

Abstract
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a poorly understood disease affecting 0.2%–2% of the global population. To gain insight into the pathophysiology of ME/CFS in New Zealand, we examined the transcriptomes of peripheral blood mononuclear cells by RNA-seq analysis in a small well-characterized patient group (10 patients), with age/gender-matched healthy controls (10 control subjects). Twenty-seven gene transcripts were increased 1.5- to sixfold and six decreased three- to sixfold in the patient group (P < 0.01). The top enhanced gene transcripts, IL8, NFΚBIA and TNFAIP3, are functionally related to inflammation, and significant changes were validated for IL8 and NFΚBIA by quantitative polymerase chain reaction (qPCR). Functional network analysis of the altered gene transcripts (P < 0.01) detected interactions between the products related to inflammation, circadian clock function, metabolic dysregulation, cellular stress responses and mitochondrial function. Ingenuity pathway analysis (P < 0.05) provided further insights into the dysfunctional physiology, highlighting stress and inflammation pathways. This analysis provides novel insights into the molecular changes in ME/CFS and contributes to the understanding of the pathophysiological mechanisms of the disease.
Keywords: chronic fatigue syndrome, circadian rhythm, inflammation, metabolic dysregulation, mitochondrial dysfunction, myalgic encephalomyelitis, oxidative stress, transcriptome
Introduction
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) affects approximately 0.2%–2% of the global population.1 All age groups and socioeconomic strata are susceptible to the syndrome, but proportionally more women are affected.1,2 As few as 5% return to a state of well-being.1 ME/CFS is a multifactorial and multisystem illness, impacting the neurological, immune, endocrine and musculoskeletal pathways, and often the gastrointestinal system.1 Often ME/CFS onset is described as ‘sudden’, following an acute infectious period, but gradual onset is also reported.2 Initiating factors such as chronic viral infection, immune activation, exposure to chemicals, stress, hypotension, lymphocyte abnormalities and neuroendocrine dysfunction have all been proposed,2 but none validated as a single unifying cause. A formal medical diagnosis of ME/CFS is made after excluding all other recognized illnesses with a similar presentation, and by self-reported symptoms that fit within one of several broad sets of clinical criteria available.1 They highlight as a key symptom disabling physical and mental fatigue, lasting over 6 months that is exacerbated by exercise, or mental or emotional exertion.2,3 While ME/CFS pathophysiology is still poorly understood, it is hypothesized that profound dysregulation of the central nervous system and immune system, dysfunction of cellular energy metabolism and cardiovascular abnormalities may play important roles in the pathophysiology of the illness.2
A number of ME/CFS immune cell gene expression studies have been published. Collectively they have identified disturbances in pathways related to immune, neuronal, mitochondrial and metabolic function, but there is little consensus on the genes that are specifically differentially expressed. High-throughput RNA sequencing technology (RNA-seq) has only recently been utilized to investigate the ME/CFS transcriptome.4 Our RNA-seq study described here focuses on peripheral blood mononuclear cells (PBMCs) in a well-characterized cohort of ME/CFS patients. PBMCs are reportedly stable, accurate reflectors of biological differences in healthy and unwell subjects, particularly for immunological or inflammatory pathways. We identified 27 gene transcripts significantly increased in the ME/CFS group when compared to healthy controls, and 6 gene transcripts significantly decreased (P < 0.01), and found enrichment in stress and inflammation pathways, as well as mitochondrial and circadian clock functions. These results expand the depth of evidence of a pathophysiological basis to ME/CFS.
Methods
PBMC collection and isolation
The study had approvals from Health and Disability Ethics Committees in New Zealand (NTY/12/06/055 and 15426). A total of 10 ME/CFS patients, diagnosed according to the canadian consensus criteria (CCC) guidelines3 by an experienced ME/CFS clinician, were recruited along with matched controls. All participants gave their informed signed consent to participate. The control group had no history of significant illness, injury or fatigue-related disorders. ME/CFS patients completed a detailed questionnaire, developed in-house, providing their clinical history and current health status (Supplementary Table S1). Within 4 h of collection, whole blood samples were diluted 1:1 with filter-sterilized phosphate-buffered saline (PBS), layered onto Ficoll-Paque PLUS (Sigma-Aldrich) and centrifuged at 400g for 40 min. The PBMC interface layer was removed and washed twice with filter-sterilized PBS (6 mL). The isolated PBMCs were then mixed with RNAlater (Thermo Fisher Scientific; 1:5 ratio) for storage.
Transcriptome analysis
The transcriptome analysis was performed in collaboration with New Zealand Genomics Ltd. RNA was isolated from PBMC samples using a MirVana™ RNA Isolation Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. All samples passed quality control RNA integrity analysis by Fragment Analyzer (RIN ⩾7). Library preparation used an Illumina TruSeq Stranded total RNA library kit (with RiboZero Human/Rat/Mouse). Total RNA libraries were pooled and sequenced by Illumina HiSeq 2x125 bp paired-end sequencing. Raw FASTQ sequences had an average per base mean quality score (Q score) of 35.84. A Q score of >30 indicates >99.9% base call accuracy and is the quality bench mark for next-generation sequencing. Raw FASTQ files were mapped and aligned to the human genome (hg38) with TopHat (tophat2) and Bowtie (bowtie2), resulting in 13,593 reads mapped to coding regions. The data were analysed in R using EdgeR (bioconductor release 3.4). A generalized linear model fit was used to normalize the data. A t-test identified significantly changed gene transcripts between the ME/CFS patients and controls, identified on the UCSC genome browser (https://genome.ucsc.edu/). The false discovery rate (FDR) reduces false positives, but at the inverse cost of increasing the number of false negatives so that the likelihood of the data to show real effects, particularly with small patient cohorts, can be easily reduced. In light of this, statistical analysis was achieved by setting the change in gene expression threshold to an un-adjusted P < 0.01 for encoded protein functional network analysis (FNA). To fully explore the dataset by ingenuity pathway analysis (IPA), the threshold was set to an unadjusted P < 0.05.
RT-qPCR validation
Taqman gene expression (RT-qPCR) assay validation was performed for the three most significantly changed gene transcripts IL8, NFKBIA and TNFAIP3, with the housekeeping genes GAPDH and 18S as stable references (#4448892 for IL8 and NFKBIA, #4453320 for TNFAIP3, GAPDH, 18S). RNA was converted to complementary DNA (cDNA) with a SuperScript™ VILO ™ cDNA Synthesis Kit (Thermo Fisher Scientific). Triplicate reactions were performed on MicroAmp Optical 96-Well reaction plates (Life Technologies) in a ViiA™ 7 Real-Time PCR System (Thermo Fisher Scientific) with TaqMan® Fast Advanced Master Mix (Thermo Fisher Scientific). The normalized relative expression levels of each gene were calculated using the 2ΔΔCt method.
FNA
Differentially expressed gene transcripts (n = 33, P < 0.01) were assessed for functional associations between encoded proteins by STRING analysis (http://string-db.org, version 10.5). STRING is an extensive database of known and predicted protein–protein interactions, centering on protein-coding gene loci, with alternative splice isoforms or post-translationally modified forms collapsed at the level of the gene locus. FDR-adjusted P-values are calculated for each enriched biological process.
IPA
A pool of 125 upregulated and 40 downregulated gene transcripts (P < 0.05, Supplementary Table S2) were analysed by IPA. IPA uses causal analytics tools to elucidate upstream biological causes and probable downstream effects on cellular biology from the dataset. Underlying these algorithms is the ingenuity knowledge base, curated from the biomedical literature or third-party databases. An enrichment score (P-value) measures the overlap of observed and predicted gene sets. A z-score assesses the match of observed and predicted regulation patterns, serving as a predictor for the activation state of the identified regulator molecule. Pathways with z-scores of >2 or <–2 are considered activated or inhibited, respectively.
Results and discussion
The concepts of Precision Medicine with appropriate statistical analysis have been applied to this small clinically well-characterized patient cohort in this RNA-seq transcriptome analysis. This approach has been successfully applied to cohorts of rare diseases, where available patients are small in number. In addition, the RNA-seq analysis itself has appropriate sequencing depth to compensate for the smaller sample size.
RNA-seq analysis revealed an altered PBMC transcriptome
A t-test of normalized RNA-seq data found 27 significantly increased gene transcripts and 6 significantly decreased gene transcripts in the ME/CFS cohort (P < 0.01, Table 1).
Table 1.
Transcripts with altered gene expression in the ME/CFS group compared to healthy controls (P < 0.01).
| Gene symbol | Gene name | P-value | Fold change |
|---|---|---|---|
| IL8 | Interleukin-8 | 2.5 × 10−6 | 5.6 |
| NFKBIA | NF-κB inhibitor alpha | 2.0 × 10−5 | 2.4 |
| TNFAIP3 | Tumour necrosis factor alpha-induced protein 3 | 1.3 × 10–4 | 3.6 |
| JUN | Jun proto-oncogene | 3.8 × 10−4 | 2.5 |
| PER1 | Period circadian protein homolog 1 | 8.8 × 10−4 | 2.3 |
| UBE2D3 | Ubiquitin conjugating enzyme E2 D3 | 2.2 × 10−3 | 2.0 |
| RBBP6 | E3 Ubiquitin-protein ligase RBBP6 | 2.3 × 10−3 | 2.1 |
| NAMPT | Nicotinamide phosphoribosyltransferase | 2.3 × 10−3 | 1.9 |
| IER3 | Immediate early response 3 | 2.7 × 10−3 | 2.0 |
| ZC3H12A | Zinc finger CCCH-type containing 12A | 2.8 × 10−3 | 1.8 |
| CREM | CAMP responsive element modulator | 2.8 × 10−3 | 1.9 |
| RIPK2 | Receptor interacting serine/threonine kinase 2 | 3.3 × 10−3 | 1.6 |
| PMAIP1 | Phorbol-12-myristate-13-acetate-inducing protein 1/immediate-early-response protein APR | 3.3 × 10−3 | 2.3 |
| PPP1R15A | Protein phosphatase 1 regulatory subunit 15A | 3.6 × 10−3 | 1.7 |
| ENC1 | Ectodermal-neural cortex 1 | 4.3 × 10−3 | 2.2 |
| PMPCB | Peptidase, mitochondrial processing beta subunit | 4.3 × 10−3 | 1.9 |
| G3BP2 | G3BP stress granule assembly factor 2 | 4.5 × 10−3 | 2.0 |
| PIGA | Phosphatidylinositol glycan anchor biosynthesis class A | 4.9 × 10−3 | 1.8 |
| CCNL1 | Cyclin L1 | 5.3 × 10−3 | 1.6 |
| SOCS3 | Suppressor of cytokine signalling 3 | 5.3 × 10−3 | 1.8 |
| CSRNP1 | Cysteine and serine rich nuclear protein 1 | 5.7 × 10−3 | 1.6 |
| ZNF274 | Zinc finger protein 247 | 6.1 × 10−3 | 1.6 |
| USP3 | Ubiquitin specific peptidase 3 | 6.2 × 10−3 | 1.7 |
| PILRB | Paired immunoglobin-like type 2 receptor beta | 7.3 × 10−3 | 1.5 |
| DDX3X | DEAD-box helicase 3, X-linked | 8.2 × 10−3 | 1.5 |
| KLHL15 | Kelch like family member 15 | 9.2 × 10−3 | 1.8 |
| ETF1 | Eukaryotic translation termination factor 1 | 1.0 × 10−2 | 1.8 |
| ANKRD46 | Ankyrin repeat domain-containing protein 46 | 2.4 × 10−3 | 0.25 |
| LPAR1 | Lysophosphatidic acid receptor 1 | 4.3 × 10−3 | 0.15 |
| PCDHGC3 | Protocadherin gamma-c3 | 5.8 × 10−3 | 0.15 |
| ZNF202 | Zinc finger protein 202 | 6.8 × 10−3 | 0.14 |
| HS3ST3B1 | Heparin sulphate glucosamine-3-o-sulfotransferase 3B1 | 8.0 × 10−3 | 0.26 |
| DCUN1D3 | DCN1-like protein 3 | 1.0 × 10−2 | 0.23 |
ME/CFS: myalgic encephalomyelitis/chronic fatigue syndrome.
qPCR validation of changes in gene expression
The top three significantly changed gene transcripts in the ME/CFS group were evaluated by RT-qPCR on the study group. The results were consistent with the RNA-seq data for IL8 and NFKBIA. As shown in the fold change graph (Figure 1(a)) and scatter plot (Figure 1(b)), IL8 (FC = 4.8; P = 0.01) and NFKBIA (FC = 2.0; P = 0.05) were both significantly increased in the patient group compared to controls. TNFAIP3 was also increased in the patient group (FC = 2.1; P = 0.19) with the change trending towards significance as is evident in the scatter plot (Figure 1(b)).
Figure 1.
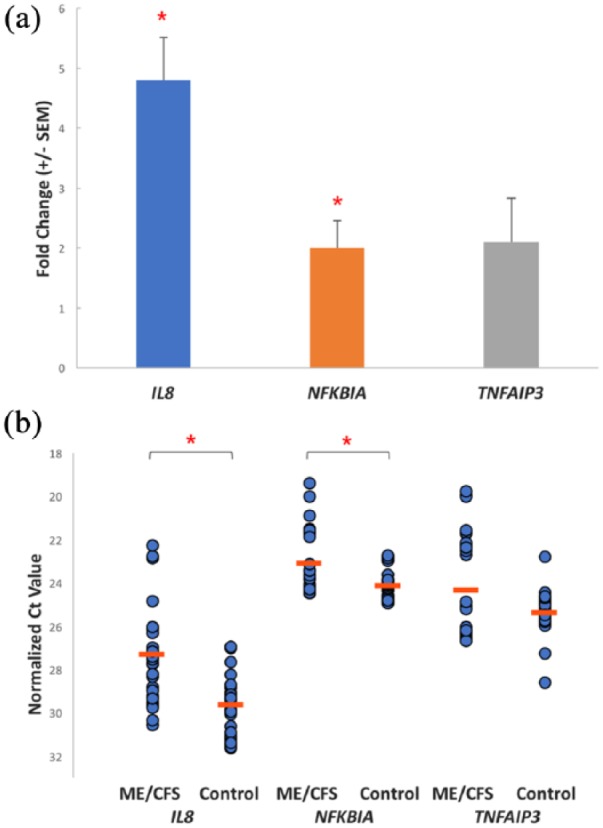
(a) Mean fold-change in expression between ME/CFS and controls of IL8, NFKBIA and TNFAIP3 after RT-qPCR assays (±SEM). (b) Scatter plot of normalized triplicate RT-qPCR assay Ct values for each gene in the ME/CFS (n = 10) and control (n = 10) cohorts. The mean Ct value for each gene in the cohorts is also shown (orange line). The Ct value is the PCR amplification cycle at which the gene transcript exceeded the individually calculated baseline threshold level for that gene.
Analysis of the transcriptome data
FNA and IPA examined the enriched functional interactions and the biochemical and physiological pathways derived from the analysis of the differentially expressed gene transcripts in ME/CFS.
FNA
The functional association networks of proteins encoded by differentially expressed gene transcripts (n = 33, P < 0.01, Table 2) were assessed using the STRING portal (http://string-db.org, version 10.5) (Figure 2). We found a significant level of network enrichment (P-value 6.1 × 10−5) in this list indicating the encoded proteins are biologically connected. Strong functional associations were observed between IL8, NFKBIA, TNFAIP3, JUN, NAMPT, CREM, PMAIP1, PPP1R15A, RBBP6, UBE2D3, SOCS3, RIPK2 and ZC3H12A (Figure 2). Several compelling functional pathways were enriched following increases in the encoded proteins (Supplementary Table S3). Collectively, these pathways reinforce significant immune and inflammatory over-activation and dysregulation occurring in the pathology of ME/CFS.
Figure 2.
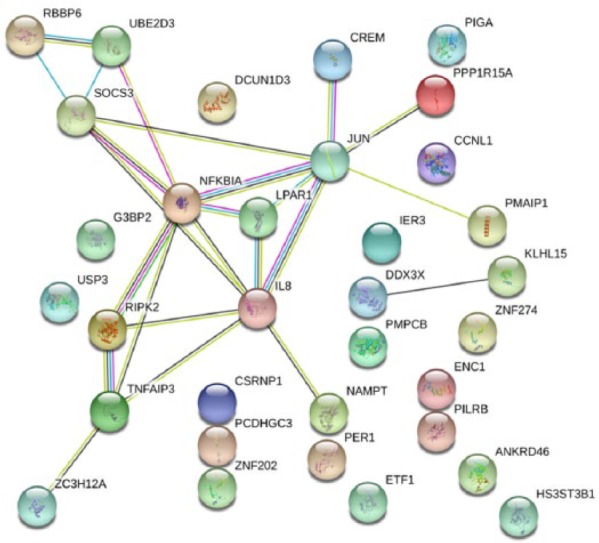
STRING analysis of significantly altered gene transcripts in the ME/CFS cohort (n = 33, P < 0.01).
Protein: protein interaction enrichment network (P = 6.1 × 10−5) of encoded proteins of differentially expressed gene transcripts in the ME/CFS group compared to controls. Twenty-four functional interactions were identified. Magenta = fusion evidence: genes fused into single open reading frames in other organisms; green = neighbourhood evidence: groups of genes frequently observed in the same chromosomal location; black = co-expression evidence: genes are observed to be correlated in expression across a large number of experiments; light blue = database evidence: known metabolic pathways, protein complexes and so on from curated databases.
IPA shows inflammation and oxidative stress in ME/CFS
IPA was carried out on the transcriptome dataset with significance set at P < 0.05, increasing the pool available for analysis to 125 increased and 40 decreased gene transcripts (Supplementary Table S2). This analysis identified a number of overlapping, functionally interlinked canonical pathways. Most pathways were directly related to inflammatory signalling and the stress response, supporting the FNA results (Supplementary Table S4). Gene networks related to oxidative stress, inflammation and nervous tissue damage (Supplementary Table S5) were also significant. These results support the hypothesis that mitochondrial dysfunction and impaired energy metabolism lead to fatigue, malaise and commonly experienced muscle pain and cognitive impairment in ME/CFS.
Anti-inflammatory responders to TNFα and NF-κB
The three top upregulated genes in our study, IL8, NFKBIA and TNFAIP3, are early responders to TNF-induced NF-κB activation.5 The proteins A20 (TNFAIP3) and IκBα (NFKBIA) are part of the two main negative feedback loops of NF-κB-driven transcription.5 Furthermore, TNFα is a potent inducer of IL-8 secretion, through a transcriptional mechanism regulated by NF-κB. Increases in IL-8 and TNFα have been identified in several cytokine and immune ME/CFS studies. Chronic inflammation is also amplified by the NF-κB signalling pathway. The increase in expression of these three gene transcripts in the ME/CFS group strongly implies an ongoing biological counter-response to unwanted excess activity of NF-κB and inflammation in ME/CFS, driven by TNFα.
Significant biological pathways enriched in the ME/CFS group, identified by FNA and IPA
The circadian rhythm
Several elevated gene transcripts in the ME/CFS group encode core proteins that regulate the circadian clock (Supplementary Table S2), the central mechanism that drives our 24-h circadian rhythm. A disturbed circadian rhythm can be linked directly to many ME/CFS symptoms, such as pain, fatigue, sleep disturbance, flu-like symptoms, cognitive impairment, post-exertional malaise and dysfunction of metabolic and immune systems.6 These results suggest that disruption of the circadian rhythm is likely in ME/CFS and could be a plausible candidate for sustaining both the symptoms and severity of ME/CFS.
Mitochondrial dysfunction
Mitochondria are essential for cellular energy production, metabolism and regulating stress responses. Results from FNA and IPA indicate mitochondrial dysfunction, characterized by signs of oxidative stress and downregulation of metabolic pathways (Supplementary Tables S3 and S4). Three significantly increased gene transcripts in the ME/CFS cohort (PMAIP1, PMPCB and JUN) play important roles in mitochondrial function and apoptotic pathways (Table 1 and Figure 2):
Respiration. Mitochondrial respiration can be inhibited by cytokine changes and oxidative stress. Regulation of cytokine production (Supplementary Table S3) and oxidative stress pathways (Supplementary Tables S3 and S5) were both enriched in the ME/CFS cohort. Recent ME/CFS studies have found key indicators of mitochondrial dysfunction, including reduced production of ATP and impairment of the oxidative phosphorylation pathways.7 Supporting our analysis, a 2017 study showed lowered maximal respiration by mitochondria in ME/CFS PBMCs.7 They found metabolic differences that showed an inability to fulfil cellular energetic demands, in particular when mitochondria were under stress.7
Downregulation of metabolism. FNA showed disturbed regulation of multiple metabolic pathways in the ME/CFS cohort, many of which involved mitochondria (Supplementary Table S3). Metabolic profiling studies of ME/CFS have linked impaired metabolism to post-exertional malaise.8 Importantly, a 2016 study investigated metabolites in ME/CFS patients and controls and discovered a homogeneous decrease of diagnostic metabolites among all ME/CFS patients, consistent with hypometabolic syndrome.8 As all of the metabolic abnormalities identified were either directly regulated by redox or by the availability of NADPH, they speculated that mitochondrial involvement was likely. Lactic acid accumulation from impaired metabolism may explain myalgia in muscles and joints, reported by ME/CFS patients. Furthermore, lactate build up in the brain’s ventricular spaces and cerebrospinal fluid of ME/CFS patients has been reported and could contribute to the neuroinflammatory-related symptoms of ME/CFS.
Stress response. FNA showed an increased cellular stress response and positive regulation of apoptotic processes in the ME/CFS group (Supplementary Table S3). IPA highlighted the production of nitric oxide (NO) and reactive oxygen species (ROS) in macrophages (Supplementary Table S4). NO and ROS production, stimulated by TNFα and NF-κβ, plays a central role in the control of infections, and ROS production is also attributed to activated NADPH oxidase, a component of the mitochondrial electron transport chain. Mitochondria densely populate cells in the CNS, providing essential energy for neurons and thereby influencing synaptic plasticity.9 Oxidative stress, caused by increased ROS in mitochondria, can lead to cognitive impairment and apoptosis – leading to mitochondrial DNA damage and contributing to cognitive disturbance by affecting both neurotransmission and Ca2+ homeostasis.9 Of relevance, many ME/CFS symptoms, such as fatigue, exercise intolerance and myalgia, are shared with primary mitochondrial diseases.10
Chronic inflammation
Sustained inflammation is emphasized in the ME/CFS transcriptome, with NF-κB and TNFα central to two primary canonical pathways predicted to be enriched by IPA (Supplementary Table S4). As mentioned previously, IL8, NFKBIA and TNFAIP3 are components of the key affected pathways. The NF-B activation pathway (Supplementary Table S4) targets a multitude of cytokine and chemokine receptors required for immune recognition, antigen presentation and adhesion receptors. The TNFR2 signalling pathway (Supplementary Table S4) regulates TNFα activity by antagonizing TNFα-induced apoptosis, especially in highly activated T cells. LPS-stimulated mitogen activated protein kinase (MAPK) (Supplementary Table S4) drives the inflammatory response in macrophage immune cells. Peroxisome Proliferator-Activated Receptors (PPAR) signalling was also identified as affected (Supplementary Table S4). PPAR signalling molecules regulate metabolic processes in heart muscle tissue, inflammation and oxidative stress and facilitate interactions between circadian, metabolic (lipid metabolism) and cardiovascular pathways. Mitochondrial dysfunction might be further influenced by increased chronic activation of immune-inflammatory stress pathways. Inflammatory mediators such as TNFα have been associated in vitro with mitochondrial dysfunction and increased ROS. Injury or infection can also lead to compromised mitochondrial integrity, triggering inflammation via NF-κB, MAPKs and interferon regulatory factors and promoting pathology in a growing number of diseases.
These results reinforce the involvement of inflammation, disturbed circadian rhythm, metabolic dysregulation and oxidative stress in ME/CFS.
Previous ME/CFS transcriptome studies support inflammation and mitochondrial involvement
To the best of our knowledge, this is the first RNA-seq that has analysed PBMCs of ME/CFS patients. This technology was, however, used in a recent whole blood study analysing adolescent ME/CFS participants by gene set enrichment.4 Our analysis was consistent with their findings, suggesting impairment of B cell differentiation and survival, enhanced innate antiviral responses and inflammation. Co-expression patterns and single gene transcripts were associated with neuroendocrine markers of altered HPA axis and autonomic nervous activity, plasma cortisol, blood monocyte and eosinophil counts. We have found significant molecular changes in our ME/CFS cohort that are consistent with those reported in larger complementary but not identical studies. Previous microarray and differential display studies of gene expression in ME/CFS have indicated disturbances in immune pathways, mitochondrial function, cell stress and apoptosis. A microarray investigation of PBMCs from a small group of post-infective ME/CFS males by Gow et al.10 identified differentially expressed genes with roles in immune modulation, oxidative stress and apoptosis and, of particular interest, also found increased TNFAIP3, in support of our results. Of significance to our study, Zhang et al.11 have identified a differentially expressed gene signature of 88 genes by microarray analysis and further substantiated by studies with other ME/CFS patient cohorts. The gene signature enabled sub-grouping of the patient cohorts, and IPA analysis of the gene interactions, disease associations and functions identified immunological disease, cancer-related, cell death, immune response and infection pathways, as well as links with Epstein–Barr and enterovirus infection indicators.
Our exploratory approach has enabled us to obtain a rich differentially expressed gene dataset to identify changed biology in ME/CFS. We have identified the circadian rhythm dysregulation pathway as a new possible underlying cause of the unrefreshing sleep, fatigue and metabolic abnormalities seen in ME/CFS. Furthermore, impaired mitochondrial function and resulting oxidative stress, coupled with chronic immune-inflammatory signalling, provides a compelling explanation for the fatigue, cognitive dysfunction and post-exertion malaise experienced in ME/CFS.
Therefore, this study is a further step towards gaining an understanding of the disease process and identifying putative biomarkers to support clinical diagnosis. The biological pathways identified offer a rational explanation of the complex and often multi-systemic nature of ME/CFS.
Supplemental Material
Supplemental material, Supplementary_Material for Changes in the transcriptome of circulating immune cells of a New Zealand cohort with myalgic encephalomyelitis/chronic fatigue syndrome by Eiren Sweetman, Margaret Ryan, Christina Edgar, Angus MacKay, Rosamund Vallings and Warren Tate in International Journal of Immunopathology and Pharmacology
Supplemental Material
Supplemental material, Supplementary_references_used_to_support_statements_in_the_text for Changes in the transcriptome of circulating immune cells of a New Zealand cohort with myalgic encephalomyelitis/chronic fatigue syndrome by Eiren Sweetman, Margaret Ryan, Christina Edgar, Angus MacKay, Rosamund Vallings and Warren Tate in International Journal of Immunopathology and Pharmacology
Acknowledgments
We express our gratitude for the support of our ME/CFS patient and control cohorts. We thank the National ME/CFS Disease Association – Associated New Zealand ME Society (ANZMES) – for their ongoing support. We especially wish to thank the patients and healthy control subjects who willingly took part in our study.
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: We gratefully acknowledge the financial contributions and grants from the ME/CFS Association of New Zealand (ANZMES), the HS & JC Anderson Charitable Trust, NZ Lottery Health, Otago Medical Research Foundation and generous private bequests towards this study.
Supplemental material: Supplemental material for this article is available online.
ORCID iD: Warren Tate  https://orcid.org/0000-0002-6971-7734
https://orcid.org/0000-0002-6971-7734
References
- 1. Shepherd C, Chaudhuri A. (2017) ME/CFS/PVFS: An Exploration of the Key Clinical Issues, 9th edn. Gawcott: The ME Association. [Google Scholar]
- 2. Lorusso L, Mikhaylova SV, Capelli E, et al. (2009) Immunological aspects of chronic fatigue syndrome. Autoimmunity Reviews 8: 287–291. [DOI] [PubMed] [Google Scholar]
- 3. Carruthers BM, Kumar Jain A, De Meirleir KL, et al. (2003) Myalgic encephalomyelitis/chronic fatigue syndrome: Clinical working case definition, diagnostic and treatment protocols. Journal of Chronic Fatigue Syndrome 11: 7–115. [Google Scholar]
- 4. Nguyen CB, Alsøe L, Lindvall JM, et al. (2017) Whole blood gene expression in adolescent chronic fatigue syndrome: An exploratory cross-sectional study suggesting altered B cell differentiation and survival. Journal of Translational Medicine 15: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Comas M, Gordon CJ, Oliver BG, et al. (2017) A circadian based inflammatory response – Implications for respiratory disease and treatment. Sleep Science and Practice 1: 18. [Google Scholar]
- 6. Tomas C, Brown A, Strassheim V, et al. (2017) Cellular bioenergetics is impaired in patients with chronic fatigue syndrome. PLoS ONE 12: e0186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naviaux RK, Naviaux JC, Li K, et al. (2016) Metabolic features of chronic fatigue syndrome. Proceedings of the National Academy of Sciences of the United States of America 113: E5472–E5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Petschner P, Gonda X, Baksa D, et al. (2018) Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience 370: 207–217. [DOI] [PubMed] [Google Scholar]
- 9. Tomas C, Newton J. (2018) Metabolic abnormalities in chronic fatigue syndrome/myalgic encephalomyelitis: A mini-review. Biochemical Society Transactions 46: 547–553. [DOI] [PubMed] [Google Scholar]
- 10. Gow JW, Hagan S, Herzyk P, et al. (2009) A gene signature for post-infectious chronic fatigue syndrome. BMC Medical Genomics 2: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang L, Gough J, Christmas D, et al. (2010) Microbial infections in eight genomic subtypes of chronic fatigue syndrome/myalgic encephalomyelitis. Journal of Clinical Pathology 63: 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplementary_Material for Changes in the transcriptome of circulating immune cells of a New Zealand cohort with myalgic encephalomyelitis/chronic fatigue syndrome by Eiren Sweetman, Margaret Ryan, Christina Edgar, Angus MacKay, Rosamund Vallings and Warren Tate in International Journal of Immunopathology and Pharmacology
Supplemental material, Supplementary_references_used_to_support_statements_in_the_text for Changes in the transcriptome of circulating immune cells of a New Zealand cohort with myalgic encephalomyelitis/chronic fatigue syndrome by Eiren Sweetman, Margaret Ryan, Christina Edgar, Angus MacKay, Rosamund Vallings and Warren Tate in International Journal of Immunopathology and Pharmacology