

Abstract
Rhabdomyosarcoma (RMS) forms in skeletal muscle and is the most common soft tissue sarcoma in children and adolescents. Current treatment is associated with debilitating side effects and treatment outcomes for patients with metastatic disease are dismal. Recently, a novel binuclear palladacycle, AJ-5, was shown to exert potent cytotoxicity in melanoma and breast cancer and to present with negligible adverse effects in mice. This study investigates the anti-cancer activity of AJ-5 in alveolar and embryonal RMS. IC50 values of ≤ 0.2 µM were determined for AJ-5 and it displayed a favourable selectivity index of >2. Clonogenic and migration assays showed that AJ-5 inhibited the ability of RMS cells to survive and migrate, respectively. Western blotting revealed that AJ-5 induced levels of key DNA damage response proteins (γH2AX, p-ATM and p-Chk2) and the p38/MAPK stress pathway. This correlated with an upregulation of p21 and a G1 cell cycle arrest. Annexin V-FITC/propidium iodide staining revealed that AJ-5 induced apoptosis and necrosis. Apoptosis was confirmed by the detection of cleaved PARP and increased levels and activity of cleaved caspases-3, -7, -8 and -9. Furthermore, AJ-5 reduced autophagic flux as shown by reduced LC3II accumulation in the presence of bafilomycin A1 and a significant reduction in autophagosome flux J. Finally, pharmacokinetic studies in mice show that AJ-5 has a promising half-life and that its volume of distribution is high, its clearance low and its intraperitoneal absorption is good. Together these findings suggest that AJ-5 may be an effective chemotherapeutic with a desirable mechanism of action for treating drug-resistant and advanced sarcomas.
Introduction
Rhabdomyosarcoma (RMS) originate in skeletal muscle and is the most common soft tissue sarcoma in children, adolescents and young adults1,2. It is classified as either embryonal (eRMS) or alveolar (aRMS), with each exhibiting distinct histological, molecular and pathological characteristics. Whereas eRMS affects predominantly infants and children, aRMS occur primarily in adolescents and young adults. Furthermore, eRMS comprises approximately 67% of RMS cases with common sites of tumorigenesis including the head, neck and genitourinary system. On the other hand, aRMS accounts for approximately 30% of RMS cases with arms and legs often being sites of primary disease3–5. RMS thus contributes to a considerable loss of years of life in comparison to other cancers as it largely affects children and adolescents.
The current standard treatment for patients with RMS is multimodal therapy consisting of local control with surgery and/or radiation therapy in conjunction with multi-agent chemotherapy6. Since the 1970s, disease control for patients with localized or completely resected RMS has been achieved with combinational chemotherapy most commonly including vincristine, dactinomycin (actinomycin D) and cyclophosphamide (VAC)7. However, this treatment regimen has had limited success for patients with regional spreading, incomplete resection or metastasis8. Furthermore, these drugs are associated with debilitating side effects including nausea, vomiting, fatigue, mouth ulcers, stomach ache, hair loss, bone marrow suppression, peripheral neuropathy and haemorrhagic cystitis9–12. Continued collaborative efforts to find more effective treatments with fewer side effects is therefore important, especially for metastatic RMS.
The use of transition metal complexes as potential therapeutics and in diagnostic medicine is of considerable importance. Platinum drugs, particularly cisplatin, are the mainstay of metal-based compounds in the treatment of cancer. However, due to dose-related adverse effects and multi-drug resistance associated with this line of therapy there has been an ongoing search for alternative metallic compounds with improved anti-cancer and pharmacokinetic properties and distinct mechanisms of action13. Palladium-based complexes have recently been reported to exert significant cytotoxicity in cancer cells14. Importantly, studies have shown that while both platinum and palladium compounds induce DNA damage, the palladium compounds exert a much greater degree of cytotoxicity in cancer cells15,16. Moreover, numerous palladium complexes exhibit strong cytotoxic effects in cells resistant to cisplatin17. Together these results suggest that palladium complexes may exert cytotoxicity through a mechanism different to that of platinum compounds and that they may prove to be less toxic and more effective for the treatment of aggressive cancers. Recently, AJ-5, a novel binuclear palladacycle complex with 1,2-bis(diphenylphosphino)ethane as co-ligand was shown to exert potent anti-tumour activity in advanced melanoma and breast cancer cells with very promising in vivo clearance of advanced melanoma18,19. In the present study we therefore explored these questions in RMS and other sarcoma subtypes. We show here for the first time that AJ-5 displays potent and selective cytotoxicity at sub-micromolar concentrations (≤0.2 µM) against aRMS and eRMS cells and inhibits their ability to survive, proliferate and migrate. We show that AJ-5 induces double-stranded DNA breaks (DSBs) which triggers the canonical DSBs ataxia telangiectasia mutated serine/threonine kinase (ATM)/checkpoint kinase 2 (Chk2) pathway as well as the p38/mitogen-activated protein kinase (MAPK) stress signalling pathway leading to a G1 cell cycle arrest, reduction in autophagic flux and induction of apoptosis. Furthermore, AJ-5 shows efficacy in a range of sarcoma subtypes and displays a promising pharmacokinetic profile in healthy mice. This study provides evidence that AJ-5 has promising therapeutic potential for the treatment of RMS and a range of other sarcoma subtypes.
Results
AJ-5 shows potent and selective cytotoxicity against RMS cells
Here we investigated the anti-tumour effects of the binuclear palladacycle complex, AJ-5, in RMS cells. To this end, MTT assays were performed on aRMS (RH30 and AX-OH-1 cell lines) and eRMS (RD and FL-OH-1 cell lines) cells treated with a range of AJ-5 concentrations (0.1–1.0 μM) for 48 h. IC50 concentrations of ≤0.2 µM were obtained for all cell lines (Fig. 1a). Light microscopy images of RMS cells treated for 24 and 48 h with their respective IC50 concentrations show the impact of AJ-5 on cell viability and morphology (Fig. 1b). To determine the selectivity of AJ-5 for RMS cells, IC50 values were determined in non-malignant fibroblasts (FG0 and DMB) and mouse myoblasts (C2C12). Results show that AJ-5 is less cytotoxic in the non-malignant cells with a selectivity index (SI) of >2 in all cases (Fig. 1c). Interestingly, there is growing evidence that mesenchymal stem cells are the cell of origin for specific sarcoma types including RMS20,21, and compared to the other non-malignant cells tested, the mesenchymal stem cells (A100021501) were the most sensitive to AJ-5.
Fig. 1. AJ-5 shows potent and selective cytotoxicity against aRMS and eRMS cells.

a MTT cell viability assays of aRMS cell lines, RH30 and AX-OH-1, and eRMS cell lines, RD and FL-OH-1, treated with a range of AJ-5 concentrations (0.1–1.0 µM) or vehicle for 48 h. Graphs show mean cell viability as a percentage of vehicle control ± SEM for each concentration of AJ-5 determined from three independent experiments performed in quadruplicate. A curve was fitted to determine the IC50 concentration of AJ-5 for each cell line. b Representative light microscopy images (×200; EVOS XL AMEX1000 Core Imaging System) of aRMS and eRMS cell lines treated with their respective IC50 concentrations of AJ-5 or vehicle for 24 and 48 h. c MTT cell viability assays of non-malignant human fibroblast cell lines, FG0 and DMB, mouse myoblast cell line, C2C12, and mesenchymal stem cell line, A10021501, treated with AJ-5 and IC50 concentrations determined as described in a above. Selectivity indices (SIs) were determined for each RMS cell line by dividing the IC50 of each non-malignant cell line by the IC50 of each RMS cell line
AJ-5 inhibits the ability of RMS cells to survive, proliferate and migrate
To explore the potential impact of AJ-5 on the long-term fate of RMS cells, clonogenic assays were performed22. Figure 2a shows representative images and quantification of colonies formed over 7–21 days after 24 h of AJ-5 treatment. It is evident that AJ-5 significantly inhibits the ability of RMS cells to survive and proliferate. Indeed, for all except one RMS cell line tested, there was a significant decrease in colony area at ¼ IC50 treatment and only a few or no RMS cell colonies formed at IC50 concentrations. Importantly, the ability of the non-malignant C2C12 myoblasts to survive and proliferate was not significantly hindered after 24 h of 0.3 µM AJ-5 (Fig. 2b). This is at least 1.5 times higher than the IC50 values obtained for the RMS cells. To further explore the anti-cancer activity of AJ-5, RMS cells were exposed for 24 h to IC50 concentrations of the drug and scratch motility assays were performed. Fig. 2c shows that AJ-5 reduced the migratory ability of all RMS cells tested. Taken together, these data suggest that AJ-5 inhibits the ability of RMS cells to survive, proliferate and migrate.
Fig. 2. AJ-5 inhibits the ability of RMS cells to survive, proliferate and migrate.

a Representative images and quantification of clonogenic assays of eRMS and aRMS cells treated with vehicle, ¼ IC50, ½ IC50 or IC50 concentrations of AJ-5 for 24 h and then replated at low densities in drug-free medium and left for 7–21 days for colonies to form. Colonies were stained with crystal violet and images from three independent repeats were quantified using the ImageJ plugin ColonyArea. The graph represents the mean colony area ± SEM of each treatment condition as a percentage of the vehicle control. b The same as a above for the non-malignant mouse myoblast cell line C2C12, but treated with vehicle, 0.15 µM, 0.3 µM, or 0.6 µM AJ-5. c Representative images (×200; EVOS XL AMEX1000 Core Imaging System) and quantification of scratch motility assays of eRMS and aRMS cells pre-treated with IC50 concentrations of AJ-5 or vehicle for 24 h and then replated at 100% confluency in drug-free medium. After cell adherence, a sterile 2 µL pipette tip was used to make a linear wound in the cell monolayer and cells were treated with 10 µg/mL mitomycin C to inhibit proliferation. Cells were imaged at 0, 3, 6, 9, 12 and 24 h post wound formation. Total area migrated was calculated by subtracting the wound area at each time point from the wound area at time 0 h, which is represented in the graphs as mean area migrated ± SEM pooled from three independent repeats. Data were analysed using GraphPad Prism 6.0 and a parametric unpaired t-test was performed where *p < 0.05, **p < 0.01, ***p < 0.001
To further explore the mechanisms by which AJ-5 exerts its cytotoxicity, the aRMS cell line RH30 and the eRMS cell line RD were used for further characterization.
AJ-5 activates the DNA damage and p38 MAPK pathways in RMS cells
γH2AX is a robust marker of DSBs23 and western blotting and immunocytochemistry revealed a dose-dependent increase in levels of γH2AX (Fig. 3a) and an accumulation of distinct γH2AX foci in the nuclei of AJ-5-treated cells (Fig. 3b), respectively. Furthermore, AJ-5 treatment led to increased levels of phosphorylated ATM and its downstream target CHK2 as well as phosphorylated p38 (Fig. 3c). AJ-5 thus activated the canonical DSB and p38 MAPK stress signalling pathways. Both of these pathways converge on p53, which ordinarily plays an important role in transcriptionally activating the cyclin-dependent kinase p2124. However, both the RMS cell lines used in this study harbour p53 mutations that result in high levels of inactive p5325. Importantly, p21 levels increased at the IC50 concentrations of AJ-5 and this response was particularly pronounced after 24 h of treatment (Fig. 3c). These results suggest that p21 levels increase in a p53-independent manner.
Fig. 3. AJ-5 activates the DNA damage and the p38 MAPK pathways.

a γH2AX protein levels detected by western blotting in RH30 (aRMS) and RD (eRMS) cells treated with vehicle (V), 0.1 µM or IC50 AJ-5 for 24 and 48 h. p38 was used as a loading control. Densitometry readings were obtained using ImageJ and protein expression levels are represented as a ratio of protein of interest/p38 normalized to the vehicle control sample (where possible). Blots are representative of at least two independent repeats. b Representative confocal immunofluorescence maximum intensity projection images (×630; Carl Zeiss LSM 510) of RH30 and RD cells treated with IC50 AJ-5 or vehicle for 24 h and γH2AX detected with a fluorophore-conjugated Cy3 secondary antibody. Nuclei of cells were stained with DAPI. Scale bar is 20 µM. Box plots represent quantification of γH2AX levels per treatment condition as mean nuclear Cy3 fluorescence from 20 fields of view from three independent repeats. Data were analysed using GraphPad Prism 6.0 and a parametric unpaired t-test was performed where *p < 0.05, **p < 0.01, ***p < 0.001. c Western blot analyses with antibodies to key DNA damage and stress signalling pathway proteins: p-ATM, p-Chk2, p-p38, p53, and p21. RH30 and RD cells were treated with AJ-5 and protein expression quantified as described above in a. Broken lines in western blots shown in this figure indicate where lanes not relevant were removed
AJ-5 triggers a G1 cell cycle arrest and induces apoptosis and necrosis
To investigate the effect of AJ-5 on cell cycle progression, FACS analyses were performed on RMS cells treated with drug for 24 and 48 h. Results show that AJ-5 induces a G1 cell cycle arrest in both cell lines at the expense of the S phase (Fig. 4a). This correlated with a decrease in levels of cyclin A and cyclin B, which are required for progression through the S and G2/M phases, respectively26 (Fig. 4b). The sub-G1 peaks seen in Fig. 4a suggested that AJ-5 treatment leads to cell death. Indeed, Annexin V-FITC assays confirm that AJ-5 induces apoptotic and necrotic cell death (Fig. 4c).
Fig. 4. AJ-5 triggers a G1 cell cycle arrest and induces apoptotic and necrotic cell death in RH30 and RD cells.

a Flow cytometry analyses of cells treated with vehicle or IC50 AJ-5 for 24 and 48 h. Graphs represent the mean proportion of cells ± SEM at each phase of the cell cycle pooled from three independent repeats. b Western blot analyses of protein harvested from cells treated as indicated and incubated with antibodies against cell cycle markers cyclin A and cyclin B1. p38 was used as a loading control. Densitometry readings were obtained using ImageJ and protein expression levels are represented as a ratio of protein of interest/p38 normalized to the vehicle control sample. Blots are representative of at least two independent repeats. Broken lines indicate where lanes not relevant were removed. c Flow cytometric analyses of cells treated with vehicle or IC50 AJ-5 and stained with Annexin V-FITC and PI. Graphs represent the mean ± SEM percentage of viable (lower left-hand quadrant), apoptotic (lower right-hand quadrant + upper right-hand quadrant) and necrotic (upper left-hand quadrant) cells from three independent experiments. For a and c data were analysed using GraphPad Prism 6.0 and a parametric unpaired t-test was performed where *p < 0.05, **p < 0.01, ***p < 0.001
AJ-5 triggers the intrinsic and extrinsic apoptosis pathways in RMS cells
To confirm that AJ-5-induces apoptosis, RD and RH30 cells were treated with AJ-5 and biological, molecular and biochemical markers of apoptosis were investigated by light microscopy, western blotting and enzymatic Caspase-Glo assays, respectively. The results show that AJ-5 induced characteristic features of apoptosis including membrane blebbing and cell shrinkage (Fig. 5a). In addition, AJ-5 treatment led to increased levels and activity of cleaved caspase-8 and caspase-9, the effectors of the extrinsic and intrinsic pathways, respectively (Fig. 5b, c). This correlated with increased levels of the active (cleaved) forms of the executioner caspases-3/7 and their substrate poly (ADP-ribose) polymerase (PARP) (Fig. 5b, c)27,28. Interestingly, we observed an ~20 kDa band above the 18 kDa active cleaved caspase-8 fragment. A similar band has also been reported by other researchers and it has been suggested that it represents an intermediate product of procaspase-8 processing29,30. Importantly, caspases-8 and -9 were more robustly activated in RD cells treated with AJ-5 than those treated with IC50 doxorubicin, a drug commonly used in the treatment of RMS and used as a positive control in apoptosis assays (Fig. 5c). Together these results show that AJ-5 induces cell death in RMS cells, in part, through the extrinsic and intrinsic apoptotic pathways.
Fig. 5. AJ-5 triggers the intrinsic and extrinsic apoptosis pathways in RMS cells.

a Representative light microcopy images (×200; EVOS XL AMEX1000 Core Imaging System) showing the morphology of RH30 and RD cells treated with vehicle, 0.1 µM or IC50 AJ-5 for 24 and 48 h. Numbered circles correspond to magnified images on the right, which highlight characteristic apoptotic morphology including membrane blebbing and cell shrinkage. b Western blot analyses of protein harvested from cells treated as in a and incubated with antibodies as indicated. p38 was used as a loading control and densitometry readings were obtained using ImageJ. Protein expression levels are represented as a ratio of protein of interest/p38 normalized to vehicle control samples (where possible). Blots are representative of at least two independent repeats. Broken lines indicate where lanes not relevant were removed. c Caspase-Glo assays showing the enzymatic activity of caspase-3/7, caspase-8 and caspase-9 for cells treated with vehicle or IC50 AJ-5. Doxorubicin was included as a positive control for caspase-8 and -9 assays at the IC50 concentration (0.5 µM) established in the cell lines tested. Graphs represent the mean fold change of caspase activity ± SEM pooled from three independent repeats. Data were analysed using GraphPad Prism 6.0 and a parametric unpaired t-test was performed where *p < 0.05, **p < 0.01, ***p < 0.001
AJ-5 induces several markers of autophagy in RMS cells
We next investigated whether AJ-5 induced autophagy because large vacuolar structures and acidic vesicles were observed in RMS cells treated with this drug (Supplementary Fig. S1A). To this end, western blotting and immunocytochemistry were performed with an antibody to microtubule-associated protein light chain 3 (LC3). Supplementary Figure S1B shows that LC3II (the autophagosomal membrane bound form of LC3) was induced in both RMS cell lines in a time- and dose-dependent manner. Furthermore, supplementary Fig. S1C shows distinct puncta, presumed to represent LC3II aggregates conjugated to autophagosomal membranes, in RD and RH30 cells from as early as 6 h after AJ-5 treatment.
AJ-5 reduces autophagic flux in RD and RH30 cells
The detection of LC3II alone does not distinguish between autophagy induction and cessation in the downstream steps of autophagy. We therefore next determined the impact of AJ-5 treatment on autophagic flux by measuring the levels of p62, a receptor of polyubiquitinated proteins. The rationale for this is based on observations that upon autophagy induction, p62 is incorporated within an autophagosome and degraded within an autolysosome31. The turnover of p62 is therefore considered an indicator of autophagic flux. Interestingly, Fig. 6a shows that whereas AJ-5 appears to reduce autophagic flux in RH30 cells, it increases autophagic flux in RD cells. To confirm the above data, levels of LC3II were determined by western blotting in the presence and absence of bafilomycin A1 which blocks the autophagy pathway by inhibiting fusion of autophagosomes with lysosomes32. An accumulation of LC3II in the presence of this inhibitor is therefore considered evidence of autophagic flux. Consistent with our p62 data in RH30 cells, AJ-5 treatment followed by bafilomycin A1 exposure led to decreased levels of LC3II (Fig. 6b). Interestingly, contrary to our p62 data in RD cells, in the presence of bafilomycin A1, AJ-5 treatment led to a negligible change in LC3II compared with vehicletreated cells (Fig. 6b). We therefore further investigated the impact of AJ-5 on autophagic flux in the RD cell line by performing single-cell autophagosome flux analyses. Our results reveal that RD cells are characterized by a predominant autolysosomal pool (nA = 83 autolysosomes/cell), with only a very minor pool of autophagosomes and lysosomes per cell (Fig. 6c). Moreover, the cells are characterized by a basal autophagosome flux J of 6.3 autophagosomes per hour per cell. Upon AJ-5 treatment, however, both autolysosome pool size as well as autophagosome flux significantly decreased. This suggests that AJ-5 negatively impacts the rate of autophagosome synthesis, which supports the data showing that in the presence of bafilomycin A1, AJ-5 treatment does not lead to LC3II accumulation (Fig. 6b). Together these data suggest that AJ-5 reduces autophagic flux in RH30 and RD cells.
Fig. 6. AJ-5 reduces autophagic flux in RD and RH30 cells.

a Western blotting of p62/SQSTM1 protein levels in RH30 and RD cells treated with vehicle (V), 0.1 µM or IC50 AJ-5 for 24 and 48 h. b Western blotting showing LC3I and LC3II protein levels in RH30 and RD cells treated with vehicle (V) or IC50 AJ-5 for 24 h followed by 2 h of treatment with 200 nM bafilomycin A1. For western blots, p38 was used as a loading control and densitometry readings were obtained using ImageJ. Protein expression levels are represented as a ratio of protein of interest/p38 normalized to vehicle control sample. Blots are representative of at least two independent repeats. c Representative single-cell fluorescence maximum intensity projection micrographs (×630; Carl Zeiss LSM 780; scale bar is 20 µM) and pool size quantification of autophagy pathway intermediates: autophagosomes (GFP-LC3, nA) (indicated with white arrows in the merged image), autolysosomes (LysoTracker Red, nAL) and lysosomes (merged, nL). Autophagosome flux J was calcuclated. Data were analysed using GraphPad Prism 6.0 and a parametric unpaired t-test was performed *p < 0.05, **p < 0.01, ***p < 0.001. # compared to untreated control, * compared to vehicle control
AJ-5 is cytotoxic in a range of sarcoma subtypes
To investigate if the therapeutic potential of AJ-5 could be extended to other sarcoma subtypes, chondrosarcoma (SW1353), liposarcoma (SW872), synovial sarcoma (SW982), fibrosarcoma (HT1080) and osteosarcoma (MG-63) cells were treated with the drug as described earlier and MTT assays were performed. Our results show that an IC50 of <0.3 µM was obtained for all the sarcoma cell lines tested (Supplementary Fig. S2A) and a favourable SI of >2 was achieved when calculated relative to the combined IC50 values for the normal fibroblasts (FG0 and DMB). However, a sub-optimal SI between 1 and 1.5 was obtained when the IC50 values for the sarcoma cells were expressed relative to the mesenchymal stem cells (A10021501) (Supplementary Fig. S2B). This raises the interesting possibility that AJ-5 may be effective against the cells of origin of these sarcoma subtypes which may be of therapeutic benefit. Furthermore, clonogenic assays reveal that as little as a ¼ IC50 concentration of AJ-5 significantly reduced the ability of cells of all sarcoma subtypes to survive and proliferate (supplementary Fig. S2C). AJ-5 therefore shows potent selective cytotoxicity against a number of diverse sarcoma subtypes and may therefore have broad therapeutic potential.
Pharmacokinetic (PK) profile of AJ-5 in healthy mice
Given its importance to the drug discovery process, we next tested the in vivo PK profile of AJ-5 in whole blood of MF1 mice following a single dose of 2 mg/kg intravenous (IV), 2 mg/kg intraperitoneal (IP) or 20 mg/kg oral (PO) for a period of 24 h. The blood concentration–time curve of AJ-5 over a 24 h period and the calculated PK parameters are shown in Supplementary Fig. S3 and Table S1. For IV administration, AJ-5 illustrated a long half-life (>10 h), which is most likely due to the low clearance (9.2 mL/min/kg) and a high volume of distribution (8.8 L/kg). The exposure of AJ-5 following the IP dose of 2 mg/kg was eight-fold higher compared to the PO dose of 20 mg/kg with an area under the curve of 88 and 11 min.µM/L, respectively. The data obtained for the IP group in healthy mice correlated well with our previously observed in vivo efficacy of AJ-5 in advanced melanoma18.
Discussion
RMS is the most common soft tissue sarcoma found in children and adolescents and while the current treatment for localized tumours results in a high overall survival rate, the chemotherapeutic agents used are associated with debilitating adverse effects10,33–36. Moreover, more than 15% of patients present with metastatic disease and there has been limited improvement in the treatment of patients with recurrent and metastatic disease37–41. The current study provides several lines of evidence that the binuclear palladacycle AJ-5 may be a promising chemotherapeutic to treat aRMS and eRMS cells as well as a range of other sarcoma subtypes. Furthermore, we provide novel and interesting mechanisms by which AJ-5 functions (see Fig. 7).
Fig. 7. Proposed model for AJ-5 in RMS.
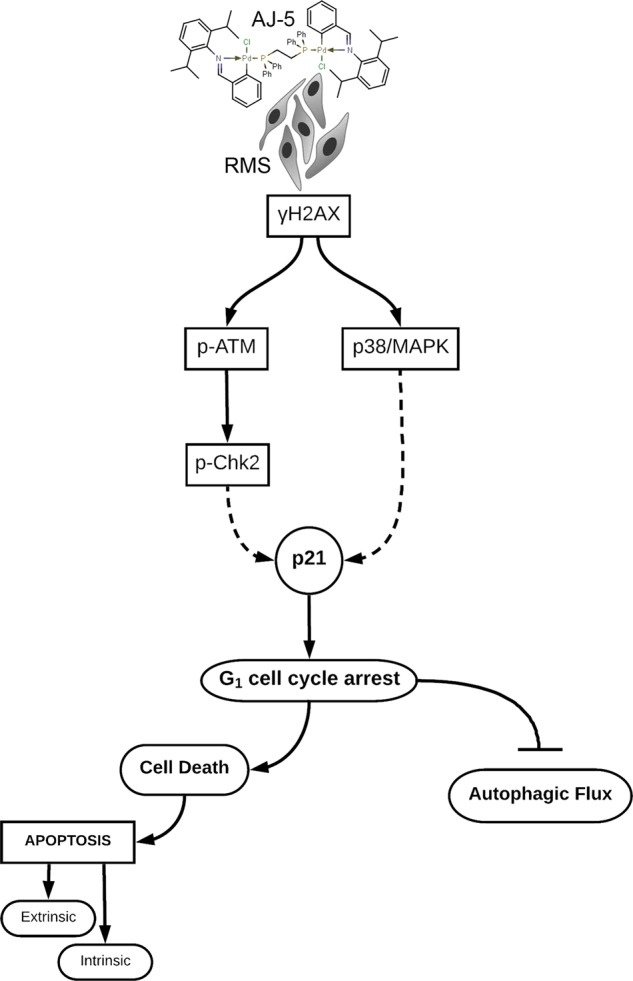
At low concentrations (≤0.2 µM) in aRMS and eRMS cells AJ-5 induces DSBs leading to increased levels of γH2AX and activation of both the canonical DSB pathway (ATM/Chk2) and the p38/MAPK stress signalling pathway. This results in a p21-induced G1 cell cycle arrest followed by the activation of the extrinsic and intrinsic apoptotic pathways and a reduction in autophagic flux
Platinum compounds such as cisplatin have been used successfully as anti-tumour agents but they have been associated with severe toxicity and tumour drug resistance42. Palladium compounds have been proposed as more effective alternatives because they appear to be more active at lower concentrations with less severe side effects43,44. Cisplatin is occasionally used to treat RMS6 and was reported to have an IC50 of >0.5 µM after 4–5 days of treatment in a range of RMS cell lines including RD and RH30 cells45,46. Our study shows that after only 2 days of AJ-5 treatment, IC50 values of ≤0.2 µM were obtained in the RD and RH30 cells. This suggests that AJ-5 displays more potent anti-cancer activity at much lower concentrations over a shorter period than cisplatin in these RMS cells. Furthermore, we show that AJ-5 displays a selectivity index of ≥2, which suggests that it has specificity for RMS cells47,48. Indeed, whereas AJ-5 treated RMS cells were unable to form any colonies, mouse myoblasts treated with 1.5–3 times higher concentrations formed colonies with areas comparable to that obtained for vehicletreated cells (Fig. 2a). Furthermore, mesenchymal stem cells from different sources are thought to represent the target cell of origin for a variety of human sarcomas and our cell viability assays (Fig. 1c) show that the A100021501 mesenchymal stem cells were the most sensitive non-malignant cell line to AJ-5 treatment20. This suggests that AJ-5 may have the ability to selectively target the sarcoma-initiating cell which could prove to be advantageous and more efficacious than current therapies. Taken together these findings support evidence that, compared to platinum compounds, palladium complexes display superior anti-cancer activity and that they may associate with fewer side effects.
While tumours initially respond to proapoptotic therapies, they frequently acquire the ability to bypass the apoptotic pathway and develop drug resistance leading to tumour recurrence49,50. It is thus predicted that drugs that can trigger alternative or multiple programmed cell death (PCD) pathways such as autophagy (PCD type II) and programmed necrosis (PCD type III) would be more efficacious51. The annexin V-FITC assays in Fig. 4c reveal that AJ-5 induces not only a significant apoptotic population but also a significant cell population which could represent necrosis, programmed necrosis (necroptosis) and/or secondary necrosis which occurs when there is insufficient clearance of apoptotic cells49,52. Interestingly, we have preliminary western blot data that show that AJ-5 treatment causes an increase in phosphorylated receptor-interacting serine/threonine protein kinase 3 (RIP3), a critical component of necroptosis, as well as increased levels of active (phosphorylated) mixed lineage kinase domain-like protein (MLKL), a key executioner of necroptosis (data not shown). The possibility that AJ-5 may induce necroptosis is exciting; however, to confirm this annexin V-FITC assays of AJ-5-treated cells in the presence of necrostatin-1 and the pan-caspase inhibitor Z-VAD-fmk will need to be performed.
Here we show for the first time that AJ-5 reduces autophagic flux in both aRMS and eRMS cells. This is an important finding because autophagy has been reported to promote cell survival and chemotherapeutic drug resistance in a number of sarcomas, including RMS, and thus autophagy has been identified as a drug target to treat sarcomas50,51,53. For example, the autophagy inhibitor, chloroquine, enhanced RMS cell death induced by ciclopirox olamine, bortezomib and 17-DMAG and stimulation of autophagy by rapamycin prevented this54,55. Consistent with our findings, palladium nanoparticles have recently been shown to block autophagic flux in Hela cells as shown by increased LC3II/I ratios, reduced degradation of p62 and autophagosome accumulation56. It is important to note that AJ-5 induces autophagy PCD in melanoma and breast cancer cells, which suggests that the effect of palladium complexes on autophagy may be cancer type specific.
In conclusion, we show that AJ-5 may be a promising chemotherapeutic to treat aRMS and eRMS because it displays potent and selective cytotoxicity at sub-micromolar concentrations and inhibits the ability of RMS cells to survive, proliferate and migrate. Importantly, AJ-5 induces apoptosis and reduces autophagic flux and it displays a favourable pharmacokinetic and safety profile.
Materials and methods
Cell culture
RH30 human aRMS cells (kindly provided by Associate Professor Judith Davie, Southern Illinois University), AX-OH-1 human aRMS and FL-OH-1 human eRMS cells (kindly provided by Professor Stefan Bath, University of Cape Town) were cultured in Roswell Park Memorial Institute Medium (RPMI)-1640 (Sigma Aldrich, Missouri, USA). RD human eRMS cells (ATCC® CCL-136™), FG0 and DMB human skin fibroblasts (kindly provided by Associate Professor Denver Hendricks, University of Cape Town), mouse myoblast cells (ATCC® CRL-1772™), human mesenchymal stem cells A10021501 (kindly provided by Professor Michael Pepper, University of Pretoria and confirmed to meet the criteria to be defined as mesenchymal stem cells as set out by the Mesenchymal and Tissue Stem Cell Committee of the International Society for Cellular Therapy57), HT1080 human fibrosarcoma cells (ATCC® CCL-120™), SW1353 human chondrosarcoma cells (ATCC® HTB-94™), SW982 human synovial sarcoma cells (ATCC® HTB-93™), SW872 human liposarcoma (ATCC® HTB-92™) and MG-63 human osteosarcoma cells (kindly provided by Associate Professor Philippa Hulley, University of Oxford) were cultured in Dulbecco’s modified Eagle’s medium (Sigma Aldrich). All culture medium was supplemented with 10% heat-inactivated foetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were maintained at 37 °C in a 95% air and 5% CO2 humidified incubator. Medium was replaced every 2 to 3 days and cells were routinely subjected to mycoplasma tests. Only mycoplasma free cells were used in experiments. Cell morphology was monitored using an Olympus CKX41 inverted microscope (MSAC Ltd, UK) and imaged with an EVOS™ XL AMEX1000 Core Imaging System (Thermo Fisher Scientific, Massachusetts, USA).
Cell treatments
AJ-5, synthesized as previously described18, was dissolved in dimethyl sulfoxide (DMSO) (D8418; Sigma Aldrich) to give a 5 mM AJ-5 stock which was stored at room temperature (RT) and used within 5 days. AJ-5 was diluted in cell culture medium to achieve the desired final concentration and a vehicle control (DMSO) of the same concentration was prepared simultaneously. Cells were treated at a confluency of 60%. For western blot experiments to determine autophagy flux, cells were treated with 200 nM bafilomycin A1 (B1793; Sigma Aldrich) for 2 h post AJ-5 treatment. Doxorubicin (z/26/0167; Teva Pharmaceutical Industries Ltd, Israel) was used as a positive control in Caspase-Glo experiments at a concentration of 0.5 µM.
Cell viability assays
Cells were seeded in 96-well plates and treated the next day with a range of AJ-5 concentrations (0.1–1.0 μM) or vehicle (1.0 μM DMSO) for 48 h. Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-trazolium bromide (MTT) assay (11465007001; Sigma Aldrich) according to the manufacturer’s instructions. Mean cell viability was calculated as a percentage of the mean vehicle control. At least three independent experiments in quadruplicate were performed from which the half maximal inhibitory concentration (IC50) was determined using GraphPad Prism version 6.0 (GraphPad Software, California, USA). The selectivity index (SI) was determined by dividing the IC50 of a normal cell line by the IC50 of a cancer cell line.
Clonogenic assays
Cells were seeded in 6-well plates and treated the following day with IC50, ½ IC50, ¼ IC50 AJ-5 and vehicle. After 24 h, 800–4000 cells were seeded in 35 mm dishes in drug-free medium. Formation of colonies were monitored and after 7–21 days cells were fixed with 3:1 methanol:acetic acid and stained with 0.5% crystal violet (Sigma Aldrich) in 100% methanol. Colonies were imaged and percentage colony area was determined using ImageJ v1.50i58 and the plugin ColonyArea59. Colony area was determined for each drug concentration and expressed as a percentage of vehicletreated control.
Scratch motility assay
Cells were seeded in 6-well plates and treated with IC50 AJ-5 and vehicle for 24 h. Cells were then collected, counted and replated to achieve 100% confluency in a 24-well plate. The following day a sterile 2 µL pipette tip was used to make a vertical scratch in the cell monolayer of each well and the cells treated with 10 µM mitomycin C (M4287; Sigma Aldrich) to inhibit proliferation. Cells were imaged at 0, 3, 6, 9, 12 and 24 h post wound formation and ImageJ v1.50i58 was used to calculate the area of the scratch. The total areas migrated was determined by subtracting the area for a specific time point from the area measured at 0h.
Western blot analyses
Cells were harvested after exposure to IC50 AJ-5, 0.1 µM AJ-5 or vehicle (DMSO) for 24 and 48 h. Cells were lysed at 4 °C in whole-cell lysis buffer (0.125 M Tris-HCl pH 6.8, 4% SDS, 0.2% glycerol, 0.1% β-mercaptoethanol and a pinch of bromophenol blue) and boiled for 10 min. Proteins were resolved by SDS-PAGE (6–15% gels) and transferred to Hybond ECL membranes (Amersham, UK). The membranes were incubated with primary antibodies: rabbit polyclonal antibodies to phospho-histone H2A.X (Ser139) (#2577), phospho-chk2 (Thr68) (#221), phospho-p38 MAPK (Thr180/Tyr182) (#9211), cleaved caspase-3 (Asp175) (#9661), PARP (#9542), caspase-9 (#9502), LC3B (#2775), p38 MAPK (#9212), rabbit monoclonal antibody to cleaved caspase-7 (Asp198) (D6H1) (#8438), mouse monoclonal antibodies to phospho-ATM (Ser1981) (D6H9) (#5883), Cyclin B1 (V152) (#4135), Caspase-8 (1C12) (#9746), SQSTM1/p62 (D5L7G) (#88588) from Cell Signaling Technology (Massachusetts, USA); mouse monoclonal antibody to p53 (DO-1) (sc-126), rabbit polyclonal antibodies to p21 (C-19) (sc-397), cyclin A (H-432) (sc-751), cyclin B1 (H-433)(sc-752), PARP-1 (H-250) (sc-7150) from Santa Cruz Biotechnology (Texas, USA); rabbit polyclonal antibody to p38 MAP kinase (M0800) from Sigma Aldrich. After primary antibody incubation, membranes were incubated with goat anti-rabbit or goat anti-mouse HRP-conjugated secondary antibodies (Bio-Rad Laboratories, California, USA). Antibody reactive proteins were visualized by enhanced chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate Kit (Thermo Fisher Scientific) or WesternBright ECL HRP Substrate Kit (Advansta, California, USA). Densitometry readings were obtained using ImageJ v1.50i58 and protein expression levels were represented as a ratio of protein of interest/p38 loading control normalized to the vehicletreated control sample where appropriate. All blots are representative of at least two independent repeats.
Immunofluorescence
Cells were plated on glass coverslips and treated with IC50 AJ-5 or vehicle. Cells were fixed with ice-cold 100% methanol at −20 °C for 5 min followed by blocking and permeabilization with 0.2% Triton-X-100/5% bovine serum albumin in phosphate-buffered saline (PBS) for 30 min at RT. Slides were incubated with antibodies against phospho-histone H2A.X (#2577) (1:500) or LC3B (#2775) (1:200) (Cell Signaling Technology) in blocking buffer overnight at 4 °C. After PBS washes coverslips were incubated with donkey anti-rabbit or donkey anti-mouse Cy3-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories Inc., Pennsylvania, USA) diluted 1:1000 for 1.5 h in blocking buffer at RT. A secondary-antibody-only control was always included in these experiments. Cells were then counterstained with Hoechst (33342; Invitrogen, California, USA) for 10 min at RT in the dark, washed with PBS and then coverslips were mounted onto glass slides using Mowiol mounting medium. Cells were imaged with an LSM 510 confocal microscope (Carl Zeiss, Germany) using a Plan-Apochromat ×63/1.40 oil DIC objective. Multiple z layers were acquired with 1 µM step width. Images were processed using ZEN 2012 imaging software (Carl Zeiss) and maximum intensity projections were generated. For quantification, mean fluorescence was measured from at least 20 fields of view per treatment condition and pooled from three independent repeats.
Cell cycle analysis
Cells were seeded and treated the following day with IC50 AJ-5 and vehicle for 24 and 48 h, trypsinized, washed with PBS, counted and permeabilized in 70% EtOH at −20 °C overnight. Cells were pelleted and treated with 50 μg/mL RNase (Fermentas, Massachusetts, USA) in PBS at 37 °C for 15 min and then stained with propidium iodide (PI). A minimum of 50 ,000 cells/sample were subjected to analysis using a Becton Dickinson FACSCalibur flow cytometer (Becton Dickinson, New Jersey, USA) with a 488 nm coherent laser. The data were acquired using CellQuest Pro version 5.2.1. software (Becton Dickinson) and the analyses were done using ModFit version 2.0. software (Verity Software House Inc, Maine, USA).
Annexin V-FITC assays
Cells were seeded, trypsinized, washed and counted as described for cell cycle analysis. The FITC Annexin V/Dead Cell Apoptosis kit (V13242; Thermo Fisher Scientific) was used according to the manufacturer's instructions. A negative control of unstained cells and positive controls of cells treated with 3% formaldehyde for 30 min on ice and stained with either PI or Annexin V were used to gate the flow cytometer for viable, early apoptotic, late apoptotic, and necrotic cell populations. Samples were subjected to FACS analyses as described for cell cycle analysis. Data were obtained and analysed using CellQuest Pro version 5.2.1. software.
Caspase activity assays
Cells were seeded and treated for 24 h with IC50 AJ-5, vehicle or 0.5 µM (IC50) doxorubicin which was included and as a positive control. Cells were trypsinized, washed and counted and caspase-8, -9, and -3/7 activity was measured using the appropriate Caspase-Glo® assay kits (Promega, Wisconsin, USA) according to the manufacturer’s instructions. Luminescence was measured 45 min after incubation with Caspase Glo® reagent using a Luminoscan Ascent luminometer (Thermo LabSystems Inc., Massachusetts, USA). Luminescence signal for treated samples were normalized to vehicle-treated samples to express a fold change increase in caspase activity.
Supravital staining with acridine orange
Cells were seeded on glass coverslips and treated for 24 h with IC50 AJ-5 or vehicle and then stained with 2 µg/mL acridine orange (Michrome No. 714; Edward Gurr Ltd, UK), which was added directly to the medium, for 15 min at RT in the dark. Coverslips were mounted on glass slides and visualized and imaged as described for immunofluorescence.
Single-cell autophagosome flux analyses
The autophagy steady-state variables were measured in RD cells treated with IC50 AJ-5 for 48 h. The autophagosome flux was calculated from the rate of autophagosome accumulation as previously described60. Briefly, this was determined after the complete inhibition of fusion between autophagosomes and lysosomes using 400 nM bafilomycin A1 (B0025; LKT Laboratories Inc., Minnesota, USA).
Cells were transfected with a GFP-LC3 construct using Lipofectamine 3000 (L3000001; Thermo Fisher Scientific) according to the manufacturer’s instructions and maintained for 2 days in order to achieve high levels of GFP-LC3 expression. Thereafter, cells were harvested by trypsinization and seeded into an eight-chamber coverslip-based dish (155411 Nunc™ Lab-Tek™; Thermo Fisher Scientific) to achieve 60% confluency after which they were treated with IC50 AJ-5 for 48 h.
Autophagosome, autolysosome and lysosome pool size was assessed by supplementing the culture media with 25 nM LysoTracker Red (L7528; Thermo Fisher Scientific) 2 h prior to image acquisition. An LSM 780 confocal with ELYRA S.1 Superresolution platform (Carl Zeiss) was used for image acquisition. The eight-chamber dish was placed in a heated chamber maintained at 5% CO2 that encased the microscope objective. An LCI Plan-Apochromat ×63/1.4 oil DIC M27 objective was used and samples were excited with an Argon multiline laser 25 mW at 488 and 514 nm with appropriate beam splitters and GaAsP detector 32+2 PMT. Multiple z layers were acquired with 1 µM step width. Laser power and electron gain were set to achieve an optimal signal to noise ratio without causing photo-toxicity. Images were processed using ZEN 2011 imaging software (Carl Zeiss) and the maximum intensity projections were exported to Fiji software61 for further analysis60.
Pharmacokinetics studies of AJ-5 in healthy mice
All studies and procedures were conducted with prior approval of the Animal Ethics Committee of the University of Cape Town (Protocol 012/049) in accordance with the South African National Standard (SANS 10386:008) for the Care and Use of Animals for Scientific Purposes62, and guidelines from the Department of Health63.
Pharmacokinetic parameters of AJ-5 were determined after IV, IP and PO dosing to MF1 nude mice randomly divided into groups. AJ-5 was prepared in the respective formulations immediately prior to dosing. For the IV group, the compound was made up in a mixture of 10% DMSO and 90% of an IV mixture, which consisted of 60% propylene glycol, 10% ethanol and 30% polyethylene glycol. For the PO group, AJ-5 was dissolved in DMSO and a hydroxypropylmethylcellulose solution (0.5% in water) containing 0.2% Tween 80 in a ratio of 1:9. For the IP group, AJ-5 was dissolved in 10% DMSO and 90% phosphate buffer solution. Mice were permitted access to food and water ad libitum. AJ-5 was administered (100 μL) by oral gavage at a dose of 20 mg/kg (n = 3), intravenously into the penile dorsal vein at a dose of 2 mg/kg (n = 2) and intraperitoneally at a dose of 2 mg/kg (n = 3).
Blood samples (20 μL per sample) were collected by tail tip bleeding in lithium heparin tubes at predetermined time points to evaluate the kinetic profile over 24 h. Samples were stored at −80 °C until analysis. Blood samples together with calibration standards and quality controls were quantified using inductively coupled plasma mass spectrometry. All results reported are for palladium (Pd). Pharmacokinetic parameters of the AJ-5 complex were determined using non-compartmental analysis in PK Solutions 2.0 (Summit Research Systems, Montrose CO, USA).
Statistical analyses
All data were obtained from at least three independent experiments (unless otherwise stated) with error bars representing standard error of the mean (SEM). Data were analysed using GraphPad Prism version 6.0 (GraphPad Software) and a parametric unpaired t-test was performed. Significance was accepted at *p < 0.05,**p < 0.01 and ***p < 0.001.
Supplementary information
Pharmacokinetic parameters of AJ-5 obtained from whole blood of healthy MF1 mice
AJ-5 induces several markers of autophagy in RMS cells
AJ-5 is cytotoxic in a range of sarcoma subtypes
Whole blood concentration of AJ-5 over 24h
Acknowledgements
This work was supported by grants from the SA Medical Research Council, the National Research Foundation (NRF), Cancer Association of South Africa (CANSA) and the University of Cape Town. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. We would like to thank Associate Professor Judith Davie, Professor Stefan Bath, Associate Professor Denver Hendricks, Professor Michael Pepper and Associate Professor Philippa Hulley who kindly provided us with cell lines used in this study.
Conflict of intererst
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41420-019-0139-9) contains supplementary material, which is available to authorized users.
References
- 1.Stout AP. Rhabdomyosarcoma of the skeletal muscles. Ann. Surg. 1946;123:447–472. doi: 10.1097/00000658-194603000-00011. [DOI] [PubMed] [Google Scholar]
- 2.Dagher R, Helman L. Rhabdomyosarcoma: an overview. Oncologist. 1999;4:34–44. [PubMed] [Google Scholar]
- 3.Perez EA, et al. Rhabdomyosarcoma in children: a SEER population based study. J. Surg. Res. 2011;170:e243–e251. doi: 10.1016/j.jss.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Wachtel M, et al. Subtype and prognostic classification of rhabdomyosarcoma by immunohistochemistry. J. Clin. Oncol. 2006;24:816–822. doi: 10.1200/JCO.2005.03.4934. [DOI] [PubMed] [Google Scholar]
- 5.Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J. Clin. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- 6.Egas-Bejar D, Huh WW. Rhabdomyosarcoma in adolescent and young adult patients: current perspectives. Adolesc. Health Med. Ther. 2014;5:115–125. doi: 10.2147/AHMT.S44582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hosoi H. Current status of treatment for pediatric rhabdomyosarcoma in the USA and Japan. Pediatr. Int. 2016;58:81–87. doi: 10.1111/ped.12867. [DOI] [PubMed] [Google Scholar]
- 8.Pappo AS, Shapiro DN, Crist WM, Maurer HM. Biology and therapy of pediatric rhabdomyosarcoma. J. Clin. Oncol. 1995;13:2123–2139. doi: 10.1200/JCO.1995.13.8.2123. [DOI] [PubMed] [Google Scholar]
- 9.Neidle, S. Cancer Drug Design and Discovery (Academic Press, San Diego, 2013).
- 10.Makin G. Principles of chemotherapy. Paediatr. Child Health (Oxf.) 2014;24:161–165. doi: 10.1016/j.paed.2013.09.002. [DOI] [Google Scholar]
- 11.Silverman, R. B. & Holladay, M. W. The Organic Chemistry of Drug Design and Drug Action (Academic Press, San Diego, 2014).
- 12.Smith, H. J. & Williams, H. Smith and Williams’ Introduction to the Principles of Drug Design and Action 4th edn (CRC Press Taylor & Francis Group, Boca Raton, 2005).
- 13.Ndagi U, Mhlongo N, Soliman ME. Metal complexes in cancer therapy—an update from drug design perspective. Drug Des. Dev. Ther. 2017;11:599–616. doi: 10.2147/DDDT.S119488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ulukaya E, et al. Anti-cancer activity of a novel palladium(II) complex on human breast cancer cells in vitro and in vivo. Eur. J. Med. Chem. 2011;46:4957–4963. doi: 10.1016/j.ejmech.2011.07.055. [DOI] [PubMed] [Google Scholar]
- 15.Kruszewski M, et al. Differential toxic effect of cis-platinum(II) and palladium(II) chlorides complexed with methyl 3,4-diamine-2,3,4,6-tetradeoxy-alpha-L-lyxo-hexopyranoside in mouse lymphoma cell lines differing in DSB and NER repair ability. Teratog. Carcinog. Mutagen. 2003;1(Suppl):1–11. doi: 10.1002/tcm.10046. [DOI] [PubMed] [Google Scholar]
- 16.Akdi K, et al. Study of the biological effects and DNA damage exerted by a new dipalladium-Hmtpo complex on human cancer cells. J. Inorg. Biochem. 2002;90:51–60. doi: 10.1016/S0162-0134(02)00370-7. [DOI] [PubMed] [Google Scholar]
- 17.Carreira M, Calvo-Sanjuán R, Sanaú M, Marzo I, Contel M. Organometallic palladium complexes with a water-soluble iminophosphorane ligand as potential anticancer agents. Organometallics. 2012;31:5772–5781. doi: 10.1021/om3006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aliwaini S, Swarts AJ, Blanckenberg A, Mapolie S, Prince S. A novel binuclear palladacycle complex inhibits melanoma growth in vitro and in vivo through apoptosis and autophagy. Biochem. Pharmacol. 2013;86:1650–1663. doi: 10.1016/j.bcp.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 19.Aliwaini S, et al. The palladacycle, AJ-5, exhibits anti-tumour and anti-cancer stem cell activity in breast cancer cells. Cancer Lett. 2015;357:206–218. doi: 10.1016/j.canlet.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez R, Rubio R, Menendez P. Modeling sarcomagenesis using multipotent mesenchymal stem cells. Cell Res. 2012;22:62–77. doi: 10.1038/cr.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hettmer S, Wagers AJ. Muscling in: uncovering the origins of rhabdomyosarcoma. Nat. Med. 2010;16:171–173. doi: 10.1038/nm0210-171. [DOI] [PubMed] [Google Scholar]
- 22.Galluzzi L, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valdiglesias V, Giunta S, Fenech M, Neri M, Bonassi S. γH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat. Res. 2013;753:24–40. doi: 10.1016/j.mrrev.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Sionov RV, Haupt Y. The cellular response to p53: the decision between life and death. Oncogene. 1999;18:6145–6157. doi: 10.1038/sj.onc.1203130. [DOI] [PubMed] [Google Scholar]
- 25.Felix CA, et al. Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res. 1992;52:2243–2247. [PubMed] [Google Scholar]
- 26.Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst.). 2016;42:63–71. doi: 10.1016/j.dnarep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321–328. doi: 10.1023/A:1016119328968. [DOI] [PubMed] [Google Scholar]
- 28.Oliver FJ. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J. Biol. Chem. 1998;273:33533–33539. doi: 10.1074/jbc.273.50.33533. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, Na B, Ou JJ, Pulliam L, Yen TSB. Hepatitis B virus alters the antioxidant system in transgenic mice and sensitizes hepatocytes to Fas signaling. PLoS ONE. 2012;7:e36818. doi: 10.1371/journal.pone.0036818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoffmann JC, Pappa A, Krammer PH, Lavrik IN. A new C-terminal cleavage product of procaspase-8, p30, defines an alternative pathway of procaspase-8 activation. Mol. Cell. Biol. 2009;29:4431–4440. doi: 10.1128/MCB.02261-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 32.Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438. doi: 10.1080/15548627.2015.1066957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C. Childhood rhabdomyosarcoma: recent advances and prospective views. J. Dent. Res. 2012;91:341–350. doi: 10.1177/0022034511421490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breneman JC, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma—a report from the Intergroup Rhabdomyosarcoma Study IV. J. Clin. Oncol. 2003;21:78–84. doi: 10.1200/JCO.2003.06.129. [DOI] [PubMed] [Google Scholar]
- 35.Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer. 2012;59:5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stevens MCG. Treatment for childhood rhabdomyosarcoma: the cost of cure. Lancet Oncol. 2005;6:77–84. doi: 10.1016/S1470-2045(05)01733-X. [DOI] [PubMed] [Google Scholar]
- 37.Oberlin O, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J. Clin. Oncol. 2008;26:2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dasgupta R, Rodeberg DA. Update on rhabdomyosarcoma. Semin. Pediatr. Surg. 2012;21:68–78. doi: 10.1053/j.sempedsurg.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 39.McDowell HP, et al. Outcomes in paediatric metastatic rhabdomyosarcoma: results of The International Society of Paediatric Oncology (SIOP) study MMT-98. Eur. J. Cancer. 2010;46:1588–1595. doi: 10.1016/j.ejca.2010.02.051. [DOI] [PubMed] [Google Scholar]
- 40.Perkins SM, Shinohara ET, DeWees T, Frangoul H. Outcome for children with metastatic solid tumors over the last four decades. PLoS ONE. 2014;9:e100396. doi: 10.1371/journal.pone.0100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shern JF, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 43.Jahromi EZ, et al. Palladium complexes: new candidates for anti-cancer drugs. J. Iran. Chem. Soc. 2016;13:967–989. doi: 10.1007/s13738-015-0804-8. [DOI] [Google Scholar]
- 44.Johnstone TC, Suntharalingam K, Lippard SJ. The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016;116:3436–3486. doi: 10.1021/acs.chemrev.5b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cocker HA, Pinkerton CR, Kelland LR. Characterization and modulation of drug resistance of human paediatric rhabdomyosarcoma cell lines. Br. J. Cancer. 2000;83:338–345. doi: 10.1054/bjoc.2000.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cocker HA, et al. High levels of the MDM2 oncogene in paediatric rhabdomyosarcoma cell lines may confer multidrug resistance. Br. J. Cancer. 2001;85:1746–1752. doi: 10.1054/bjoc.2001.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Badisa RB, et al. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009;29:2993–2996. [PMC free article] [PubMed] [Google Scholar]
- 48.Koch A, Tamez P, Pezzuto J, Soejarto D. Evaluation of plants used for antimalarial treatment by the Maasai of Kenya. J. Ethnopharmacol. 2005;101:95–99. doi: 10.1016/j.jep.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 49.Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis. 2017;22:1189–1204. doi: 10.1007/s10495-017-1413-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11:342–357. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Viola G, et al. MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochem. Pharmacol. 2012;83:16–26. doi: 10.1016/j.bcp.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–4499. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 53.Min L, et al. Autophagy as a potential target for sarcoma treatment. Biochim. Biophys. Acta Rev. Cancer. 2017;1868:40–50. doi: 10.1016/j.bbcan.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 54.Zhou H, et al. Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway. Oncotarget. 2014;5:10140–10150. doi: 10.18632/oncotarget.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peron M, Bonvini P, Rosolen A. Effect of inhibition of the ubiquitin-proteasome system and Hsp90 on growth and survival of rhabdomyosarcoma cells in vitro. BMC Cancer. 2012;12:233. doi: 10.1186/1471-2407-12-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang L, et al. Palladium nanoparticles induce autophagy and autophagic flux blockade in Hela cells. RSC Adv. 2018;8:4130–4141. doi: 10.1039/C7RA11400A. [DOI] [Google Scholar]
- 57.Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 58.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guzmán C, et al. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE. 2014;9:e92444. doi: 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.du Toit, A., Hofmeyr, J.-H. S., Gniadek, T. J. & Loos, B. Measuring autophagosome flux. Autophagy 1–12 (2018). 10.1080/15548627.2018.1469590 [DOI] [PMC free article] [PubMed]
- 61.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.SABS Standards Division. South African Bureau of Standards, South African National Standard: The Care and Use of Animals for Scientific Purposes (SANS 10386:2008) 1st edn (SABS Standards Division, Pretoria, 2015).
- 63.Department of Health. Ethics in Health Research: Principles, Processes and Structures 2nd edn (Department of Health, Pretoria, 2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pharmacokinetic parameters of AJ-5 obtained from whole blood of healthy MF1 mice
AJ-5 induces several markers of autophagy in RMS cells
AJ-5 is cytotoxic in a range of sarcoma subtypes
Whole blood concentration of AJ-5 over 24h