

Abstract
Herpes simplex virus type-1 (HSV-1) is a ubiquitous pathogen that infects a large majority of the human population worldwide. It is also a leading cause of infection-related blindness in the developed world. HSV-1 infection of the cornea begins with viral entry into resident cells via a multistep process that involves interaction of viral glycoproteins and host cell surface receptors. Once inside, HSV-1 infection induces a chronic immune-inflammatory response resulting in corneal scarring, thinning and neovascularization. This leads to development of various ocular diseases such as herpes stromal keratitis, resulting in visual impairment and eventual blindness. HSV-1 can also invade the central nervous system and lead to encephalitis, a relatively common cause of sporadic fetal encephalitis worldwide. In this review, we discuss the pathological processes activated by corneal HSV-1 infection and existing antiviral therapies as well as novel therapeutic options currently under development.
keywords: Herpes simplex virus, Immune response, Anti-viral therapy, Pathological process, Ocular herpes
Introduction
Herpes simplex virus-1 (HSV-1) is a neurotropic double-stranded DNA virus belonging to the Alphaherpesvirinae family, a sub-family of Herpesviridae. It has been recognized as one of the most adapted human pathogens, infecting 50–90% of the world population [88, 96]. HSV-1 is a ubiquitous virus causing a wide range of innocuous diseases including herpes labialis (cold sores), gingivostomatitis, herpetic whitlow, genital herpes, epithelial and/or stromal keratitis. Spread of the virus to central nervous system (CNS) initiates a rapidly progressive, necrotizing, and potentially fatal encephalitis in humans. Similar to the infection of CNS, HSV-1 in the eye can result in serious episodes of blepharitis, conjunctivitis, retinitis, and epithelial keratitis [61]. Herpetic stromal keratitis (HSK) is caused by recurrent herpetic eye disease and is regarded as the most vision-threatening form of HSV-1-induced pathology.
Primary infection of the corneal epithelium by HSV-1 or HSV-2 results in productive lytic replication of the virus and establishment of latency that lasts for the lifetime of the host. Progeny virions enter the axons of sensory neurons innervating the affected area and travel in a retrograde fashion to neuronal cell bodies of the trigeminal ganglion (TG), which becomes the site for HSV latency [111]. During latency, the viral genome is transcriptionally silenced except for latency-associated transcripts (LATs) and some micro-RNAs. The virus periodically reactivates from latency resulting in replication and production of infectious virus. Upon reactivation, progeny virions are produced and subsequently transported to the initial site of inoculation [79]. In case of the cornea, reactivation of latent HSV-1 can result in HSK, which can turn into a chronic inflammatory disease. Herpetic lesions, associated immune responses and neovascularization cause scarring and corneal damage leading to vision loss during HSK [16].
In this review, we first describe our latest understanding of the molecular basis of HSV-1 invasion of corneal cells. We then focus on the pathological process activated by HSV-1 infection of the cornea and explain how it contributes to development of specific ocular and neurological diseases. Finally, we discuss most recent advances in controlling HSV-associated morbidities and corneal pathologies.
Molecular basis of HSV-1 entry and spread in the cornea
The mature HSV-1 virion is composed of a large DNA genome encased in an icosadeltahedral protein capsid, surrounded by proteins and mRNAs, a layer which is referred to as the tegument, and finally a lipid bilayer envelope composed of about a dozen different glycoproteins, several of which, including gB, gC, gD, gH and gL, are required for viral entry (Fig. 1) [33]. The basic mechanism of HSV-1 entry into host cell primarily involves the interaction of the viral entry glycoproteins with various cell surface receptors that facilitates virion envelop fusion with the plasma membrane of the host cell causing capsid penetration into the cytoplasm (Fig. 2). The capsid is then transported by cellular motors on microtubules to the nuclear membrane for uncoating and the release of HSV-1 DNA into the nucleus.
Fig. 1.
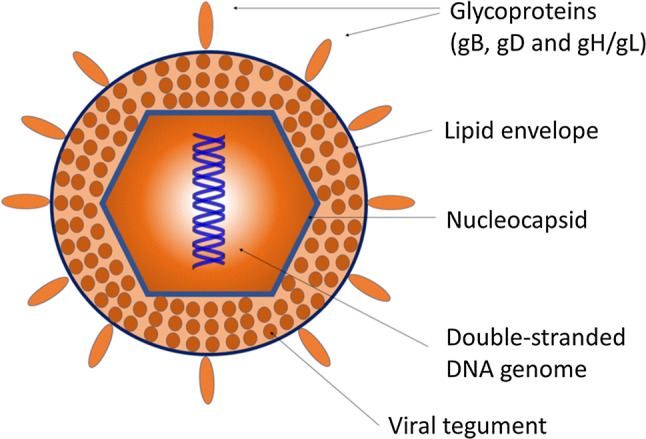
Herpes simplex virus-1 structure. The glycoproteins essential for viral entry into host cells are listed
Fig. 2.

HSV-1 entry glycoproteins and their known receptors. Initial interactions of HSV envelope glycoproteins gB and/or gC with heparan sulfate proteoglycans (HSPG) occur on the surface of a host cell, which facilitate viral attachment and surfing on membrane projections to reach the plasma membrane. This is followed by specific interactions of HSV gD and gB with their corresponding receptors leading to membrane fusion and delivery of the viral nucleocapsid and tegument proteins into the cytoplasm
HSV-1 can also enter directly into the cellular cytoplasm through the formation of a membrane fusion pore or via endocytosis and/or phagocytosis-like uptake mechanism [26]. Either way, the initial interaction occurs between viral glycoproteins, gB and/or gC, and heparan sulfate proteoglycan on the cell surfaces [95]. This interaction results in lateral viral movement along the length of F-actin-rich membrane protrusions called filopodia, termed as ‘viral surfing’, to migrate towards the cell body [76, 95]. Following the initial interaction, binding of gD by a gD receptor on the surface of host membrane, such as nectin-1, herpes virus entry mediator (HVEM) or 3-O-sulfated HS (3-OS-HS), causes a conformational change that transmits an activation signal to recruit the fusion complex which comprises gB, gH and gL [21, 95]. gB is also recognized by one of its receptors: paired immunoglobulin-like 2 (PILRα), n-muscle myosin heavy chain IIA (NMHC-IIA) or myelin-associated glycoprotein (MAG). The glycoprotein gB is a membrane fusion protein required for the penetration and delivery of the viral nucleocapsid into the cytoplasm. The nucleocapsid is then transported along microtubules to the nuclear membrane, where viral DNA is released for replication in the nucleus.
The entry of HSV into ocular cells serves as a key step in pathogenesis and is both cell type and viral subtype specific [33]. For example, nectin-1 is the primary receptor for entry of HSV-1 in various cell types that lead to the development of HSV-1-induced pathologies such as conjunctivitis, epithelial keratitis and acute retinal keratitis [3, 110]. Similarly, 3-OS-HS serves as the primary receptor for HSV-1 entry into stromal fibroblasts and is, therefore, implicated as a host factor in stromal keratitis and conjunctivitis [108]. Understanding key features of viral entry and how it relates to corneal disease development can provide better direction for designing antiviral therapeutics.
Initial host response to viral invasion of the cornea
Several key events are induced by HSV infection in the cornea that subsequently result in the development of ocular pathologies and drive the infection into a chronic immuno-inflammatory disease. Following virus infection of the corneal epithelium, detection of the pathogen evokes a sequential cascade of immunological responses [77]. The innate immune system induces the first of these responses, resulting in the production of a variety of cytokines and infiltration of leukocytes, as well as other cells. This condition is also accompanied by the release of pro-angiogenic factors that cause neovascularization, which in turn contributes to HSV disease of the cornea. Specific host factors that drive both conditions are discussed below.
Virus-induced inflammation
A robust response elicited by the innate immune system acts as the first line of defense against the pathogen and also the trigger for chronic inflammatory conditions. The innate immune cells that respond to the infection of the cornea include neutrophils, plasmacytoid dendritic cells (pDCs), natural killer (NK) cells and macrophages. Neutrophils are clearly instrumental in controlling replication and dissemination of the virus as depletion of neutrophils in mouse models resulted in an apparent increase of viral replication and shedding [107]. Similarly, pDCs have demonstrated to be vital for viral clearance in the cornea by attracting natural killer cells through the release of cytokines and inflammatory monocytes to the site of HSV-1 lesions [20]. NK cells could directly kill HSV-infected cells and indirectly inhibit HSV proliferation by IFN-γ secretion [35]. Macrophages secrete pro-inflammatory cytokines and other factors to restrict viral growth, recruit other immune cells and enhance T cell-mediated adaptive immune response [23]. They are also the predominant producers of IFNs during viral infection. Regardless of the precise recruitment mechanism, it is well established that the presence of innate immune cells in the cornea promotes neovascularization, neolymphangiogenesis, opacification and scarring of the cornea.
Recognition of viral ligands by pattern recognition receptors (PRRs) in innate immune cells leads to downstream signaling that ultimately converges on the activation of either TANK-binding kinase 1 (TBK-1) or IκB kinase (IKK) complex. TBK1 leads to the activation of IFN regulatory factor 3 (IRF-3), a transcription factor whose role in the expression of type I IFN and downstream pathways is thought to be of paramount importance. IKK complex, composed of NEMO (IKKγ), IKKα and IKKβ, phosphorylates IκB, allowing nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) to translocate to the nucleus and increases the transcription of various interleukins (IL) and tissue necrosis factor (TNF) [4, 5]. The regulation of NFκB in the presence of infection is complicated in that the virus both activates NFκB which facilitates its replication but also inhibits NFκB to limit production of antiviral and inflammatory cytokines [64].
Immune cells including those residing in the cornea express functional Toll-like receptors (TLRs), a type of PPR, that detect and bind pathogen-associated molecular patterns (PAMPs), such as those associated with HSV-1 [71]. This triggers the dimerization of TLRs which subsequently activates signaling pathways that provoke the production of pro-inflammatory cytokines such as IFNs, TNF, and various interleukins and upregulation of NF-κB [5]. The host adaptive response is then primed through further signaling pathways. Several TLRs have been implicated as important mediators of viral containment and/or destructive inflammation in response to HSV-1 infection of the CNS. Currently, 10 TLR family members are expressed differently among immune cells, each responding to different stimuli. At least six TLRs (TLR1, 2, 3, 7, 8, and 9) are known to be involved in HSV PAMP detection [89].
One of the most important TLRs to mediate host cell response to HSV-1 is TLR-2. TLR2 resides on the cell surface and recognizes viral envelope proteins and lipopeptides, including specific HSV glycoproteins that serve as PAMPs [4, 5]. TLR2 signals through a myeloid differentiation factor 88 (MyD88) or Toll-interleukin 1 receptor domain containing adaptor protein (TIRAP)-dependent cascade, resulting in the recruitment of IR-1R-associated protein kinase (IRAK1) and TNF receptor-associated factor 6 (TRAF6), both of which are upstream of the IKK complex and NF-κB [104] (Fig. 3). HSV-1 activation of NF-κB induces upregulation of pro-inflammatory cytokines like IL-15 and IL-6 [2, 56]. Consequently, TLR2 knockout mice showed considerable resistance to disease expression, fewer clinical lesions, reduced levels of neovascularization and stromal inflammatory reactions at both early and peak times of viral response, proving to play a pivotal role in mediating an inflammatory response upon viral infection. Reduced inflammatory reactions in the TG were also observed in another study [56]. Ultimately, TLR2-deficient mice were protected from the lethal effects of HSV infection [56, 68].
Fig. 3.

TLR2, TLR3 and TLR9 regulate a robust immune response following HSV-1 infection. TLR2 resides on the cell surface and recognizes viral proteins. TLR2 signals through a myeloid differentiation factor 88 (MyD88) or Toll-interleukin 1 receptor domain containing adaptor protein (TIRAP)-dependent cascade, resulting in the recruitment of IR-1R-associated protein kinase (IRAK1) and TNF receptor-associated factor 6 (TRAF6), both of which are upstream of the IKK complex and NF-κB. TLR9 is expressed in the endosome and recognizes the virus by rich CpG sequences and signals through MyD88-dependent pathway that induces NF-κB activation. TLR3 is also expressed in the endosome and recognizes dsRNA. TLR3 signals through MyD88-dependent pathway, activating NF-κB, as well as TIR-domain-containing adapter-inducing interferon-β (TRIF), which is upstream of TBK-1, and leads to the activation of IRF-3
HSV-1 has also been shown to serve as a ligand for TLR9, which is expressed in the endosome and recognizes the virus by rich CpG sequences and signal through MyD88-dependent pathway that induces NF-κB activation (Fig. 3) [4, 43, 77]. In vitro studies have shown that inhibition of TLR9 using oligonucleotides prevented the expression of essential immediate early herpes gene products, and as a result also reduced NFκB activity in nuclear extracts [90]. In the absence of TLR9, other compensatory mechanisms are involved in producing both type I interferon and cytokines, showing that TLR9 only partially contributes to the response against the virus [46, 55]. TLR3 are also expressed in the endosomes and recognize viral dsRNA [5]. TLR3 signals through MyD88-dependent pathway, activating NF-κB, as well as TIR-domain-containing adapter-inducing interferon-b (TRIF), which is upstream of TBK-1, and leads to the activation of IRF-3 (Fig. 3). This signaling cascade results in the primary response of antiviral cytokines, primarily type I IFNs [41]. TLR3 deficiency has been observed in various pathologies such as HSV-1 encephalitis (HSE) and shown to play a protective role against HSV-1 [120]. In these cases, increased viral infection and replication is detected due to reduced interferon production, proving TLR3-mediated immunity to be crucial in protection against HSV-1 infection of the central nervous system.
HSV-1 DNA recognition by TLR induces an immune defense mechanism aiming at virus clearance from the cornea; however, the initiation of an inflammatory response has shown to contribute to pathogenesis leading to cornea destruction rather than to protection. While the absence of TLRs does result in less infection, MyD88 knockout mice showed increased susceptibility to lethal HSV-1 infection showing that the immunity plays an important role in host resistance to viral infection and pathogenesis [68]. The host inflammatory response to ocular infection with herpes simplex virus (HSV) can be either protective and fight against infection, or it can promote diseases.
Ocular neovascularization
Corneal neovascularization (CNV) is induced by various stimuli, such as HSV-1 infection, and results in the formation of new vascular structures in the cornea, a region previously regarded as being avascular. CNV is a sight-threatening condition associated with inflammation upon infection of the ocular surface that mediates the pathogenesis of stromal keratitis [9, 122]. Two main processes may be involved in NV: angiogenesis, the physiological process through which new blood vessels form from pre-existing vessels, and vasculogenesis, the de novo production of endothelial cells from precursor angioblasts and formation of primitive vascular networks [47].
Though angiogenesis is a normal and vital process in growth and development, wound healing and the formation of granulation tissue, it proves to be detrimental when observed in the human cornea. Angiogenesis is a common mechanism involved in corneal and retinal disorders and is known to be a major cause of visual impairment [66]. In normal ocular function, maintaining the passage of photons through ocular tissues to the retina with minimal resistance is required. This maintenance is characterized by a fine balance between angiogenic and anti-angiogenic factors in the cornea [9, 24]. Therefore, the upregulation of angiogenic factors and development of blood vessels can damage vision by blocking and diffracting light from entering the retina and can cause deformation of the structural integrity of the cornea. Pro-angiogenic factors include vascular endothelial growth factors, fibroblast growth factor, and matrix metalloproteinases, hepatocyte growth factor, interleukin-6 and tumor necrosis factor alpha, all of which promote continued genesis and maintenance of CNV during virus infection and following virus clearance [22, 28, 54, 105]. Neutrophils also participate in CNV since they can be a source of angiogenic factors as well as proteases that facilitate blood vessel growth through the corneal matrix [103].
Several reports illustrate the mechanisms behind increased level of VEGF-A during HSV-1 virus infection and its downstream effects in CNV and developing pathologies [6]. VEFG-A is present and bound to sVR-1 (soluble form of the VEGF receptor 1) in normal ocular tissue. HSV-1 infection disrupts this interaction and results in increased levels of VEGF-A and decreased levels of sVR-1, inducing angiogenesis and subsequently CNV [103]. VEGF-A transcription is directly activated by an HSV-1-encoded immediate early gene product, infected cell protein 4 (ICP-4), upon its binding to guanine cytosine-enriched sites within the VEGF-A promoter region [115]. Transcriptional activation of VEGF-A promotes several steps of angiogenesis, endothelial cell proliferation, migration, and capillary tube formation. VEGF-A also increases levels of microRNA-132 (miR-132) via cAMP response element-binding protein (CREB) transcription factor. Ras inhibitor is deactivated by miR-132, therefore, leading to Ras activity and to the development of CNV. Silencing miR-132 by antagomir-132 nanoparticles found to increase levels of Ras-GAP, thereby, inhibiting angiogenic Ras activity [75]. The requirement for VEGF-A in promoting these processes became evident in an experiment that showed the inhibition of neovascularization in rat model following stromal implantation of an anti-VEGF-A blocking antibody [6]. This was further shown in a GF blocking peptide in a rabbit corneal model resulting in the inhibition of VEGF-induced angiogenesis [15].
Another angiogenic factor, fibroblast growth factor-2 (FGF-2), has also been shown to play a role in CNV following infection. FGF-2 is a heparin-binding peptide that not only induces angiogenesis, but also stimulates mitogenesis, cellular differentiation and proliferation in various cell types. It controls the expression of many different cytokines including IL-6, VEGF-A, HGF, MMP-9, and angiopoietin-2 [29, 62, 63, 86, 92]. Neutralization of FGF-2 blocked progressive CNV by reducing the expression of the angiogenic factors including HGF, IL-6, VEGF-A and, therefore, resulted in improved visual outcome. Anti-FGF-2 antibody treatment was also found to partially preserve visual acuity in HSV-1-infected mice but had no effect on corneal sensitivity [42]. Thus, FGF-2 may serve as a master regulator for other pro-angiogenic factors expressed during HSV-1 infection.
Matrix metalloproteinases (MMPs) have also been observed to be upregulated during the angiogenesis caused by ocular infection with HSV-1 [13, 57]. However, their presence is ambiguous and may serve as precursors to anti-angiogenic or pro-angiogenic factors. Studies by Lee el al. show that neutrophil infiltration following HSV-1 infection secretes MMP-9 which contributes to CNV. Angiogenesis was significantly diminished upon inhibition of MMP-9 and in MMP-9 knockout mice [58]. However, most recently a study shows no difference in WT and MMP-9-deficient mice suggesting a dispensable role of MMP-9 and neutrophils in angiogenesis following viral clearance [42].
The infiltrating inflammatory cells including neutrophils, monocytes, macrophages, and T cells can also be a tremendous source of growth factors and cytokines that can induce pro-angiogenic factors due to which neovascularization prolongs. Neutrophil infiltration has been shown to be a result of NV and lead to corneal destruction [10]. On the contrary, a recent study shows that leukocytes including neutrophils and T cells have not been found to have major contribution to corneal neovascularization, as antibody-mediated depletion of these cells did not show significant impact on lymphatic or blood vessel genesis [42].
At the molecular level, a new trigger for corneal inflammation and NV was recently identified. In the late 2017, we reported that heparanase (HPSE), a cornea resident enzyme, acts as the molecular trigger for multiple pathologies associated with HSV-1 infection [1]. Heparanase is an endoglycosidase which is upregulated during HSV infection by infected cell protein 34.5 (ICP34.5). HPSE has been rigorously studied in cancer biology, where it is well known to play a role in cancer metastasis, angiogenesis, and stimulation of both extracellular signal-regulated kinase (ERK) and MMP-9 [36, 81]. Simultaneous increase in levels of HPSE and MMP-9 is the major cause of degrading extracellular matrix (ECM) and basement membrane (BM) allowing pro-angiogenic factors’ release and thus playing a key role in angiogenesis [25, 106]. Further evidence of the involvement of HPSE in ocular angiogenesis comes from Xian-Jun et al. who observed the increased mRNA and protein levels of HPSE and VEGF-A in the retina of mice having oxygen-induced retinopathy (OIR) [59]. Furthermore, the treatment of OIR neovascularization mouse model with the sulfated oligosaccharide phosphomannopentaose sulfate (PI-88) suppressed angiogenesis by downregulating the HPSE and VFGF-A. HPSE is also known to orchestrate FGF-2-mediated cell signaling [69]. The FGF-2 operates through stable activation of PI3 K/AKT pathway which is only transient in heparanase-silenced cells. In support of these, Vishnu et al. explains involvement of two different HPSE functional domains (the enzyme active site and a separate site) in driving HGF signaling by enhancing HGF expression and its activity [86]. In the light of heparanase pathophysiologic importance in induction of FGF-2 and VEGF-A (important regulators of herpetic angiogenesis), development of therapeutics targeting heparanse could be a promising approach for current and future treatment of HSV-1-induced ocular neovascularization (Fig. 4).
Fig. 4.

Induction of corneal neovascularization and inflammation by HSV-1 infection: HSV-1 infection results in the increased expression of heparanase (HPSE) which causes heparan sulfate proteoglycan (e.g., syndecan) shedding. HPSE-degraded heparan sulfate releases VEGF and FGF-2, which signal ERK phosphorylation and cause pro-inflammatory cytokine induction. Production of pro-inflammatory factors (IL-6, MMP-9, FGF-2, HGF) then contributes to inflammation and infiltration of macrophages, T cells, and Nk cells in the cornea. In addition, HSV-1-infected corneas predominantly produce VEGF-A that leads to angiogenic sprouting
Therefore, understanding the mechanistic factors mediating the development of CNV can provide a clear therapeutic target in inhibiting the development of HSK. Few studies have suggested that controlling the levels of VEGF-A or inhibiting its receptor can alleviate HSV-1-induced neovascularization [10, 121]. An experiment using TIMP-1, a specific MMP9 inhibitor, has also shown to reduce CNS symptoms and was recommended as a therapeutic agent against herpes keratitis [58]. It is likely that future drug to treat HSK will target virus as well as CNV and inflammation.
HSV-associated ocular pathologies and systemic diseases: blepharitis and conjunctivitis
HSV-1 infection of the eye is manifested in many different ways. More superficially it can cause oculodermal diseases such as blepharitis and conjunctivitis. Blepharitis is most often seen as inflammation of the eyelid, whereas inflammation of the conjunctiva commonly known as pinkeye is the most common manifestation of HSV conjunctivitis. Both viral and host factors play a role in the pathogenesis of inflammatory ocular and dermal lesions. Reports indicate that similar broad spectrum of immune reactions that occur in the development of corneal lesions in response to HSV-1 infection also occurs in the eyelid and conjunctiva [67], namely rapid infiltration of neutrophils and cytokine production in the cornea and eyelids were similar in nature and development of pathology [100]. Both blepharitis and conjunctivitis can be annoying diseases of the eye but they often self-resolve and do not cause blindness.
Herpes stromal keratitis
A potentially devastating outcome of HSV-1 infection is herpes stromal keratitis (HSK), a vision-threatening syndrome in which recurrent reactivation of latent virus from the TG initiates chronic inflammation and results in increased corneal damage [16]. HSK is the second leading cause of blindness, after cataract, in developed countries, mainly due to its recurrent nature.
The success of HSV and dominance over the host’s immune system is attributed to the establishment of latency, HSV-1 latency [63].
Although the functions of host innate and adaptive immunity, such as type I IFN, innate immune cell activity, and CD8 + T cell activity are needed to suppress HSV-1 replication and infection, they are also shown to be detrimental to the host. CD4 + T cells are expanded in the draining lymph nodes and re-stimulated in infected cornea, mediating the production of Th1 and Th17 cytokines and orchestrating chronic immune-inflammatory lesions, resulting in ocular HSK [37, 101]. Elevated levels of neutrophils and dendritic cells contribute to inflammation and, therefore, development of ocular pathologies [16, 27]. Studies observed significantly diminished SK lesion severity and neovascularisation by decreasing the influx of CD4 + cells and innate cells such as neutrophils and, therefore, decreasing the production of pro-inflammatory cytokines [101].
Encephalitis
HSV-1 encephalitis (HSE) is the most commonly diagnosed form of sporadic viral encephalitis in humans caused by primary or secondary infections. It is the most serious neurological complication caused by HSV-1. Invasion and establishment of infection in the central nervous system is a rare and life-threatening event that is still without a foolproof treatment option [114]. Acyclovir (ACV) is the most commonly used antiviral drug to treat HSV-1 corneal infection and is sufficient in blocking viral replication. However, studies indicate that treatment with ACV after the virus had already reached the brain stem resulted in fatal cases of encephalitis in mouse models, showing that fatality is a result of brain stem inflammation rather than viral replication [65].
HSE results from widespread damage in the brain stem caused by destructive inflammatory responses initiated early in infection by massive infiltration of innate cells. Consequently, studies have also shown enhanced survival in mice with depletion of either macrophages or neutrophils, as a result of reduced hyperinflammation initiated by early infiltrating innate cells [65]. Stimulation of the innate immune response and activation of TLR2-induced inflammatory cytokines have shown to be associated with lethal viral encephalitis upon HSV-1 infection [56]. Similarly, administration of a TLR9 inhibitory oligonucleotide after HSV-1 infection of mice decreased mortality by controlling the inflammatory response in the brain [17]. Other studies have indicated that mice lacking the adapter molecule MyD88, which is downstream of TLR, were resistant to lesion development, but were unable to control infection and succumbed to lethal encephalitis [68]. Inflammation caused by the immune response is a major cause of development of central nervous system (CNS) pathology resulting in severe pathological diseases and fatality.
On the contrary, deficiencies in the immune response to HSV may leave the host susceptible to herpes simplex virus-1 encephalitis. While the inflammatory response mediated by the innate immune system can be destructive and result in neurologic sequelae, the host immune response is critical for viral control and protection. Both the innate and adaptive immune system determine disease severity, the extent of lesions, latency and recurrence. Recurrent infections, corneal scarring and mortality were shown to be more significant in immunocompromised patients [18]. Reduced antiviral cytokine production in TLR3-deficient patients resulted in the pathogenesis of encephalitis [120]. In accordance with these findings, IRF-3 knockout mice showed increased viral replication and inflammatory cytokine production in the brain tissue as well as a deficit in the production of both IFN-β and IFN-α [72]. Recent clinical studies identified that patients with genetic defects in IRF-3 exhibited increased susceptibility to viral infection in the CNS, due to blunted IFN production upon TLR3 stimulation by HSV-1 [7]. Antiviral cytokine secretion as a result of immune activity is crucial for virus clearance and control even though the inflammatory response it elicits may lead to innocuous pathologies.
Existing and investigational therapies to control HSV-induced ocular pathologies
Various antiviral agents are already in use to prevent infection and treat already existing diseases caused by HSV-1 infection. The first FDA-approved over-the-counter topical medication for orolabial herpes was docosanol, a drug that inhibits the fusion between the plasma membrane and the HSV envelope thus exhibiting anti-HSV activity [73, 80]. The administration of docosanol, therefore, resulted in inhibition of viral genome entry to the nucleus and subsequently lack of replicative events [80]. However, this drug fails to eliminate viral genomes of already infected cells and latent infection. The current treatment modality for ocular HSV-1 includes systematic administration of the following nucleoside analogs: acyclovir (ACV), valacyclovir and famciclovir as well as topical administration of ganciclovir and trifluridine (TFT) [112]. These antiviral drugs limit the viral DNA replication and prevent viral recurrences, therefore, suppressing disease symptoms. However, therapeutic drugs currently used to treat HSV suffer from many limitations including the development of antiviral resistance, induction of ocular disorders with prolonged use of TFT and nephrotoxicity caused by prolonged use of ACV [32, 34, 50, 70, 118]. A clinical study reported 34 (44.7%) of 76 patients suffering from HK as ACV resistant and also identified a higher prevalence of resistance among immunocompromised patients [113]. Also, these drugs fail to prevent permanent establishment of viral latency, allowing the virus to persist and cause clinical diseases at later times. For these reasons, alternative methods are needed to provide more effective treatment at earlier phases of the viral infection and to prevent the development of drug resistance by the virus (Table 1, Fig. 5) [8, 51].
Table 1.
Antiviral drugs under research as well as current modes of treatment
| Drugs | Mechanism of action | References |
|---|---|---|
| Targeting host factors | ||
| BX795 | Targets Akt phosphorylation in infected cells, leading to the blockage of viral protein synthesis | [49, 102] |
| OGT2115, phosphomannopentaose sulfate (PI-88) | Inactivates heparanase and vascular endothelial growth factor | [1, 59] |
| Src kinase inhibitor (TG100572) | Block IL-8/CXCL1 involved in inflammatory cell recruitment that are a source of VEGF, diminishes cellular infiltration in the cornea, and reduces proliferation and migration of CD4(+) T cells into the corneas | [94] |
| Targeting host entry receptors | ||
| G2-C peptide | Prevents entry of HSV-1 by blocking heparan sulfate receptors | [48] |
| Apolipoprotein E peptide | Inhibited migration, invasion, endothelial cell proliferation and capillary tube formation | [14] |
| Targeting viral glycoproteins | ||
| DNA aptamer | Restricts viral entry and replication by disrupting the binding of gD to its cognate host receptors | [116] |
| Retrocylin 2 | Prevents entry of the virus by binding gB | [19] |
| Humanized monoclonal antibody (mAb hu2c) | Targets viral glycoproteins and neutralizes the virus particle, restricting viral entry | [12, 53] |
| Other antiviral therapy | ||
| Receptor Robo4 (sR4) protein | Activates an anti-angiogenic pathway that counteracts VEGF downstream signaling | [39] |
| Soluble FasL (sFasL) | Fas is a member of the tumor necrosis factor-R family. sFasL induces apoptosis of Fas+ vascular endothelial cells and thereby restricts inflammatory responses | [87] |
| Resolvin E1 (RvE1) and aspirin-triggered resolvin D1 (AT-RvD1) | Decreases pro-angiogenic factors. Decreases the influx of neutrophils and effector CD4þ T cells (both TH1 cells and TH17 cells), pro-inflammatory mediators (IL-1β, IL-6, IL-12, CXCL1, MCP-1, MIP-2, vascular endothelial growth factor (VEGF)-A, matrix metalloproteinase 9 (MMP-9)) pro-inflammatory miRNA, such as miR-155, miR-132, and miR-223. RvE1 increased levels of the anti-inflammatory cytokine IL-10. | [82, 84, 93] |
| Anti-miR-132 (antagomir-132) nanoparticles | Controls miR-132 expression during ocular HSV-1 infection and reduces angiogenic Ras activity in corneal CD31-enriched cells | [75] |
| Galectin-1 (gal-1) and anti-miR-155 | Suppress the functions of Th1 and Th17 cells which primarily orchestrates the inflammatory lesions | [85] |
| 2-Deoxy-glucose | A glucose inhibitor that limits the differentiation and functionality of CD4 T cells which are more active in developing lesion | |
| Monoamine oxidase inhibitors | Prevents viral transcription of the IE genes, thus preventing the virus from reactivating from latency | [60] |
| BAY 57-1293 | Helicase–primase complex inhibitor | [52] |
| Cyclosporin A | Inhibits T-cell activation by blocking IL-2 production | [119] |
| Rigid amphipathic fusion inhibitors (RAFIs) | Inhibit the increased negative curvature required for the initial stages of fusion | [97] |
| Current regimens | ||
| Acyclovir, Valaciclovir, Famciclovir, Ganciclovir, Cidofovir, Foscarnet, Vidarabine, Trifluridine | Inactivates HSV-specified DNA polymerases preventing further viral DNA synthesis | [31, 111] |
| Cyclooxygenase inhibitors (lornoxicam) | Downregulates nuclear factor kappa β | [38] |
| Docosanol | Inhibits the fusion between the plasma membrane and the HSV envelope | [73, 80] |
Fig. 5.

Virus replication cycle and the steps which are targeted by antiviral drugs currently in use or under investigation
Recent development in new antivirals: targeting host factors
The current understanding of the molecular basis of HSV-1 infection highlights a few host molecules that serve as essential mediators in productive virus infection. A recent study by our lab published in 2018 identified BX795, a commonly used inhibitor of TANK-binding kinase 1 (TBK1), as a potent antiviral suppressing infection of multiple strains of HSV-1, including an ACV-resistant HSV-1 strain. BX795’s antiviral activity was observed in transformed and primary human cells, cultured human and animal corneas, and a murine model of ocular infection. In this study, BX795 was shown to target Akt, which in turn, blocks viral protein synthesis. BX795 demonstrates its antiviral activity by reducing Akt phosphorylation in infected cells, leading to reduced mTORC1 activity and the loss of hyperphosphorylation of 4E-BP1. However, a more precise antiviral mechanism of action induced by BX795 requires further investigation as many additional targets [49, 102].
Heparanase (HPSE) is another host factor that mediates HSV-1 infection and has been reported to drive HSV-1-induced pathogenesis [1]. HPSE is a HS-degrading enzyme of the host that has been shown to increase in expression upon HSV infection and, therefore, dramatically decreased surface HS levels of infected cells. The study concludes that active HPSE is required for efficient viral egress and subsequently cell-to-cell spread [44]. Several HPSE-blocking agents are currently being used in phase-1 and -2 clinical trials for cancers and, therefore, targeting HPSE in infection may result in a promising therapy. Studies in our lab using a pharmacological inhibitor of HPSE, OGT2115, showed decreased expression of pro-inflammatory factors IL-1β, IL-6 and TNF-α and loss of viral cell-to-cell spread in human corneal epithelial cells [1]. In ex vivo porcine cornea infection model, OGT2115 also showed an antiviral effect where it arrested the spread of infectious virus and resulted in eventual clearance of virus. Thus, by blocking the active site of the HPSE enzyme, OGT2115 provides a promising therapeutic agent against HSV-1.
Targeting ocular angiogenesis and inflammation can also provide an effective treatment option against HSK. Specifically, Src family of tyrosine kinases are responsible for VEGF-mediated vascular permeability and are involved in signaling other angiogenic factors resulting in pathological angiogenesis [30, 91]. Studies show that topical or systemic administration of Src kinase inhibitor, TG100572, on infected mice resulted in diminished angiogenesis by blocking VEGF signaling pathway [94]. The use of TG100572 resulted in the inhibition of CXCL1, a pro-angiogenic and pro-inflammatory chemokine, resulting in diminished cellular infiltration and migration of CD4 + T cells into the corneas. Thereby TG100572 inhibits downstream molecules involved in the VEGF signaling pathway and results in diminished levels of HSV-induced angiogenesis and significantly reduces the severity of HSK lesions.
Targeting host entry receptors
Antiviral peptides inhibiting viral entry into host cells have proven to be effective in eliminating infection and, therefore, suppressing development of ocular diseases. Recently, studies in our lab have identified antiviral activity of two small cationic peptides, G1 and G2, by binding specifically to HS and a modified form of the HS polysaccharide, 3-O-sulfated HS (3-OS-HS) and, therefore, inhibiting HSV-1 entry and spread [109]. G2 peptide also showed efficacy in combined therapy with ACV in both in vitro and in vivo studies [78]. Continued studies with this antiviral peptide showed prolonged release in commercially available contact lenses demonstrated antiviral activity in in vitro, ex vivo, and in vivo models of corneal HSV-1 infection, providing a new and effective way to control corneal herpes [48].
Another study targeted apolipoprotein E (ApoE) which is well known to contribute to the pathogenesis of HSV-1 in ocular infection [45]. Human host-derived peptidomimetic apoE (apoEdp) has been reported to block the development and progression of ocular herpes by competing with HSV-1 for the binding site of the entry receptor HS. Treatment on the eye with apoEdp has reported to reduce pro-inflammatory cytokines IL-1α, IL-1β, IL-6, TNFα, IFN-γ, and a pro-angiogenic cytokine, VEGF [14].
Targeting viral glycoproteins
Our lab has also recently developed a novel approach targeting viral glycoprotein gD, proving to be an effective therapy against ocular herpes both in vitro and in vivo. Topical application of the 45-nt-long DNA aptamer that has high affinity to HSV-1 gD resulted in disruption of gD binding to its cognate host receptor, resulting in significant restriction of viral entry due to the requirement of gD for viral entry. The use of this DNA aptamer resulted in 50–80% reduction in viral entry and an EC50 of 2 μM [116]. Furthermore, DNA aptamer showed to inhibit cell fusion and restrict intracellular viral spread without inducing any toxic effects.
Another peptide shown to regulate HSV-1 infection is synthetic theta-defensin, specifically retrocyclin 2 [117]. Retrocyclin 2 binds viral gB protein and blocks viral attachment, restricting viral entry without inducing cytotoxicity. However, these results were only significant when retrocyclin 2 was preincubated with HSV-1 KOS in vitro, proving to be effective only if used prophylactically [117].
Antiviral antibodies have also been identified as promising tools to prevent HSV infection by targeting viral glycoproteins and neutralizing the virus particle. A study published by Krawczyk et al. generated a novel humanized monoclonal antibody (mAb hu2c) that neutralizes clinical HSV isolates, including multi-drug-resistant variants both in vitro and in immunocompromised mice [53]. Similarly, a more recent study showed that humanized HSV-1/2-gB antibody neutralizes the virus and protects mice from ocular disease by blocking the neuronal spread of HSV [12]. These studies suggest that mAb hu2c may be used as a potent therapeutic option for severe ocular HSV infections, immunocompromised patients, and patients with multi-drug-resistant HSV isolates.
Other antiviral therapy
The endothelial receptor Robo4 (R4) has been shown to directly influence the development of angiogenesis by enhancing anti-angiogenic factors that counteract VEGF downstream signaling. Furthermore, R4 knockout mice showed significantly higher angiogenesis after HSV-1 ocular infection than did infected wild-type (WT) controls while the administration of soluble R4 (sR4) showed to rescue this phenotype. Therefore, sR4 has been proposed as a useful therapeutic antiviral drug to control angiogenesis and lessen the severity of SK [39]. However, further mechanistic studies are ongoing.
Rigid amphipathic fusion inhibitors (RAFIs) have been shown to inhibit viral infectivity of several enveloped viruses, including HSV-1 and -2, by inhibiting the increased negative curvature required for the initial stages of fusion [97]. These unique inhibitors also showed antiviral activity towards otherwise drug-resistant HSV-1 mutants. However, while RAFIs inhibit entry of HSV-1 tegument proteins into the target cells, they do not prevent primary attachment, secondary binding or viral DNA replication and, therefore, are not suitable remedies for all cases of HSV infection.
The therapeutic use of soluble Fas ligand (sFasL) has also been investigated and shown to reduce inflammatory infiltrate and neovascularization in primary and recurrent forms of HSK. In this study, BALB/c mice treated with sFasL following acute and recurrent HSV-1 infection showed a decrease in corneal opacity and neovascularization [87]. The interaction of Fas, a member of the tumor necrosis factor-R family, with FasL, a ligand of Fas, is an important factor in controlling HSK during acute infection of the cornea [74]. Accordingly, FasL expressed on ocular tissue serves as an important barrier for inflammatory cells and formation of new blood vessels maintain immune privilege of the eye [40, 98, 99].
Other therapies may include specialized pro-resolving mediators (SPM) such as lipoxins, resolvins, protectins and maresins which exhibit potent anti-inflammatory and pro-resolving actions [11, 83, 93]. In vivo studies using SPM showed decreased production of inflammatory mediators, pro-angiogenic factors and stimulating IL-10, anti-inflammatory cytokine [82, 84]. A recent study showed treatment with aspirin-triggered resolving D1 (AT-RvD1) diminished corneal neovascularization and severity of SK lesions in mice with HSV-1 due to the detection of fewer numbers of inflammatory cells including neutrophils, Th1 and Th17 cells in the infected cornea [82]. Moreover AT-RvD1 has also been reported to decrease levels of pro-inflammatory mediators such as micro-RNA (miRNA) (miR-155, miR-132 and miR-223) [82].
Conclusion
While current modes of therapy exist to alleviate symptoms of HSV-1 infection, an effective antiviral drug that eliminates HSV-1 infection without limitations is still desired. Latency established after HSV-1 infection is able to evade the antiviral activity of the immune system and the virus remains dormant in sensory neurons, making it difficult to target and dismantle. Reactivation of HSV-1 from this state results in pathogenesis of HSK, the leading cause of blindness. Recent developments in the field have generated an immense opportunity to target both the virus and the host responses, especially the CNV and the corneal inflammation. It is likely that the most successful drug of the future will not only retard the virus growth but will also stop the host triggers and mediators of inflammation from going out of control.
Footnotes
Equal contribution authors: Lulia Koujah and Rahul K. Suryavanshi.
References
- 1.Agelidis AM, Hadigal SR, Jaishankar D, Shukla D. Viral activation of heparanase drives pathogenesis of herpes simplex virus-1. Cell Rep. 2017;20:439–450. doi: 10.1016/j.celrep.2017.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmad R, El Bassam S, Cordeiro P, Menezes J. Requirement of TLR2-mediated signaling for the induction of IL-15 gene expression in human monocytic cells by HSV-1. Blood. 2008;112:2360–2368. doi: 10.1182/blood-2008-02-137711. [DOI] [PubMed] [Google Scholar]
- 3.Akhtar J, Tiwari V, Oh MJ, Kovacs M, Jani A, Kovacs SK, Valyi-Nagy T, Shukla D. HVEM and nectin-1 are the major mediators of herpes simplex virus 1 (HSV-1) entry into human conjunctival epithelium. Invest Ophthalmol Vis Sci. 2008;49:4026–4035. doi: 10.1167/iovs.08-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 5.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 6.Amano S, Rohan R, Kuroki M, Tolentino M, Adamis AP. Requirement for vascular endothelial growth factor in wound- and inflammation-related corneal neovascularization. Invest Ophthalmol Vis Sci. 1998;39:18–22. [PubMed] [Google Scholar]
- 7.Andersen LL, Mork N, Reinert LS, Kofod-Olsen E, Narita R, Jorgensen SE, Skipper KA, Honing K, Gad HH, Ostergaard L, Orntoft TF, Hornung V, Paludan SR, Mikkelsen JG, Fujita T, Christiansen M, Hartmann R, Mogensen TH. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med. 2015;212:1371–1379. doi: 10.1084/jem.20142274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antoine T, Park PJ, Shukla D. Glycoprotein targeted therapeutics: a new era of anti-herpes simplex virus-1 therapeutics. Rev Med Virol. 2013;23:194–208. doi: 10.1002/rmv.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azar DT. Corneal angiogenic privilege: angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing (an american ophthalmological society thesis) Trans Am Ophthalmol Soc. 2006;104:264–302. [PMC free article] [PubMed] [Google Scholar]
- 10.Azher TN, Yin X, Stuart PM. Understanding the role of chemokines and cytokines in experimental models of herpes simplex keratitis. J Immunol Res. 2017 doi: 10.1155/2017/7261980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basil MC, Levy BD. Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol. 2015;16:51–67. doi: 10.1038/nri.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bauer D, Alt M, Dirks M, Buch A, Heilingloh CS, Dittmer U, Giebel B, Görgens A, Palapys V, Kasper M, Eis-Hübinger AM, Sodeik B, Heiligenhaus A, Roggendorf M, Krawczyk A. A Therapeutic Antiviral Antibody Inhibits the Anterograde Directed Neuron-to-Cell Spread of Herpes Simplex Virus and Protects against Ocular Disease. 2017;8:2115. doi: 10.3389/fmicb.2017.02115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharjee PS, Neumann DM, Foster TP, Clement C, Singh G, Thompson HW, Kaufman HE, Hill JM. Effective treatment of ocular HSK with a human apolipoprotein E mimetic peptide in a mouse eye model. Invest Ophthalmol Vis Sci. 2008;49:4263–4268. doi: 10.1167/iovs.08-2077. [DOI] [PubMed] [Google Scholar]
- 15.Binetruy-Tournaire R, Demangel C, Malavaud B, Vassy R, Rouyre S, Kraemer M, Plouet J, Derbin C, Perret G, Mazie JC. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000;19:1525–1533. doi: 10.1093/emboj/19.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biswas PS, Rouse BT. Early events in HSV keratitis—setting the stage for a blinding disease. Microbes Infect. 2005;7:799–810. doi: 10.1016/j.micinf.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Boivin N, Menasria R, Piret J, Boivin G. Modulation of TLR9 response in a mouse model of herpes simplex virus encephalitis. Antiviral Res. 2012;96:414–421. doi: 10.1016/j.antiviral.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 18.Bradshaw MJ, Venkatesan A. Herpes simplex virus-1 encephalitis in adults: pathophysiology. Diag Manag. 2016;13:493–508. doi: 10.1007/s13311-016-0433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandt CR, Akkarawongsa R, Altmann S, Jose G, Kolb AW, Waring AJ, Lehrer RI. Evaluation of a theta-defensin in a Murine model of herpes simplex virus type 1 keratitis. Invest Ophthalmol Vis Sci. 2007;48:5118–5124. doi: 10.1167/iovs.07-0302. [DOI] [PubMed] [Google Scholar]
- 20.Buela K-G, Hendricks RL. Cornea-infiltrating and lymph node dendritic cells contribute to CD4 + T cell expansion after herpes simplex virus-1 ocular infection. J Immunol. 2015;194:379–387. doi: 10.4049/jimmunol.1402326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell. 2001;8:169–179. doi: 10.1016/S1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- 22.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 23.Cathcart HM, Zheng M, Covar JJ, Liu Y, Podolsky R, Atherton SS. Interferon-gamma, macrophages, and virus spread after HSV-1 injection. Invest Ophthalmol Vis Sci. 2011;52:3984–3993. doi: 10.1167/iovs.10-6449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang JH, Gabison EE, Kato T, Azar DT. Corneal neovascularization. Curr Opin Ophthalmol. 2001;12:242–249. doi: 10.1097/00055735-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Chen Y, Huang L, Yu J. Evaluation of heparanase and matrix metalloproteinase-9 in patients with cutaneous malignant melanoma. J Dermatol. 2012;39:339–343. doi: 10.1111/j.1346-8138.2011.01441.x. [DOI] [PubMed] [Google Scholar]
- 26.Clement C, Tiwari V, Scanlan PM, Valyi-Nagy T, Yue BY, Shukla D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J Cell Biol. 2006;174:1009–1021. doi: 10.1083/jcb.200509155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conrady CD, Drevets DA, Carr DJJ. Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol. 2010;220:1–9. doi: 10.1016/j.jneuroim.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cursiefen C, Chen L, Borges LP, Jackson D, Cao J, Radziejewski C, D’Amore PA, Dana MR, Wiegand SJ, Streilein JW. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. 2004;113:1040–1050. doi: 10.1172/JCI20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delrieu I, Arnaud E, Ferjoux G, Bayard F, Faye JC. Overexpression of the FGF-2 24-kDa isoform up-regulates IL-6 transcription in NIH-3T3 cells. FEBS Lett. 1998;436:17–22. doi: 10.1016/S0014-5793(98)01086-2. [DOI] [PubMed] [Google Scholar]
- 30.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. doi: 10.1016/S1097-2765(00)80221-X. [DOI] [PubMed] [Google Scholar]
- 31.Elion GB. Mechanism of action and selectivity of acyclovir. Am J Med. 1982;73:7–13. doi: 10.1016/0002-9343(82)90055-9. [DOI] [PubMed] [Google Scholar]
- 32.Englund JA, Zimmerman ME, Swierkosz EM, Goodman JL, Scholl DR, Balfour HH., Jr Herpes simplex virus resistant to acyclovir. A study in a tertiary care center. Ann Intern Med. 1990;112:416–422. doi: 10.7326/0003-4819-76-3-112-6-416. [DOI] [PubMed] [Google Scholar]
- 33.Farooq AV, Valyi-Nagy T, Shukla D. Mediators and mechanisms of herpes simplex virus entry into ocular cells. Curr Eye Res. 2010;35:445–450. doi: 10.3109/02713681003734841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Field AK, Biron KK. “The end of innocence” revisited: resistance of herpesviruses to antiviral drugs. Clin Microbiol Rev. 1994;7:1–13. doi: 10.1128/CMR.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frank GM, Buela K-G, Maker DM, Harvey SAK, Hendricks RL. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J Immunol. 2012;188:1350–1359. doi: 10.4049/jimmunol.1101968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuster MM, Wang L. Endothelial heparan sulfate in angiogenesis. Prog Mol Biol Transl Sci. 2010 doi: 10.1016/s1877-1173(10)93009-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gangappa S, Deshpande SP, Rouse BT. Bystander activation of CD4 + T cells accounts for herpetic ocular lesions. Invest Ophthalmol Vis Sci. 2000;41:453–459. [PubMed] [Google Scholar]
- 38.Gebhardt BM, Varnell ED, Kaufman HE. Inhibition of cyclooxygenase 2 synthesis suppresses Herpes simplex virus type 1 reactivation. J Ocul Pharmacol Ther. 2005;21:114–120. doi: 10.1089/jop.2005.21.114. [DOI] [PubMed] [Google Scholar]
- 39.Gimenez F, Mulik S, Veiga-Parga T, Bhela S, Rouse BT. Robo 4 counteracts angiogenesis in herpetic stromal keratitis. Plos One. 2015;10:e0141925. doi: 10.1371/journal.pone.0141925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 41.Guo Y, Audry M, Ciancanelli M, Alsina L, Azevedo J, Herman M, Anguiano E, Sancho-Shimizu V, Lorenzo L, Pauwels E, Philippe PB, Perez de Diego R, Cardon A, Vogt G, Picard C, Andrianirina ZZ, Rozenberg F, Lebon P, Plancoulaine S, Tardieu M, Valerie D, Jouanguy E, Chaussabel D, Geissmann F, Abel L, Casanova JL, Zhang SY. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011;208:2083–2098. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gurung HR, Carr MM, Bryant K, Chucair-Elliott AJ, Carr DJJ. Fibroblast growth factor-2 drives and maintains progressive corneal neovascularization following HSV-1 infection. Mucosal Immunol. 2018;11:172–185. doi: 10.1038/mi.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hacker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H. Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF)6. J Exp Med. 2000;192:595–600. doi: 10.1084/jem.192.4.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hadigal SR, Agelidis AM, Karasneh GA, Antoine TE, Yakoub AM, Ramani VC, Djalilian AR, Sanderson RD, Shukla D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat Commun. 2015;6:6985. doi: 10.1038/ncomms7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill JM, Bhattacharjee PS, Neumann DM. Apolipoprotein E alleles can contribute to the pathogenesis of numerous clinical conditions including HSV-1 corneal disease. Exp Eye Res. 2007;84:801–811. doi: 10.1016/j.exer.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H. Herpes simplex virus type-1 induces IFN-a production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2004;101:11416–11421. doi: 10.1073/pnas.0403555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Isner JM, Asahara T. Angiogenesis and vasculogenesis as therapeutic strategies for postnatal neovascularization. J Clin Invest. 1999;103:1231–1236. doi: 10.1172/JCI6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaishankar D, Buhrman JS, Valyi-Nagy T, Gemeinhart RA, Shukla D. Extended release of an anti-heparan sulfate peptide from a contact lens suppresses corneal herpes simplex virus-1 infection. Invest Ophthalmol Vis Sci. 2016;57:169–180. doi: 10.1167/iovs.15-18365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaishankar D, Yakoub AM, Yadavalli T, Agelidis A, Thakkar N, Hadigal S, Ames J, Shukla D. An off-target effect of BX795 blocks herpes simplex virus type 1 infection of the eye. Sci Transl Med. 2018 doi: 10.1126/scitranslmed.aan5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jayamanne DG, Vize C, Ellerton CR, Morgan SJ, Gillie RF. Severe reversible ocular anterior segment ischaemia following topical trifluorothymidine (F3T) treatment for herpes simplex keratouveitis. Eye (Lond) 1997;11(Pt 5):757–759. doi: 10.1038/eye.1997.193. [DOI] [PubMed] [Google Scholar]
- 51.Jiang YC, Feng H, Lin YC, Guo XR. New strategies against drug resistance to herpes simplex virus. Int J Oral Sci. 2016;8:1–6. doi: 10.1038/ijos.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaufman HE, Varnell ED, Gebhardt BM, Thompson HW, Atwal E, Rubsamen-Waigmann H, Kleymann G. Efficacy of a helicase-primase inhibitor in animal models of ocular herpes simplex virus type 1 infection. J Ocul Pharmacol Ther. 2008;24:34–42. doi: 10.1089/jop.2007.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krawczyk A, Arndt MAE, Grosse-Hovest L, Weichert W, Giebel B, Dittmer U, Hengel H, Jäger D, Schneweis KE, Eis-Hübinger AM, Roggendorf M, Krauss J. Overcoming drug-resistant herpes simplex virus (HSV) infection by a humanized antibody. Proc Natl Acad Sci USA. 2013;110:6760. doi: 10.1073/pnas.1220019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kreuger J, Phillipson M. Targeting vascular and leukocyte communication in angiogenesis, inflammation and fibrosis. Nat Rev Drug Discov. 2016;15:125–142. doi: 10.1038/nrd.2015.2. [DOI] [PubMed] [Google Scholar]
- 55.Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Nat Commun. 2004;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- 56.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kvanta A, Sarman S, Fagerholm P, Seregard S, Steen B. Expression of matrix metalloproteinase-2 (MMP-2) and vascular endothelial growth factor (VEGF) in inflammation-associated corneal neovascularization. Exp Eye Res. 2000;70:419–428. doi: 10.1006/exer.1999.0790. [DOI] [PubMed] [Google Scholar]
- 58.Lee S, Zheng M, Kim B, Rouse BT. Role of matrix metalloproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J Clin Invest. 2002;110:1105–1111. doi: 10.1172/JCI200215755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang X, Yuan L, Hu J, Yu H, Li T, Lin S, Tang S. Phosphomannopentaose sulfate (PI-88) suppresses angiogenesis by downregulating heparanase and vascular endothelial growth factor in an oxygen-induced retinal neovascularization animal model. Mol Vis. 2012;18:1649–1657. [PMC free article] [PubMed] [Google Scholar]
- 60.Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med. 2009;15:1312–1317. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liesegang TJ. Herpes simplex virus epidemiology and ocular importance. Cornea. 2001;20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 62.Liu J, Crepin M, Liu J-, Barritault D, Ledoux D. FGF-2 and TPA induce matrix metalloproteinase-9 secretion in MCF-7 cells through PKC activation of the Ras/ERK pathway. Biochem Biophys Res Commun. 2002;293:1174–1182. doi: 10.1016/S0006-291X(02)00350-9. [DOI] [PubMed] [Google Scholar]
- 63.Liu T, Khanna KM, Chen X, Fink DJ, Hendricks RL. Cd8(+) T cells can block herpes simplex virus Type 1 (HSV-1) reactivation from latency in sensory neurons. J Exp Med. 2000;191:1459–1466. doi: 10.1084/jem.191.9.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu X, Fitzgerald K, Kurt-Jones E, Finberg R, Knipe DM. Herpesvirus tegument protein activates NF-κB signaling through the TRAF6 adaptor protein. Proc Natl Acad Sci USA. 2008;105:11335–11339. doi: 10.1073/pnas.0801617105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lundberg P, Ramakrishna C, Brown J, Tyszka JM, Hamamura M, Hinton DR, Kovats S, Nalcioglu O, Weinberg K, Openshaw H, Cantin EM. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J Virol. 2008;82:7078–7088. doi: 10.1128/JVI.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maddula S, Davis DK, Maddula S, Burrow MK, Ambati BK. Horizons in therapy for corneal. Angiogenesis. 2011;118:591–599. doi: 10.1016/j.ophtha.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maggs DJ, Chang E, Nasisse MP, Mitchell WJ. Persistence of herpes simplex virus type 1 DNA in chronic conjunctival and eyelid lesions of mice. J Virol. 1998;72:9166–9172. doi: 10.1128/jvi.72.11.9166-9172.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mansur DS, Kroon EG, Nogueira ML, Arantes RME, Rodrigues SCO, Akira S, Gazzinelli RT, Campos MA. Lethal encephalitis in myeloid differentiation factor 88-deficient mice infected with herpes simplex virus 1. Am J Pathol. 2005;166:1419–1426. doi: 10.1016/S0002-9440(10)62359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Masola V, Gambaro G, Tibaldi E, Brunati AM, Gastaldello A, D’Angelo A, Onisto M, Lupo A. Heparanase and syndecan-1 interplay orchestrates fibroblast growth factor-2-induced epithelial-mesenchymal transition in renal tubular cells. J Biol Chem. 2012;287:1478–1488. doi: 10.1074/jbc.M111.279836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maudgal PC, Van Damme B, Missotten L. Corneal epithelial dysplasia after trifluridine use. Graefes Arch Clin Exp Ophthalmol. 1983;220:6–12. doi: 10.1007/BF02307009. [DOI] [PubMed] [Google Scholar]
- 71.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 72.Menachery VD, Pasieka TJ, Leib DA. Interferon regulatory factor 3-dependent pathways are critical for control of herpes simplex virus type 1 central nervous system infection. J Virol. 2010;84:9685–9694. doi: 10.1128/JVI.00706-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Modi S, Van L, Gewirtzman A, Mendoza N, Bartlett B, Tremaine AM, Tyring S. Single-day treatment for orolabial and genital herpes: a brief review of pathogenesis and pharmacology. Ther Clin Risk Manag. 2008;4:409–417. doi: 10.2147/TCRM.S1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morris JE, Zobell S, Yin XT, Zakeri H, Summers BC, Leib DA, Stuart PM. Mice with mutations in Fas and Fas ligand demonstrate increased herpetic stromal keratitis following corneal infection with HSV-1. J Immunol. 2012;188:793–799. doi: 10.4049/jimmunol.1102251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mulik S, Xu J, Reddy PBJ, Rajasagi NK, Gimenez F, Sharma S, Lu PY, Rouse BT. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am J Pathol. 2012;181:525–534. doi: 10.1016/j.ajpath.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oh MJ, Akhtar J, Desai P, Shukla D. A role for heparan sulfate in viral surfing. Biochem Biophys Res Commun. 2010;391:176–181. doi: 10.1016/j.bbrc.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Paludan SR, Bowie AG, Horan KA, Fitzgerald KA. Recognition of herpesviruses by the innate immune system. Nat Rev Immunol. 2011;11:143–154. doi: 10.1038/nri2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park PJ, Antoine TE, Farooq AV, Valyi-Nagy T, Shukla D. An investigative peptide-acyclovir combination to control herpes simplex virus type 1 ocular infection. Invest Ophthalmol Vis Sci. 2013;54:6373–6381. doi: 10.1167/iovs.13-12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perng GC, Jones C. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis. 2010;2010:262415. doi: 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pope LE, Marcelletti JF, Katz LR, Lin JY, Katz DH, Parish ML, Spear PG. The anti-herpes simplex virus activity of n-docosanol includes inhibition of the viral entry process. Antiviral Res. 1998;40:85–94. doi: 10.1016/S0166-3542(98)00048-5. [DOI] [PubMed] [Google Scholar]
- 81.Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rajasagi NK, Bhela S, Varanasi SK, Rouse BT. Frontline Science: aspirin-triggered resolvin D1 controls herpes simplex virus-induced corneal immunopathology. J Leukoc Biol. 2017;102:1159–1171. doi: 10.1189/jlb.3HI1216-511RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rajasagi NK, Reddy PB, Mulik S, Gjorstrup P, Rouse BT. Neuroprotectin D1 reduces the severity of herpes simplex virus-induced corneal immunopathology. Invest Ophthalmol Vis Sci. 2013;54:6269–6279. doi: 10.1167/iovs.13-12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rajasagi NK, Reddy PB, Suryawanshi A, Mulik S, Gjorstrup P, Rouse BT. Controlling herpes simplex virus-induced ocular inflammatory lesions with the lipid-derived mediator resolvin E1. J Immunol. 2011;186:1735–1746. doi: 10.4049/jimmunol.1003456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rajasagi NK, Suryawanshi A, Sehrawat S, Reddy PB, Mulik S, Hirashima M, Rouse BT. Galectin-1 reduces the severity of herpes simplex virus-induced ocular immunopathological lesions. J Immunol. 2012;188:4631–4643. doi: 10.4049/jimmunol.1103063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ramani VC, Yang Y, Ren Y, Nan L, Sanderson RD. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011;286:6490–6499. doi: 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rogge M, Yin X, Godfrey L, Lakireddy P, Potter CA, Del Rosso CR, Stuart PM. Therapeutic use of soluble fas ligand ameliorates acute and recurrent herpetic stromal keratitis in mice. Invest Ophthalmol Vis Sci. 2015;56:6377–6386. doi: 10.1167/iovs.15-16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roizman B, Knipe DM, Whitley RJ. Herpes simplex viruses. Clin Infect Dis. 2007;1:2503–2602. doi: 10.1086/514600. [DOI] [PubMed] [Google Scholar]
- 89.Sarangi PP, Kim B, Kurt-Jones E, Rouse BT. Innate recognition network driving herpes simplex virus-induced corneal immunopathology: role of the toll pathway in early inflammatory events in stromal keratitis. J Virol. 2007;81:11128–11138. doi: 10.1128/JVI.01008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sauter MM, Gauger JJL, Brandt CR. Oligonucleotides designed to inhibit TLR9 block herpes simplex virus type 1 infection at multiple steps. Antiviral Res. 2014 doi: 10.1016/j.antiviral.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scheppke L, Aguilar E, Gariano RF, Jacobson R, Hood J, Doukas J, Cao J, Noronha G, Yee S, Weis S, Martin MB, Soll R, Cheresh DA, Friedlander M. Retinal vascular permeability suppression by topical application of a novel VEGFR2/Src kinase inhibitor in mice and rabbits. J Clin Invest. 2008;118:2337–2346. doi: 10.1172/JCI33361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seghezzi G, Patel S, Ren CJ, Gualandris A, Pintucci G, Robbins ES, Shapiro RL, Galloway AC, Rifkin DB, Mignatti P. Fibroblast growth factor-2 (FGF-2) induces vascular endothelial growth factor (VEGF) expression in the endothelial cells of forming capillaries: an autocrine mechanism contributing to angiogenesis. J Cell Biol. 1998;141:1659–1673. doi: 10.1083/jcb.141.7.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Serhan CN. Novel pro-resolving lipid mediators in inflammation are leads for resolution physiology. Nature. 2014;510:92–101. doi: 10.1038/nature13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma S, Mulik S, Kumar N, Suryawanshi A, Rouse BT. An anti-inflammatory role of VEGFR2/Src kinase inhibitor in herpes simplex virus 1-induced immunopathology. J Virol. 2011;85:5995–6007. doi: 10.1128/JVI.00034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shukla D, Spear PG. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest. 2001;108:503–510. doi: 10.1172/JCI13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith JS, Robinson NJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis. 2002;186(Suppl 1):3. doi: 10.1086/343739. [DOI] [PubMed] [Google Scholar]
- 97.St Vincent MR, Colpitts CC, Ustinov AV, Muqadas M, Joyce MA, Barsby NL, Epand RF, Epand RM, Khramyshev SA, Valueva OA, Korshun VA, Tyrrell DL, Schang LM. Rigid amphipathic fusion inhibitors, small molecule antiviral compounds against enveloped viruses. Proc Natl Acad Sci USA. 2010;107:17339–17344. doi: 10.1073/pnas.1010026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stuart PM, Griffith TS, Usui N, Pepose J, Yu X, Ferguson TA. CD95 ligand (FasL)-induced apoptosis is necessary for corneal allograft survival. J Clin Invest. 1997;99:396–402. doi: 10.1172/JCI119173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stuart PM, Pan F, Plambeck S, Ferguson TA. FasL-Fas interactions regulate neovascularization in the cornea. Invest Ophthalmol Vis Sci. 2003;44:93–98. doi: 10.1167/iovs.02-0299. [DOI] [PubMed] [Google Scholar]
- 100.Stumpf TH, Shimeld C, Easty DL, Hill TJ. Cytokine production in a murine model of recurrent herpetic stromal keratitis. Invest Ophthalmol Vis Sci. 2001;42:372–378. [PubMed] [Google Scholar]
- 101.Stumpf TH, Case R, Shimeld C, Easty DL, Hill TJ. Primary herpes simplex virus type 1 infection of the eye triggers similar immune responses in the cornea and the skin of the eyelids. J Gen Virol. 2002;83:1579–1590. doi: 10.1099/0022-1317-83-7-1579. [DOI] [PubMed] [Google Scholar]
- 102.Su AR, Qiu M, Li YL, Xu WT, Song SW, Wang XH, Song HY, Zheng N, Wu ZW. BX-795 inhibits HSV-1 and HSV-2 replication by blocking the JNK/p38 pathways without interfering with PDK1 activity in host cells. Acta Pharmacol Sin. 2017;38:402–414. doi: 10.1038/aps.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Suryawanshi A, Mulik S, Sharma S, Reddy PBJ, Sehrawat S, Rouse BT. Ocular neovascularization caused by herpes simplex virus type 1 infection results from breakdown of binding between vascular endothelial growth factor A and its soluble receptor. J Immunol. 2011;186:3653–3665. doi: 10.4049/jimmunol.1003239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 105.Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 106.Tang D, Piao Y, Zhao S, Mu X, Li S, Ma W, Song Y, Wang J, Zhao W, Zhang Q. Expression and correlation of matrix metalloproteinase-9 and heparanase in patients with breast cancer. Med Oncol. 2014 doi: 10.1007/s12032-014-0026-4. [DOI] [PubMed] [Google Scholar]
- 107.Thomas J, Gangappa S, Kanangat S, Rouse BT. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J Immunol. 1997;158:1383–1391. [PubMed] [Google Scholar]
- 108.Tiwari V, Clement C, Xu D, Valyi-Nagy T, Yue BY, Liu J, Shukla D. Role for 3-O-sulfated heparan sulfate as the receptor for herpes simplex virus type 1 entry into primary human corneal fibroblasts. J Virol. 2006;80:8970–8980. doi: 10.1128/JVI.00296-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tiwari V, Liu J, Valyi-Nagy T, Shukla D. Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J Biol Chem. 2011;286:25406–25415. doi: 10.1074/jbc.M110.201103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tiwari V, Oh MJ, Kovacs M, Shukla SY, Valyi-Nagy T, Shukla D. Role for nectin-1 in herpes simplex virus 1 entry and spread in human retinal pigment epithelial cells. FEBS J. 2008;275:5272–5285. doi: 10.1111/j.1742-4658.2008.06655.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Toma HS, Murina AT, Areaux RG, Jr, Neumann DM, Bhattacharjee PS, Foster TP, Kaufman HE, Hill JM. Ocular HSV-1 latency, reactivation and recurrent disease. Semin Ophthalmol. 2008;23:249–273. doi: 10.1080/08820530802111085. [DOI] [PubMed] [Google Scholar]
- 112.Tsatsos M, MacGregor C, Athanasiadis I, Moschos MM, Hossain P, Anderson D. Herpes simplex virus keratitis: an update of the pathogenesis and current treatment with oral and topical antiviral agents. Clin Exp Ophthalmol. 2016;44:824–837. doi: 10.1111/ceo.12785. [DOI] [PubMed] [Google Scholar]
- 113.van Velzen M, van de Vijver DA, van Loenen FB, Osterhaus AD, Remeijer L, Verjans GM. Acyclovir prophylaxis predisposes to antiviral-resistant recurrent herpetic keratitis. J Infect Dis. 2013;208:1359–1365. doi: 10.1093/infdis/jit350. [DOI] [PubMed] [Google Scholar]
- 114.Whitley RJ, Lakeman F. Herpes simplex virus infections of the central nervous system: therapeutic and diagnostic considerations. Clin Infect Dis. 1995;20:414–420. doi: 10.1093/clinids/20.2.414. [DOI] [PubMed] [Google Scholar]
- 115.Wuest T, Zheng M, Efstathiou S, Halford WP, Carr DJJ. The herpes simplex virus-1 transactivator infected cell protein-4 drives VEGF-A dependent Neovascularization. PLoS Pathog. 2011 doi: 10.1371/journal.ppat.1002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yadavalli T, Agelidis A, Jaishankar D, Mangano K, Thakkar N, Penmetcha K, Shukla D. Targeting herpes simplex virus-1 gD by a DNA aptamer can be an effective new strategy to curb viral infection. Mol Ther Nucleic Acids. 2017;9:365–378. doi: 10.1016/j.omtn.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yasin B, Wang W, Pang M, Cheshenko N, Hong T, Waring AJ, Herold BC, Wagar EA, Lehrer RI. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J Virol. 2004;78:5147–5156. doi: 10.1128/JVI.78.10.5147-5156.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yildiz C, Ozsurekci Y, Gucer S, Cengiz AB, Topaloglu R. Acute kidney injury due to acyclovir. CEN Case Rep. 2013;2:38–40. doi: 10.1007/s13730-012-0035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yoon KC, Heo H, Kang IS, Lee MC, Kim KK, Park SH, Cho KO. Effect of topical cyclosporin A on herpetic stromal keratitis in a mouse model. Cornea. 2008;27:454–460. doi: 10.1097/ICO.0b013e318160602d. [DOI] [PubMed] [Google Scholar]
- 120.Zhang S, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, Segal D, Sancho-Shimizu V, Lorenzo L, Puel A, Picard C, Chapgier A, Plancoulaine S, Titeux M, Cognet C, Von Bernuth H, Ku C, Casrouge A, Zhang X, Barreiro L, Leonard J, Hamilton C, Lebon P, Hron B, Valle L, Quintana-Murci L, Hovnanian A, Rozenberg F, Vivier E, Geissmann F, Tardieu M, Abel L, Casanova J. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 121.Zheng M, Deshpande S, Lee S, Ferrara N, Rouse BT. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J Virol. 2001;75:9828–9835. doi: 10.1128/JVI.75.20.9828-9835.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zheng M, Schwarz MA, Lee S, Kumaraguru U, Rouse BT. Control of stromal keratitis by inhibition of neovascularization. Am J Pathol. 2001;159:1021–1029. doi: 10.1016/S0002-9440(10)61777-4. [DOI] [PMC free article] [PubMed] [Google Scholar]