

Abstract
Spliceosomal introns are ubiquitous features of eukaryotic genomes, but the mechanisms responsible for their loss and gain are difficult to identify. Microsporidia are obligate intracellular parasites that have significantly reduced genomes and, as a result, have lost many if not all of their introns. In the microsporidian Encephalitozoon cuniculi, a relatively long intron was identified and was spliced at higher levels than the remaining introns. This long intron is part of a set of unique introns in two unrelated genes that show high levels of sequence conservation across diverse microsporidia. The introns possess a unique internal conserved region, which overlaps with a shared, predicted stem–loop structure. The unusual similarity and retention of these long introns in reduced microsporidian genomes could indicate that these introns function similarly, are homologous, or both. Regardless, the significant genome reduction in microsporidia provides a rare opportunity to understand intron evolution.
Keywords: microsporidia, spliceosomal introns, genome reduction
Introduction
Spliceosomal introns are intervening sequences in eukaryotic genomes that are removed from premRNA by a ribonucleoprotein complex called the spliceosome. The spliceosome is composed of five small nuclear RNAs (snRNAs; U1, U2, U4, U5, and U6) and a large number of associated proteins. The spliceosome facilitates interactions between snRNAs and intron motifs to catalyze the process. Two key interactions are between the U1 snRNA and the intron’s 5′ splice site, and between the U2 snRNA and the branchpoint motif located near the 3′ end of the intron.
Noncoding DNA, including introns, evolves more quickly than protein-coding sequences because of limited selective pressures, resulting in higher levels of sequence divergence. Hence, predicting homologous introns based on sequence similarity can be extremely difficult, or impossible. Indeed, when homologous genes possess introns at conserved positions, these introns are inferred to be homologous, even in the absence of sequence conservation (Knowles and McLysaght 2006; Wilkerson et al. 2009). Studies identifying conserved intron positions are helpful in determining rates of intron gain and loss over evolutionary time, but struggle to identify mechanisms behind gain and loss events (Hoy et al. 2016). Clear cases where mechanisms can be identified are exceedingly rare, and are restricted to those resulting from recent intron gain events in populations or in closely related species (Sharpton et al. 2008; Zhu and Niu 2013; Li et al. 2014). Li et al. (2014) observed specific introns gained via repair of staggered, double-strand breaks in populations of Daphnia pugnax. Other intron gain models have been proposed, including transposon insertion and tandem genomic duplication (Yenerall and Zhou 2012; Huff et al. 2016; Lee and Stevens 2016), but currently lack experimental support.
Mechanisms underlying intron loss are better understood (Niu et al. 2005; Coulombe-Huntington and Majewski 2007; Cohen et al. 2012). Introns are predicted to be primarily lost via a reverse transcriptase mediated process. In this model, a mature mRNA molecule (with introns spliced) is reverse transcribed, and the subsequent cDNA replaces the intron-containing genomic copy through recombination. This model not only describes intron loss, but also predicts that introns at the 5′ ends of transcripts may escape loss due to incomplete reverse transcription (Niu et al. 2005; Cohen et al. 2012; Ma et al. 2015). Introns are also lost through genomic deletions. As these deletions affect coding and noncoding sequences, they are less precise than RT-mediated intron loss, causing introns to be fragmented or lost entirely along with their surrounding exons (Yang et al. 2013). Obviously, other types of genomic mutations also affect intron abundance. For example, point mutations can result in loss or gain through changes to spliceosomal motifs in the intron.
Regardless of the mechanism of intron loss, the process has had a distinct impact on genome size. Lineages with reduced genomes have dispensed with much non-coding material, including introns (Kämper et al. 2006; Lane et al. 2007; Hu et al. 2011). Microsporidia are a group of fungal intracellular parasites with some of the smallest eukaryotic genomes known. The 2.9-Mb genome of Encephalitozoon cuniculi was sequenced in 2001, and only 16 introns were described (Katinka et al. 2001). These introns, found primarily in ribosomal protein-coding genes, are extremely short (only ∼25 nt) with standard GT–AG boundaries and an extreme 5′ bias, usually starting in the codon directly after the initiator ATG. In 2010, we identified and verified an additional 20 E. cuniculi introns (Lee et al. 2010). Although similar in size to the original set of E. cuniculi introns, these introns do not have a strong 5′ bias in position and are not primarily found in ribosomal protein-coding genes (Lee et al. 2010). However, a unique intron in the new set was found in a predicted Poly-A binding protein gene and this intron stands out by being 76 nt long: Approximately three times longer than the other miniature E. cuniculi introns. We assessed levels of splicing in E. cuniculi, and again we found that the long Poly-A intron was distinct. Whereas average splicing levels for the short introns are <20%, the long Poly-A intron was spliced at levels similar to those seen in Saccharomyces introns (∼80%; Grisdale et al. 2013). Similar long introns have been annotated in the microsporidia Spraguea lophii (Campbell et al. 2013), Trachipleistophora hominis (Watson et al. 2015), and Vavraia culicis (Desjardins et al. 2015) in the unrelated 40S ribosomal protein S23 gene as well as in an ORF that appears homologous to the Poly-A ORF in E. cuniculi (Grisdale et al. 2013). As in E. cuniculi, the long introns in these taxa were distinct from other annotated introns. Beyond the difference in size, the long introns were shown to be spliced at higher levels, with many of the other annotated introns showing no transcriptomic evidence of splicing (Campbell et al. 2013; Desjardins et al. 2015; Watson et al. 2015).
Here, we propose that the unusual Poly-A and S23 introns are part of an intron family found in these two independent genes and possess unusual sequence conservation across distantly related microsporidia. We discuss implications for intron gain and functional retention in otherwise reduced genomes.
Results and Discussion
To examine why the long intron in the Poly-A ORF was retained against a background of reduction in microsporidia, we sought to determine the extent that long introns are conserved across microsporidia. We assessed the available microsporidian data, currently limited to 27 genomes. Fortunately, these cover a phylogenetically diverse range of microsporidia and hosts (Pombert et al. 2015). We identified unusual long introns in 15 microsporidia (fig. 1 ), including an intron predicted based on conserved position and sequence in an apparent intron-lacking taxon, Pseudoloma neurophilia (Ndikumana et al. 2017). Indeed, the only microsporidia that lack the long introns appear to have lost introns entirely. Seven of the intron-possessing microsporidia not only have a long intron in the Poly-A binding gene as was first identified in E. cuniculi, but also possess an intron of similar length and sequence in the 40S ribosomal protein S23 as seen in other microsporidia (Campbell et al. 2013; Watson et al. 2015; Desjardins et al. 2015; fig. 1). These long introns, found in two unrelated genes, across diverse microsporidia, are remarkably similar in size and have high levels of sequence conservation (fig. 1). The branchpoint appears to be conserved in the long introns beyond what was previously described as an extended branchpoint in yeast (Berglund et al. 1997). Furthermore, it is extended in a manner that could increase the base pairing potential of the branchpoint with the U2 snRNA (fig. 2 ). Indeed, these microsporidian introns are unique in possessing the most extended branchpoint-U2 snRNA interaction known. This is relevant because the increased levels of splicing of the Poly-A intron we previously identified for the E. cuniculi Poly-A intron (Grisdale et al. 2013) could be the result of a higher affinity of the U2 snRNA for this hyperextended branchpoint. Assuming that this is the case, we predict elevated splicing levels for the Poly-A and S23 introns of other microsporidia.
Fig. 1.

—Alignment of the Poly-A (top) and S23 long introns (bottom). Spliceosomal motifs are represented by bars between the alignments: 5′ and 3′ splice sites (black); typical eukaryotic branchpoint (blue); extension of S. cerevisiae branchpoint (yellow); hyper-extension of long intron branchpoints (red). Green bars represent conserved stem–loop structures of long introns: Loop (solid); stem (hatched). Outside the alignment the large bracket indicates the internal conserved region. Smaller brackets represent potential C/D box snoRNA motifs (see text for details). Poly-A ORF IDs are as follows: Encephalitozoon romaleae, EROM_090870; Encephalitozoon hellem, EHEL_091190; Encephalitozoon cuniculi, ECU09_1470; Encephalitozoon intestinalis, Eint_091220; Ordospora colligata, M896_021830; Nosema apis, NAPIS_ORF02048; Nosema ceranae, NCER_100552; Nosema bombycis, NBO_41g0035; Anncaliia algerae, H312_02562; Trachipleistophora hominis, THOM_0860/THOM_0861; Vavraia culicis, VCUG_02155; Pseudoloma neurophilia, M153_1793000463; Spraguea lophii, SLOPH_1788. S23 ORF IDs are as follows: A. algerae, H312_01000; T. hominis, THOM_3123; V. culicis, VCUG_00403; P. neurophilia, M153_1640006413; S. lophii, SLOPH_2680. IDs from Sporanauta perivermis, Antonospora locustae, and Hamiltosporidium tvaerminnensis are unavailable due to a lack of annotation files.
Fig. 2.
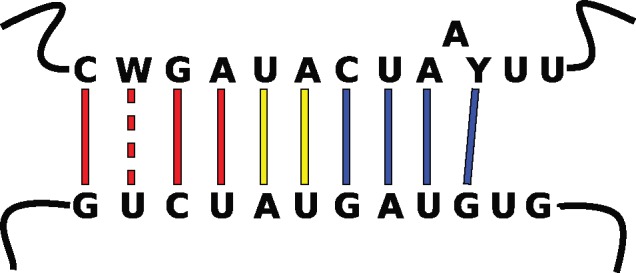
—Predicted basepairing interaction between the U2 snRNA (bottom) and the extended branchpoint of the long introns (top). Color of base-pairings is as in figure 1.
Transcriptomes from intron-possessing microsporidia are available for V. culicis (Desjardins et al. 2015), S. lophii (Campbell et al. 2013), Nosema bombycis (Liu et al. 2016), and T. hominis (Watson et al. 2015). Desjardins et al. (2015) identified 34 introns in V. culicis, but did not include the Poly-A intron we predicted bioinformatically. However, our analysis of the V. culicis transcriptome confirms splicing of the Poly-A intron, and corroborates splicing of the remaining introns discussed in Desjardins et al. (2015). In fact, the two long introns are the highest spliced introns in V. culicis. Of the eight annotated introns in S. lophii (Campbell et al. 2013), our analysis confirms that only the two long introns in the Poly-A and S23 ORFs are spliced. The lack of evidence for the splicing of the remaining six short introns could be the result of levels of splicing so low that they escape detection, although we cannot rule out limitations of the data or an inability of the mapping software to map the extremely 5′ biased introns. In N. bombycis we were only able to find evidence for splicing of the Poly-A intron and could identify no split reads for the shorter annotated introns (bearing in mind similar data limitations to S. lophii). On the basis of the presence of conserved E. cuniculi spliceosomal motifs, 85 introns were predicted in the genome of T. hominis (Heinz et al. 2012). Heinz et al. (2012) identified the T. hominis Poly-A ORF in the genome (orf_860) but did not annotate an intron in the gene. The transcriptome of T. hominis suggested the presence of introns in 14 ORFs including S23 and Poly-A (where the latter was not identified); furthermore, the connection between orf_860 and the long Poly-A intron was not made in this study (Watson et al. 2015). Indeed, Watson et al. (2015) only predict high levels of splicing for these two long introns. Our own analysis of the T. hominis transcriptome also confirms the presence of these highly spliced long introns, but we believe the remaining junctions called by the mapping software are false positives likely caused by repeat regions in the genome (supplementary table 1, Supplementary Material online, T hominis Junctions tab). Furthermore, none of the remaining 83 introns predicted from the T. hominis genome could be validated in our analysis, suggesting that they are not, in fact, introns.
To summarize, the long introns identified in the V. culicis data showed higher levels of splicing than the remaining introns, which parallels splicing patterns observed for the long and short introns in E. cuniculi (Grisdale et al. 2013). Without direct comparisons to shorter introns in the other three transcriptomes, we cannot determine if the long introns differ quantifiably in terms of splicing. However, the observation that these long introns are the only two introns with evidence of splicing in the transcriptomes T. hominis, N. bombycis, and S. lophii, coupled with the possession of conserved extended branchpoints, strongly indicates that their splicing is exceptional compared with other microsporidian introns.
The unusual observation of shared sequence similarity in the two sets of long introns across diverse microsporidia (fig. 1) caused us to consider whether these introns could be homologous. To assess this possibility, we examined the distribution of the long introns across microsporidia and their close relatives. We inspected the genomes of Amphiamblys sp. WSBS2006, Mitosporidium daphniae (Haag et al. 2014), and the rozellomycotan parasites, Paramicrosporidium saccamoebae (Quandt et al. 2017) and Rozella allomycis (James et al. 2013), four recently sequenced members of lineages that branch as sisters to the microsporidia. Presence or absence of the long introns in these taxa could allow us to better understand the timeline of gain and loss of these unique introns and assess their homology. The metchnikovellid, Amphiamblys sp. WSBS2006 appears devoid of introns (Mikhailov et al. 2017), whereas R. allomycis contains two putative introns in S23 that are short (∼30 nt), introduce premature stop codons, and lack the conserved 5′ splice site and branchpoint motifs of even the short E. cuniculi introns. There are also two introns in the S23 ORFs of M. daphniae and P. saccamoebae. Unlike the introns in R. allomycis, they are long (152 and 120 bp, respectively) and the M. daphniae intron lacks the conserved spliceosomal motifs of E. cuniculi introns. These introns may provide some hints to the evolution of the long microsporidian introns. The Poly-A and S23 introns of microsporidia both have internal conserved regions (fig. 1), the introns in the M. daphniae and P. saccamoebae S23 genes appear to have this same internal region although the M. daphniae intron appears to lack the hyperextended branchpoint motif seen in microsporidia (supplementary fig. 1, Supplementary Material online). The presence of the S23 intron in M. daphniae and P. saccamoebae complicates the evolutionary timeline of loss of these introns in the genomes of microsporidia (fig. 3 ). Molecular data from other groups related to rozellomycota and/or early diverging microsporidia, such as Nucleophaga spp. (Corsaro et al. 2014; Corsaro et al. 2016) will need to be assessed to address this issue more extensively. These sampling issues hinder conclusive assessments of homology, though all microsporidia that possess introns have the Poly-A intron (fig. 3), whereas only seven have the S23 intron, clearly indicating that events of loss have been prevalent (fig. 3).
Fig. 3.

—Distribution of long introns across microsporidia. Presence of the Poly-A (blue) and S23 (red) long introns are indicated by predicted folding of the released intron. Absence of the structure indicates absence of the intron in the genome. The stem–loop structure in the internal conserved region is highlighted. Microsporidian relationships follow Pombert et al. (2015) with more recent modifications suggested by Luallen et al. (2016), Ndikumana et al. (2017), Mikhailov et al. (2017), and Quandt et al. (2017). Enterocytozoonidae/Vittaforma and Nematocida have no long introns and have been collapsed. Nematocida spp. and Edhazardia aedis do not have identified ORFs homologous to the Poly-A ORF of Encephalitozoon cuniculi. Enterocytozoonidae and Vittaforma have Poly-A ORFs without introns (supplementary fig. 3, Supplementary Material online).
The Poly-A gene exhibits low levels of conservation outside of the intron (supplementary fig. 2, Supplementary Material online). None of the Nematocida spp. has an identifiable Poly-A ORF. Whether the ORF was lost or was never present is unclear. We were able to identify divergent Poly-A homologs by looking at conserved gene order (Corradi et al. 2007; supplementary fig. 3, Supplementary Material online) in Vittaforma corneae and the Enterocytozoonidae. However, the ORFs we identified in these intron-lacking taxa do not contain the same RNA recognition domains of the intron-containing genes, likely indicating that the genes have a different function (supplementary fig. 2, Supplementary Material online). In metozoa, a set of homologous, poorly conserved genes harbors expressed ncRNAs (Tanaka-Fujita et al. 2007), and it is worthwhile to consider the possibility that the long introns in S23 and Poly-A are functional.
Indeed, in T. hominis the Poly-A ORF is the 3rd most abundant transcript (Watson et al. 2015) and in E. cuniculi codes the 25th most abundant protein (Brosson et al. 2006). The relatively high levels at which the Poly-A ORF is expressed in microsporidia that possess long Poly-A introns, and loss of functional domains in the ORF after introns are lost, both in the ORF itself and throughout the genome, leads us to speculate that the ORF could be highly expressed in order to express the intron. Considering this possibility, along with the unusual retention of these long introns despite reductive pressures, and low levels of conservation of the genes in which they reside, leads us to predict these long introns could be functional.
Conservation of a function could explain the unusual sequence similarity of these long introns. Spliced introns have been known to encode noncoding RNAs: One major group of these intronic RNAs, the small nucleolar RNAs (snoRNAs), functions in the maturation of ribosomes. A previous study failed to identify snoRNAs in microsporidia based on sequence similarity predictions (Gardner et al. 2010). More recently, Belkorchia et al. (2017) identified snoRNAs in Encephalitozoon and Nosema species but noted that the presence of intronic snoRNAs is unlikely due to very short intron lengths. Provocatively, C/D box snoRNAs (Makarova and Kramerov 2009) have similar motifs (CUGA and UGAUGA) as those found in the internal conserved region of the long introns (fig. 1), raising the possibility that these introns could encode highly divergent snoRNAs. We also carried out structural predictions to determine if all the introns shared any similar structures that could be linked to function. Surprisingly, there appears to be a stem–loop of similar size in the conserved regions of all the long introns (fig. 3). Although the stem–loop structure does not look like typical snoRNA structures, the structure is consistent in both sets of long introns, overlaps with the internal conserved region, and is outside of the spliceosomal motifs. We cannot rule out the possibility that this internal conserved loop binds a spliceosomal protein, however, to the best of our knowledge this is not a recognized motif in other eukaryotic introns. The predicted stem also contains compensatory point mutations, which would not change the base-pairing potential of the stem as a result of G•U wobble base-pairing and furthermore support the presence of the stem (Varani and McClain 2000). These data suggest that the RNA released upon splicing could possess a function in the parasite. Taken together, a functional role for theses introns is supported by a conserved secondary structure, increased levels of splicing, loss of functional domains in the Poly-A ORF, and unusually high levels of intron sequence conservation across taxa. The potential function makes the loss of the long, microsporidian introns in seven taxa all the more intriguing.
In conclusion, the retention of the stem–loop structure, the maintenance of strict spliceosomal motifs, and the unusual relative size of these long introns has afforded us the unique opportunity to identify a family of introns we predict are functional, and hence retained across microsporidia despite reductive pressures. Conserved function does not preclude the possibility that these microsporidian introns are homologous and could offer rare insights into aspects of intron evolution that could be masked in larger, more complex genomes.
Materials and Methods
We retrieved the publicly available microsporidian genomes from the NCBI and Broad Microsporidian databases (E. cuniculi, Encephalitozoon romaleae, Encephalitozoon intestinalis, Encephalitozoon hellem, Ordospora colligata, Nosema apis, Nosema ceranae, N. bombycis, Antonospora locustae, Anncaliia algerae, Hamiltosporidium tvaerminnensis, T. hominis, V. culicis, S. lophii, Nematocida parisii, Nematocida sp. 1, Nematocida displodere, Enterocytozoon bieneusi, Enterocytozoon hepatopenaei, Enterospora canceri, Hepatospora eriocheir, V. corneae, P. neurophilia, R. allomycis, M. daphniae, P. saccamoebae, Amphiamblys sp. WSBS2006) and the genome of Sporanauta perivermis (unpublished: PolyA Accession MK190414). Long introns were identified based on sequence similarity to the E. cuniculi Poly-A intron using reciprocal BlastN (e < 1 × 10 − 10). To search for Poly-A ORFs that did not contain a long intron we used TBlastN with a cutoff of e < 1 × 10 − 1 and a BLOSUM62 substitution matrix and the NCBI default gap costs. To determine if candidate ORFs in intron-lacking taxa are homologous with the Poly-A ORFs, TBlastN was used to identify the ORFs surrounding the candidate Poly-A ORF and gene order was compared among Poly-A intron containing microsporidia and Poly-A intron-lacking microsporidia (supplementary fig. 3, Supplementary Material online). TBlastN (e < 1 × 10 − 10) was also used to analyze the presence of the S23 ORF across microsporidia. InterProScan 5.30-v69.0 (Jones et al. 2014) was used to determine if candidate ORFs shared similar protein domains in the Poly-A candidate genes across microsporidia. As an example, Edhazardia aedis has an ORF with an RRM-domain but based on a reciprocal BLAST is homologous with a different polyadenylated-binding protein in E. cuniculi and as a result has been eliminated as a candidate for the Poly-A ORF (supplementary table 1, Supplementary Material online, PolyA BLAST tab).
Using MUSCLE, the long intron, host genes, and U2 snRNA sequences were aligned and then alignments were refined by eye (supplementary fig. 4, Supplementary Material online). Sfold Srna algorithms v. 2.2 (Ding et al. 2004) were used to predict the minimum free energy structure of the long introns at 37 °C and 1 M NaCl with no divalent ions. The RNA folding predictions were then manually refined by drawing out potential folding patterns of the conserved region. To map the publicly available transcriptome data we used STAR v. 2.5.3a (Dobin et al. 2013) with a minimum intron size of 10 nt and a maximum intron size of 300 nt. To help reduce false positive junctions in repetitive regions of the genomes, we filtered out multimapping reads. Junctions predicted by STAR were verified visually using Interactive Genome Viewer (IGV; Robinson et al. 2011) to determine if the junction could have been called erroneously as a result of mismapping due to short repeats in the genome (supplementary table 1, Supplementary Material online, T. hominis Junctions tab).
To compare splicing levels among predicted junctions in V. culicis, custom Python scripts (available upon request) were used to count the coverage of mapped reads at the boundary the junction. Using read counts for split and unsplit reads, percent spliced reads was calculated by dividing the number or split reads by the total reads at the boundary of the predicted junction (supplementary table 1, Supplementary Material online; V. culicis Junctions tab). Any junction that had <10 mapped reads was excluded from analysis.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We acknowledge funding support from the Natural Sciences and Engineering Research Council of Canada Discovery Grant (Grant Number: 262988 to N.M.F.); and a grant from the Centre for Microbial Diversity and Evolution from the Tula Foundation. We also acknowledge D. Tack (formerly of the University of British Columbia and currently of Pennsylvania State University) for his custom scripts used in the analysis of the RNA-Seq data.
Footnotes
Data deposition: We have submitted the Sporanauta perivermis Poly-A Binding sequence to GenBank under the accession KC172651
Literature Cited
- Belkorchia A, et al. 2017. Comparative genomics of microsporidian genomes reveals a minimal non-coding RNA set and new insights for transcription in minimal eukaryotic genomes. DNA Res. 243:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund JA, Chua K, Abovich N, Reed R, Rosbash M. 1997. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 895:781–787. [DOI] [PubMed] [Google Scholar]
- Brosson D, et al. 2006. Proteomic analysis of the eukaryotic parasite Encephalitozoon cuniculi (microsporidia): a reference map for protein expressed in late sporogonial stages. Proteomics 612:3625–3635. [DOI] [PubMed] [Google Scholar]
- Campbell SE, et al. 2013. The genome of Spraguea lophii and the basis of host–microsporidian interactions. PLOS Genet. 98:e1003676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NE, Shen R, Carmel L. 2012. The role of reverse transcriptase in intron gain and loss mechanisms. Mol Biol Evol. 291:179–186. [DOI] [PubMed] [Google Scholar]
- Corsaro D, et al. 2014. Rediscovery of Nucleophaga amoebae, a novel member of the Rozellomycota. Parasitol Res. 113:4491–4498. [DOI] [PubMed] [Google Scholar]
- Corsaro D, et al. 2016. Molecular identification of Nucleophaga terricolae sp. nov. (Rozellamycota), and new insights on the origin of the microsporidia. Parasitol Res. 1158:3003–3011. [DOI] [PubMed] [Google Scholar]
- Corradi N, et al. 2007. Patterns of genome evolution among the microsporidian parasite Encephalitozoon cuniculi, Antonospora locustae and Enterocytozoon bieneusi. PLoS One 212:e1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe-Huntington J, Majewski J. 2007. Characterization of intron loss events in mammals. Genome Res. 171:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins CA, et al. 2015. Contrasting host–pathogen interactions and genome evolution in two generalist and specialist microsporidian pathogens of mosquitoes. Nat Commun. 2015(6):7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Chan CY, Lawrence CE. 2004. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 32(Web Server issue):W135–W141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, et al. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Bateman A, Poole AM. 2010. SnoPatrol: how many snoRNA genes are there? J Biol. 2010. 91:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisdale CJ, Bowers LC, Didier ES, Fast NM. 2013. Transcriptome analysis of the parasite Encepalitozoon cuniculi: an in-depth examination of pre-mRNA splicing in a reduced eukaryote. BMC Genomics. 14:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag KT, et al. 2014. Evolution of a morphological novelty occurred before genome compaction in a lineage of extreme parasites. Proc Natl Acad Sci U S A. 11143:15480–15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz E, et al. 2012. The genome of the obligate intracellular parasite Trachipleistophora hominis: new insights into microsporidian genome dynamics and reductive evolution. PLoS Pathog. 810:e1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy MA, et al. 2016. Genome sequencing of the phytoseiid predatory mite Metaseiulus occidentalis reveals completely atomized Hox genes and superdynamic intron evolution. Genome Biol Evol. 86:1762–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu TT, et al. 2011. The Arabidopsis lyrata genome sequence and the basis of the rapid genome size change. Nat Genet. 435:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff JT, Zilberman D, Roy SW. 2016. Mechanism for DNA transposons to generate introns on genomic scales. Nature 5387626:533–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TY, et al. 2013. Shared signatures of parasitism and phylogenomics unite Cryptomycota and Microsporidia. Curr Biol. 2316:1548–1553. [DOI] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 309:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kämper J, et al. 2006. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 4447115:97–101. [DOI] [PubMed] [Google Scholar]
- Katinka M, et al. 2001. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 4146862:450–453. [DOI] [PubMed] [Google Scholar]
- Knowles DG, McLysaght A. 2006. High rate of recent intron gain and loss in simultaneously duplicated Arabidopsis genes. Mol Biol Evol. 238:1548–1557. [DOI] [PubMed] [Google Scholar]
- Lane C, et al. 2007. Nucleomorph genome of Hemiselmis andersenii reveals complete intron loss and compaction as a driver of protein structure and function. Proc Natl Acad Sci U S A. 10450:19908–199913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RCH, Gill EE, Roy SW, Fast NM. 2010. Constrained intron structures in a microsporidian. Mol Biol Evol. 279:1979–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Stevens SW. 2016. Spliceosomal intronogenesis. Proc Natl Acad Sci U S A. 11323:6514–6519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Kuzoff R, Wong C, Tucker A, Lynch M. 2014. Characterization of newly gained introns in Daphnia populations. Genome Biol Evol. 69:2218–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, et al. 2016. Transcriptome sequencing and characterization of ungerminated and germinated spores of Nosema bombycis. Acta Biochim Biophys Sin. 483:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luallen RJ, et al. 2016. Discovery of a natural pathogen with broad tissue tropism in Caenorhabditis elegans. PLoS Pathog. 126:e1005724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MY, Che XR, Porceddu A, Niu DK. 2015. Evaluation of the mechanisms of intron loss and gain in the social amoeba Dictyostelium. BMC Evol Biol. 15:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova JA, Kramerov DA. 2009. Analysis of C/D box snoRNA genes in vertebrates: the number of copies decreases in placental mammals. Genomics 941:11–19. [DOI] [PubMed] [Google Scholar]
- Mikhailov KV, Simdyanov TG, Aleoshin VV. 2017. Genomic survey of a hyperparasitic microsporidian Amphiamblys sp. (Metchnikovellidae). Genome Biol Evol. 93:454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndikumana S, et al. 2017. Genome analysis of Pseudoloma neurophilia: a microsporidian parasite of zebrafish (Danio rerio). J Eukaryot Microbiol. 641:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu D, Hou W, Li S. 2005. mRNA-mediated intron losses: evidence from extraordinary large exons. Mol Biol Evol. 226:1475–1481. [DOI] [PubMed] [Google Scholar]
- Pombert JF, Haag KL, Beidas S, Ebert D, Keeling PJ. 2015. The Ordospora colligata genome; evolution of extreme reduction in microsporidia and host-to-parasite horizontal gene transfer. mBio 6:e02400–e02414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt CA, et al. 2017. The genome of an intracellular parasite, Paramicrosporidium saccamoebae, reveals alternative adaptations to obligate intracellular parasitism. eLife 6:e29594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, et al. 2011. Interactive genome viewer. Nat Biotechnol. 291:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpton TJ, Neafsey DE, Galagan JE, Taylor JW. 2008. Mechanisms of intron gain and loss in Cryptococcus. Genome Biol. 91:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka-Fujita R, Soeno Y, Satoh H, Nakamura Y, Mori S. 2007. Human and mouse protein-noncoding snoRNA host genes with dissimilar nucleotide sequences show chromosomal synteny. RNA 136:811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani G, McClain WH. 2000. The G•U wobble base pair. EMBO Rep. 11:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AK, et al. 2015. Transcriptomic profiling of host-parasite interactions in the microsporidian Trachipleistophora hominis. BMC Genomics 16:983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson MD, Ru Y, Brendel VP. 2009. Common introns with orthologous genes: software and applications in plants. Brief Bioinformatics. 106:631–644. [DOI] [PubMed] [Google Scholar]
- Yang YF, Zhu T, Niu DK. 2013. Association of intron loss with high mutation rates in Arabidopsis: implications for genome size evolution. Genome Biol Evol. 54:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenerall P, Zhou L. 2012. Identifying the mechanisms of intron gain: progress and trends. Biol Direct. 7:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Niu DK. 2013. Mechanisms of intron loss and gain in the fission yeast Schizosaccharomyces. PLoS One 84:e61683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.