Epub 1993/01/01. [DOI] [PubMed] [Google Scholar]
- 83.Schneider H, Malek A. Lack of permeability of the human placenta for erythropoietin. Journal of perinatal medicine 1995;23(1–2):71–6. Epub 1995/01/01. [DOI] [PubMed] [Google Scholar]
- 84.Juul SE, Yachnis AT, Christensen RD. Tissue distribution of erythropoietin and erythropoietin receptor in the developing human fetus. Early Hum Dev 1998;52(3):235–49. Epub 1998/11/10. [DOI] [PubMed] [Google Scholar]
- 85.Juul SE, Yachnis AT, Rojiani AM, Christensen RD. Immunohistochemical localization of erythropoietin and its receptor in the developing human brain. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 1999;2(2):148–58. Epub 1999/02/09. [DOI] [PubMed] [Google Scholar]
- 86.Fairchild Benyo D, Conrad KP. Expression of the erythropoietin receptor by trophoblast cellsin the human placenta. Biol Reprod 1999;60(4):861–70. Epub 1999/03/20. [DOI] [PubMed] [Google Scholar]
- 87.Toth B, Fischl A, Scholz C, Kunze S, Friese K, Jeschke U. Erythropoietin and erythropoietin receptor expression in normal and disturbed pregnancy. European Journal of Obstetrics Gynecology and Reproductive Biology 2008;140(2):192–200. [DOI] [PubMed] [Google Scholar]
- 88.Bodor GS, Porterfield D, Voss EM, Smith S, Apple FS. Cardiac troponin-I is not expressed in fetal and healthy or diseased adult human skeletal muscle tissue. Clinical chemistry 1995;41(12 Pt 1):1710–5. Epub 1995/12/01. [PubMed] [Google Scholar]
- 89.Apple FS. Tissue specificity of cardiac troponin I, cardiac troponin T and creatine kinase-MB. Clin Chim Acta 1999;284(2):151–9. Epub 1999/08/18. [DOI] [PubMed] [Google Scholar]
- 90.Shastri AT, Samarasekara S, Muniraman H, Clarke P. Cardiac troponin I concentrations in neonates with hypoxic-ischaemic encephalopathy. Acta Paediatrica 2012;101(1):26–9. [DOI] [PubMed] [Google Scholar]
- 91.Turker G, Babaoglu K, Duman C, Gokalp A, Zengin E, Arisoy AE. The effect of blood gas and Apgar score on cord blood cardiac troponin I. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2004;16(5):315–9. Epub 2004/12/29. [DOI] [PubMed] [Google Scholar]
- 92.Turker G, Sarper N, Babaoglu K, Gokalp AS, Duman C, Arisoy AE. Early prognostic significance of umbilical cord troponin I in critically ill newborns. Prospective study with a control group. Journal of perinatal medicine 2005;33(1):54–9. Epub 2005/04/22. [DOI] [PubMed] [Google Scholar]
- 93.Matturri L, Lavezzi AM. Unexplained stillbirth versus SIDS: common congenital diseases of the autonomic nervous system--pathology and nosology. Early Hum Dev 2011;87(3):209–15. Epub 2011/01/26. [DOI] [PubMed] [Google Scholar]
- 94.Widness JA, Clemons GK, Garcia JF, Oh W, Schwartz R. Increased immunoreactive erythropoietin in cord serum after labor. American journal of obstetrics and gynecology 1984;148(2):194–7. [DOI] [PubMed] [Google Scholar]
- 95.Committee on Obstetric Practice tAIoUiM, the Society for Maternal-Fetal M. Committee Opinion No 700: Methods for Estimating the Due Date. Obstetrics and gynecology 2017;129(5):e150–e4. Epub 2017/04/21. [DOI] [PubMed] [Google Scholar]
- 96.Macdorman MF, Kirmeyer S. The challenge of fetal mortality. NCHS Data Brief 2009(16):1–8. Epub 2009/04/25. [PubMed] [Google Scholar]
- 97.ACOG Committee on Obstetric Practice. ACOG practice bulletin. Diagnosis and management of preeclampsia and eclampsia. Number 33, January 2002. American College of Obstetricians and Gynecologists. International journal of gynaecology and obstetrics: the official organ of the International Federation of Gynaecology and Obstetrics 2002;77(1):67. [PubMed] [Google Scholar]
- 98.ACOG. Hypertension in pregnancy. Report of the ACOG Task Force on Hypertension in Pregnancy. Obstet Gynecol 2013;122:1122–31. [DOI] [PubMed] [Google Scholar]
- 99.Alexander GR, Himes JH, Kaufman RB, Mor J, Kogan M. A United States national reference for fetal growth. Obstetrics and gynecology 1996;87(2):163–8. Epub 1996/02/01. [DOI] [PubMed] [Google Scholar]
- 100.ADA. Standards of Medical Care in Diabetes-2017: Summary of Revisions. Diabetes Care 2017;40(Suppl 1):S4–S5. Epub 2016/12/17. [DOI] [PubMed] [Google Scholar]
- 101.Ananth CV, Lavery JA, Vintzileos AM, Skupski DW, Varner M, Saade G, et al. Severe placental abruption: clinical definition and associations with maternal complications. American journal of obstetrics and gynecology 2016;214(2):272 e1-e9. Epub 2015/09/24. [DOI] [PubMed] [Google Scholar]
- 102.Fishman SG, Chasen ST, Maheshwari B. Risk factors for preterm delivery with placenta previa. Journal of perinatal medicine 2011;40(1):39–42. Epub 2011/11/17. [DOI] [PubMed] [Google Scholar]
- 103.Yoon BH, Romero R, Moon JB, Shim SS, Kim M, Kim G, et al. Clinical significance of intra-amniotic inflammation in patients with preterm labor and intact membranes. American journal of obstetrics and gynecology 2001;185(5):1130–6. [DOI] [PubMed] [Google Scholar]
- 104.Chaiyasit N, Romero R, Chaemsaithong P, Docheva N, Bhatti G, Kusanovic JP, et al. Clinical chorioamnionitis at term VIII: a rapid MMP-8 test for the identification of intra-amniotic inflammation. Journal of perinatal medicine 2017;45(5):539–50. Epub 2017/07/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Romero R, Chaemsaithong P, Chaiyasit N, Docheva N, Dong Z, Kim CJ, et al. CXCL10 and IL-6: Markers of two different forms of intra-amniotic inflammation in preterm labor. American journal of reproductive immunology 2017;78(1). Epub 2017/05/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Romero R, Chaemsaithong P, Docheva N, Korzeniewski SJ, Kusanovic JP, Yoon BH, et al. Clinical chorioamnionitis at term VI: acute chorioamnionitis and funisitis according to the presence or absence of microorganisms and inflammation in the amniotic cavity. Journal of perinatal medicine 2016;44(1):33–51. Epub 2015/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Romero R, Chaemsaithong P, Docheva N, Korzeniewski SJ, Tarca AL, Bhatti G, et al. Clinical chorioamnionitis at term IV: the maternal plasma cytokine profile. Journal of perinatal medicine 2016;44(1):77–98. Epub 2015/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Romero R, Chaemsaithong P, Korzeniewski SJ, Tarca AL, Bhatti G, Xu Z, et al. Clinical chorioamnionitis at term II: the intra-amniotic inflammatory response. Journal of perinatal medicine 2016;44(1):5–22. Epub 2015/05/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Romero R, Chaemsaithong P, Korzeniewski SJ, Kusanovic JP, Docheva N, Martinez-Varea A, et al. Clinical chorioamnionitis at term III: how well do clinical criteria perform in the identification of proven intra-amniotic infection? Journal of perinatal medicine 2016;44(1):23–32. Epub 2015/04/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chaemsaithong P, Romero R, Korzeniewski SJ, Martinez-Varea A, Dong Z, Yoon BH, et al. A point of care test for interleukin-6 in amniotic fluid in preterm prelabor rupture of membranes: a step toward the early treatment of acute intra-amniotic inflammation/infection. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2016;29(3):360–7. Epub 2015/03/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chaemsaithong P, Romero R, Korzeniewski SJ, Martinez-Varea A, Dong Z, Yoon BH, et al. A rapid interleukin-6 bedside test for the identification of intra-amniotic inflammation in preterm labor with intact membranes. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2016;29(3):349–59. Epub 2015/03/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Romero R, Miranda J, Kusanovic JP, Chaiworapongsa T, Chaemsaithong P, Martinez A, et al. Clinical chorioamnionitis at term I: microbiology of the amniotic cavity using cultivation and molecular techniques. Journal of perinatal medicine 2015;43(1):19–36. Epub 2015/02/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Romero R, Mazor M, Wu YK, Sirtori M, Oyarzun E, Mitchell MD, et al. Infection in the pathogenesis of preterm labor. Seminars in perinatology 1988;12(4):262–79. Epub 1988/10/01. [PubMed] [Google Scholar]
- 114.Romero R, Sirtori M, Oyarzun E, Avila C, Mazor M, Callahan R, et al. Infection and labor. V. Prevalence, microbiology, and clinical significance of intraamniotic infection in women with preterm labor and intact membranes. American journal of obstetrics and gynecology 1989;161(3):817–24. Epub 1989/09/01. [DOI] [PubMed] [Google Scholar]
- 115.Romero R, Miranda J, Chaiworapongsa T, Chaemsaithong P, Gotsch F, Dong Z, et al. A novel molecular microbiologic technique for the rapid diagnosis of microbial invasion of the amniotic cavity and intra-amniotic infection in preterm labor with intact membranes. American journal of reproductive immunology 2014;71(4):330–58. Epub 2014/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pacora P, Romero R, Erez O, Maymon E, Panaitescu B, Kusanovic JP, et al. The diagnostic performance of the beta-glucan assay in the detection of intra-amniotic infection with Candida species. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2017:1–18. Epub 2017/12/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mostoufi-zadeh M, Driscoll SG, Biano SA, Kundsin RB. Placental evidence of cytomegalovirus infection of the fetus and neonate. Archives of pathology & laboratory medicine 1984;108(5):403–6. Epub 1984/05/01. [PubMed] [Google Scholar]
- 118.Stanek J ST, Coffman. Focal segmental lymphoplasmacytic villitis of cytomegalovirus infection:The most omminous diffuse pattern of palcental injury. Placenta 2016;45:90 Abstract P1.56. [Google Scholar]
- 119.Romero R, Kim YM, Pacora P, Kim CJ, Benshalom-Tirosh N, Jaiman S, Bhatti G,Kim JS, Qureshi F, Jacques SM,Jung EJ, Yeo L, Panaitescu B, Maymon E, Hassan S,Hsu AD,Erez O. The frequency and type of placental histologic lesions in term pregnancies with normal outcome. J Perinatal Med 2018;46:613–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khong TY, Mooney EE, Ariel I, Balmus NC, Boyd TK, Brundler MA, et al. Sampling and definitions of placental lesions: Amsterdam Placental Workshop Group Consensus Statement. Archives of pathology & laboratory medicine 2016;140(7):698–713. Epub 2016/05/26. [DOI] [PubMed] [Google Scholar]
- 121.Seidmann L, Suhan T, Kamyshanskiy Y, Nevmerzhitskaya A, Gerein V, Kirkpatrick CJ. CD15 -a new marker of pathological villous immaturity of the term placenta. Placenta 2014;35(11):925–31. Epub 2014/08/26. [DOI] [PubMed] [Google Scholar]
- 122.Morgan TK, Tolosa JE, Mele L, Wapner RJ, Spong CY, Sorokin Y, et al. Placental villous hypermaturation is associated with idiopathic preterm birth. The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstet 2013;26(7):647–53. Epub 2012/11/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Treacy A, Higgins M, Kearney JM, McAuliffe F, Mooney EE. Delayed villous maturation of the placenta: quantitative assessment in different cohorts. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 2013;16(2):63–6. Epub 2012/11/10. [DOI] [PubMed] [Google Scholar]
- 124.Genest DR. Estimating the time of death in stillborn fetuses: II. Histologic evaluation of the placenta; a study of 71 stillborns. Obstetrics and gynecology 1992;80(4):585–92. Epub 1992/10/01. [PubMed] [Google Scholar]
- 125.Gardosi J, Kady SM, McGeown P, Francis A, Tonks A. Classification of stillbirth by relevant condition at death (ReCoDe): population based cohort study. Bmj 2005;331(7525):1113–7. Epub 2005/10/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Smith GC, Yu CK, Papageorghiou AT, Cacho AM, Nicolaides KH. Maternal uterine artery Doppler flow velocimetry and the risk of stillbirth. Obstetrics and gynecology 2007;109(1):144–51. Epub 2007/01/02. [DOI] [PubMed] [Google Scholar]
- 127.Vogel M Pathologie der Plazenta: Spatz Schwangerschaft und fetoplazentare Einheit. In: Klöppel G, Kreipe H, Remmele W, editors. Pathologie Heidelberg:Springer-Verlag; 2013. p. 519–39. [Google Scholar]
- 128.Kim CJ, Romero R, Chaemsaithong P, Chaiyasit N, Yoon BH, Kim YM. Acute chorioamnionitis and funisitis: definition, pathologic features, and clinical significance. American journal of obstetrics and gynecology 2015;213(4 Suppl):S29–52. Epub 2015/10/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Froen JF, Pinar H, Flenady V, Bahrin S, Charles A, Chauke L, et al. Causes of death and associated conditions (Codac): a utilitarian approach to the classification of perinatal deaths. BMC pregnancy and childbirth 2009;9:22 Epub 2009/06/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Flenady V, Froen JF, Pinar H, Torabi R, Saastad E, Guyon G, et al. An evaluation of classification systems for stillbirth. BMC pregnancy and childbirth 2009;9:24 Epub 2009/06/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Teramo K, Kari MA, Eronen M, Markkanen H, Hiilesmaa V. High amniotic fluid erythropoietin levels are associated with an increased frequency of fetal and neonatal morbidity in Type 1 diabetic pregnancies. Diabetologia 2004;47(10):1695–703. [DOI] [PubMed] [Google Scholar]
- 132.Jazayeri A, Tsibris JC, Spellacy WN. Fetal erythropoietin levels in growth-restricted and appropriately grown neonates with and without abnormal fetal heart rate tracings: a comparison with cord blood gases and Apgar scores. Journal of perinatology : official journal of the California Perinatal Association 1999;19(4):255–9. [DOI] [PubMed] [Google Scholar]
- 133.Lamont K, Scott NW, Jones GT, Bhattacharya S. Risk of recurrent stillbirth: systematic review and meta-analysis. Bmj 2015;350:h3080 Epub 2015/06/26. [DOI] [PubMed] [Google Scholar]
- 134.Malacova E, Regan A, Nassar N, Raynes-Greenow C, Leonard H, Srinivasjois R, et al. Risk of stillbirth, preterm delivery, and fetal growth restriction following exposure in a previous birth: systematic review and meta-analysis. BJOG : an international journal of obstetrics and gynaecology 2018;125(2):183–92. Epub 2017/09/01. [DOI] [PubMed] [Google Scholar]
- 135.Gibbs RS. The origins of stillbirth: infectious diseases. Seminars in perinatology 2002;26(1):75–8. Epub 2002/03/06. [DOI] [PubMed] [Google Scholar]
- 136.Copper RL, Goldenberg RL, DuBard MB, Davis RO. Risk factors for fetal death in white, black, and Hispanic women. Collaborative Group on Preterm Birth Prevention. Obstetrics and gynecology 1994;84(4):490–5. Epub 1994/10/01. [PubMed] [Google Scholar]
- 137.Howard J, Hall B, Brennan LE, Arbuckle S, Craig ME, Graf N, et al. Utility of newborn screening cards for detecting CMV infection in cases of stillbirth. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology 2009;44(3):215–8. Epub 2009/01/31. [DOI] [PubMed] [Google Scholar]
- 138.Syridou G, Spanakis N, Konstantinidou A, Piperaki ET, Kafetzis D, Patsouris E, et al. Detection of cytomegalovirus, parvovirus B19 and herpes simplex viruses in cases of intrauterine fetal death: association with pathological findings. Journal of medical virology 2008;80(10):1776–82. Epub 2008/08/21. [DOI] [PubMed] [Google Scholar]
- 139.Shi TL, Huang LJ, Xiong YQ, Zhong YY, Yang JJ, Fu T, et al. The risk of herpes simplex virus and human cytomegalovirus infection during pregnancy upon adverse pregnancy outcomes: A meta-analysis. Journal of clinical virology : the official publication of the Pan American Society for Clinical Virology 2018;104:48–55. Epub 2018/05/08. [DOI] [PubMed] [Google Scholar]
- 140.Mayhew TM. A stereological perspective on placental morphology in normal and complicated pregnancies. Journal of anatomy 2009;215(1):77–90. Epub 2009/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Serra AE, Lemon LS, Mokhtari NB, Parks WT, Catov JM, Venkataramanan R, et al. Delayed villous maturation in term placentas exposed to opioid maintenance therapy: a retrospective cohort study. American journal of obstetrics and gynecology 2017;216(4):418 e1-e5. Epub 2016/12/28. [DOI] [PubMed] [Google Scholar]
- 142.Stallmach T, Hebisch G, Meier K, Dudenhausen JW, Vogel M. Rescue by birth: defective placental maturation and late fetal mortality. Obstetrics and gynecology 2001;97(4):505–9. Epub 2001/03/29. [DOI] [PubMed] [Google Scholar]
- 143.Seidmann L, Suhan T, Unger R, Gerein V, Kirkpatrick CJ. Imbalance of expression of bFGF and PK1 is associated with defective maturation and antenatal placental insufficiency. European journal of obstetrics, gynecology, and reproductive biology 2013;170(2):352–7. Epub 2013/07/31. [DOI] [PubMed] [Google Scholar]
- 144.Al-Adnani M, Marnerides A, George S, Nasir A, Weber MA. “Delayed Villous Maturation” in Placental Reporting: Concordance among Consultant Pediatric Pathologists at a Single Specialist Center. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society 2015;18(5):375–9. Epub 2015/11/26. [DOI] [PubMed] [Google Scholar]
- 145.Seidmann L, Anspach L, Roth W. The embryo-placental CD15-positive “vasculogenic zones” as a source of propranolol-sensitive pediatric vascular tumors. Placenta 2016;38:93–9. Epub 2016/02/26. [DOI] [PubMed] [Google Scholar]
- 146.Seidmann L, Kamyshanskiy Y, Martin SZ, Fruth A, Roth W. Immaturity for gestational age of microvasculature and placental barrier in term placentas with high weight. European journal of obstetrics, gynecology, and reproductive biology 2017;215:134–40. Epub 2017/06/19. [DOI] [PubMed] [Google Scholar]
- 147.Seidmann L, Suhan T, Unger R, Gerein V, Kirkpatrick CJ. Transient CD15-positive endothelial phenotype in the human placenta correlates with physiological and pathological fetoplacental immaturity. European journal of obstetrics, gynecology, and reproductive biology 2014;180:172–9. Epub 2014/07/22. [DOI] [PubMed] [Google Scholar]
- 148.Page JM, Christiansen-Lindquist L, Thorsten V, Parker CB, Reddy UM, Dudley DJ, et al. Diagnostic Tests for Evaluation of Stillbirth: Results From the Stillbirth Collaborative Research Network. Obstetrics and gynecology 2017;129(4):699–706. Epub 2017/03/24. [DOI] [PubMed] [Google Scholar]
- 149.Silver RM. Examining the link between placental pathology, growth restriction, and stillbirth. Best practice & research Clinical obstetrics & gynaecology 2018;49:89–102 [DOI] [PubMed] [Google Scholar]
- 150.Man J, Hutchinson JC, Ashworth M, Heazell AE, Levine S, Sebire NJ. Effects of intrauterine retention and postmortem interval on body weight following intrauterine death: implications for assessment of fetal growth restriction at autopsy. Ultrasound Obstet Gynecol 2016. November;48(5):574–578. Epub 2016 Oct 25. [DOI] [PubMed] [Google Scholar]
- 151.Llanos AJ, Riquelme RA, Sanhueza EM, Hanson MA, Blanco CE, Parer JT, et al. The fetal llama versus the fetal sheep: different strategies to withstand hypoxia. High Alt Med Biol 2003;4(2):193–202. Epub 2003/07/12. [DOI] [PubMed] [Google Scholar]
- 152.Jensen A, Garnier Y, Berger R. Dynamics of fetal circulatory responses to hypoxia and asphyxia. European journal of obstetrics, gynecology, and reproductive biology 1999;84(2):155–72. Epub 1999/07/31. [DOI] [PubMed] [Google Scholar]
- 153.Rudolph AM. Distribution and regulation of blood flow in the fetal and neonatal lamb. Circ Res 1985;57(6):811–21. Epub 1985/12/01. [DOI] [PubMed] [Google Scholar]
- 154.Giussani DA, Spencer JA, Moore PJ, Bennet L, Hanson MA. Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. The Journal of physiology 1993;461:431–49. Epub 1993/02/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Giussani D SJ, Hanson M. Fetal cardiovascular reflex responses to hypoxaemia. Fetal and Maternal Medicine Review 1994;6(1):17–37. [Google Scholar]
- 156.Burton GJ, Jauniaux E. Pathophysiology of placental-derived fetal growth restriction. American journal of obstetrics and gynecology 2018;218(2s):S745–s61. Epub 2018/02/10. [DOI] [PubMed] [Google Scholar]
- 157.McCowan LM, Figueras F, Anderson NH. Evidence-based national guidelines for the management of suspected fetal growth restriction: comparison, consensus, and controversy. American journal of obstetrics and gynecology 2018;218(2S):S855–S68. Epub 2018/02/10. [DOI] [PubMed] [Google Scholar]
- 158.Gardosi J, Francis A, Turner S, Williams M. Customized growth charts: rationale, validation and clinical benefits. American journal of obstetrics and gynecology 2018;218(2S):S609–S18. Epub 2018/02/10. [DOI] [PubMed] [Google Scholar]
- 159.Tarca AL, Romero R, Gudicha DW, Erez O, Hernandez-Andrade E, Yeo L, et al. A new customized fetal growth standard for African American women: the PRB/NICHD Detroit study. American journal of obstetrics and gynecology 2018;218(2S):S679–S91 e4. Epub 2018/02/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Papageorghiou AT, Kennedy SH, Salomon LJ, Altman DG, Ohuma EO, Stones W, et al. The INTERGROWTH-21(st) fetal growth standards: toward the global integration of pregnancy and pediatric care. American journal of obstetrics and gynecology 2018;218(2S):S630–S40. Epub 2018/02/10. [DOI] [PubMed] [Google Scholar]
- 161.Kiserud T, Benachi A, Hecher K, Perez RG, Carvalho J, Piaggio G, et al. The World Health Organization fetal growth charts: concept, findings, interpretation, and application. American journal of obstetrics and gynecology 2018;218(2S):S619–S29. Epub 2018/02/10. [DOI] [PubMed] [Google Scholar]
- 162.Deter RL, Lee W, Yeo L, Erez O, Ramamurthy U, Naik M, et al. Individualized growth assessment: conceptual framework and practical implementation for the evaluation of fetal growth and neonatal growth outcome. American journal of obstetrics and gynecology 2018;218(2S):S656–S78. Epub 2018/02/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Figueras F, Caradeux J, Crispi F, Eixarch E, Peguero A, Gratacos E. Diagnosis and surveillance of late-onset fetal growth restriction. American journal of obstetrics and gynecology 2018;218(2S):S790–S802 e1. Epub 2018/02/10. [DOI] [PubMed] [Google Scholar]
- 164.Crispi F, Miranda J, Gratacos E. Long-term cardiovascular consequences of fetal growth restriction: biology, clinical implications, and opportunities for prevention of adult disease. American journal of obstetrics and gynecology 2018;218(2S):S869–S79. Epub 2018/02/10. [DOI] [PubMed] [Google Scholar]
- 165.Gunn AJ, Bennet L. Fetal hypoxia insults and patterns of brain injury: insights from animal models. Clin Perinatol 2009;36(3):579–93. Epub 2009/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Giussani DA, Camm EJ, Niu Y, Richter HG, Blanco CE, Gottschalk R, et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PloS one 2012;7(2):e31017 Epub 2012/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Giussani DA, McGarrigle HH, Spencer JA, Moore PJ, Bennet L, Hanson MA. Effect of carotid denervation on plasma vasopressin levels during acute hypoxia in the late-gestation sheep fetus. The Journal of physiology 1994;477(Pt 1):81–7. Epub 1994/05/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Giussani DA, McGarrigle HH, Moore PJ, Bennet L, Spencer JA, Hanson MA. Carotid sinus nerve section and the increase in plasma cortisol during acute hypoxia in fetal sheep. The Journal of physiology 1994;477(Pt 1):75–80. Epub 1994/05/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Thakor AS, Giussani DA. Effects of acute acidemia on the fetal cardiovascular defense to acute hypoxemia. American journal of physiology Regulatory, integrative and comparative physiology 2009;296(1):R90–9. Epub 2008/10/17. [DOI] [PubMed] [Google Scholar]
- 170.Morrison S, Gardner DS, Fletcher AJ, Bloomfield MR, Giussani DA. Enhanced nitric oxide activity offsets peripheral vasoconstriction during acute hypoxaemia via chemoreflex and adrenomedullary actions in the sheep fetus. The Journal of physiology 2003;547(Pt 1):283–91. Epub 2003/02/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Thakor AS, Giussani DA. Calcitonin gene-related peptide contributes to the umbilical haemodynamic defence response to acute hypoxaemia. The Journal of physiology 2005;563(Pt 1):309–17. Epub 2004/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Herrera EA, Kane AD, Hansell JA, Thakor AS, Allison BJ, Niu Y, et al. A role for xanthine oxidase in the control of fetal cardiovascular function in late gestation sheep. The Journal of physiology 2012;590(8):1825–37. Epub 2012/02/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kane AD, Hansell JA, Herrera EA, Allison BJ, Niu Y, Brain KL, et al. Xanthine oxidase and the fetal cardiovascular defence to hypoxia in late gestation ovine pregnancy. The Journal of physiology 2014;592(3):475–89. Epub 2013/11/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Gardner DS, Fowden AL, Giussani DA. Adverse intrauterine conditions diminish the fetal defense against acute hypoxia by increasing nitric oxide activity. Circulation 2002;106(17):2278–83. Epub 2002/10/23. [DOI] [PubMed] [Google Scholar]
- 175.Herrera EA, Rojas RT, Krause BJ, Ebensperger G, Reyes RV, Giussani DA, et al. Cardiovascular function in term fetal sheep conceived, gestated and studied in the hypobaric hypoxia of the Andean altiplano. The Journal of physiology 2016;594(5):1231–45. Epub 2015/09/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gardner DS, Giussani DA. Enhanced umbilical blood flow during acute hypoxemia after chronic umbilical cord compression: a role for nitric oxide. Circulation 2003;108(3):331–5. Epub 2003/07/02. [DOI] [PubMed] [Google Scholar]
- 177.Gardner DS, Fletcher AJ, Bloomfield MR, Fowden AL, Giussani DA. Effects of prevailing hypoxaemia, acidaemia or hypoglycaemia upon the cardiovascular, endocrine and metabolic responses to acute hypoxaemia in the ovine fetus. The Journal of physiology 2002;540(Pt 1):351–66. Epub 2002/04/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wibbens B, Bennet L, Westgate JA, De Haan HH, Wassink G, Gunn AJ. Preexisting hypoxia is associated with a delayed but more sustained rise in T/QRS ratio during prolonged umbilical cord occlusion in near-term fetal sheep. American journal of physiology Regulatory, integrative and comparative physiology 2007;293(3):R1287–93. Epub 2007/07/27. [DOI] [PubMed] [Google Scholar]
- 179.Yaffe H, Parer JT, Block BS, Llanos AJ. Cardiorespiratory responses to graded reductions of uterine blood flow in the sheep fetus. J Dev Physiol 1987;9(4):325–36. Epub 1987/08/01. [PubMed] [Google Scholar]
- 180.Wassink G, Bennet L, Booth LC, Jensen EC, Wibbens B, Dean JM, et al. The ontogeny of hemodynamic responses to prolonged umbilical cord occlusion in fetal sheep. J Appl Physiol (1985) 2007;103(4):1311–7. Epub 2007/07/28. [DOI] [PubMed] [Google Scholar]
- 181.Poudel R, McMillen IC, Dunn SL, Zhang S, Morrison JL. Impact of chronic hypoxemia on blood flow to the brain, heart, and adrenal gland in the late-gestation IUGR sheep fetus. American journal of physiology Regulatory, integrative and comparative physiology 2015;308(3):R151–62. Epub 2014/11/28. [DOI] [PubMed] [Google Scholar]
- 182.Thakor AS, Giussani DA. Nitric oxide reduces vagal baroreflex sensitivity in the late gestation fetus. Pediatr Res 2009;65(3):269–73. Epub 2009/04/25. [DOI] [PubMed] [Google Scholar]
- 183.Reller MD, Burson MA, Lohr JL, Morton MJ, Thornburg KL. Nitric oxide is an important determinant of coronary flow at rest and during hypoxemic stress in fetal lambs. Am J Physiol 1995;269(6 Pt 2):H2074–81. Epub 1995/12/01. [DOI] [PubMed] [Google Scholar]
- 184.Miller TE, Estrella E, Myerburg RJ, Garcia de Viera J, Moreno N, Rusconi P, et al. Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome. Circulation 2004;109(24):3029–34. Epub 2004/06/09. [DOI] [PubMed] [Google Scholar]
- 185.Cuneo BF, Etheridge SP, Horigome H, Sallee D, Moon-Grady A, Weng HY, et al. Arrhythmia phenotype during fetal life suggests long-QT syndrome genotype: risk stratification of perinatal long-QT syndrome. Circ Arrhythm Electrophysiol 2013;6(5):946–51. Epub 2013/09/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Remme CA. Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. The Journal of physiology 2013;591(17):4099–116. Epub 2013/07/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Adsit GS, Vaidyanathan R, Galler CM, Kyle JW, Makielski JC. Channelopathies from mutations in the cardiac sodium channel protein complex. J Mol Cell Cardiol 2013;61:34–43. Epub 2013/04/06. [DOI] [PMC free article] [PubMed] [Google Scholar]