

Abstract
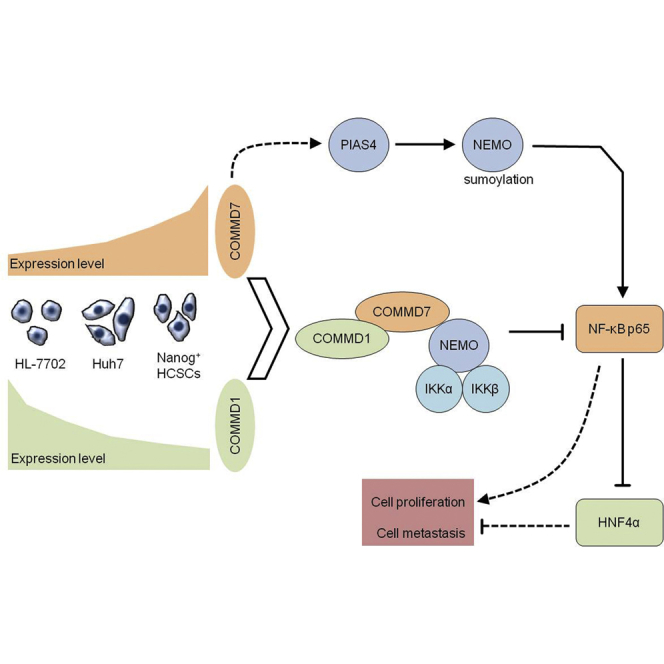
Previous studies showed that the COpper Metabolism gene MURR1 Domain (COMMD) family of proteins was abnormally expressed in hepatocellular carcinoma (HCC). This study aimed to explore the roles of COMMD1 and COMMD7 in regulating nuclear factor κB (NF-κB) signaling in HCC stem cells (HCSCs). In vivo, the expression of COMMD7 and COMMD1 was determined in 35 pairs of HCC cancer tissues and adjacent tissues, and the effect of COMMD7 silencing on xenograft tumor growth was evaluated. In vitro, the effects of COMMD7 silencing and COMMD1 overexpression on HCSC function were assessed. Results found that the expression levels of COMMD7 were higher, whereas COMMD1 levels were lower in HCC tissues and HCSCs. COMMD7 silencing or COMMD1 overexpression inhibited cell proliferation, migration, and invasion through suppression of NF-κB p65. Furthermore, COMMD7 positively regulated NF-κB by upregulating protein inhibitor for activated stat 4 (PIAS4). This study demonstrates that COMMD7 has a dual regulatory role in the NF-κB signaling pathway in Nanog+ HCSCs.
Keywords: hepatocellular carcinoma, cancer stem cells, tumorigenicity
Graphical Abstract
Introduction
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related death worldwide and has a strikingly high mortality rate in China.1, 2 Until now, surgical liver resection and liver transplantation were effective treatments for early-stage HCC but were unsuitable for the majority of patients at advanced stages of the disease.3, 4 For patients at the most advanced stages of HCC, the clinical outcome and prognosis following conventional chemotherapy or radiotherapy remain poor because of development of resistance.5, 6 The presence of HCC stem cells (HCSCs) is the primary cause of failure of traditional treatments for HCC.7
Carcinoma stem cells (CSCs) are a minority cell population within tumors and have been proven to possess the abilities of self-renewal and differentiation into heterogeneous lineages of cancer cells; these characteristics confer rapid tumor growth and enhanced resistance to chemotherapy or radiotherapy.8, 9 Increasing evidence has suggested that HCSCs exhibit higher expression levels of oncogenes and lower expression levels of tumor suppressors involved in the initiation, recurrence, and drug resistance of HCC.10, 11, 12, 13 Therefore, understanding the molecular mechanism of HCSCs in promoting metastasis and relapse of HCC might facilitate the development of novel therapies to improve the clinical treatment and survival rate of HCC.
Nuclear factor κB (NF-κB) belongs to a family of ubiquitous transcription factors and is widely considered a key regulator in physiological cellular functions, including cell survival, differentiation, and inflammation.14 Aberrant activation of NF-κB has been observed in various types of human cancer, such as colon cancer15 and pancreatic cancer.16 Thus, inhibition of NF-κB has been considered a promising strategy to improve anti-cancer therapies by negatively affecting the gene expression profile of the NF-κB signaling pathway.17 The COpper Metabolism MURR1 Domain (COMMD) protein family, which is composed of 10 ubiquitously expressed proteins all sharing a structurally conserved C-terminal motif, was recently identified as a new class of proteins that facilitate the assembly of crucial molecular components involved in regulating NF-κB activity.18, 19 Among the members of the COMMD family, COMMD1 has been the most extensively characterized and operates as a terminator of NF-κB signaling;20 COMMD1 reduction is associated with cancer progression caused by constitutive NF-κB activation.21, 22 Another member of the COMMD family, COMMD7 has been identified as a novel NF-κB essential modulator (NEMO)-interacting protein and was reported to function as molecular target in several types of cancers including pancreatic ductal adenocarcinoma23 and HCC24, 25 in our previous studies. Moreover, we previously found that COMMD7 positively correlated with the expression of NF-κB, and that the NF-κB signaling pathway could mediate the proliferative and anti-apoptotic effects of COMMD7 in HCC cells.
Thus, the aims of this study were to further investigate the function of COMMD7 and the underlying mechanisms mediating NF-κB activity in HCSCs. For this purpose, the expression of COMMD7, COMMD1, and NF-κB was determined in HCC surgical tissues and HCSCs. Loss-of-function and gain-of-function assay were performed to investigate whether COMMD7 alone or together with COMMD1 is involved in the proliferation and invasion of HCSCs in vitro. Moreover, the in vivo HCSC models were established to further study the potential use of targeting COMMD7 against HCC progression. Furthermore, bioinformatics analysis of gene expression patterns was performed to explore the downstream molecular mechanisms of COMMD7 involved in regulating the NF-κB signaling pathway of HCSCs.
Results
COMMD7 Was Negatively Correlated with COMMD1 and HNF4α in HCCs
Using real-time qPCR analysis, we found that both Nanog and COMMD7 were significantly elevated in 35 pairs of HCC tissues compared with adjacent para-carcinoma tissues (Figure 1A). On the contrary, low expressions of COMMD1 and HNF4α were observed. Subsequently, we further illustrated the correlation between COMMD7 and COMMD1 or HNF4α. As shown in Figure 1B, COMMD7 was negatively correlated with COMMD1 (r = −0.6247, p < 0.001) and HNF4α (r = −0.4642, p = 0.006). Western blot and immunohistochemistry (IHC) were subsequently performed to further validate these findings. The protein expressions of Nanog, COMMD7, and the p65 subunit of NF-κB were all elevated, but COMMD1 and HNF4α were both reduced in representative four fresh HCC tumors compared with their paired adjacent normal tissues (Figure 1C). Furthermore, IHC revealed that the staining of COMMD7 and COMMD1 were differentially distributed between HCC and para-carcinoma tissues (Figure 1D).
Figure 1.

Comparison of Nanog, COMMD7, COMMD1, NF-κB, and HNF4α Expression between HCC and Adjacent Tissues
(A) Expression of Nanog, COMMD7, COMMD1, and HNF4α in hepatocellular carcinoma (HCC) (cancer tissues [CTs]), compared with matched adjacent tissues (ATs). (B) Correlation analysis revealed a significant negative correlation between COMMD7 and COMMD1, and between COMM7 and HNF4α. (C) Western blot assays showed upregulation of Nanog, COMMD7, and NF-κB, and downregulation of COMMD1 and HNF4α in CT, compared with AT samples. (D) COMMD7 and COMMD1 expression in CT and AT samples using immunohistochemistry. Representative images from independent experiments are shown (×200 original magnification; scale bars, 500 μm.). ***p < 0.001 versus AT.
High Expressed COMMD7 and Low Expressed COMMD1 in Nanog+ HCSCs
Next, the expression levels of Nanog, COMMD7, COMMD1, NF-κB, and HNF4α were further detected among Huh7, HL-7702, and Nanog+ HCSCs. As shown in Figure 2A, the expression of COMMD7 in Nanog+ HCSCs was significantly higher than that in both Huh7 and HL-7702, whereas COMMD1 and HNF4α were low expressed. The corresponding protein levels of Nanog, COMMD7, COMMD1, and HNF4α presented similar trends using western blot analysis (Figure 2B). In addition, the expression of the p65 subunit of NF-κB protein was elevated in Nanog+ HCSCs compared with Huh7 and HL-7702 cells (Figure 2B). Furthermore, immunofluorescence (IFC) further confirmed these results (Figures 2C and 2D) and supported a positive correlation between COMMD7 and NF-κB p65, but a negative correlation between COMMD7 and COMMD1 or HNF4α.
Figure 2.

Expression of COMMD7, COMMD1, NF-κB, and HNF4α in Huh7, HL-7702, and Nanog+ HCSCs
(A) Expression of Nanog, COMMD7, COMMD1, and HNF4α in Huh7, HL-7702, and Nanog+ HCSCs. (B) Protein level of Nanog, COMMD7, COMMD1, NF-κB p65, and HNF4α. (C) Co-localization of COMMD7 and COMMD1. (D) Co-localization of COMMD7 and NF-κB p65. Scale bars, 50 μm. **p < 0.01; ***p < 0.001.
Silencing of COMMD7 Inhibited Nanog+ HCSC Proliferation and Metastasis
Considering COMMD7 was significantly elevated in HCC tissues and Nanog+ HCSCs, we then performed loss-of-function assays to investigate the function role of COMMD7 in Nanog+ HCSCs. As shown in Figure 3A, short hairpin RNA (shRNA)-targeted COMMD7 was stably transfected in Nanog+ HCSCs, which downregulated the expression of NF-κB p65 and upregulated the expression of COMMD1 and HNF4α (Figures 3B–3D). Besides, knockdown of COMMD7 significantly reduced the proliferation of Nanog+ HCSCs (p < 0.05; Figure 3E). Hoechst 33258 staining and flow cytometry showed that sh-COMMD7 transfection effectively increased cell apoptotic rate (Figures 3F and 3G). In addition, the effect of COMMD7 on Nanog+ HCSCs mobility was determined. As shown in Figure 3H, representative images of the wound healing assay and transwell Matrigel invasion assay show that the migration and invasion of the sh-COMMD7 transfection group was significantly inhibited.
Figure 3.

The Effect of Stable Transfection of COMMD7 shRNA on Cell Proliferation, Apoptosis, Migration, and Invasion in Nanog+ HCSCs
(A and B) The expression of COMMD7 (A) and COMMD1 (B) mRNA was measured in Nanog+ HCSCs after COMMD7 knockdown using real-time qPCR analysis. (C) Western blot assays analysis of Nanog, COMMD1, NF-κB, and HNF4α. (D) Immunofluorescence images of COMMD7 and COMMD1. (E) CCK-8 assay was performed to evaluate proliferative ability. (F and G) Hoechst 33258 staining (F) and flow cytometry assay (G) were performed to quantify the apoptotic rate. (H) Cell migration and invasion ability determined by wound healing and transwell invasion assay. (I) The subcutaneous xenograft murine model and representative images of tumors formed in the mice implanted with sh-COMMD7 or blank lentivirus-transfected Nanog+ HCSCs. (J) Changes in the tumor volume every 2 days from day 10 until day 26 after the cells were implanted subcutaneously (n = 10). (K) Expression analysis of COMMD7, COMMD1, NF-κB, and HNF4α in tumor tissues collected from mice. Scale bars, 50 μm. *p < 0.05; ***p < 0.001 versus vector.
To confirm the above findings, stable COMMD7-silenced Nanog+ HCSCs were injected separately into nude mice. Results showed that the volume of tumors was significantly suppressed in mice inoculated with COMMD7-silenced Nanog+ HCSCs compared with control groups (Figures 3I and 3J). In addition, the expression levels of COMMD7 and p65 in tumors in the sh-COMMD7 group were lower, whereas the expression levels of COMMD1 and HNF4α were higher than those in the control group (Figure 3K), suggesting that the expression of COMMD7 is correlated with the tumor growth of Nanog+ HCSCs.
COMMD1 and COMMD7 Co-regulated Level of NF-κB p65
It has been demonstrated that COMMD1 and COMMD7 are involved in the termination of NF-κB signaling,26 but our previous study found that COMMD7 could promote HCC growth by activating NF-κB.24 To investigate their exact roles, Nanog+ HCSCs were transfected with pcDNA3.1-COMMD1 alone or together with pcDNA3.1-COMMD7 or sh-COMMD7, respectively. The expression of COMMD1 was significantly elevated after single pcDNA3.1-COMMD1 transfection, but reduced after co-transfection with COMMD1 and COMMD7, which were confirmed by real-time qPCR (Figure 4A) and western blotting analysis (Figure 4B). Additionally, we found the expression of HNF4α was induced by COMMD1 (Figure 4B). Furthermore, we detected the expression of p65 in the nucleus and cytoplasm after COMMD1 overexpression or co-transfection with COMMD1 and COMMD7. As shown in Figure 4C, following COMMD1 increase, the entry of p65 to the nucleus was inhibited, and there was an accumulation of p65 in the cytoplasm following co-expression of COMMD1 and COMMD7 in Nanog+ HCSCs. Similar trends were also confirmed by IFC assays (Figure 4D), suggesting that the expression of p65 could be suppressed after co-expression of COMMD1 and COMMD7, but enhanced after single COMMD7 overexpression.
Figure 4.

Co-expression of COMMD1 and COMMD7 Suppressed the Expression of the NF-κB Signaling Pathway
Nanog+ HCSCs were transfected with empty vector, pcDNA3.1-COMMD1 alone, pcDNA3.1-COMMD1 + pcDNA3.1-COMMD7, or pcDNA3.1-COMMD1 + sh-COMMD7, respectively. (A) The expression of COMMD1 and COMMD7 was determined using real-time qPCR analysis. Western blotting analysis was performed to detect the expression of (B) COMMD7, COMMD1, and HNF4α, as well as (C) NF-κB p65 in the nucleus and cytoplasm, respectively. (D and E) Immunofluorescence images of HNF4α (D) and NF-κB (E). Scale bars, 50 μm. ***p < 0.001.
Of note, we found sole COMMD1 overexpression significantly suppressed cell proliferation, promoted cell apoptosis, and inhibited cell migration and invasion compared with empty vector group in Nanog+ HCSCs, whereas co-transfection with COMMD1 and sh-COMMD7 partially reversed the effects of sole COMMD1 overexpression on the above cell functions, as determined by CCK-8 assay (Figure 5A), flow cytometry (Figure 5B), wound healing assay, and transwell Matrigel invasion assay (Figure 5C), respectively. These results suggested that COMMD1 and COMMD7 co-regulate activation of NF-κB p65, and that COMMD1 overexpression inhibits proliferation and metastasis of Nanog+ HCSCs, which requires the involvement of COMMD7.
Figure 5.

Overexpression of COMMD1 Partially Reversed the Function of COMMD7 in Nanog+ HCSCs
Nanog+ HCSCs were transfected with empty vector, pcDNA3.1-COMMD1 alone, pcDNA3.1-COMMD1 + pcDNA3.1-COMMD7, or pcDNA3.1-COMMD1 + sh-COMMD7, respectively. (A) CCK-8 assay was performed to evaluate proliferative ability. (B) Flow cytometry assay was performed to quantify the apoptotic rate. (C) Wound healing and transwell invasion assay were used to determine cell migration and invasion ability. *p < 0.05; **p < 0.01; ***p < 0.001.
Transcriptional Expression Profile after COMMD7 Knockdown in Nanog+ HCSCs
Based on the above findings, we realized that COMMD7 may play a dual role in regulation of NF-κB signaling. To identify critical genes closely related to the NF-κB signaling pathway involved in Nanog+ HCSCs, we used cDNA microarray to analyze the gene expression profiling of the Nanog+ HCSCs with or without COMMD7 knockdown. Within the total set of 1,017 differentially expressed genes (DEGs) analyzed, 560 were downregulated and 457 were upregulated in Nanog+ HCSCs after COMMD7 knockdown with p value <0.05 and |log fold change (FC)| value >2.0 or <0.5 as cutoff criterion. In Figure 6A, a heatmap based on clustering analysis of upregulated or downregulated genes is shown. The top 10 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for target downregulated genes and upregulated genes are presented in Figures 6B and 6C, respectively. DEGs involved in the above pathways are summarized in Table S2. Notably, a total of seven DEGs were identified in the downregulated NF-κB signaling pathway, including chemokine C-X-C motif ligand 12 (CXCL12), CXCL2, intercellular adhesion molecule 1 (ICAM1), lymphocyte antigen 96 (LY96), nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha (NFKBIA), protein inhibitor for activated stat 4 (PIAS4), and tumor necrosis factor alpha-induced protein 3 (TNFAIP3), which may play important roles in COMMD7-mediated regulation of the NF-κB signaling pathway.
Figure 6.

Bioinformatics Analysis of Gene Expression Patterns Involved in NF-κB Signaling Pathway in Nanog+ HCSCs
(A) Heatmap generated by hierarchical clustering of differentially downregulated or upregulated expressed genes in Nanog+ HCSCs after COMMD7 knockdown. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of DEGs. (B) For upregulated DEGs, the top 10 enriched pathways are shown. (C) For downregulated DEGs, the top 10 enriched pathways are shown.
COMMD7 Regulation of the NF-κB Signaling Pathway via Elevated PIAS4 Expression
The identification of several downregulated genes involved in the NF-κB signaling pathway was validated by real-time qPCR and western blotting. The results indicated that the expression levels of PIAS4, CXCL12, and CXCL2 were all downregulated in Nanog+ HCSCs after COMMD7 knockdown (Figures 7A and 7B). As a previous study showed that PIAS4 could mediate NEMO sumoylation and NF-κB activation,27 we speculated that PIAS4 might be a key gene in the COMMD7-mediated regulation of the NF-κB signaling pathway. To validate this hypothesis, the expression of PIAS4 was overexpressed in Nanog+ HCSCs with COMMD7 knockdown and confirmed by real-time qPCR assay and western blotting (Figure 7C). PIAS4 overexpression significantly upregulated the expression of p65, but downregulated the expression of NEMO and HNF4α (Figure 7D).
Figure 7.

Overexpression of PIAS4 Influenced the Effect of COMMD7 Knockdown in Nanog+ HCSCs
(A and B) The expression of PIAS4 was determined using real-time qPCR (A) and western blotting (B) analysis of the protein level of CXCL12, CXCL2, and PIAS4 in Nanog+ HCSCs after COMMD7 knockdown. Then the COMMD7-silenced Nanog+ HCSCs were transfected with empty vector or pcDNA3.1-PIAS4. (C) The mRNA expression of PIAS4 after PIAS4 overexpression. (D) Western blotting analysis of the protein expression of NEMO, p65, HNF4α, and PIAS4. The proliferative ability, apoptotic rate, migration, and invasion were evaluated using (E) CCK-8 assay, (F) flow cytometry assay, and (G) wound healing and transwell invasion assay in Nanog+ HCSCs, respectively. *p < 0.05; ***p < 0.001 versus vector or sh-COMMD7.
Subsequently, the in vitro experiments showed that PIAS4 overexpression significantly reversed the suppressive effects of COMMD7 knockdown on cell proliferation, migration, and invasion in Nanog+ HCSCs, as detected by cell counting kit 8 (CCK-8) assay (Figure 7E), flow cytometry (FCM) assay with Annexin V-fluorescein-5-isothiocyanate (FITC) and PI staining (Figure 7F), wound healing assay, and transwell Matrigel invasion assay (Figure 7G), respectively. These findings further demonstrated that COMMD7 knockdown could suppress the activation of p65, possibly through downregulation of PIAS4-mediated NEMO sumoylation.
Discussion
Our previous research has shown that COMMD7 is correlated with a novel NF-κB-positive feedback loop in HCC cells. Moreover, NF-κB directly binds to the COMMD7 promoter and serves as an activator for COMMD7 transcription.25 Inconsistent with our previous findings, reports indicated that COMMD7 binds to the IκB kinase alpha (IKK) complex through NEMO and induces degradation of p65 leading to the termination of NF-κB signaling.19, 26 Based on these facts, we further investigated the function of COMMD7 and the underlying mechanisms mediating NF-κB activity in HCCs using Nanog+ HCSCs, a pivotal therapeutic target in the eradication of HCCs. Similarly, our data have shown that COMMD7 and p65 are overexpressed in HCCs with a negative correlation to COMMD1. COMMD7, p65, and Nanog were also aberrantly overexpressed in the human HCSC line, compared with the HCC cell line, Huh7, and normal liver cell line, HL-7702. These results strongly suggest that COMMD7, COMMD1, and NF-κB may play key roles in Nanog+ HCSCs biological function, participating in the development and progression of HCC.
Given the relatively higher expression of COMMD7 in Nanog+ HCSCs, we performed loss-of-function assays to investigate its function in HCSCs. The results showed that knockdown of COMMD7 significantly suppressed cell proliferation, migration, and invasion, but promoted cell apoptosis in HCSC models. Moreover, COMMD7 knockdown inhibited tumorigenesis in HCSC mouse models. Our results were consistent with the outcome of COMMD7 silencing in HCC cells through regulation of CXCL1028 and in pancreatic ductal adenocarcinoma cells, in which cell proliferation was suppressed through cell-cycle arrest and apoptosis.23 Further analysis showed that HCSCs and mouse models expressed elevated amounts of COMMD1 and HNF4α, but reduced expression of COMMD7 and p65 after COMMD7 knockdown. Nanog is a well-studied transcription factor that has been reported to be overexpressed in human tumor tissues29, 30 and is a biomarker of progenitor or stem cells in HCC.31, 32 HNF4α, a highly conserved member of the nuclear receptor superfamily of ligand-dependent transcription factors, is expressed at relatively low levels in cancers of different organs, including colon cancer33 and HCC.34 There is also growing evidence that HNF4α acts as a master regulator of liver morphogenesis and hepatocyte differentiation, and has anti-proliferation and tumor suppression functions in the liver.35, 36 In disagreement with a previous study, we have found that COMMD7 knockdown inhibits NF-κB activity.19 But the signaling pathway underlying COMMD7 regulating NF-κB activity in HCSCs still remains unclear.
Currently, a possible explanation has been put forward that COMMD7 works in concert with COMMD1 to inhibit NF-κB signaling.26 Considering the relatively lower expression of COMMD1, we thought it would be unlikely to detect the complex of COMMD1 and COMMD7 in Nanog+ HCSCs. To validate this hypothesis, COMMD1 was overexpressed in Nanog+ HCSCs. The in vitro experiments indicated that overexpression of COMMD1 alone could significantly suppress cell proliferation, promote cell apoptosis, as well as inhibit cell migration and invasion in Nanog+ HCSCs, whereas co-transfection of both COMMD1 and COMMD7 could partially reverse these effects of single COMMD1 overexpression. Surprisingly, the entry of p65 to the nucleus was gradually inhibited following an increase in COMMD7 expression, causing an accumulation of p65 in the cytoplasm during the co-expression of COMMD1 and COMMD7 in Nanog+ HCSCs. Based on these findings, we reasoned that overexpression of COMMD7 alone could enhance the accumulation of p65 in the cytoplasm, but co-overexpression of both COMMD7/COMMD1 terminates NF-κB signaling.
Furthermore, we performed bioinformatics analysis to identify critical genes underlying COMMD7-mediated promotion of the NF-κB signaling pathway in Nanog+ HCSCs. Among several downregulated DEGs participating in the NF-κB signaling pathway, PIAS4 was screened as a potential key gene in Nanog+ HCSCs after COMMD7 knockdown because it has been reported to mediate NEMO sumoylation and NF-κB activation.27 In agreement with this, PIAS4 overexpression significantly reversed the inhibitory effects of COMMD7 knockdown on cell proliferation, cell apoptosis, as well as migration and invasion in Nanog+ HCSCs. The data reported here demonstrate that silencing of COMMD7 results in a selective mechanism of suppressing PIAS4 expression and PIAS2-mediated NEMO sumoylation, causing the downregulation of p65.
In summary, this study demonstrates that COMMD7 mediates positive and negative regulation of the NF-κB signaling pathway in Nanog+ HCSCs. During positive regulation, overexpression of COMMD7 alone increases the expression of NF-κB p65 by elevating PIAS4 mediating NEMO sumoylation. During negative regulation, co-overexpression of both COMMD7/COMMD1 terminates NF-κB signaling. Although the exact mechanism underlying COMMD7 regulation of NF-κB signaling requires further investigation, these preliminary findings identify COMMD7 as a potentially useful therapeutic target for the improvement of HCC outcomes.
Materials and Methods
Cancer Specimens and Tissue Microarray
A total of 35 matched pairs of primary HCC tissue (cancer tissue [CT]) specimens and adjacent tissue (AT) specimens, as well as HCC tissue microarrays from patients with HCC who underwent curative resection, were provided by the Department of Hepatobiliary Surgery in the Second Affiliated Hospital of Army Medical University. HCC tissue specimens and corresponding paired AT tissue specimens were snap frozen in liquid nitrogen and stored at −80°C before use. All specimens were confirmed by pathological examination. None of the patients had received chemotherapy or radiotherapy before surgery. Clinical staging was performed according to the International Union for Cancer Control (UICC). The clinicopathological data of all samples were well documented including age, sex, tumor-node-metastasis (TNM) stage, and lymph node metastasis.
Ethics
Written informed consent was obtained for all patient samples. This study was approved by the institutional review board of the Second Affiliated Hospital of Army Medical University.
Immunohistochemistry
The relevant immunohistochemical staining was performed on arrayed tissues as previously reported.37 In brief, the sections were subsequently rinsed in PBS for 5 min and then incubated with primary antibody against COMMD7 (1:150; Abcam) or COMMD1 (1:100; Abcam) at 4°C overnight. Sections incubated without the primary antibody were used as negative controls. The sections were then incubated with horseradish peroxidase (HRP)-labeled secondary antibody (1:5,000; Cell Signaling, USA). Immunostaining was scored independently by two pathologists in a blinded manner.
Cell Lines and Culture
The human HCC cell line Huh7 and HL-7702 hepatocytes were purchased from Cell Bank of Shanghai Institute, Chinese Academy of Sciences. Huh7 and HL-7702 cells were cultured in RPMI-1640 medium (Invitrogen, USA) containing 10% heat-inactivated fetal bovine serum (FBS; GIBCO), 1% penicillin and streptomycin. The Nanog+ HCSCs were obtained by performing FCM to sort HCC cell lines after infection with Lv-PNanog-GFP as described previously.38 Nanog+ HCSCs were cultured in DMEM/F12 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with B27 (catalog no. 17504-044; GIBCO, Grand Island, NY, USA). All cell lines were maintained in a humidified incubator at 37°C under 5% CO2.
Cell Transfection
For knockdown of COMMD7, three pre-designed shRNAs targeting COMMD7 (sh-COMMD7-1, sh-COMMD7-2, sh-COMMD7-3) and the corresponding negative control (NC) were lentivirus packaged by Shanghai GenePharma (Shanghai, China). Nanog+ HCSCs were plated in a six-well plate (2 × 105/well) and cultured for 2 days, then 20 μL of virus with a titer of 108 TU/mL (contains at least 1 × 108 biologically active virus particles per milliliter of virus solution) was added for 48 h. To analyze the effects of the combination of COMMD7 with COMMD1, COMMD1, or COMMD7 overexpression, plasmids (pcDNA3.0-COMMD1 or pcDNA3.1-COMMD7) and their corresponding vector were purchased from GenePharma. Subsequently, Nanog+ HCSCs were seeded in a six-well plate (2 × 105/well) and transfected with NC vector or pcDNA3.1-COMMD1, respectively. Moreover, Nanog+ HCSCs transfected with pcDNA3.1-COMMD1 were further classified into two groups as follows: COMMD1 + COMMD7 group (HCSCs transfected with pcDNA3.1-COMMD1 were transfected with pcDNA3.1-COMMD7) and COMMD1 + sh-COMMD7 group (HCSCs transfected with pcDNA3.1-COMMD1 were transfected with sh-COMMD7). For PIAS4 overexpression, PIAS4 overexpression plasmid (pcDNA3.1-PIAS4) was also purchased from GenePharma. Then Nanog+ HCSCs transfected with sh-COMMD7 were transfected with pcDNA3.1-PIAS4.
All cell transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Following transfection, HCSCs were examined for the downregulation of COMMD7 and the upregulation of COMMD1, COMMD7, or PIAS4 by real-time qPCR, western blotting, and immunofluorescence, and cells were used for further experiments.
Real-Time qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen, USA). Reverse transcription reaction and cDNA synthesis were performed using the M-MLV Reverse Transcriptase (Promega) according to the manufacturer’s instructions. The real-time qPCR analysis was performed on Stratagene Mx3000P Real-time PCR (Agilent) with 20 μL of DBI Bestar SybrGreen qPCR Master Mix. The primers used in this study are listed in Table S1. The 2-ΔΔCt method39 was used to calculate relative gene expression relative to GAPDH. The experiment was performed three times, in triplicate each time.
Western Blotting
Fresh HCC tissues or cell samples were lysed in 2× SDS lysis buffer at 4°C and boiled for 5 min. After centrifuging at 12,000 × g for 10 min at 4°C, the supernatant was collected and the protein was quantified using a bicinchoninic acid (BCA) protein assay kit. Subsequently, proteins were separated by 10% SDS-PAGE gel electrophoresis and transferred onto polyvinylidene fluoride (PVDF) membranes. Next, the membranes were blocked in 5% nonfat milk dissolved in Tris-buffered saline with Tween 20 (TBST) solution for 1 h at room temperature followed by incubation with primary antibodies against Nanog (1:1,000, ab153419; Abcam), COMMD7 (1:2,000, #9742; Cell Signaling), COMMD1 (1:1,000, #24640; SAB), NF-κB p65 (1:1,500, 19553-1-AP; Proteintech), HNF4α (1:1,000, #2947; Cell Signaling), NEMO (1:3,000, #23870; SAB), CXCL12 (1:1,000, #36120; SAB), CXCL2 (1:4,000, #24681; SAB), PIAS4 (1:2,500, #8709; Cell Signaling), histone H3.1 (1:2,000, #9265; Cell Signaling), β-actin (1:1,000, #5762; Cell Signaling), and GAPDH (1:10,000, 10494-1-AP; Proteintech) at 4°C overnight. After washing three times, the membranes were then incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. The signals of proteins were detected with enhanced chemiluminescence (ECL) as described by the manufacturer (Beyotime, China). GAPDH or β-actin protein was used as an internal control in cytoplasm. Histone H3.1 was used as the internal control in the nucleus.
Immunofluorescence
For immunofluorescence, cells were fixed in 4% paraformaldehyde and then washed with PBS for 5 min. Next, cells were incubated with primary antibody against COMMD7, COMMD1, p65, or HNF4α at 4°C overnight, followed by incubation with FITC-labeled goat against secondary antibody (Beyotime Institute of Biotechnology). Cells were counterstained with DAPI to label the nucleus. Samples were observed using an inverted fluorescence microscope (Olympus Corporation, Japan).
Cell Proliferation Assay
A CCK-8 assay was performed to evaluate the proliferative ability of Nanog+ HCSCs. In brief, cells were seeded at a density of 2 × 104 cells per well in 96-well tissue culture plates and incubated for 24, 48, and 72 h. Then, 10 μL of CCK-8 (5 g/L; Dojindo, China) was added to each well, and cells were incubated for 1 h. The optical density (OD) value was read at 450 nm using a microplate reader (Bio-Rad, USA).
Hoechst 33258 Staining Assay
Following different treatments, Nanog+ HCSCs were seeded at 4 × 105 cells per well in six-well plates for 24 h. After washing twice with PBS, cells were incubated with 5 μM Hoechst 33258 (Sigma, St. Louis, MO, USA) for 30 min in the dark at room temperature. Cells were washed with 0.5% Triton X-100 in PBS and then examined for changes in nuclear morphology using a fluorescence microscope (Olympus, Tokyo, Japan).
Cell Apoptotic Assay of HCSCs by FCM
Cell apoptosis was further confirmed by FCM using an Annexin V-FITC and PI double-staining kit. In brief, Nanog+ HCSCs were harvested by trypsinization and washed with PBS. After centrifuging at 1,500 × g for 3 min at room temperature, the apoptotic cells were resuspended in Annexin V binding buffer. Then, a total of 5 μL of Annexin V-FITC and 5 μL of PI (KeyGen, China) were successively added to the cells and incubated for 15 min in the dark at room temperature. After incubation, the stained HCSCs were analyzed by FCM (FACSCalibur; BD Biosciences, USA). The percentage of Annexin V+ cells divided by the total number of cells was calculated and expressed as the overall apoptosis rate in the gated region. All experiments were performed three times in triplicate.
Wound Healing Assay
Wound healing assay was performed to assess cell migration in Nanog+ HCSCs. In brief, cells were seeded onto six-well plates until they reached 80%–90% confluence. The medium was then discarded and a wound field was made using a sterile 200-μL pipette tip through the cell layer in each well. Subsequently, the wound closure was quantified at 0 and 24 h post-wound, respectively. The rate of migration was calculated by measuring the remaining un-migrated area using ImageJ.
Transwell Matrigel Invasion Assay
For the invasion assay, transwell chambers with 8.0-μm pore size (Costar, USA) were pre-coated with 30 μL of Matrigel (BD Biosciences, USA), placed into 24-well plates, and incubated for 30 min. After 48-h transfection, Nanog+ HCSCs were trypsinized and placed into the upper chamber, whereas 10% FBS was added into the lower chambers as a chemoattractant. Twenty-four hours later, non-migratory cells were removed using cotton swabs. The migrated cells on the lower surface were fixed and stained with crystal violet for 20 min, followed by quantification under a light microscope in five randomly selected fields.
Tumorigenicity Assay in Nude Mice
Six-week-old nude mice were provided by Vital River Laboratory Animal Technology (Beijing, China). Prior to inoculation, two groups of Nanog+ HCSCs were prepared: NC lentivirus-transfected Nanog+ HCSCs and sh-COMMD7 lentivirus-transfected Nanog+ HCSCs. A total of 20 mice were randomized into two groups with 10 mice in each group. Each mouse was subcutaneously injected in the right flank with the above two types of cells (2 × 106) in 0.1 mL of PBS. Once tumors were formed, the measurement of the tumor volume (V) was performed by caliper daily using the formula V = (L × W2)/2, where L was the length and W was the width of the tumor every 2 days from day 10. Growth curves were plotted using average tumor volume within each experimental group. After 26 days, the mice were euthanized, and the dissected tumors were prepared for subsequent analyses. All animal experiments were approved by the Institutional Committee for Animal Research and performed according to the Guide for the Care and Use of Laboratory Animals.
Microarray Expression Analysis
Total RNAs were isolated from cells using the QIAGEN RNeasy Mini Kit (Valencia, CA, USA) and purified using the RNeasy Mini kit (QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions. For microarray analysis, total RNA was amplified and transcribed into fluorescent cRNA with Agilent Quick Amp Labeling protocol (version 5.7; Agilent Technologies), and then hybridized to the Agilent SureHyb microarray chip (Agilent Technologies). The microarrays were then automatically washed and scanned using Agilent DNA Microarray Scanner. The Agilent Feature Extraction software (v11.0.0.1) was used to analyze the acquired array images and obtain scaled quantitative information. Data were subsequently normalized, and the DEGs were analyzed using Agilent GeneSpring GX v12.1 software (Agilent Technologies). The DEGs were screened using cutoff criteria of p value <0.05 and |log FC| value >1.5 or <0.5. All DEGs were further analyzed with unsupervised hierarchical clustering based on the standard correlation of logarithmic transformed data.
KEGG Pathway Enrichment Analysis
The KEGG is a database used to assign sets of DEGs to specific pathways.40 KEGG pathway enrichment analysis of DEGs was performed using DAVID, with gene counts ≥5 and p < 0.05 set as threshold values to find critical genes closely related to the NF-κB signaling pathway involved in HCSCs initiation and progression.
Statistical Analysis
All statistical calculations were carried out using SPSS version 19 (IBM Corporation, USA). All results were expressed as the mean ± SD. The differences in Nanog, COMMD7, COMMD1, or HNF4α expression between HCC tissues and ATs were analyzed using Wilcoxon signed rank test. Pearson’s correlation coefficient was determined between COMMD7 and COMMD1 or HNF4α expression of HCC tissues using GraphPad Prism 5.0 software. Statistical comparisons for continuous variables were performed using Student’s t test between two groups and one-way ANOVA in more than two groups. Each experiment was repeated at least three times. The p value <0.05 was considered statistically significant.
Author Contributions
L.Z. and J.L. conceived and designed the experiments. N.Y., X.H., H.G., and N.M. performed the experiments and analyzed the data. L.Z. and J.L. wrote the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by National Natural Science Foundation of China (contract grants 81672902 and 81372561).
Footnotes
Supplemental Information includes five figures and two tables and can be found with this article online at https://doi.org/10.1016/j.omto.2018.12.006.
Supplemental Information
References
- 1.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Mittal S., El-Serag H.B. Epidemiology of hepatocellular carcinoma: consider the population. J. Clin. Gastroenterol. 2013;47(Suppl):S2–S6. doi: 10.1097/MCG.0b013e3182872f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mir N., Jayachandran A., Dhungel B., Shrestha R., Steel J.C. Epithelial-to-mesenchymal transition: a mediator of sorafenib resistance in advanced hepatocellular carcinoma. Curr. Cancer Drug Targets. 2017;17:698–706. doi: 10.2174/1568009617666170427104356. [DOI] [PubMed] [Google Scholar]
- 4.Llovet J.M. Liver cancer: time to evolve trial design after everolimus failure. Nat. Rev. Clin. Oncol. 2014;11:506–507. doi: 10.1038/nrclinonc.2014.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang C., Long J., Liu B., Xu M., Wang W., Xie X., Wang X., Kuang M. miR-500a-3p promotes cancer stem cells properties via STAT3 pathway in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2017;36:99. doi: 10.1186/s13046-017-0568-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Serag H.B., Rudolph K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 7.Visvader J.E., Lindeman G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 8.Clarke M.F., Dick J.E., Dirks P.B., Eaves C.J., Jamieson C.H., Jones D.L., Visvader J., Weissman I.L., Wahl G.M. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 9.Dean M., Fojo T., Bates S. Tumour stem cells and drug resistance. Nat. Rev. Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 10.Yang Z.F., Ho D.W., Ng M.N., Lau C.K., Yu W.C., Ngai P., Chu P.W., Lam C.T., Poon R.T., Fan S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–166. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Xu Q., Xu H.X., Li J.P., Wang S., Fu Z., Jia J., Wang L., Zhu Z.F., Lu R., Yao Z. Growth differentiation factor 15 induces growth and metastasis of human liver cancer stem-like cells via AKT/GSK-3β/β-catenin signaling. Oncotarget. 2017;8:16972–16987. doi: 10.18632/oncotarget.15216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song K., Wu J., Jiang C. Dysregulation of signaling pathways and putative biomarkers in liver cancer stem cells (Review) Oncol. Rep. 2013;29:3–12. doi: 10.3892/or.2012.2082. [DOI] [PubMed] [Google Scholar]
- 13.Huo X., Han S., Wu G., Latchoumanin O., Zhou G., Hebbard L., George J., Qiao L. Dysregulated long noncoding RNAs (lncRNAs) in hepatocellular carcinoma: implications for tumorigenesis, disease progression, and liver cancer stem cells. Mol. Cancer. 2017;16:165. doi: 10.1186/s12943-017-0734-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayden M.S., Ghosh S. NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hassanzadeh P. Colorectal cancer and NF-κB signaling pathway. Gastroenterol. Hepatol. Bed Bench. 2011;4:127–132. [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., Zhou Y., Jia G., Han B., Liu J., Teng Y., Lv J., Song Z., Li Y., Ji L. Shikonin suppresses tumor growth and synergizes with gemcitabine in a pancreatic cancer xenograft model: involvement of NF-κB signaling pathway. Biochem. Pharmacol. 2014;88:322–333. doi: 10.1016/j.bcp.2014.01.041. [DOI] [PubMed] [Google Scholar]
- 17.Lin Y., Bai L., Chen W., Xu S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets. 2010;14:45–55. doi: 10.1517/14728220903431069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartuzi P., Hofker M.H., van de Sluis B. Tuning NF-κB activity: a touch of COMMD proteins. Biochim. Biophys. Acta. 2013;1832:2315–2321. doi: 10.1016/j.bbadis.2013.09.014. [DOI] [PubMed] [Google Scholar]
- 19.Burstein E., Hoberg J.E., Wilkinson A.S., Rumble J.M., Csomos R.A., Komarck C.M., Maine G.N., Wilkinson J.C., Mayo M.W., Duckett C.S. COMMD proteins, a novel family of structural and functional homologs of MURR1. J. Biol. Chem. 2005;280:22222–22232. doi: 10.1074/jbc.M501928200. [DOI] [PubMed] [Google Scholar]
- 20.Geng H., Wittwer T., Dittrich-Breiholz O., Kracht M., Schmitz M.L. Phosphorylation of NF-kappaB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009;10:381–386. doi: 10.1038/embor.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zoubeidi A., Ettinger S., Beraldi E., Hadaschik B., Zardan A., Klomp L.W., Nelson C.C., Rennie P.S., Gleave M.E. Clusterin facilitates COMMD1 and I-kappaB degradation to enhance NF-kappaB activity in prostate cancer cells. Mol. Cancer Res. 2010;8:119–130. doi: 10.1158/1541-7786.MCR-09-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernández Massó J.R., Oliva Argüelles B., Tejeda Y., Astrada S., Garay H., Reyes O., Delgado-Roche L., Bollati-Fogolín M., Vallespí M.G. The antitumor peptide CIGB-552 increases COMMD1 and inhibits growth of human lung cancer cells. J. Amino Acids. 2013;2013:251398. doi: 10.1155/2013/251398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.You N., Li J., Gong Z., Huang X., Wang W., Wang L., Wu K., Zheng L. COMMD7 functions as molecular target in pancreatic ductal adenocarcinoma. Mol. Carcinog. 2017;56:607–624. doi: 10.1002/mc.22520. [DOI] [PubMed] [Google Scholar]
- 24.Zheng L., Liang P., Li J., Huang X.B., Liu S.C., Zhao H.Z., Han K.Q., Wang Z. ShRNA-targeted COMMD7 suppresses hepatocellular carcinoma growth. PLoS ONE. 2012;7:e45412. doi: 10.1371/journal.pone.0045412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng L., Deng C.L., Wang L., Huang X.B., You N., Tang Y.C., Wu K., Liang P., Mi N., Li J. COMMD7 is correlated with a novel NF-κB positive feedback loop in hepatocellular carcinoma. Oncotarget. 2016;7:32774–32784. doi: 10.18632/oncotarget.9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esposito E., Napolitano G., Pescatore A., Calculli G., Incoronato M.R., Leonardi A., Ursini M.V. COMMD7 as a novel NEMO interacting protein involved in the termination of NF-κB signaling. J. Cell. Physiol. 2016;231:152–161. doi: 10.1002/jcp.25066. [DOI] [PubMed] [Google Scholar]
- 27.Mabb A.M., Wuerzberger-Davis S.M., Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat. Cell Biol. 2006;8:986–993. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- 28.You N., Li J., Huang X., Wu K., Tang Y., Wang L., Li H., Mi N., Zheng L. COMMD7 promotes hepatocellular carcinoma through regulating CXCL10. Biomed. Pharmacother. 2017;88:653–657. doi: 10.1016/j.biopha.2017.01.046. [DOI] [PubMed] [Google Scholar]
- 29.Meng H.M., Zheng P., Wang X.Y., Liu C., Sui H.M., Wu S.J., Zhou J., Ding Y.Q., Li J. Over-expression of Nanog predicts tumor progression and poor prognosis in colorectal cancer. Cancer Biol. Ther. 2010;9:295–302. doi: 10.4161/cbt.9.4.10666. [DOI] [PubMed] [Google Scholar]
- 30.Jeter C.R., Badeaux M., Choy G., Chandra D., Patrawala L., Liu C., Calhoun-Davis T., Zaehres H., Daley G.Q., Tang D.G. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27:993–1005. doi: 10.1002/stem.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang Y., Kitisin K., Jogunoori W., Li C., Deng C.X., Mueller S.C., Ressom H.W., Rashid A., He A.R., Mendelson J.S. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc. Natl. Acad. Sci. USA. 2008;105:2445–2450. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliva J., French B.A., Qing X., French S.W. The identification of stem cells in human liver diseases and hepatocellular carcinoma. Exp. Mol. Pathol. 2010;88:331–340. doi: 10.1016/j.yexmp.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka T., Jiang S., Hotta H., Takano K., Iwanari H., Sumi K., Daigo K., Ohashi R., Sugai M., Ikegame C. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4alpha in the pathogenesis of human cancer. J. Pathol. 2006;208:662–672. doi: 10.1002/path.1928. [DOI] [PubMed] [Google Scholar]
- 34.Mizuguchi T., Mitaka T., Hirata K., Oda H., Mochizuki Y. Alteration of expression of liver-enriched transcription factors in the transition between growth and differentiation of primary cultured rat hepatocytes. J. Cell. Physiol. 1998;174:273–284. doi: 10.1002/(SICI)1097-4652(199803)174:3<273::AID-JCP1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 35.Saha S.K., Parachoniak C.A., Ghanta K.S., Fitamant J., Ross K.N., Najem M.S., Gurumurthy S., Akbay E.A., Sia D., Cornella H. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walesky C., Apte U. Role of hepatocyte nuclear factor 4α (HNF4α) in cell proliferation and cancer. Gene Expr. 2015;16:101–108. doi: 10.3727/105221615X14181438356292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee J.M., Yang J., Newell P., Singh S., Parwani A., Friedman S.L., Nejak-Bowen K.N., Monga S.P. β-Catenin signaling in hepatocellular cancer: implications in inflammation, fibrosis, and proliferation. Cancer Lett. 2014;343:90–97. doi: 10.1016/j.canlet.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shan J., Shen J., Liu L., Xia F., Xu C., Duan G., Xu Y., Ma Q., Yang Z., Zhang Q. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012;56:1004–1014. doi: 10.1002/hep.25745. [DOI] [PubMed] [Google Scholar]
- 39.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 40.Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.