

Abstract
Prader–Willi and Angelman syndromes are often referred to as a sister pair of neurodevelopmental disorders, resulting from different genetic and epigenetic alterations to the same chromosomal region, 15q11-q13. Some of the primary phenotypes of the two syndromes have been suggested to be opposite to one another, but this hypothesis has yet to be tested comprehensively, and it remains unclear how opposite effects could be produced by changes to different genes in one syndrome compared to the other. We evaluated the evidence for opposite effects on sleep and eating phenotypes in Prader–Willi syndrome and Angelman syndrome, and developed physiological–genetic models that represent hypothesized causes of these differences. Sleep latency shows opposite deviations from controls in Prader–Willi and Angelman syndromes, with shorter latency in Prader–Willi syndrome by meta-analysis and longer latency in Angelman syndrome from previous studies. These differences can be accounted for by the effects of variable gene dosages of UBE3A and MAGEL2, interacting with clock genes, and leading to acceleration (in Prader–Willi syndrome) or deceleration (in Angelman syndrome) of circadian rhythms. Prader–Willi and Angelman syndromes also show evidence of opposite alterations in hyperphagic food selectivity, with more paternally biased subtypes of Angelman syndrome apparently involving increased preference for complementary foods (“baby foods”); hedonic reward from eating may also be increased in Angelman syndrome and decreased in Prader–Willi syndrome. These differences can be explained in part under a model whereby hyperphagia and food selectivity are mediated by the effects of the genes SNORD-116, UBE3A and MAGEL2, with outcomes depending upon the genotypic cause of Angelman syndrome. The diametric variation observed in sleep and eating phenotypes in Prader–Willi and Angelman syndromes is consistent with predictions from the kinship theory of imprinting, reflecting extremes of higher resource demand in Angelman syndrome and lower demand in Prader–Willi syndrome, with a special emphasis on social–attentional demands and attachment associated with bedtime, and feeding demands associated with mother-provided complementary foods compared to offspring-foraged family-type foods.
Keywords: Angelman syndrome, Prader–Willi syndrome, evolutionary medicine, genomic imprinting, hyperphagia, sleep
Introduction
Prader–Willi syndrome (PWS) and Angelman syndrome (AS) are often referred to as a sister pair of genetic disorders, both resulting in cognitive and neurological impairments, along with unique physiological and behavioral phenotypes.1 Similarly, both AS and PWS show some degree of comorbidity with autism spectrum disorders (ASDs) as compared to the general population.2–5 However, expression levels of several traits related to early childhood development, such as birth weight, interest toward suckling and somnolence have been suggested to be opposite between the syndromes.6 The behavioral phenotypes of the syndromes have also been noted to contrast each other, as individuals with AS typically show a sociable disposition, with constant smiling and laughter, while individual PWS tends to show considerable negative affect, with frequent temper tantrums and obsessive–compulsive behavior.1,7 While both syndromes are due to multiple types of different mutations in the 15q11-q13 chromosome region, the specific nature of each mutation is critical to the epigenetic alterations involved in each syndrome as shown in Figures 1 and 2 and Table 1. AS is due to the absence of the maternal copy of the 15q11-q13 chromosome region (the UBE3A gene in particular), while PWS is due to the absence of the paternal copy of the same chromosome region. The resulting genetic and epigenetic alterations lead to losses of expression for separate sets of genes in each syndrome, which presents an apparent paradox. Are the phenotypes of these syndromes truly opposite of each other and, if so, what underlying factors could explain the opposite nature of the phenotypes despite the different nature of the genetic alterations involved in each syndrome?
Figure 1.

A schematic of the relevant genes in the 15q11-q13 chromosome region. The paternally expressed genes involved in PWS are marked in blue, while the maternally expressed genes involved in AS are marked in red. Genes marked in gray are silenced by an imprinting mechanism, while the genes in green are expressed from both parental copies. The arrow in blue shows the region specific to a long, non-coding antisense transcript that contains a sequence complimentary to UBE3A (UBE3A-ATS). This transcript is transcribed only from the paternal allele and thought to regulate the silencing of the paternal copy of UBE3A in neurons.8
Figure 2.

Illustration of the different genotypes involved in PWS and AS. In typical development, paternally imprinted genes are expressed only from the maternally derived chromosome and vice versa. In PWS and AS, de novo mutations lead to a lack of expression for paternally or maternally expressed genes in the 15q11q-q13 chromosome region. However, the dosage of paternally and maternally expressed genes in the chromosome region varies depending on the genotype as shown above.
Table 1.
A comparison of the different mutations and the effects on the imprinted and non-imprinted genes in the Prader-Willi and Angelman syndromes. Note that while loss-of-function mutations for UBE3A show the full phenotype of Angelman syndrome, no single gene mutation has been shown to reproduce the full phenotype of Prader-Willi syndrome.
| Deletion | Uniparental disomy | Imprinting defect | Loss-of-function mutations | |
|---|---|---|---|---|
| PWS | ||||
| Frequency | 65%–75% of affected individuals5 | 20%–30% of affected individuals5 | 1%–3% of affected individuals5 | Not applicable |
| Effect on imprinted genes | No expression of paternally expressed genes in the 15q11-q13 chromosome region5 | No expression of paternally expressed genes in the 15q11-q13 chromosome region, predicted increases in dosage for maternally expressed genes5 | No expression of paternally expressed genes in the 15q11-q13 chromosome region, predicted increases in dosage for maternally expressed genes5 | Not applicable |
| Effect on non-imprinted genes | One copy of the GABRB3, GABRB5, GABRG3, OCA2 and HERC2 genes, additional loss of TUBGC5, CYFIP1, NIPA1 and NIPA2 in Class I deletions5 | None | None | Not applicable |
| AS | ||||
| Frequency | ~70% of affected individuals9 | ~2% of affected individuals9 | ~2%–3% of affected individuals9 | ~25% of affected individuals9 |
| Effect on imprinted genes | No expression of UBE3A in neurons10 | No expression of UBE3A in neurons10 | No expression of UBE3A in neurons10 | No expression of UBE3A in neurons10 |
| Effect on non-imprinted genes | One copy of the GABRB3, GABRB5, GABRG3, OCA2 and HERC2 genes, additional loss of TUBGC5, CYFIP1, NIPA1 and NIPA2 in Class I deletions5 | None | None | None |
PWS: Prader–Willi syndrome; AS: Angelman syndrome.
Both PWS and AS involve a number of imprinted genes, which are expressed in a manner dependent on their parental origin. According to the kinship theory, genomic imprinting may evolve from intragenomic conflict when genes of different kin benefit from different phenotypes of the same trait.11 In particular, genes that may increase the inclusive fitness of the mother by exerting effects that lead to more equal distribution of resources among offspring are expected to be expressed only when maternally inherited. In contrast, maternally imprinted (paternally expressed) genes are expected to exert effects that lead to increased demands imposed on the mother, increasing the fitness of the child’s paternal genes. Consequently, the phenotype of PWS has been argued to reflect extreme, pathological development of phenotypes associated with fitness benefits to the mother.4 Thus, as children with PWS fail to express paternally derived genes, the resulting phenotype displays low birth weight, sleepiness and low activity, poor sucking ability and a failure to thrive,7 followed by gradual development of a voracious appetite, which coincides with the timing of early adrenarche, around the age of 8–9 years.12,13 As maternally and paternally derived genes tend to disagree over the allocation of maternal resources, it has been further argued that sucking presents a cost to the mother’s inclusive fitness through nutritional value and conversely the transition to family and self-foraged foods after weaning presents comparably reduced maternal costs.4
In contrast to PWS, subjects with AS fail to express maternally derived genes and show comparatively high birth weight14 and have been argued to sleep less than their peers.15 In the context of the kinship theory and parent–offspring conflict, the behavioral phenotype of AS has been argued to reflect an extreme pathological development of phenotypes related to affect signaling.16,17 Smiling and laughter are hypothesized as signals of positive affection, which have fitness benefits to the child, sending the signal, and fitness costs to the mother, receiving the signal and providing increased parental attention.
Based on the lines of evidence and theory described above, core phenotypes of PWS and AS have been considered to result from disruptions of genetic conflict, where the disappearance of one side of opposing developmental influences leads to an extreme response disadvantageous to both the mother’s and the child’s inclusive fitness. However, the opposite nature of the phenotypes of these two syndromes has never been evaluated in any detail.
In this article, we focus on reviewing two aspects of the behavioral phenotypes of PWS and AS, sleeping and eating behavior.
First, we provide relevant background on the genetic and epigenetic causes of PWS and AS, focusing on the different dosages of paternally and maternally expressed genes in each syndrome. Second, we use currently available research to determine the degree to which the observed phenotypes of sleep and eating behavior in PWS and AS are opposite to one another and in what respects. Third, we review current research on mouse models of PWS and AS to assess the roles of imprinted genes in sleep and behavior in the two syndromes and to develop genetic and physiological hypotheses to explain how the different genetic alterations in the two syndromes could produce the observed phenotypes. Finally, we evaluate whether the phenotypic patterns observed in PWS compared to AS, and our hypotheses, are consistent with the kinship theory of genomic imprinting.
Methods
Comprehensive searches of the literature were conducted for each aspect of the review, with both review and research articles utilized. The search was conducted with the Web of Science database using both term- and reference-based search strategies. Search terms included Prader-Willi and Angelman, coupled with relevant terms including gene names and general terms related to traits and characteristics including obesity, hyperphagia, sleep, sleep pressure, duration, latency, REM, apnea, daytime sleepiness, somnolence, circadian, melatonin, food-related behavior, food preferences, food refusal, eating, feeding, homeostatic feeding hedonic feeding, serotonin and dopamine.
Literature search for meta-analysis of sleep latency and duration in PWS
We searched both PubMed and Web of Science databases for peer-reviewed scientific articles up to May 2018. Search terms were “Sleep AND Prader-Willi,” which brought up 208 articles at PubMed and 371 articles at Web of Science, which were all screened manually. The search was further supplemented by manual searching strategies such as articles cited in the literature relevant to PWS. Studies were included in the meta-analysis based on the following criteria: (1) the study included a measurement of sleep onset latency or sleep duration on PWS individuals and a comparison to a control group of typically developing individuals; (2) sufficient data were available for estimating mean and standard deviation of the relevant sleep parameters. The application of these criteria yielded a total of eight studies for sleep onset latency as well as seven studies for sleep duration. The selection process is detailed in full with a Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) checklist provided in the Supplementary File and summarized in a flow diagram (see Supplemental Figure 3.)
Statistical procedures
As a few of the studies involved in our analyses only provided ranges and medians for the relevant sleep traits, the mean and the standard deviation of these parameters were estimated based on the procedures described in Hozo et al.10 The meta-analysis was conducted with the R software (version 3.5.0 “Joy in playing,”9 using the metafor package).18 We chose a fixed-effect model for our approach and Hedge’s G was used as a measurement of the effect size. Heterogeneity among the studies was measured using the Q test, while publication bias was evaluated using a weighted regression test with multiplicative dispersion to test for funnel plot asymmetry between the estimate of effect size and within-study standard error.
Results
Genetic, genomic and epigenetic causes of PWS and AS
The 15q11-q13 chromosome region contains a number of both paternally and maternally imprinted genes as shown in Figure 1. AS has been shown to be caused by a lack of expression for the maternally derived copy of UBE3A.19,20 More specifically, the paternal copy of UBE3A is uniquely silenced in neurons21 and so deletions or epigenetic alterations affecting the maternal copy of the gene lead to a complete lack of UBE3A expression in neurons. By contrast, no single mutation of a gene has been shown to display the full phenotype of PWS, although lack of expression for the SNORD116 snoRNA has been shown to produce several central aspects of the phenotype of PWS, indicating an important role in the development of the disorder.22
As both PWS and AS are ultimately due to de novo mutations of the same chromosome region, it follows that deletions involve a loss of one parental copy of the 15q11-q13 region, while in uniparental disomy both chromosomes are derived from the same parent. The imprinting mechanisms involved in both PWS and AS are further controlled by the imprinting center of the 15q11-q13 chromosome region, which is defined by the shortest mutations known to produce the full phenotype of each syndrome. The paternal copy of the imprinting center is transcribed as a part of a long, non-coding antisense transcript, which spans across the imprinting center and the SNURF-SNRPN gene to the end of the opposite strand of UBE3A and is thought to regulate the silencing of the paternal copy of UBE3A in neurons.8 In contrast, the maternal copy of the imprinting center is typically methylated, preventing the expression of the antisense transcript.23 Thus, imprinting defects result from either microdeletions which span the shortest region of overlap for each syndrome or varied epigenetic causes. These genotypes demonstrate either a fully paternal or a fully maternal methylation pattern despite the presence of both parental alleles, as is also shown in Figure 2.
The different mechanisms behind the genotypes of the two syndromes lead to the corresponding differences in dosage for the imprinted genes involved, as also shown in Figure 2 and Table 1. Maternal uniparental disomy (matUPD) in PWS involves two maternally expressed copies of UBE3A and ATP10A, and conversely the genotype of paternal uniparental disomy (patUPD) in AS involves two copies of the paternally expressed genes in the locus. Deletions involve additional losses of non-imprinted genes which may result in haploinsufficiency for a varying number of genes depending on the size of the deletion (see Table 1).
Sleep in PWS and AS
Sleep phenotypes and the regulation of circadian rhythms
Sleep phenotypes are notably altered in both AS and PWS and have been well studied in both.
By way of background, the sleep–wake cycle is regulated by two major processes: first, homeostatic sleep pressure which is defined as the gradual accumulation of sleep factors (i.e. peptides, hormones and neurotransmitters) promoting sleep in the brain, which slowly inhibit the function of wake-promoting neural pathways.24 Second, circadian rhythms can be viewed as a network between the internal time-keeping mechanisms based on the self-maintaining feedback loop of the core time-keeping genes Per, Cry, CLOCK and BMAL1 and their target genes, which in turn mediate circadian timing in physiological processes such as sleep, feeding and energy balance.25 In vertebrates, circadian rhythms form a hierarchical network between the suprachiasmatic nucleus (SCN) and a number of peripheral circadian clocks in different tissues and cells.26 The secretion of melatonin from the pineal gland is controlled by a projection from the SCN.27 However, the circadian integration of sleep, activity and feeding further depends on a reciprocal connection between the SCN and the arcuate nucleus (ARC).28 Recent work has also revealed that there may be a higher than expected number of imprinted genes expressed in the hypothalamus.29 Given the crucial role of the hypothalamus in the regulation of sleep, it can be seen that imprinted genes similarly influence the regulation of sleep via the hypothalamus.30 Similarly, a number of these imprinted genes are also central to the genotypes of the PWS and AS,9,16 influencing the specific sleep phenotypes typical to each syndrome.
The quality of sleep has been traditionally measured with subjective observations, but a more precise study of the sleep phenotype and structure can be conducted with a combination of electrophysiological recordings (i.e. polysomnography). While research on sleep relies on the simultaneous measurement of electrical brain activity (i.e. electroencephalography (EEG)) and physiological parameters such as heart rate and respiration, these are only used as biomarkers for the underlying neural and physiological states.31 The aforementioned states of sleep can be broadly divided into rapid eye movement (REM) sleep, defined by a high degree of brain activity and its counterpart, and non-rapid eye movement (NREM) sleep, conversely defined by a lack of such activity. NREM sleep is maintained by the neural circuitry controlling homeostatic sleep pressure, while REM sleep is promoted by the subdorsolateral nucleus.24 Together, the two opposing processes produce the typical cycle of NREM and REM sleep states.31
A range of qualitative and quantitative sleep phenotypes are altered in both PWS and AS.15,32–34 In this section, we describe research on sleep phenotypes that have been studied in both AS and PWS, to address whether or not they exhibit opposite or similar features, and how these features are associated with the genetic and epigenetic alterations that underlie the two syndromes. In particular, we will discuss six main sleep phenotypes in the context of both syndromes:
Sleep onset latency and difficulties with falling asleep: sleep onset latency can be defined as the quantification of time from sleep attempt to initiation, and difficulties with falling asleep refer to any occurrence where the subject experiences difficulties with falling asleep in the evening, regardless of the underlying cause.
Sleep duration, which refers to the total time spent asleep during a 24-h period.
Sleep efficiency, commonly defined as a part of an overnight sleep study, calculated as the time spent asleep compared to the time spent in bed during the measurement period.
Sleep architecture, referring to the durations and total percentages of the various sleep stages such as REM sleep and slow-wave sleep, commonly measured during an overnight sleep study with a polysomnographic recording.
Daytime sleepiness, referring to increased sleepiness during the day, quantified either with a subjective estimate as a part of a caretaker survey or with a study measuring the subject’s proneness to falling asleep during the day (e.g. the multiple sleep latency test (MSLT)).
Associations of sleep traits with melatonin, a hormone involved in the regulation of the sleep–wake cycle. Namely, melatonin secretion from the pineal gland is controlled directly by the suprachiasmatic nucleus,27 so the secretion of melatonin may be altered in AS and PWS, influencing their sleep phenotypes.
The overall features of sleep phenotypes in AS and PWS
Extreme sleep disturbances, abnormal sleep–wake cycles and diminished need for sleep are typically described as features of the sleep phenotype in AS.15,35,36 The abnormal sleep–wake cycle refers to the common observation that individuals with AS often have difficulties in initiating and maintaining sleep and sleep less than their age-matched peers as a result. The feature is especially prominent in childhood, but continues to improve toward adulthood with a moderate prevalence.35,36 While the lack of sleep is often described to “not affect the alertness or activity level of the subject or even their personal quality of life,”15 daytime sleepiness has also been reported in a number of studies.35–38 However, a recent meta-analysis later inferred that the trait was not significantly affected in AS.32 Nevertheless, as the sleep phenotype of AS is highly disturbed, the genotype of the syndrome may also be involved with the regulation of sleep.
The sleep phenotype of PWS is characterized most broadly by excessive daytime sleepiness (EDS),33 apparent problems with organization of REM sleep patterns39 and obstructive sleep apnea (OSA).40 While daytime sleepiness is often attributed to OSA, daytime sleepiness in PWS is often described to be unrelated to the quantity and quality of nocturnal sleep.41
As noted above, previous studies have shown that sleep phenotypes are prominently altered in both PWS and AS. However, no study to date has systematically compared these syndromes with regard to their sleep phenotypes.
Sleep onset latency and difficulties with falling asleep
Extreme variation of sleep onset emerges as a prominent phenotype in numerous studies concerning the sleep phenotypes of both AS and PWS. Several questionnaire studies concerning sleep and sleep-related behaviors have addressed difficulties with falling asleep and bedtime resistance (behavioral avoidance of bedtime), while a number of physiological overnight sleep studies (polysomnography) have further quantified the trait as sleep onset latency, defined as the time spent between settling down in bed and falling asleep, based on the detection of the first stage of sleep in the subject’s electroencephalogram pattern.
The notion of prolonged sleep onset latency in AS is supported by a recent meta-analysis of sleep studies, which showed a moderate but significant effect size for difficulties with falling asleep across relevant questionnaire studies concerning AS subjects.32 In closer detail, all three questionnaire studies reviewed for the meta-analysis state that about 50%–60% of AS subjects are reported to show frequent difficulties with falling asleep35–37 indicating that the sleep phenotype of AS may involve increased sleep onset latency as compared to typically developing individuals. Similarly, a recent meta-analysis of relevant overnight sleep studies indicated that sleep onset latency is significantly increased in AS across the relevant studies.32 In contrast, a study comparing sleep problems in groups of infants with different neurogenetic disorders found that infants with AS showed marginally shortened sleep onset latency, as compared to the control group of typically developing infants.42 However, the authors mention that the AS group also showed atypically longer night wakings as compared to the control group and showed a high amount of variation for night waking frequency (a range of 0–10 awakenings in a night) which suggests that problems with maintaining and initiating sleep may emerge in early infancy in AS.
Numerous behavioral studies have characterized behaviors with further connections to bedtime and falling asleep. First, behaviors indicating high sensitivity to the sleep environment, characterized by regular complaints on feeling uncomfortable in bed, were estimated as significantly more common among AS subjects as compared to the control subjects.32 Second, fear or anxiety regarding sleep has been shown to be higher in AS than in matched controls, by meta-analysis.32 Third, regular use of both medical and non-medical sleep aids (such as light in bedroom or security objects) has also been estimated to be significantly higher among AS subjects compared to typically developing controls.32 Finally, several studies note a particular tendency for bedtime resistance and insistence on particular bedtime routines among individuals with AS. A questionnaire study on sleep problems on individuals with AS notes that reluctance to go to bed was reported in a significantly higher proportion of AS subjects (approximately 60% of individuals under the age of 15 years) compared to typically developing controls.36 Insistence on particular bedtime routines has similarly been reported in 63% of the subjects.34 Furthermore, a small-scale trial study found that behavioral treatment for children with AS, including regular sleep schedules and adequate parent–child bedtime interactions, led to shortening of sleep onset latency and the children also showed significant improvements in bedtime behavior and were able to fall asleep independently, as opposed to baseline results prior to treatment.43 A questionnaire study on parental stress and the sleep habits of children with AS found that the sleep time variability of the child was positively correlated with parental stress. The children with AS also showed a higher level of concerning sleep habits including bedtime resistance, sleep anxiety, night waking and difficulties in falling asleep, compared to a group of typically developing children.44 In summary, the increased bedtime resistance, insistence of particular bedtime routines and wide use of both medical and non-medical sleeping aids suggest that the increased sleep onset latency of AS subjects may be caused by their greater restlessness around bedtime.
In PWS, numerous sleep studies show opposite results in comparison to AS, with questionnaire studies showing a lack of problems related to sleep onset, while overnight sleep studies offer mixed support for shortened sleep onset latency. In particular, an early questionnaire study on sleep and behavioral problems in PWS found that none of the PWS subjects were reported to suffer from regular sleep settling problems.45 Similarly, a later questionnaire study on sleep and behavioral problems in PWS found that only 1 of the 79 subjects was reported to have a regular sleep settling problem.46
As overnight sleep studies in PWS provide conflicting evidence on sleep onset latency, we conducted a meta-analysis of the relevant studies39,42,47–52 to further quantify sleep onset latency in PWS, as compared to typically developing individuals.
We chose a fixed-effect model for our approach, as a relatively small number of studies fit with our inclusion criteria and fixed-effect models are known to perform better when the number of studies included is low.16 Furthermore, the measured trait was defined in a similar fashion across the studies, and as all of the studies involve a comparison between PWS individuals and age-matched controls, we would expect the true effect of the condition on sleep onset latency to be rather similar across the studies.
Our findings with the meta-analysis of sleep onset latency in PWS were threefold. First, we found that sleep onset latency in PWS shows a small but significant negative effect in comparison to typically developing individuals, indicating evidence of reduced sleep onset latency in PWS. Second, our analysis indicates that there is significant heterogeneity across the studies. This finding mirrors the recent large-scale meta-analysis on sleep traits in AS32 and likely reflect the variation across the relevant studies with other factors potentially influencing sleep traits, such as age, and medication and OSA. The scale of the effects shown in each study and the overall effect across the studies is also shown in Figure 3. Finally, we conducted an analysis of publication bias by fitting the relevant studies into a funnel plot and found no significant evidence of publication bias with respect to sleep onset latency.
Figure 3.

The effect of PWS in relation to sleep onset latency. The effect size for each study is shown, measured with Hedge’s G, along with upper and lower limits of the effect sizes. The overall effect is the weighted average of the effect sizes.
In summary, there is a diametric pattern to sleep onset latency in AS compared to PWS individuals. Both questionnaire studies and overnight sleep studies offer support for the notion of increased sleep onset latency in AS. In comparison, the opposite pattern is shown in PWS, as both questionnaire studies and overnight sleep studies support the notion of reduced sleep onset latency in relation to typically developing individuals.
Sleep duration in AS and PWS
Extreme phenotypes of sleep duration have been characterized in both AS and PWS, and, in direct comparison, infants with AS showed a significantly shortened sleep duration as compared to typically developing controls, while the PWS group showed a longer sleep duration, though only approaching significance compared to controls.42
Questionnaire studies on sleep problems in AS have featured questions on whether the caretaker felt that the child slept less than children of their age, and this item has been endorsed consistently across studies with a prevalence of about 40%–50% among AS patients.35,37 Furthermore, a questionnaire study on sleep problems in AS found that a significantly greater percentage of the AS subjects compared to the control group of typically developing children was reported to regularly sleep less than 8 h in a day.36 Similarly, a recent meta-analysis of currently available overnight sleep studies shows a significantly shortened sleep duration (of about 7 h) in AS individuals, as compared to controls.32
Sleep duration in PWS has been primarily with overnight sleep studies, while questionnaires have focused mainly on sleep problems.45,46 A recent sleep study by Ghergan et al.53 found that approximately 58% of adult PWS subjects were reported to sleep more than 9 h in day. While 8% of the participants were found to sleep over 11 h in a 24-h sleep recording session, the mean sleep duration of PWS subjects was below 8 h. As earlier overnight sleep studies have provided similarly conflicting results on sleep duration, we conducted a meta-analysis of all previous overnight sleep studies which featured a comparison between PWS subjects and a typically developing control group.
A total of seven studies fulfilled our criteria.42,47–52 First, our analysis with a fixed-effect model indicated that sleep duration in PWS subjects did not show a significant effect in either direction as compared to typically developing individuals. Second, our analysis indicated significant heterogeneity across the studies. Finally, the funnel plot analysis did not indicate a significant publication bias with respect to sleep duration.
In summary, relevant studies have shown support for significantly shortened sleep duration in AS, but an opposite pattern of increased sleep duration cannot be seen in studies with PWS. Our analysis of currently available studies indicates that despite relatively high self-reported levels of sleepiness and long sleep durations in PWS, sleep duration in PWS as a whole does not differ significantly from typically developing subjects.
Sleep efficiency in AS and PWS
Sleep efficiency reflects the time spent awake during the night, which is affected by both sleep latency and wakefulness after sleep onset and can be induced by factors such as apnea or restlessness. In AS, four overnight sleep studies have included measurement of sleep efficiency and were later compiled into a meta-analysis, which showed that sleep efficiency is significantly reduced in AS compared to control subjects.32 A particular overnight sleep study also notes that AS subjects showed higher wakefulness after sleep onset compared to patients with varied intellectual disabilities.54
In PWS, sleep efficiency has been especially studied in connection to apnea. OSA has been assessed as a co-occurring condition with PWS, with a meta-analysis reporting an 80% prevalence among 224 patients from a total of 14 studies.40 Apnea in PWS has traditionally been associated with obesity and, in support of this notion, the meta-analysis study also reported that greater body mass index (BMI) was positively correlated with OSA. However, a later collaborative study of a large cohort of PWS patients found no correlation between obstructive apnea; high BMI and facial dysmorphic features or hypotonia were instead suggested to play a role in the occurrence of OSA.55
While OSA could be expected to affect the sleep efficiency significantly, results from other overnight sleep studies in PWS are somewhat inconclusive. Two overnight sleep studies have reported a reduced sleep efficiency as compared to normative values,49,56 while three other studies found no significant difference in sleep efficiency between the PWS and control groups or normative values.48,50,57
In summary, sleep efficiency is reduced in both PWS and AS; however, current studies suggest that the underlying reasons are different. In AS, longer sleep latency and wakefulness after sleep onset are likely to affect sleep efficiency negatively, whereas the high prevalence of obstructive apnea is likely to affect sleep efficiency in PWS. While obesity has been shown to correlate positively with apnea, other factors may also affect sleep efficiency in PWS.
Variation in sleep architecture
Several polysomnographic studies have noted differences in sleep structure in AS and PWS patients. First, a significantly reduced overall amount of REM sleep as compared to control subjects has been found in AS.54,58 Similarly, a meta-analysis of overnight sleep studies in PWS found that 55% of PWS patients analyzed in a total of 20 sleep studies showed a reduced percentage (REM% < 20) of REM sleep.33 While two later sleep studies found that overall REM sleep percentage did not differ significantly from normative values,56,57 an overnight sleep study comparing PWS subjects and typically developing individuals found that PWS subjects had a significantly reduced overall REM sleep percentage as compared to controls.49
Second, an opposite pattern between PWS and AS is suggested with regard to the timing of onset for REM sleep. Typically, the onset of REM sleep first occurs approximately 90 min after the onset of sleep.31 However, AS patients show a considerably increased REM latency, though a comparison with a control group only approached statistical significance.54,58 Conversely, in PWS REM sleep is often present during sleep onset. A meta-analysis of sleep studies in PWS found that 27% of PWS subjects showed the presence of REM during sleep onset at night, while a further 34% showed an onset of REM during a daytime nap. Similarly, a shortened REM latency (>70 min) was reported in 17% of PWS subjects.33 Intrusions of REM sleep during daytime naps have similarly been found in later studies concerning PWS.53,57 Fragmentation of sleep architecture is also evident in PWS, as multiple sleep studies have reported either a significant increase in sleep stage shifts49 or an increased amount of REM sleep periods.39,48
In summary, reduced overall amount of REM sleep has been shown in both AS and PWS, while an opposite pattern is suggested in REM latency, with reduced REM latency in PWS and increased REM latency in AS. As the hypothalamus is known to regulate sleep through both neural mechanisms31 and circadian rhythmicity,27 the opposite dysregulations of REM onset could also be indicative of disrupted regulation of sleep onset in PWS and AS.
Daytime sleepiness
EDS is a common concern with PWS and has been studied in both questionnaire and overnight sleep studies, while several questionnaire studies have also addressed the condition in AS.
First, daytime sleepiness among AS subjects has been reported in several questionnaire studies. A recent meta-analysis of these studies showed a small but significant effect size, indicating a higher prevalence of daytime sleepiness in AS compared to controls.32
Second, two questionnaire studies have addressed daytime sleepiness in connection to behavioral problems in PWS. A questionnaire study on sleep and behavioral problems noted that daytime sleepiness was reported in PWS subjects with significantly greater prevalence (about 35%) compared to a control group, and that daytime sleepiness was correlated positively with behavioral problems.45 A later questionnaire study similarly found that daytime sleepiness is shown with about 35% of PWS subjects, but no significant correlations between sleep problems and behavioral problems were found.46 A recent large-scale sleep study found that about 50% of PWS subjects showed a questionnaire result indicative of daytime sleepiness, while about 65% of the subjects reported sleepiness as a problem.53
Several sleep studies have also quantified daytime sleepiness in PWS with MSLTs. Commonly used as a quantitative measure of EDS and narcolepsy, MSLT measures the onset of sleep during multiple 20-min daytime nap periods. A mean sleep onset time lower than 5 min is usually considered to indicate that the condition is affecting the patient’s daytime activities. A meta-analysis on sleep studies in PWS33 lists a total population of 72 patients across eight different studies, with 40% of the subjects showing an MSLT result indicative of being severely affected.
Daytime sleepiness is commonly attributed to a deficiency of sleep due to OSA, which is a common concern in PWS.33 However, while apnea has been suggested as a co-occurring condition in PWS and apnea has also been shown to correlate positively with the BMI among PWS subjects, support for the causation between EDS and OSA in PWS is not entirely conclusive. Several studies have indeed reported a positive correlation between apnea or BMI and a measure of sleepiness with PWS subjects,33,46 but a number of conflicting studies found no significant correlations between BMI or reports of apnea and sleepiness in PWS.45,53,57
In summary, daytime sleepiness shows a significantly increased prevalence as compared to controls in both PWS and AS. In PWS, the trait may be partially attributable to OSA, but the underlying causes are likely to be multifactorial. In AS, the condition is likely to be connected to reduced sleep duration and frequent waking at night.
Associations with melatonin secretion
Circadian rhythms regulate sleep patterns, and the secretion of melatonin is directly regulated by the circadian period in the SCN.27 As a disruption of sleep patterns is evident in both AS and PWS, an imbalance in the secretion of melatonin may be affecting the sleep phenotypes of the two syndromes.
Melatonin and its associations to sleep phenotype have been studied in AS using sleep studies and treatment trials. A study on circadian rhythms and sleep traits in AS reported that circadian sleep disorders were diagnosed with a prevalence of about 50% among AS subjects, and they found that nighttime serum levels of melatonin were significantly reduced in AS subjects as compared to a control group.59 A later overnight sleep study similarly found that AS subjects showed a significant delay in the pattern of nighttime melatonin secretion as compared to the control group.60 Furthermore, treatment trials among AS subjects have shown that treatment with melatonin improves sleep duration and reduces sleep latency.61,62
While melatonin and its connection to sleep traits have not been studied as extensively in PWS as in AS, a study on endocrinal traits found that melatonin levels of PWS subjects did not differ significantly from typical controls.63 A later study on morning melatonin levels in PWS subjects similarly reported that serum melatonin levels did not differ from the control group.64
In conclusion, current evidence supports a reduction in nighttime melatonin secretion in AS. However, an opposite pattern is not seen in PWS, as melatonin secretion has not been shown to differ significantly from controls in currently available studies.
Effect of genotype and gene dosage on sleep phenotypes in AS and PWS
Given the different dosages of paternally and maternally expressed genes between the different genetic subtypes of PWS and AS (as shown in Figure 2), variation in sleep phenotypes between the genetic subtypes could be expected in both syndromes. However, several studies of sleep phenotypes in both AS and PWS have independently reported that no significant associations between genetic subtype and specific sleep phenotypes were found in their results.36,46,49,53,56,57,65 The only exception is that the paternal UPD genotype of AS has been associated with an increased tendency toward expressive sleeping disturbances, such as nightmares,35 though a later questionnaire study was unable to replicate this result.37 The lack of variation in sleep phenotypes within each syndrome may indicate that the mechanisms for the alterations of sleep phenotypes apparent in each syndrome are not dependent on gene dosage effects, but rather the lack of expression for the imprinted genes in both syndromes.
Genetic bases of sleep phenotypes and circadian rhythms in PWS and AS
Recent mouse model studies have highlighted the roles of imprinted genes in the regulation of circadian rhythms and sleep.30 Several mouse models of both PWS and AS have shown that imprinted genes take part in the regulation of circadian rhythms.
Circadian clock mechanisms regulate the daily cycle of activity and rest through a transcriptional feedback loop of the core clock genes, which in turn coordinate the daily oscillation of thousands of diurnally regulated genes.66 Diurnally regulated genes also show periodic changes in methylation, which further corresponds to rhythmic changes in expression patterns of these genes.67 The transcriptional feedback loop of the core circadian clock is composed of the activators Clock and BMAL1 (ARNTL in human), which act as transcription factors for both other clock genes and diurnally regulated genes, and the repressors, Period (Per1, Per2 and Per3) and Cryptochrome (Cry1, Cry2), which regulate the activators, eventually suppressing their own transcription. The expression of the core clock genes follows a roughly 24-h circadian rhythm, which, by convention, is indicated as circadian time (CT), where CT 0 stands for the beginning of the subjective day and CT 12 stands for the beginning of the subjective night.25 The rhythmic expression of the core clock genes shows a 3- to 9-h delay between the master pacemaker in the SCN and peripheral tissues.26 In the mouse SCN, the relative level of gene expression for Bmal1 starts to rise after the subjective midday (CT 8) and peaks during the subjective evening (CT 12). The CLOCK and BMAL1 proteins accumulate in the nucleus and in turn activate the transcription of the Per and Cry genes around the midpoint of subjective night (CT 18), further reaching the relative peak of their expression around subjective midday. The PER and CRY proteins accumulate over time and interact with CLOCK and BMAL1, effectively repressing their own transcription.25 The relative protein levels of both the repressors and the activators are further regulated by ubiquitination mechanisms, and genetic alterations to these mechanisms have been characterized to alter the length of the circadian period and phenotypes of sleep.25 The circadian rhythm of gene expression has been characterized in humans via the blood transcriptome, where the expression of the core clock genes follows a different phase, with transcription of Per genes reaching its relative peak during the subjective night, whereas the expression of Arntl (Bmal1) peaks during the day.68
Sleep and regulation of circadian rhythms in mouse models of AS
Several studies have highlighted the role of UBE3A in the regulation of sleep and circadian rhythms, suggesting that epigenetic mechanisms involving this gene could provide specific cues to the central circadian clock in the SCN.
Circadian rhythmicity has been studied in the mouse models of AS with somewhat conflicting results, due to the nature of the Ube3a imprinting mechanism in the SCN. A mouse model study by Ehlen et al.69 found that Ube3a imprinting is uniquely relaxed in the SCN, allowing the AS mouse model to largely maintain its circadian rhythmicity. The model mice showed no significant differences in their ability to maintain the circadian rhythms in constant darkness or aberrant lighting in comparison to controls. However, the model mice would skip a rest period typical to the wild-type mice, and they showed a blunted response to sleep deprivation, with a significant decrease in the overall amount of REM sleep during a 24-h period. The authors concluded that Ube3a regulates the accumulation of sleep pressure, the homeostatic mechanism in the regulation of sleep.
Contrary to these results, another mice model study by Shi et al.70 found that maternal deletion of Ube3a lengthens the circadian period. In similarity to Ehlen et al.,69 circadian rhythmicity was measured with running wheel activity in varied lighting and the AS model mice were found to show a significantly longer circadian period in constant darkness compared to controls, while the lengthened circadian period was even more pronounced in the mouse model with a larger deletion spanning from Ube3a to Gabrb3, which is similar to the Class 1 deletion of AS. Since Bmal1 has been identified as a target protein of the UBE3A ubiquitin ligase,71 the authors further suggested that Ube3a may regulate the turnover of Bmal1, and so the deficiency for expression of Ube3a would in turn lead to an excess of activators in the nucleus as opposed to the repressors. This imbalance would further lengthen the 24-h circadian period. As the secretion of melatonin is controlled by the input of the SCN,27 lengthening of the circadian rhythm would offer a direct explanation for the reduced melatonin levels and increased sleep onset latency in AS.
Molecular studies of the expression of clock genes in AS model mice have similarly produced contrasting results. Ehlen et al.69 initially reported that the model mice showed no significant differences in the expression levels of the clock genes or the protein levels of Per2 in SCN tissues, as compared to controls. Similarly, Jones et al.72 ascertained the expression of paternally derived Ube3a in the SCN by comparing protein levels of Ube3a in mice with maternal deletions, deletions of both copies and controls. As opposed to mice with deletions of both copies, Ube3a was detected uniquely in the SCN tissues of the AS model mice, implying that the imprinting of the paternal copy must be relaxed in the SCN. In contrast, Shi et al. ascertained the expression of the clock genes with a Per2:LUC reporter gene and compared the rhythmicity of Per2 expression between SCN slices and spleen and lung tissues. The lack of expression for Ube3a was found to significantly increase the length of the luminescence period in SCN tissues but not in the lung or spleen tissues, indicating that lack of expression for Ube3a may alter feedback loops of core clock genes.70 Furthermore, treatment with topotecan, a topoisomerase inhibitor, which may inhibit the gene silencing mechanisms, significantly shortened the luminescence period of Per2 in SCN slices of the model mice but not in the controls or other tissues, indicating that Ube3a expression from the paternal copy may have shortened the circadian period in the treated SCN slices, while the treatment leaves the circadian period unaltered in other tissues, where Ube3a is expressed from both parental copies.70
Current mouse model studies may thus indicate that circadian rhythmicity is mosaic in AS: imprinting of the paternal copy is relaxed in the peripheral tissues and the SCN, while other brain regions and neurons experience a lengthened circadian period. While the secretion of melatonin depends on an indirect projection from the SCN,27 the circadian integration of activity and rest further depends on a reciprocal connection between the SCN and the ARC.28 Reduced levels of melatonin secretion and a relatively high incidence of circadian sleep disorders have been shown in studies concerning AS.59,60 However, it remains unclear if the mosaic nature of circadian rhythmicity can fully explain the alterations to the sleep phenotypes of AS.
Sleep in mouse models of PWS
Recent mouse model studies have also linked the genotype of PWS to circadian rhythmicity. To date, two of the maternally imprinted genes affected in the syndrome, MAGEL2 and SNORD116, have been shown to be involved in the regulation of circadian rhythms.
A study of a PWS mouse model found that Magel2 shows a pattern of circadian expression in the SCN and that loss of Magel2 expression confers a phenotype of fragmented activity and rest.73 The length of the circadian period in constant darkness did not differ significantly from controls. However, the model mice showed significantly less nighttime activity compared to controls and a fragmentation for periods of activity and rest, evident as a significantly greater number of bouts of activity and a significantly shortened average bout duration. The expression of Magel2 in the SCN was shown to peak late in the day, with a marked decrease in levels of expression during the subjective night. The fragmentation of activity and rest and the circadian pattern of gene expression in the SCN thus indicate that Magel2 may be regulating the circadian clock mechanism in the SCN.
Further molecular studies have shown that the expression of Magel2 may further regulate the transcriptional feedback loop of the circadian clock mechanism.74 In a cell-based model, the co-expression of Magel2 with Clock and Bmal1 was shown to repress the transcription of Per2, as compared to the level of expression with only Clock and Bmal1 present. The effect was intermediate in strength, compared to the more repressive Cry which is known to regulate the negative feedback loop of the circadian clock mechanism. Several possible mechanisms for the repressing effect of Magel2 were investigated, and Magel2 was shown to promote the cytoplasmic accumulation of Clock. While Clock is expressed continuously in the SCN, the subcellular localization of the Clock protein shows circadian variation. Clock is primarily cytoplasmic by itself and starts to accumulate in the nucleus only as the relative levels of gene expression of Bmal1 start to rise. Therefore, Magel2 may further regulate a programmed delay in the circadian feedback loop period through post-translational modification of the Clock protein.74 As the expression of diurnally regulated genes is regulated by the nuclear accumulation of both Clock and Bmal1,25 the lack of a programmed delay in the circadian period due to lack of expression for Magel2 could offer a direct mechanism for the dysregulation of sleep and activity evident in PWS.
Loss of expression for Snord116 has also been shown to alter the expression of diurnally regulated genes. Powell et al.75 identified dysregulation of about 6000 diurnally regulated genes in the mouse cortex, including dysregulation of the pacemaker genes such as Cry1, Clock, Per2 and Mtor as well as increased expression of Ube3a, which regulates the oscillatory pattern of Bmal1 via ubiquitination.71 Coulson et al.76 further showed that loss of expression for Snord116 led to a pattern of shifted diurnal methylation characterized by losses during the light phase and increased diurnal methylation during the dark phase. The authors suggest that the gene expression of epigenetic and circadian regulators is increased in the model mice during the light phase, which may lead to prolonged accumulation of these proteins into the dark phase, resulting in the shifted methylation pattern. As circadian rhythms regulate the daily cycle of activity and rest by further regulating the gene expression of thousands of diurnally regulated genes, the dysregulation of these gene expression patterns through a shift in the diurnal methylation cycle due to lack of expression for SNORD116 could explain the dysregulation of activity and rest which is evident in PWS.
In conclusion, recent mouse model studies of sleep and circadian rhythmicity show that both MAGEL2 and SNORD116 are involved in the regulation of circadian rhythms and diurnally regulated gene expression. However, unlike UBE3A, none of the genes have been shown to alter the length of the circadian rhythm directly. Instead, MAGEL2 may alter the regulation of diurnal gene expression by regulating the circadian feedback loop in the SCN73,74 while SNORD116 may alter the expression of genes critical to the oscillatory pattern of the circadian rhythm, further affecting the expression of diurnally regulated genes.75,76
Model for explaining opposite sleep phenotypes in PWS and AS
Since both AS and PWS show evidence of opposite phenotypes for sleep onset latency, and relevant mouse model studies indicate that both the maternally expressed UBE3A and the paternally expressed MAGEL2 and SNORD116 may be involved in the regulation of circadian rhythmicity in the SCN, we propose a hypothetical model for explaining the opposite sleep phenotypes based on the known interactions of these genes with the circadian clock mechanisms. First, as a baseline, assuming that the gene expression pattern of the core clock genes in peripheral tissues is delayed by approximately 6 h relative to the circadian rhythm of the SCN,25 we estimate that the transcription of ARNTL would reach its peak in the subjective morning, leading to nuclear accumulation of CLOCK and ARNTL proteins during the day and transcription of the Per and Cry genes reaching its peak early in the evening.68 As the PER and CRY proteins heterodimerize and accumulate to the nucleus, the expression of PER and CRY is suppressed completely by the midpoint of the subjective night.
Second, the maternally expressed UBE3A, as well as the paternally expressed MAGEL2 and SNORD116, has been shown to interact with the clock genes in the SCN, which may alter the length of the circadian period and the rhythmic expression of diurnally regulated genes. The maternally expressed UBE3A regulates the turnover of BMAL1 (ARNTL) via ubiquitination. As the duration of the circadian period is determined by rhythmic variation of abundance of the core clock proteins, lack of expression for UBE3A and the reduced turnover of BMAL1 may lengthen the circadian period.70 Our model shows that the imbalance in the protein levels of BMAL1 (ARNTL) may also alter the timing of sleep onset at the subjective evening in AS. In contrast, the paternally expressed MAGEL2 may promote the cytoplasmic accumulation of CLOCK or regulate the expression of PER in the SCN through other molecular interactions, while SNORD116 has been shown to regulate the expression of UBE3A and several circadian pacemaker genes.75 An overexpression of UBE3A would be expected to accelerate the oscillatory pattern of BMAL1, while MAGEL2 may mediate a programmed delay in the feedback loop of the circadian rhythm.74,75 Thus, a lack of expression for MAGEL2 and SNORD116 may lead to a shortened circadian rhythm and dysregulation of diurnally regulated gene expression as well as a further dysregulation of sleep and activity.73,75,76 As also shown in Figure 4, these interactions produce opposite alterations to the circadian period in both syndromes due to the variable dosages of paternally and maternally expressed genes. In AS, both the expression of MAGEL2 and lack of expression for UBE3A may therefore contribute to a lengthened circadian period, while in PWS both lack of expression for MAGEL2 and dysregulation in the expression of UBE3A due to loss of expression for SNORD116 are expected to contribute toward a shortened circadian rhythm and dysregulation of diurnally regulated gene expression.
Figure 4.
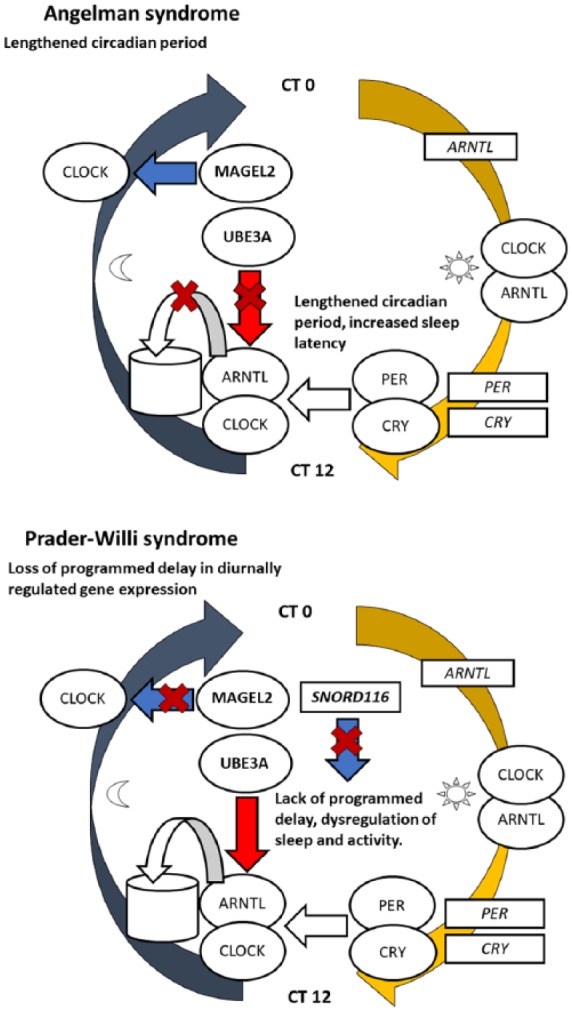
The circadian clock mechanism and its interactions with the genes involved in the Prader–Willi and Angelman syndromes. The Clock and Arntl (Bmal1) proteins, marked as round shapes, start to accumulate in the nucleus after midday and activate the transcription of the Per and Cry genes (marked as rectangles) early in the evening. Per and Cry proteins accumulate in the nucleus by nighttime and repress the transcriptional activity of Clock and Arntl, simultaneously preventing their own transcription. Imprinted genes are also involved in the regulation of circadian rhythms. UBE3A regulates the turnover of BMAL1 via ubiquitination (shown as a simplified diagram of ubiquitination and protein recycling), while MAGEL2 has been hypothesized to mediate the cytoplasmic accumulation of Clock prior to nuclear accumulation of both Clock and Arntl.
These opposite alterations of the circadian rhythm may further explain opposite alterations to the timing of sleep onset in both syndromes, as is also shown in Figure 4. AS involves a phenotype of increased sleep latency along with reduced levels of melatonin secretion and increased bedtime resistance. As secretion of melatonin from the pineal gland is directly dependant of input from the SCN, a misaligned rhythm could explain the reduced levels of melatonin secretion in AS. However, the imprinting of Ube3a may be uniquely relaxed in the SCN and so it has been further argued that the increased sleep onset latency in AS may alternatively be due to reduced accumulation of sleep pressure.69 In contrast to AS, our proposed model suggests that PWS may involve a dysregulation of diurnal gene expression and a relatively shorter subjective day due to the lack of programmed delay in the circadian period, which may further regulate neural and physiological regulation of sleep and wakefulness, which would similarly help explain the reduced sleep onset latency and EDS in the phenotype of PWS.
The evolutionary significance of regulatory mechanisms for sleep and wakefulness can be further understood in the context of human life histories by considering bedtime interactions, which involve numerous soothing routines and can be viewed to represent an important time for maternal bonding. The timing of sleep onset and difficulties with falling asleep may reflect the importance these interactions. Thus, maternal bonding and the regulation of sleep and wakefulness may be subject to an evolutionary tug-of-war between paternally and maternally imprinted genes. Paternally expressed genes may have been selected to favor an innate tendency for increased sleep onset latency and more frequent waking to solicit more maternal resources, while the opposite may be true for maternally expressed genes. Evidently, both AS and PWS involve extreme phenotypes in the regulation of sleep and wakefulness as well as opposite imbalances in dosages of paternally and maternally imprinted genes.
Eating in PWS and AS
Feeding behavior and the development of hyperphagia
For our purposes, feeding behavior can be understood in two overlapping contexts: (1) the evolutionary bases of feeding behavior and life history in human childhood and (2) neural and endocrine mechanisms for the regulation of appetite. Human life histories feature two major transitions of feeding behavior: first, weaning from maternally provided breast milk involves the gradual introduction of complementary foods approximately from the age of 6 months and onwards; the second major transition involves a further nutritional shift from specially prepared complementary foods toward more diverse family foods, coinciding with the development of adequate dentition between ages of 6 and 8 years.77,78 The age period of complementary feeding coincides with a phase characterized by consistent refusal of new foods (food neophobia),79 and the tendency for refusal of new foods has also been shown to be highly heritable among humans.80 Modern practices of complementary feeding can be interpreted to involve specially prepared “baby foods” such as porridge, purees and other foods with constant, soft and smooth textures. Ethnographical records of existing hunter–gatherer societies also indicate that ancestral complementary foods may have consisted mainly of a diverse selection of premasticated foods.81
The second, gradual transition toward an adult diet can also be viewed to involve a reduced burden of maternal investment. In ancestral human societies, children would begin to contribute to their own nutrition at the ages of about 5–7 years by collecting edibles such as fruit or berries (foraging), which coincides with the transition toward more diverse family foods in modern societies.82 The relatively early weaning, as compared to ancestors and other great apes, typical of human childhood, and the introduction of complementary foods can be further interpreted as unique evolutionary adaptations for shorter interbirth intervals in humans.78
The regulation of feeding behaviors in the context of neural and genetic mechanisms, which regulate food intake, is largely based on mouse model studies. The neurocircuits that regulate feeding behavior are thought to be disrupted in both hyperphagia and hypophagia. The neural circuits that regulate feeding can be divided into homeostatic feeding mechanisms, which maintain the energy balance of the body, and hedonic feeding mechanisms, which are driven by neural signals of reward.83 The regulation of homeostatic feeding is maintained by peripheral short-term signals of satiety and hunger, as well as long-term signals of energy balance, which are produced in the body and processed by the hypothalamus. Short-term signals of satiety and hunger are produced in the gut and include ghrelin, which stimulates hunger, and cholecystokinin (CCK), glucagon-like peptide-1 and peptide YY, which signal satiety.84 Long-term signals of energy balance, such as insulin and leptin, are produced in proportion to the levels of adipose tissue in the body and enter the brain through blood circulation.85 The peripheral signals of satiety, hunger and energy balance converge in the hypothalamus, which regulates food intake and energy expenditure through two opposite neural mechanisms in the ARC. Neuropeptide Y (NPY)-expressing neurons and agouti-related peptide (AgRP)-expressing neurons thus promote food intake, while neurons expressing peptides derived from pro-opiomelanocortin (Pomc) limit food intake.84,85
The regulation of hedonic eating is based on the rewarding aspect of feeding, as both the consumption and sensory representations of food induce responses in the neural reward circuitry. The neural reward circuitry involves reciprocal connections between monoaminergic, intermediate and ventricular nuclei. Monoaminergic systems are driven by neurotransmitters including serotonin and dopamine, which mediate the motivation for rewarding behaviors such as feeding or mating. In the regulation of feeding, monoaminergic nuclei further project to intermediate nuclei in the lateral hypothalamic area and other brain regions similarly connected to the ARC, which in turn governs feeding via the hypothalamus. The reciprocal connections of the reward circuitry have been shown to promote feeding and to play a particular role in the development of food preferences and increased consumption of palatable foods.83
Genetic mouse models of PWS and AS show opposite alterations in both dopamine and serotonin levels in the brain.86,87 Furthermore, dysfunction of the hypothalamus is central to several physiological phenotypes in PWS.88 Thus, it can be hypothesized that the specific eating behaviors related to these syndromes may involve opposite dysfunctions in the regulation of hedonic feeding mechanisms.
Eating behavior phenotypes in AS and PWS
In this section, we review the evidence from empirical human studies and genetic mouse models on the phenotypes of eating behaviors of AS and PWS, and evaluate if certain traits could be defined as opposites of each other between the two syndromes. In particular, we will focus on the following traits:
Hyperphagia, that is, significantly increased consumption of food as compared to healthy individuals, regardless of the underlying etiology or associated behaviors.
A comparison of selective and unselective eating, and related behaviors such as food refusal or marked interests for certain types of foods.
Food-seeking behavior, that is, independent behaviors driven by the condition of hyperphagia including stealing, storing or taking food without approval.
A comparison of hyperphagia in PWS and AS
PWS involves a gradually developing condition of hyperphagia, manifested by low birth weight and an early restriction of growth, followed by rapid weight gain after weaning, and the development of hyperphagic behavior and obesity, consistent across all genotypes.11,12,89 The rapid weight gain after weaning has been traditionally associated with overeating. However, Miller et al.13 showed that the changes in weight gain precede the changes in appetite, implying that the development of hyperphagic behavior is preceded by metabolic changes. The authors reviewed complete growth and nutritional records of 58 PWS subjects involved in a longitudinal study to characterize the development of hyperphagia in PWS. The condition was found to follow a gradual progression through several nutritional phases, distinguishable by significant changes in weight gain and dietary intake, as compared to each of the previous phases. The progression and phenotypical changes involved with the nutritional phases can be summarized as follows.
First, infancy is characterized by low birth weight and significant growth restrictions and accompanied by feeding difficulties and overall failure to thrive. Similarly, the infants who did not receive tube feeding also showed metabolic rates indicative of underfeeding. However, the first changes in appetite and weight become apparent approximately at the age of 9 months. At this point, the infant is taking adequate nutrition and the weight gain follows a growth curve similar to typical development. It is notable that the first changes in appetite occur around the age when complementary foods are first introduced to an infant’s diet. Furthermore, the infants who began to receive growth hormone treatment at early age also showed a significantly faster development of appetite in infancy, compared to individuals who began to receive growth hormone treatment later, implying a role for endocrine changes in the development of appetite. Second, the rapid weight gain that precedes the gradual rise of appetite and the development of hyperphagia becomes apparent at approximately 2 years of age. This increase in weight gain is also associated with a significant increase in the serum levels of insulin-like growth factor (IGF-1), implying a role for endocrine changes in the development of hyperphagia. As PWS involves a hypothalamic dysfunction, which has been further associated with the growth hormone deficiency typical to the syndrome,89 it can be further postulated that the metabolic changes involved in the development of hyperphagia in PWS may be caused by the hypothalamic dysfunction. Third, the gradual rise of interest toward food and further changes in appetite can be recognized at approximately 4–5 years of age, while the development of independent food-seeking behaviors and visible hyperphagia become apparent at 8–9 years of age.
The concept of distinct nutritional phases has been criticized by Kotler et al.12 who performed a retrospective review of clinical records for 55 individuals with PWS and found that routinely collected clinical records contained inadequate information for assigning an individual to one of the nutritional phases defined earlier by Miller et al.13 Furthermore, the identification of the later nutritional phases relies on changes in appetite and behavior, but as pointed out by Kotler et al. the analyses applied by Miller and colleagues did not control for changes in appetite with age, effects of psychiatric medication or any restrictions in the availability of food. Kotler et al. note that PWS involves incomplete pubertal development, but the progression of early stages of puberty is accelerated, as compared to typical development.90 Thus, it can be postulated that the imprinted genes involved in the development of PWS may affect the comparably earlier onset of adrenarche in PWS, and that the onset of extreme hyperphagic behavior approximately at the age of 8–9 years coincides with the beginning of this juvenile phase.12,78
In comparison to PWS, few studies have characterized the development of hyperphagic behavior in AS. Berry et al.91 note that behaviors indicative of hyperphagia were reported in AS individuals in significantly greater proportions compared to a control group of children with intellectual disabilities. Approximately one third of the AS individuals in the study were reported to steal or gorge on foods regularly, indicating a tendency for hyperphagic behavior. However, no significant differences in reported behaviors were found between different genotypes. A comparative questionnaire study on food-related behaviors among five genetic neurodevelopmental syndromes similarly found that their group of AS subjects displayed a significantly higher degree of behaviors indicating impaired satiety, compared to subjects with Cornelia de Lange syndrome but also a significantly lower degree of impaired satiety compared to PWS subjects.92
In contrast to the results of Berry et al. and the development of hyperphagia in PWS, phenotypes indicative of hyperphagic behavior in AS have been strongly associated with imprinting defects and paternal disomy, which involve increased dosage for the paternally expressed genes in the 15q11-q13 locus.93,94 In particular, Mertz et al. found that AS individuals with patUPD had significantly increased birth weights and also showed a significant increase in BMI at approximately 3 years of age and afterward, as compared to AS subjects with the deletion genotype or UBE3A mutations. Similarly, a study on early childhood development in AS also found that AS individuals with imprinting defects or the patUPD genotypes developed a disproportionally high BMI within the first 4 years of age, in comparison to individuals with UBE3A mutations.94 In addition, Mertz et al.93 found that AS individuals with the patUPD genotype showed significantly higher degrees of hyperphagic behavior, drive and severity, compared to AS individuals with the deletion genotype. However, the study design of Mertz et al. did not enable precise assessments on the age of onset for the hyperphagic behaviors. Thus, it is not possible to estimate if the hyperphagic behavior in AS develops at an early age, as suggested by the early change in BMI, or if the condition involves a more gradual development of appetite, similar to that of PWS.
In conclusion, PWS involves a gradually developing condition of hyperphagia consistent across all genotypes. In comparison, hyperphagic behaviors are reported with approximately one third of AS individuals, while rapid weight gain at an early age and significantly increased degrees of hyperphagic behavior are further associated with genotypes showing relatively more paternal imprinted gene biases. Furthermore, while patUPD and imprinting defects each account for approximately 2%–3% of all AS cases,9 we note that relatively more paternal genotypes were disproportionally represented (18% UPD, 6% imprinting defect, 5% abnormal methylation) in the study of Berry et al.14 However, it is currently unclear if hyperphagic behaviors are exclusively associated with relatively more paternal genotypes in AS.
Selective and unselective eating in PWS and AS
Due to the central role of hyperphagia in the behavioral phenotype of PWS, food preferences have been studied extensively in this syndrome.95 Individuals with PWS have been noted to show a consistent preference for sweet foods over other tastes.96–100 Kotler et al.12 also note that about one third of their participants (17 out of 55) were described as “picky eaters” during their clinical visits. An avoidance of meat and chunky or non-pureed foods was shown at an age of 1–3 years, while preferences for starchy foods and avoidance of meat were common throughout all age groups. However, as food neophobia is a consistent feature of typical childhood development at early ages, it is difficult to estimate if these food preferences are consistently narrower or broader compared to typically developing individuals. While tendencies for particular food preferences are present in PWS, two behavioral studies further suggest that the amount of food available is consistently more important compared to taste. Glover et al.99 showed that PWS subjects would consistently choose a larger amount of a less preferred food over a smaller amount of their favorite food, while obese control subjects instead showed a tendency toward choosing their preferred foods. Similarly, Joseph et al.101 showed that adult PWS subjects consistently chose larger amounts of food regardless of any preference in taste or any delay in presentation. In addition, behavioral studies also suggest that PWS subjects are more likely to accept contaminated or inappropriately placed foods. Dykens102 used photos of food items to assess acceptance of different foods and found that PWS subjects were significantly more likely to endorse contaminated or highly unusual foods compared to both typically developing controls and intelligent quotient (IQ)-matched controls with varied intellectual disabilities. Similarly, Young et al.103 found that both PWS subjects and children with varied intellectual disabilities were significantly more likely to express acceptance of inappropriately placed foods, such as on food the floor or in a trash can compared to typically developing individuals, indicating a consistent tendency for unselective eating. Furthermore, a group of three individuals with a mean age of approximately 12 years was found to actively seek and consume inappropriately placed food in an experimental setting, while older individuals (mean age of approximately 20 years) did not show a similar tendency.
In comparison to PWS, both narrow food preferences and marked interests for certain types of foods have been reported in studies of AS. Clarke and Marston conducted a caretaker questionnaire on problematic behaviors, comparing a group of AS subjects aged 5–33 years to previously studied groups with varied intellectual disabilities. The authors noted that a range of varied food-related problem behaviors, including overeating or a narrow range of food preferences, were reported in 64% of the participants.104 Similarly, according to Berry et al.,14 narrow food preferences are reported with a prevalence between 33% and 100% in the relevant literature concerning AS. Behaviors concerning narrow food preferences were also reported in significantly greater proportions among AS subjects (aged between 1 and 40 years, with a mean age of 13.6 years), compared to a control group of children with intellectual disabilities, with a prevalence of approximately 70% among the AS individuals.14 Finally, AS has been noted to involve specific interests toward certain foods. In particular, “marked preference for certain foods, particularly those that do not require much chewing such as bread, pasta or banana” has been noted.105 Hence, while the evidence is limited in nature, studies concerning AS support the notion of a consistent tendency for relatively selective eating in AS, along with an exaggerated interest in foods resembling specially prepared complementary foods in texture.
Although relevant studies characterizing narrow or limited food preferences are few in AS, behavioral tendencies for narrow food preferences are well documented in subjects with ASDs (reviewed in Marí-Bauset et al.106). Given the relatively high degree of comorbidity between autism and AS,2–4 a certain resemblance of feeding behavior patterns and food preferences between the two conditions may be assumed. Children with ASDs display a consistent tendency for significantly increased selectivity toward food, as compared to typically developing children: Raiten and Massaro107 compared food preferences among children with ASDs and typically developing children with a 7-day food diary and a parental questionnaire and found that children with ASDs showed a significantly higher degree of food selectivity compared to typically developing children. Similar results have been further shown in several behavioral studies.108–115 While the association of narrow food preferences and ASDs is consistently reported across studies, it is less clear if the selectivity is based on taste, difficulty in consumption or other aspects such as visual representation. For example, Schreck et al.108 found that children with ASDs were significantly more likely to require specific utensils or particular presentation of food items, compared to typically developing children, and that children with ASDs were also more likely to accept foods with constant texture, such as purees or mashed potatoes. Similarly, Hubbard et al.116 noted that children with ASDs were significantly more likely to refuse foods based on their texture, smell and taste compared to typically developing children.
In conclusion, while the evidence is limited, AS may involve a tendency for a narrow range of preferred foods, which resemble complementary foods in texture. In comparison, PWS subjects tend to choose larger amounts of food over preferred foods and may endorse both contaminated and inappropriately placed foods. These behaviors suggest a tendency for unselective eating, which develops gradually along with the gradual rise in appetite at the age of 4–5 years.
Food-seeking behaviors
The hyperphagic condition of PWS has been characterized to involve an exaggerated preoccupation with food and the development of independent food-seeking behaviors, including stealing or taking food without approval as well as bargaining for food and snacks with their caretakers.88,117 As food-seeking behaviors have also been reported in studies of AS,14,92 it should be assessed whether any quantifiable differences in hyperphagic behavior can be found between AS and PWS.
Two large-scale questionnaire studies have documented a wide range of hyperphagic behaviors in PWS. Russell and Oliver designed a questionnaire for food-related behaviors based on structured interviews with parents and caretakers, and derived subscales for behavioral traits to further characterize preoccupation with food, impaired satiety and negative food-related behaviors, which include taking or stealing food, eating inappropriate items (pica) and reacting inappropriately when food is taken away. The PWS subjects scored consistently higher in all subscales, compared to a control group of children with intellectual disabilities living in a similar community setting, indicating a consistent tendency for food-seeking behaviors and an exaggerated preoccupation with food.117
Dykens et al. assessed hyperphagic behaviors in PWS with a specifically designed questionnaire. Hyperphagic behaviors were categorized into different factors and a principal components analysis was performed to further characterize which of the factors best explained the variance in the results. While the drive for food (hyperphagic drive) was found to be consistent across all age groups, extremely obese individuals with PWS displayed significantly greater drive for food, compared to other over- or normal-weight PWS subjects. In addition, PWS subjects above the age of 10 were found to show a significantly greater variety of hyperphagic behaviors compared to younger children with PWS. Hyperphagic severity, indicating the individual’s preoccupation with food, was found to be similar in all other age groups, while the oldest age group (30 years and above) showed significantly lower scores. In other words, hyperphagic problem behaviors only become evident in late childhood, while the preoccupation with food diminishes as the individual matures.88 In contrast to the results of Mertz et al.93 concerning AS subjects, no significant correlations were found between genotypes and any degree of hyperphagic behavior, drive or severity, further indicating that the condition of hyperphagia is consistent across all the genotypes in PWS.88
Food-seeking behaviors have been described in three studies concerning food-related behaviors in AS. As noted earlier, stealing food or overeating has been reported in a significantly larger proportion of AS subjects (approximately one third) compared to control subjects with intellectual disabilities.14 Furthermore, in a direct comparison of food-related problem behaviors between five genetic neurodevelopmental syndromes, both AS and PWS individuals were reported to show greater degrees of food-seeking behaviors, compared to the groups of CdLs and 1p36 deletion syndrome individuals. However, individuals with PWS also showed significantly greater preoccupation with food compared to individuals with AS.92 Similarly, as noted earlier, the patUPD genotype has been characterized to display a significantly greater degree of food-seeking behaviors and a greater degree of hyperphagic drive and severity, compared to AS subjects with the deletion genotype.93
In summary, both AS and PWS show a significant tendency for food-seeking behaviors, but while food-seeking behaviors are consistent across all genotypes in PWS, in AS the patUPD genotype shows a higher degree of food-seeking behaviors. In addition, behaviors indicating a preoccupation with food are reported to a greater degree in PWS.
Developing models of eating behavior for PWS and AS
To further characterize the behavioral phenotypes of feeding behavior in PWS and AS, we review current research and relevant mouse model studies on the mechanisms of hyperphagia and food preferences and use this information to develop models of feeding behavior for both PWS and AS.
Human studies on the regulation of hunger and satiety in PWS
Several behavioral and physiological human studies have characterized the hyperphagic condition of PWS as involving an impairment of satiety. First, Holland et al.118 showed that PWS subjects consumed significantly higher amounts of calories during a meal session, compared to typical control subjects. As expected, the blood levels of CCK, associated with regulation of satiety, were also significantly higher in the PWS subjects compared to controls. The increase in blood CCK levels during the meal session thus indicates that the hyperphagic behavior is not associated with a failure in the release of peripheral satiety signals. Similarly, physiological studies have indicated that two other peripheral signals of satiety, leptin and peptide YY, are also significantly elevated in PWS subjects.119 While significantly elevated levels of circulating ghrelin, a peptide involved in the peripheral signaling of hunger, have been noted in several studies,120,121 it is unlikely that hyperghrelinemia would be causal to the hyperphagia in PWS, as the levels of fasting ghrelin are also elevated in infants and children in early nutritional phases before the onset of hyperphagic behavior.122 As the elevated levels of peripheral signals of satiety would be expected to regulate meal size, these results suggest that the apparent dysregulation of food intake may instead be connected to the hypothalamic dysfunction central to PWS.
Neuroimaging studies have provided evidence that impaired satiety in PWS is connected to a hypothalamic dysfunction and a failure in neural recognition of peripheral satiety signals. Hinton et al.100 measured neural activation in response to an overnight fast followed by a high-energy breakfast and found that the PWS subjects showed a comparable lack of neural activation in brain regions previously associated with satiety after meal consumption, indicating a dysfunction in neural regulation of food intake.
Recently, a post-mortem transcriptional analysis of brain tissues in PWS subjects has confirmed that PWS subjects display an imbalance in the expression of hypothalamic neurotransmitters involved in the regulation of hunger and satiety.123 Comparing gene expression, some ~3600 genes were found to be differentially expressed in the hypothalamic tissues of PWS subjects, as compared to controls with comparable BMI. Furthermore, the expression of AgRp and other genes predominantly expressed in AgRp-expressing neurons was found to be significantly upregulated in PWS subjects, while the expression of Pomc and other genes predominantly expressed in Pomc-expressing neurons was found to be significantly downregulated. Comparisons with the gene expression profiles of fasted animals indicated that the genes upregulated in PWS subjects also represented genes commonly upregulated in response to hunger. In addition, a dysregulation of genes involved in the regulation of energy homeostasis and adipocyte tissues is also implied. Finally, using a targeted deletion of SNORD116 in a human cell model and analysis of predicted splicing targets for the SNORD116 SnoRNA, the researchers showed that lack of expression for SNORD116 may lead to marked neurodegeneration and reduced neuronal differentiation through dysregulation of alternative splicing of several genes previously implied in neuron development and synaptic plasticity.123 Taken together, these results indicate that the pleiotropic effects of neuronal loss and reduced neuronal differentiation may also lead to a dysregulation of hunger and satiety in the hypothalamus in PWS.
The complex nature of the genetic mechanism for hyperphagia and obesity in PWS is further highlighted in studies of Schaaf–Yang syndrome. Schaaf–Yang syndrome is caused by truncating mutations or deletions of the paternally derived copy of the MAGEL2 gene, and so the genotype of Schaaf–Yang syndrome has partial overlap with the genotypes of PWS. While both Schaaf–Yang syndrome and PWS involve intellectual disability, hypotonia and feeding problems during infancy, hyperphagia is described in only 35% of subjects with Schaaf–Yang syndrome, whereas excessive weight gain has been reported in 47% of the subjects.124 In a partially overlapping study, McCarthy et al.125 reported that all nine subjects with Schaaf–Yang syndrome showed elevated levels of fasting ghrelin, while hyperphagia had not been reported in any of the patients involved in the study. The consistent features between the phenotypes of PWS and Schaaf–Yang syndrome indicate that the lack of expression for MAGEL2 may play a role in the development of early feeding restrictions and the later development of obesity. However, the partial penetrance of hyperphagia in Schaaf–Yang syndrome indicates that other paternally expressed genes of the 15q11-q13 locus further affect the phenotype of hyperphagia in PWS.
Genetic mouse model studies of the regulation of hunger and satiety in PWS
Several mouse model studies have investigated the genetic mechanisms for the central features in the phenotype of PWS: an early growth restriction followed by the subsequent development of hyperphagia and obesity.
Bischof et al.126 showed that mice with two inactivated copies of Magel2 exhibited an early growth deficit from the first week after birth until weaning, which was followed by rapid weight gain and obesity after weaning. However, the mutant mice showed a ~10% reduction in food intake and were also less active compared to controls. These results resemble the phenotype of PWS closely, which may not only indicate a central role for Magel2 in the development of both early growth restrictions and later changes in weight gain but also indicate that losses of expression for other paternally expressed may be responsible for the development of hyperphagia in PWS.
The role of MAGEL2 in the development of obesity has been shown to involve interactions with leptin in the hypothalamus: Mercer et al.127 showed that model mice with deletions of the Magel2 gene lack the anorexigenic response to leptin, which induced restrictions in food intake in the control mice. Based on the observations of genetic markers for neural activation, it was shown that leptin fails to activate the Pomc-expressing neurons in the cells of MAGEL2-null mice, indicating a neural dysregulation of food intake. As also shown in Figure 5, leptin regulates food intake via the hypothalamus. Thus, the authors hypothesized that Magel2 may have a role in regulating intracellular leptin responses in hypothalamic neurons. Recently, another mouse model study confirmed that Magel2 interacts with necdin and three ubiquitin pathway proteins (Rnf41, Usp8 and Stam1) to regulate the lysosomal degradation of the leptin receptor.128 As Magel2 was found to regulate the stability of Rnf41 and Usp8, the authors postulated that Magel2 may regulate the abundance of leptin receptors in the hypothalamus indirectly through ubiquitination pathways. Together, these results indicate that MAGEL2 may regulate long-term energy homeostasis via its interactions with leptin, and lack of expression for MAGEL2 may be the central mechanism for the obesity characteristic to the phenotype of PWS.
Figure 5.
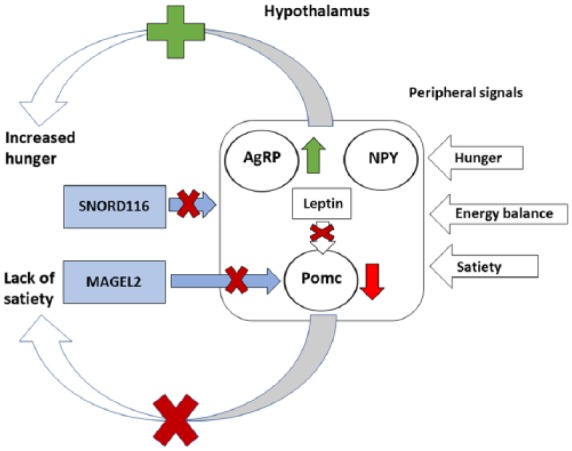
The dysregulation of the homeostatic feeding mechanism in PWS. Neural stimulation of neuropeptide Y (NPY)- and Agouti-like peptide (AgRP)-expressing neurons promotes feeding in response to peripheral signals of hunger, while neural stimulation of neurons expressing pro-opiomelanocortin (Pomc) inhibits feeding in response to peripheral signals of satiety. Long-term signals of energy balance such as leptin induce a fasting response and limit food intake when energy balance is favorable, preventing obesity. In PWS, lack of expression for SNORD116 may lead to neuronal degeneration and imbalance in the expression of AgRP and Pomc in the hypothalamus leading to increase in feeding. Similarly, MAGEL2 has been shown to be required for the leptin-mediated activation of Pomc-expressing neurons as MAGEL2 regulates the abundance of leptin receptors through ubiquitination pathways. Hence, both regulation of hunger and satiety and long-term regulation of energy balance are disrupted in Prader–Willi syndrome, leading to the impaired satiety and obesity typical of the syndrome.
Several mouse model studies have suggested that the mechanism for the early growth restrictions and late development of hyperphagia in PWS may be dependent on the lack of expression for SNORD116 snoRNA. Ding et al.129 showed that mice with paternally inherited deletions of Snord116 develop normally in prenatal stages but show a growth reduction at early ages, followed by the development of hyperphagia at a later age. However, the model mice did not replicate the phenotype of PWS in full; despite their hyperphagic condition, the model mice would stay lean and showed altered metabolism, with higher rates of oxygen consumption compared to control mice.
Several mouse model studies have addressed the role of SNORD116 in the development of hyperphagia in PWS with partly contradicting results, but none of the mouse models have replicated the full phenotype of hyperphagia and obesity in PWS. Qi et al.130 showed that lack of expression for Snord116 may alter the regulation of food intake via the NPY-expressing neurons in the hypothalamus. Model mice with a selective deletion of Snord116 in the NPY-expressing neurons replicated the phenotype of early growth restriction and late development of hyperphagia closely. Thus, Snord116 may be critical for regulating the expression of Pomc and NPY, as the model mice also showed a significant upregulation of both NPY and Pomc mRNA in the hypothalamus. In particular, the anorexigenic response of Pomc-expressing neurons was hypothesized to play a role in the growth reductions at an early age, while the drive for increased food intake induced by the NPY-expressing neurons would take hold later in life.130
However, another recent mouse model study by Polex-Wolf et al.131 contradicted the results of previous studies129,130 as the authors found that a mouse model with a paternal deletion of Snord116 did not develop hyperphagia at a later age. However, mice with a selective deletion of Snord116 in the mediobasal hypothalamus induced at adult age showed the development of hyperphagia 10 weeks after the procedure. The model mice also showed a significantly greater weight gain in comparison to controls, though only a small subset (5 out of 21) of the mice would develop significant obesity and increased fat mass. However, the expression of Pomc, NPY and leptin receptor mRNAs during a fast was not shown to be significantly different from controls. Similarly, the expression of prohormone convertase 1 (Pcsk1) and its upstream regulator did not differ significantly from controls, indicating that the increase in food intake could not be explained by an imbalance in the homeostatic regulation of feeding and that an alternative explanation for the mechanism of hyperphagia would be required.
Although mouse model studies have implied that the lack of expression for both SNORD116 and MAGEL2 may play roles in the development of hyperphagia and obesity in PWS, the analysis of these studies is complicated by a recent study which notes the paradoxical leanness of model mice with a deletion of the PWS imprinting center. The model mice showed an early reduction of growth and failure to thrive in infancy, with later development of food hoarding behaviors. However, significantly increased food intake as compared to controls would only develop with model mice on high-fat diets.132 As shown in the results of Ding et al.129 and Qi et al.,130 the model mice also showed a significant reduction in body weight and fat mass. In further investigation, the model mice did not show an elevation of white fat mass in a thermoneutral environment, indicating that increased energy usage in maintaining body temperature is unlikely to cause the leanness of PWS model mice. As the model mice similarly failed to gain weight on a high-fat diet, the authors postulated that the leanness of the model mice may result from a failure of lipid accumulation in white adipocytes, which may further indicate that the model mice failed to model the phenotype of PWS due to metabolic differences between humans and mice.
In summary, mouse model studies have shown that the lack of expression for MAGEL2 may be connected to the development of obesity due to dysregulation of leptin-induced anorexigenic responses in the hypothalamus. Furthermore, while all of the currently reviewed mouse model and human studies indicate that the development of hyperphagia in PWS is associated with lack of expression for SNORD116 in the hypothalamus, further research is necessary for understanding the precise mechanisms of how SNORD116 and alternative splicing mechanisms might alter the regulation of both neuronal development123 and homeostatic feeding as regulated by the hypothalamus.130
A model for hypothalamic dysregulation of homeostatic feeding in PWS
Currently available studies on both humans and mouse models suggest that the hyperphagia typical to PWS results from a dysregulation of homeostatic feeding in the hypothalamus. Based on these findings, we have developed a model for the dysregulation of homeostatic feeding mechanisms in PWS. By this model, peripheral signals of satiety and long-term energy balance are produced at elevated levels, but the processing of these signals in the hypothalamus is disrupted due to an imbalance in hypothalamic neurotransmitters, ultimately due to the lack of expression for the paternally expressed genes MAGEL2 and SNORD116.
First, we note that the regulation of long-term energy balance in the hypothalamus is dependent on the anorexigenic response induced by leptin. However, lack of expression for Magel2 leads to dysregulation in the ubiquitination of leptin receptors,129 so leptin fails to induce the fasting response in the hypothalamus due to an accelerated turnover of leptin receptors at Pomc-expressing neurons, contributing to both increase in food intake and the development of obesity.
Second, SNORD116 has been shown to be involved in the regulation of food intake via the hypothalamus,130,131 and transcriptional analysis of hypothalamic tissues of PWS subjects suggests that the lack of expression for SNORD116 may alter the regulation of neuronal development123 leading to an imbalance in the regulation of the hypothalamic feeding mechanism. The expression of AGRP, previously implied in the regulation of hunger, is significantly upregulated in the hypothalamus, while the expression of POMC, previously implied in the regulation of satiety, is significantly downregulated. Thus, the impaired satiety central to phenotype of eating behaviors in PWS may be induced by continuous signaling of hunger and diminished signaling of satiety in the hypothalamus.
As PWS involves a gradual transition from poor feeding in infancy to the development of hyperphagia in late childhood, further research is necessary to understand how a lack of expression for paternally expressed genes would gradually alter the regulation of food intake from infancy to early childhood and further from childhood to the juvenile phase. As the development of hyperphagia coincides with early adrenarche, it has been suggested that the changes in appetite may reflect changes in the expression of adrenal androgens during childhood development.12
A model of the central hypothalamic mechanisms in the regulation of food intake and their currently known interactions with the paternally expressed genes, MAGEL2 and SNORD116, is shown in Figure 5.
Mouse models on the role of palatability in food intake
In addition to the homeostatic mechanisms regulated by hunger and satiety, appetite can also be driven by the reward circuitry of the brain. While mouse model studies of PWS have focused primarily on the development of hyperphagia and the mechanism of homeostatic feeding, the role of palatability in the regulation of feeding behavior has been highlighted in a number of mouse model studies.
Exploring the role of hedonic eating in PWS, Davies et al.133 characterized the feeding behavior in a mouse model with a paternally inherited deletion corresponding to the imprinting center of the PWS–AS locus. The model mice showed significantly increased food consumption after an overnight fast as compared to wild-type controls, with both regular chow and high-sugar content food. In order to dissociate the impact of nutrition and taste, the researchers studied the licking behavior. The mice were accustomed and given limited access to a lick-measuring device which would dispense sugared water or alternatively a solution with saccharin, an artificial sweetener with no nutritional value. With sucrose, no significant differences between the model mice and wild-type controls were found in comparisons of repeated licking behavior. However, with saccharin, the model mice showed a significant reduction in the number of total licks as compared to wild-type controls. In addition, when trained with a treat-dispensing device, the model mice again matched the behavior of the wild-type controls with a sugar-based treat but showed a significant reduction in the number of responses when rewarded with a saccharin-based treat instead. The authors concluded that the PWS model mice appeared to be particularly sensitive to the calorific content of palatable food, rather than taste, thus resembling the lack of satiety and the tendency for unselective eating among PWS subjects.
The role of dopamine in the development of food preferences has been shown in a multitude of studies. For example, Cooper and Al-Naser134 found that dopamine may influence the development of preference for palatable foods. Food intake of fasted rats was measured in experimental settings, with comparisons based on the palatability of the food offered. While highly palatable food was consistently consumed in greater amounts compared to regular food pellets, treatment with a selective D1 dopamine receptor agonist significantly increased the consumption of the palatable food as compared to the control, while treatment with a selective D2/D3 dopamine receptor agonist significantly decreased the consumption of the highly palatable food and increased the consumption of regular chow. Hence, preference for highly palatable foods is at least partly driven by the dopamine reward system.
The consumption of palatable foods thus shows a definite connection to the dopamine reward system. Furthermore, opposite alterations to both dopamine and serotonin neurochemistry have been shown in mouse models of both AS and PWS. Farook et al. studied mice models with either duplications or deletions of Ube3a. Mice with a maternally inherited deletion of Ube3a resembling the genotype of AS had elevated levels of dopamine in the striatum and frontal cortex and elevated levels of serotonin in the striatum and cortex, as compared to controls.86 Conversely, mice with a maternal duplication of Ube3a, resembling the matUPD genotype of PWS, had elevated levels of dopamine in the midbrain and the striatum. However, Mercer et al.87 showed that Magel2-null mice had significantly decreased levels of serotonin in the cortex, prefrontal cortex and hypothalamus as well as significantly decreased levels of dopamine in the hypothalamus. Thus, studies of mice models indicate that AS may involve highly elevated levels of dopamine in brain regions critical to feeding. In contrast, PWS may involve decreased levels of dopamine in multiple brain regions critical to feeding.
A model for the development of hyperphagia and food preferences in AS and PWS
Considering the existing evidence from both empirical studies in AS and PWS and findings from relevant mouse model studies, we have developed a model to explain the development of hyperphagia and food preferences in both AS and PWS. First, we assume that the hyperphagic condition of PWS is caused by impaired satiety due to a disruption of the homeostatic feeding mechanism, as shown in Figure 5. Furthermore, we predict that PWS confers a consistent tendency for unselective eating, as suggested by the mouse model study of Davies et al.133 as well as the observation that PWS patients consistently choose larger amounts of food regardless of preference in taste or delay in presentation.100,101 Second, both increased selectivity toward food and marked preferences for certain foods, in particular foods with consistent and soft texture, are prominent in AS. Thus, we have hypothesized that the hyperphagic phenotype of AS is connected to an increased interest toward complementary foods. In contrast to the unselective eating in PWS, this behavior can be hypothesized to be driven by an influence of paternally expressed genes, as the hyperphagic phenotype of AS is strongly associated to the patUPD genotype, which confers a higher dosage of the paternally expressed genes in the 15q11-q13 chromosome region.9
It can be predicted that complementary foods, which differ from adult diets and could be more difficult to obtain and prepare, may involve increased maternal costs. Paternally expressed genes may thus have been selected to favor an increased preference for complementary foods and prolonged parental care in juvenile stages, so a disruption of the genetic conflict could further result in extreme behaviors such as overeating and limited preferences for specific foods, as seen with the phenotypes of both AS and autism.
To illustrate our hypothesis, we have developed a framework around hedonic feeding mechanisms and the known alterations of dopamine neurochemistry in AS and PWS, as shown in Figure 6. First, food selectivity similar to ASD may contribute to increased selectivity toward food across all genotypes. Second, both a lack of expression for UBE3A and an increased dosage of paternally expressed genes such as MAGEL2 are expected to contribute toward elevated levels of dopamine in the brain, so the more paternal patUPD and imprinting defect genotypes of AS are expected to show both elevated hedonic value of food and tendency for the development of hyperphagia with selective eating. In contrast, all genotypes of PWS lack expression of MAGEL2, so asymmetrical alterations to hedonic feeding can be expected between AS and PWS, as shown in Figure 6. However, the exact mechanism of hyperphagia in AS does not need to depend on hedonic feeding mechanisms to fulfill our initial expectations on the influence of paternally expressed genes in the development of hyperphagia and selective eating.
Figure 6.

A hypothetical mechanism involving dysfunctions of hedonic feeding and the development of hyperphagia phenotypes in AS and PWS. PWS and AS may involve opposite dysfunctions of dopaminergic pathways due to losses and gains in dosages of imprinted genes. The effect is more pronounced with uniparental disomies and imprinting defects due to an increased dosage of paternally (or maternally) expressed genes. The increased dosage of paternally expressed genes and the expected increases in dopamine levels may explain the tendency for selective eating and the early development of hyperphagia in AS associated with patUPD and imprinting defects.
Currently available evidence on the timing of the development of hyperphagic behavior in PWS indicates that the development of hyperphagia coincides approximately with the timing of early adrenarche, as well as the development of adult dentition and simultaneous transition toward an adult diet of diverse family foods.77 While exact information on the timing of the development of hyperphagia in AS is currently lacking, the rapid increase of BMI between 2 and 5 years of age93 suggests that the development of hyperphagic behavior in AS may coincide with the usual period of dependence on complementary foods, which are the primary source of nutrition for the child after weaning until the development of mature dentition at the age of 6–8 years.78 Thus, we have further complemented our model with predictions on the timing of the development of hyperphagia and food preferences in both AS and PWS. As shown in Figure 7, our model predicts increased interest toward complementary foods and early development of hyperphagia in AS and conversely a gradual rise of interest toward food and late development of hyperphagia and indiscriminate “foraging” of diverse foods in PWS in accordance to earlier work.78
Figure 7.
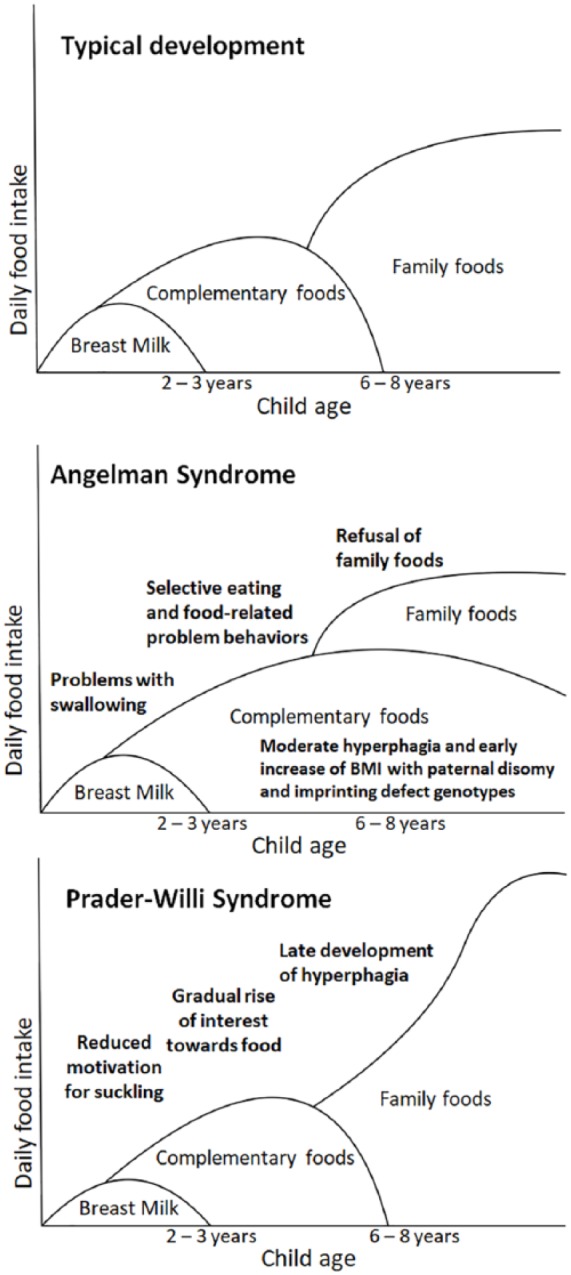
The development food preferences and hyperphagia in AS and PWS as compared to typical childhood development. Maternally provided breast milk is the primary source of nutrition for the infant until the age of weaning at the age of 2–3 years, while specially prepared complementary foods are gradually introduced from the age of 6 months and onwards. Diverse family foods resembling an adult diet replace complementary foods by the age of 6–8 years. Poor feeding during infancy is prominent in both AS and PWS, although for different reasons. A gradual rise of interest toward food and late development of hyperphagia can be seen in PWS, whereas AS may involve comparably earlier development of hyperphagia and a specific interest for complementary foods and prolonged refusal of family foods.
Conclusion
In this article, we have compared sleeping and eating behavior phenotypes of PWS and AS and evaluated, for the phenotypes with sufficient data, which phenotypes are opposite to one another and which are not. Furthermore, we have assessed these behavioral phenotypes in the context of relevant mouse model studies and developed genetic and physiological models for sleeping and eating behavior to help explain how the different genetic alterations of these syndromes could produce opposite phenotypes, especially from alterations to dosages of different imprinted genes. Finally, we have evaluated our findings in the context of human childhood development and the kinship theory of imprinting. Our main findings are as follows:
First, relevant articles and our meta-analysis showed evidence of opposite phenotypes for sleep onset latency between AS and PWS, and partially opposite phenotypes for sleep duration, while other traits of interest showed relatively similar phenotypes in both syndromes. As relevant mouse model studies have indicated that both paternally and maternally expressed genes may regulate circadian rhythms and sleep, we suggested a model (Figure 4) to explain how variable dosages of paternally and maternally expressed genes inherent to each syndrome could produce opposite phenotypes of sleep onset latency in AS and PWS. Thus, in PWS, both the increased expression of UBE3A due to lack of expression for SNORD116 and a lack of expression for MAGEL2 may contribute to a lack of programmed delay and acceleration of the circadian period. In contrast, both a lack of expression for UBE3A and the expression of MAGEL2 are expected to contribute to a deceleration of the circadian period in AS. These opposite alterations to the circadian rhythm and diurnally regulated gene expression patterns may lead to opposite alterations to the timing of sleep onset as also shown in the results of the relevant studies reviewed here. In AS, a lengthened circadian period may lead to a phase delay in the 24-h circadian rhythm and a longer subjective day and increased sleep onset latency. In PWS, the opposite pattern is shown, with a lack of programmed delay leading to a shorter subjective day and reduced sleep latency.
Second, we describe evidence that that AS and PWS show a shared tendency for overeating and food-seeking behaviors,14,88,93 but apparent opposite tendencies for selective and unselective eating preferences.14,100–102 However, while the hyperphagic phenotype of PWS is consistent across all genotypes,13,88 in AS hyperphagic phenotypes are strongly associated with the patUPD and imprinting defect genotypes93,94 which are further characterized by increased dosages of paternally expressed genes as also shown in Figure 2. Since relevant human and mouse model studies indicate that PWS may involve both a hypothalamic dysregulation of the homeostatic feeding mechanism and diminished hedonic value of food due to lack of expression for maternally imprinted genes, we have also suggested a model (Figure 6) to explain how selective versus unselective food preferences in AS and PWS, as well as the association of hyperphagia with relatively “more paternal” genotypes in AS, may be explained by opposite alterations to dopamine reward circuitry and hedonic feeding mechanisms.
As human life history can be interpreted to involve a unique evolutionary adaptation to shortened birth intervals with early weaning and the introduction of complementary feeding with specially prepared “baby foods,”77,78 we hypothesize that the development of hyperphagia and food selectivity in both AS and PWS may reflect nutritional shifts in human childhood as shown in Figure 7. In AS, increased selectivity toward foods may result from a prolonged interest in mother-provided complementary foods, while hyperphagic behavior is driven by a further exaggeration of this interest. Conversely, as described previously,12 PWS involves a gradual rise in interest toward food, leading to the development of hyperphagia around the age of adrenarche, which coincides with a transition from the mother-provided complementary foods to diverse family foods. This hypothesis is readily testable, as it further predicts that the timing and development of both selective food preferences and hyperphagia in AS should coincide with the period of complementary feeding in early childhood.
Third, we have assessed how varying dosages of the paternally expressed MAGEL2 and SNORD116 as well as the maternally expressed UBE3A may affect phenotypes of both sleep and eating behavior. Due to the wide-reaching roles of MAGEL2 and UBE3A in regulating numerous gene networks through ubiquitination pathways,135,136 both UBE3A and MAGEL2 may affect several behavioral phenotypes including both the regulation of circadian rhythms and the regulation of long-term energy balance and feeding through a variety of different molecular mechanisms. Furthermore, UBE3A and MAGEL2 as well as SNORD116 are expressed in the same brain region, the hypothalamus. Numerous studies have indeed shown that the hypothalamus plays a dual role in both sleep–wake regulation and the regulation of feeding. For example, lesions of the NPY-expressing neurons in the mediobasal hypothalamus have been shown to cause hyperphagia and a lack of circadian variation for the distribution of non-REM sleep in mouse model studies,137 and orexin neuropeptides expressed by neurons in the lateral hypothalamic area play a role in the regulation of both sleep and eating.138 Lack of expression for SNORD116 in the mediobasal hypothalamus is also involved in the development of hyperphagia in PWS,129–131 while a lack of expression for Magel2 has been connected to reduced levels of orexin and orexin-expressing neurons in the lateral hypothalamus, along with fragmentation in circadian regulation of activity and rest73 as well as reductions in growth followed by later development of obesity.126,127
The role of the hypothalamus in the development of AS is currently understudied. As alterations to the expression levels of MAGEL2, SNORD116 and UBE3A may affect the phenotypes of both eating and sleeping via the hypothalamus, our hypothesis predicts that other human neurogenetic syndromes that resemble AS or PWS phenotypically139,140 or result from alterations to the same genes124,136 may also exhibit joint effects on sleeping, eating and other hypothalamus-mediated phenotypes.
The opposite alterations to sleep onset latency described here can be interpreted to follow from extreme manifestations of paternally and maternally expressed genes, as also previously discussed by Kotler and Haig.78 Since settling to sleep often involves separation of the mother and the infant, bedtime can be seen as an important time for maternal bonding.141 Furthermore, as the behavioral phenotype of AS has been argued to reflect an extreme development of phenotypes related to affect signaling,16,17 paternally expressed genes may have been selected to favor increased bedtime resistance and more frequent waking in solicitation of both nutrition and social interaction from the mother, which may also lead to increased sleep onset latency and difficulties in falling asleep later in life. In contrast, maternally expressed genes may have been selected to favor reduced bedtime resistance and less frequent waking to reduce maternal stress during early infancy, which may further lead to a consistent tendency for reduced sleep onset latency and excessive sleepiness in later life.
Supplemental Material
Supplemental material, PRISMA_2009_checklist_1 for Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes by Iiro Ilmari Salminen, Bernard J Crespi and Mikael Mokkonen in SAGE Open Medicine
Supplemental Material
Supplemental material, PRISMA_2009_flow_diagram_1 for Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes by Iiro Ilmari Salminen, Bernard J Crespi and Mikael Mokkonen in SAGE Open Medicine
Footnotes
Declaration of conflicting interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval: We confirm that our work is solely a scientific review, and thus ethics approval was not applicable.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The The research reported in this article was funded through the NSERC grant 2014-06505.
Informed consent: We confirm that our work is solely a scientific review, and thus informed consent was not applicable.
Supplemental material: Supplemental material for this article is available online.
ORCID iD: Iiro Ilmari Salminen  https://orcid.org/0000-0003-0411-3160
https://orcid.org/0000-0003-0411-3160
References
- 1. Cassidy SB, Dykens E, Williams CA. Prader-Willi and Angelman syndromes: sister imprinted disorders. Am J Med Genet 2000; 97: 136–146. [DOI] [PubMed] [Google Scholar]
- 2. Peters S, Beaudet A, Madduri N, et al. Autism in Angelman syndrome: implications for autism research. Clin Genet 2004; 66(6): 530–536. [DOI] [PubMed] [Google Scholar]
- 3. Bonati MT, Russo S, Finelli P, et al. Evaluation of autism traits in Angelman syndrome: a resource to unfold autism genes. Neurogenetics 2007; 8(3): 169–178. [DOI] [PubMed] [Google Scholar]
- 4. Trillingsgaard A, Østergaard JR. Autism in Angelman syndrome: an exploration of comorbidity. Autism 2004; 8(2): 163–174. [DOI] [PubMed] [Google Scholar]
- 5. Dykens EM, Roof E, Hunt-Hawkins H, et al. Diagnoses and characteristics of autism spectrum disorders in children with Prader-Willi syndrome. J Neurodev Disord 2017; 9(1): 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haig D, Wharton R. Prader-Willi syndrome and the evolution of human childhood. Am J Hum Biol 2003; 15(3): 320–329. [DOI] [PubMed] [Google Scholar]
- 7. Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genet Med 2012; 14(1): 10–26. [DOI] [PubMed] [Google Scholar]
- 8. Runte M, Hüttenhofer A, Gross S, et al. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet 2001; 10(23): 2687–2700. [DOI] [PubMed] [Google Scholar]
- 9. R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing, 2018. [Google Scholar]
- 10. Hozo SP, Djulbegovic B, Hozo I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Meth 2005; 5: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haig D. Coadaptation and conflict, misconception and muddle, in the evolution of genomic imprinting. Heredity 2014; 113: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kotler J, Balko K, Berall G, et al. Nutritional phases in Prader-Willi syndrome: evolutionary and clinical interpretations. J Evol Med 2016; 4: 1–7. [Google Scholar]
- 13. Miller JL, Lynn CH, Driscoll DC, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet A 2011; 155(5): 1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Byars SG, Stearns SC, Boomsma JJ. Opposite risk patterns for autism and schizophrenia are associated with normal variation in birth size: phenotypic support for hypothesized diametric gene-dosage effects. Proc Biol Sci 2014; 281(1794): 20140604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pelc K, Cheron G, Boyd SG, et al. Are there distinctive sleep problems in Angelman syndrome? Sleep Med 2008; 9(4): 434–441. [DOI] [PubMed] [Google Scholar]
- 16. Brown WM, Consedine NS. Just how happy is the happy puppet? An emotion signaling and kinship theory perspective on the behavioral phenotype of children with Angelman syndrome. Med Hypotheses 2004; 63(3): 377–385. [DOI] [PubMed] [Google Scholar]
- 17. Oliver C, Demetriades L, Hall S. Effects of environmental events on smiling and laughing behavior in Angelman syndrome. Am J Ment Retard 2002; 107(3): 194–200. [DOI] [PubMed] [Google Scholar]
- 18. Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw 2010; 36(3): 1–48. [Google Scholar]
- 19. Sadikovic B, Fernandes P, Zhang VW, et al. Mutation update for UBE3A variants in Angelman syndrome. Hum Mutat 2014; 35: 1407–1417. [DOI] [PubMed] [Google Scholar]
- 20. Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 1997; 15: 70–73. [DOI] [PubMed] [Google Scholar]
- 21. Hillman PR, Christian SGB, Doan R, et al. Genomic imprinting does not reduce the dosage of UBE3A in neurons. Epigenetics Chromatin 2017; 10(1): 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duker AL, Ballif BC, Bawle EV, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet 2010; 18(11): 1196–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perk J, Makedonski K, Lande L, et al. The imprinting mechanism of the Prader-Willi/Angelman regional control center. EMBO J 2002; 21(21): 5807–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown RE, Basheer R, McKenna JT, et al. Control of sleep and wakefulness. Physiol Rev 2012; 92: 1087–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet 2016; 18: 164–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol 2001; 63: 647–676. [DOI] [PubMed] [Google Scholar]
- 27. Saper CB, Lu J, Chou TC, et al. The hypothalamic integrator for circadian rhythms. Trends Neurosci 2005; 28: 152–157. [DOI] [PubMed] [Google Scholar]
- 28. Buijs FN, Guzmán-Ruiz M, León-Mercado L, et al. Suprachiasmatic nucleus interaction with the arcuate nucleus; essential for organizing physiological rhythms. eNeuro 2017; 4(2): ENEURO.0028-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ivanova E, Kelsey G. Imprinted genes and hypothalamic function. J Mol Endocrinol 2011; 47: R67–R74. [DOI] [PubMed] [Google Scholar]
- 30. Tucci V. Genomic imprinting: a new epigenetic perspective of sleep regulation. PLoS Genet 2016; 12(5): e1006004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Scammell TE, Arrigoni E, Lipton JO. Neural circuitry of wakefulness and sleep. Neuron 2017; 93: 747–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spruyt K, Braam W, Curfs LM. Sleep in Angelman syndrome: a review of evidence. Sleep Med Rev 2018; 37: 69–84. [DOI] [PubMed] [Google Scholar]
- 33. Camfferman D, McEvoy RD, O’Donoghue F, et al. Prader-Willi syndrome and excessive daytime sleepiness. Sleep Med Rev 2008; 12(1): 65–75. [DOI] [PubMed] [Google Scholar]
- 34. Wharton RH, Levine K, Hobson JA. Abnormalities of sleep and arousal in Prader-Willi syndrome. Am J Med Genet 1992; 42: 261. [Google Scholar]
- 35. Walz NC, Beebe D, Byars K. Sleep in individuals with Angelman syndrome: parent perceptions of patterns and problems. Am J Ment Retard 2005; 110(4): 243–252. [DOI] [PubMed] [Google Scholar]
- 36. Bruni O, Ferri R, D’Agostino G, et al. Sleep disturbances in Angelman syndrome: a questionnaire study. Brain Dev 2004; 26(4): 233–240. [DOI] [PubMed] [Google Scholar]
- 37. Conant KD, Thibert RL, Thiele EA. Epilepsy and the sleep-wake patterns found in Angelman syndrome. Epilepsia 2009; 50: 2497–2500. [DOI] [PubMed] [Google Scholar]
- 38. Didden R, Korzilius H, Smits MG, et al. Sleep problems in individuals with Angelman syndrome. Am J Ment Retard 2004; 109: 275–284. [DOI] [PubMed] [Google Scholar]
- 39. Vgontzas AN, Bixler EO, Kales A, et al. Daytime sleepiness and REM abnormalities in Prader-Willi syndrome: evidence of generalized hypoarousal. Int J Neurosci 1996; 87: 127–139. [DOI] [PubMed] [Google Scholar]
- 40. Sedky K, Bennett DS, Pumariega A. Prader-Willi syndrome and obstructive sleep apnea: co-occurrence in the pediatric population. J Clin Sleep Med 2014; 10(4): 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nixon GM, Brouillette RT. Sleep and breathing in Prader-Willi syndrome. Pediatr Pulmonol 2002; 34: 209–217. [DOI] [PubMed] [Google Scholar]
- 42. Abel EA, Tonnsen BL. Sleep phenotypes in infants and toddlers with neurogenetic syndromes. Sleep Med 2017; 38: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Allen KD, Kuhn BR, DeHaai KA, et al. Evaluation of a behavioral treatment package to reduce sleep problems in children with Angelman syndrome. Res Dev Disabil 2013; 34: 676–686. [DOI] [PubMed] [Google Scholar]
- 44. Goldman SE, Bichell TJ, Surdyka K, et al. Sleep in children and adolescents with Angelman syndrome: association with parent sleep and stress. J Intellect Disabil Res 2011; 56: 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Richdale AL, Cotton S, Hibbit K. Sleep and behavior disturbance in Prader-Willi syndrome: a questionnaire study. J Intellect Disabil Res 1999; 43(5): 380–392. [DOI] [PubMed] [Google Scholar]
- 46. Maas APHM, Sinnema M, Didden R, et al. Sleep disturbances and behavioural problems in adults with Prader-Willi syndrome. J Intellect Disabil Res 2010; 54: 906–917. [DOI] [PubMed] [Google Scholar]
- 47. Cotton SM, Richdale AL. Sleep patterns and behaviour in typically developing children and children with autism, Down syndrome, Prader-Willi syndrome and intellectual disability. Res Autism Spect Dis 2010; 4(3): 490–500. [Google Scholar]
- 48. Priano L, Grugni G, Miscio G, et al. Sleep cycling alternating pattern (CAP) expression is associated with hypersomnia and GH secretory pattern in Prader-Willi syndrome. Sleep Med 2006; 7(8): 627–633. [DOI] [PubMed] [Google Scholar]
- 49. Verrillo E, Bruni O, Franco P, et al. Analysis of NREM sleep in children with Prader-Willi syndrome and the effect of growth hormone treatment. Sleep Med 2009(6): 646–650. [DOI] [PubMed] [Google Scholar]
- 50. Gibbs S, Wiltshire E, Elder D. Nocturnal sleep measured by actigraphy in children with Prader-Willi syndrome. J Pediatr 2013; 162(4): 765–776. [DOI] [PubMed] [Google Scholar]
- 51. Yee BJ, Buchanan PR, Mahadev S, et al. Assessment of sleep and breathing in adults with Prader-Willi syndrome: a case control series. J Clin Sleep Med 2007; 3(7): 713–718. [PMC free article] [PubMed] [Google Scholar]
- 52. Joo E, Hong SB, Sohn YB, et al. Plasma adiponectin level and sleep structures in children with Prader-Willi syndrome. J Sleep Res 2010; 19: 248–254. [DOI] [PubMed] [Google Scholar]
- 53. Ghergan A, Coupaye M, Leu-Semenescu S, et al. Prevalence and phenotype of sleep disorders in 60 adults with Prader-Willi syndrome. Sleep 2017; 40(12): zsx162. [DOI] [PubMed] [Google Scholar]
- 54. Miano S, Bruni O, Elia M, et al. Sleep breathing and periodic leg movement pattern in Angelman Syndrome: a polysomnographic study. Clin Neurophysiol 2005; 116(11): 2685–2692. [DOI] [PubMed] [Google Scholar]
- 55. Pavone M, Caldarelli V, Khirani S, et al. Sleep disordered breathing in patients with Prader-Willi syndrome: a multicenter study. Pediatr Pulmonol 2015; 50: 1354–1359. [DOI] [PubMed] [Google Scholar]
- 56. Lin H-Y, Lin S-P, Lin C-C, et al. Polysomnographic characteristics in patients with Prader-Willi syndrome. Pediatr Pulmonol 2007; 42: 881–887. [DOI] [PubMed] [Google Scholar]
- 57. Williams K, Scheimann A, Sutton V, et al. Sleepiness and sleep disordered breathing in Prader-Willi syndrome: relationship to genotype, growth hormone therapy, and body composition. J Clin Sleep Med 2008; 4(2): 111–118. [PMC free article] [PubMed] [Google Scholar]
- 58. Miano S, Bruni O, Leuzzi V, et al. Sleep polygraphy in Angelman syndrome. Clin Neurophysiol 2004; 115(4): 938–945. [DOI] [PubMed] [Google Scholar]
- 59. Takaesu Y, Komada Y, Inoue Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med 2012; 13: 1164–1170. [DOI] [PubMed] [Google Scholar]
- 60. Paprocka J, Kijonka M, Wojcieszek P, et al. Melatonin and Angelman syndrome: implications and mathematical model of diurnal secretion. Int J Endocrinol 2017; 2017: 5853167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhdanova IV, Wurtman RJ, Wagstaff J. Effects of a low dose of melatonin on sleep in children with Angelman syndrome. J Pediatr Endocrinol Metab 1999; 12: 57–67. [DOI] [PubMed] [Google Scholar]
- 62. Braam W, Didden R, Smits MG, et al. Melatonin for chronic insomnia in Angelman syndrome: a randomized placebo-controlled trial. J Child Neurol 2007; 23(6): 649–654. [DOI] [PubMed] [Google Scholar]
- 63. Willig RP, Braun W, Commentz JC, et al. Circadian fluctuation of plasma melatonin in Prader-Willi’s syndrome and obesity. Acta Endocrinol Suppl 1986; 279: 411–415. [DOI] [PubMed] [Google Scholar]
- 64. Butler MG, Brandau DT, Theodoro MF, et al. Morning melatonin levels in Prader-Willi syndrome. Am J Med Genet A 2009; 149A(8): 1809–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Manni R, Politini L, Nobili L, et al. Hypersomnia in the Prader-Willi Syndrome: clinical-electrophysiological features and underlying factors. Clin Neurophysiol 2011; 112: 800–805. [DOI] [PubMed] [Google Scholar]
- 66. Yan J, Wang H, Liu Y, et al. Analysis of gene regulatory networks in the mammalian circadian rhythm. PLoS Comput Biol 2008; 4: e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lim ASP, Srivastava GP, Yu L, et al. 24-Hour rhythms of DNA methylation and their relation with rhythms of RNA expression in the human dorsolateral prefrontal cortex. PLoS Genet 2014; 10: e1004792. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68. Moller-Levet CS, Archer SN, Bucca G, et al. Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. Proc Natl Acad Sci U S A 2013; 110: E1132–E1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ehlen JC, Jones KA, Pinckney L, et al. Maternal UBE3a loss disrupts sleep homeostasis but leaves circadian rhythmicity largely intact. J Neurosci 2015; 35(40): 13587–13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shi S, Bichell TJ, Ihrie RA, et al. UBE3a imprinting impairs circadian robustness in Angelman syndrome models. Curr Biol 2015; 25(5): 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gossan NC, Zhang F, Guo B, et al. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acid Res 2014; 42(9): 5765–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jones KA, Han JE, DeBruyne JP, et al. Persistent neuronal Ube3a expression in the suprachiasmatic nucleus of Angelman syndrome model mice. Sci Rep 2016; 6: 28238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kozlov SV, Bogenpohl JW, Howell MP, et al. The imprinted gene MAGEL2 regulates normal circadian output. Nat Genet 2007; 39(10): 1266–1272. [DOI] [PubMed] [Google Scholar]
- 74. Devos J, Weselake SV, Wevrick R. Magel2, a Prader-Willi syndrome candidate gene, modulates the activities of circadian rhythm proteins in cultured cells. J Circadian Rhythms 2011; 9: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Powell WT, Coulson RL, Crary FK, et al. A Prader-Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum Mol Genet 2013; 22(21): 4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Coulson RL, Yasui DH, Dunaway KW, et al. Snord116-dependent diurnal rhythm of DNA methylation in mouse cortex. Nat Commun 2018; 9: 1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sellen DW. Evolution of infant and young child feeding: implications for contemporary public health. Ann Rev Nutr 2007; 27: 123–148. [DOI] [PubMed] [Google Scholar]
- 78. Kotler J, Haig D. The tempo of human childhood: a maternal foot on the accelerator, a paternal foot on the brake. Evol Anthropol 2018; 27: 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nicklaus S. Development of food variety in children. Appetite 2009; 52: 253–255. [DOI] [PubMed] [Google Scholar]
- 80. Cooke LJ, Haworth CM, Wardle J. Genetic and environmental influences on children’s food neophobia. Am J Clin Nutr 2007; 86(2): 428–433. [DOI] [PubMed] [Google Scholar]
- 81. Pelto GH, Zhang Y, Habicht J-P. Premastication: the second arm of infant and young child feeding for health and survival? Matern Child Nutr 2010; 6: 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Haig D. Transfers and transitions: parent-offspring conflict, genomic imprinting, and the evolution of human life history. Proc Natl Acad Sci U S A 2010; 107: 1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rossi MA, Stuber GD. Overlapping brain circuits for homeostatic and hedonic feeding. Cell Metab 2018; 27(1): 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Huda MSB, Wilding JPH, Pinkney JH. Gut peptides and the regulation of appetite. Obes Rev 2006; 7(2): 163–182. [DOI] [PubMed] [Google Scholar]
- 85. Schwartz MW, Woods SC, Porte D, et al. Central nervous system control of food intake. Nature 2000; 404(6778): 661–671. [DOI] [PubMed] [Google Scholar]
- 86. Farook MF, DeCuypere M, Hyland K, et al. Altered serotonin, dopamine and norepinepherine levels in 15q duplication and Angelman syndrome mouse models. PLoS ONE 2012; 7(8): e43030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mercer RE, Kwolek EM, Bischof JM, et al. Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal behavior in mice null for the circadian rhythm output gene Magel2. Am J Med Genet B 2009; 150B: 1085–1099. [DOI] [PubMed] [Google Scholar]
- 88. Dykens EM, Maxwell MA, Pantino E, et al. Assessment of hyperphagia in Prader-Willi syndrome. Obesity 2007; 15(7): 1816–1826. [DOI] [PubMed] [Google Scholar]
- 89. Heksch R, Kamboj M, Anglin K, et al. Review of Prader-Willi syndrome: the endocrine approach. Transl Pediatr 2017; 6: 274–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Siemensma EP, de Lind van Wijngaarden RF, Otten BJ, et al. Pubarche and serum dehydroepiandrosterone sulphate levels in children with Prader-Willi syndrome. Clin Endocrinol 2011; 75(1): 83–89. [DOI] [PubMed] [Google Scholar]
- 91. Berry RJ, Leitner RP, Clarke AR, et al. Behavioral aspects of Angelman syndrome: a case control study. Am J Med Genet A 2005; 132A(1): 8–12. [DOI] [PubMed] [Google Scholar]
- 92. Welham A, Lau JKL, Moss J, et al. Are Angelman and Prader-Willi syndromes more similar than we thought? Food-related behavior problems in Angelman, Cornelia de Lange, Fragile X, Prader-Willi and 1p36 deletion syndromes. Am J Med Genet A 2015; 167(3): 572–578. [DOI] [PubMed] [Google Scholar]
- 93. Mertz LGB, Christensen R, Vogel I, et al. Eating behavior, prenatal and postnatal growth in Angelman syndrome. Res Dev Disabil 2014; 35(11): 2681–2690. [DOI] [PubMed] [Google Scholar]
- 94. Brennan M-L, Adam MP, Seaver LH, et al. Increased body mass in infancy and early toddlerhood in Angelman syndrome patients with uniparental disomy and imprinting center defects. Am J Med Genet A 2015; 167A: 142–146. [DOI] [PubMed] [Google Scholar]
- 95. Michel LM, Haqq AM, Wismer WV. A review of chemosensory perceptions, food preferences and food-related behaviours in subjects with Prader-Willi syndrome. Appetite 2016; 99: 17–24. [DOI] [PubMed] [Google Scholar]
- 96. Caldwell ML, Taylor RL, Bloom SR. An investigation of the use of high- and low-preference food as a reinforcer for increased activity of individuals with Prader-Willi syndrome. J Ment Defic Res 1986; 30: 347–354. [DOI] [PubMed] [Google Scholar]
- 97. Taylor RL, Caldwell ML. Type and strength of food preferences of individuals with Prader-Willi syndrome. J Ment Defic Res 1985; 29: 109–112. [DOI] [PubMed] [Google Scholar]
- 98. Fieldstone A, Zipf WB, Schwartz HC, et al. Food preferences in Prader-Willi syndrome, normal weight and obese controls. Int J Obes Relat Metab Disord 1997; 21: 1046–1052. [DOI] [PubMed] [Google Scholar]
- 99. Glover D, Maltzman I, Williams C. Food preferences among individuals with and without Prader-Willi syndrome. Am J Ment Retard 1996; 101: 195–205. [PubMed] [Google Scholar]
- 100. Hinton EC, Holland AJ, Gellatly MS, et al. Neural representations of hunger and satiety in Prader-Willi syndrome. Int J Obes 2006; 30: 313–321. [DOI] [PubMed] [Google Scholar]
- 101. Joseph B, Egli M, Koppekin A, et al. Food choice in people with Prader-Willi syndrome: quantity and relative preference. Am J Ment Retard 2002; 107: 128–135. [DOI] [PubMed] [Google Scholar]
- 102. Dykens EM. Contaminated and unusual food combinations: what do people with Prader-Willi syndrome choose? Ment Retard 2000; 38: 163–171. [DOI] [PubMed] [Google Scholar]
- 103. Young J, Zarcone J, Holsen L, et al. A measure of food seeking in individuals with Prader-Willi syndrome. J Intellect Disabil Res 2006; 50: 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Clarke DJ, Marston G. Problem behaviors associated with 15q- Angelman syndrome. Am J Ment Retard 2000; 105(1): 25–31. [DOI] [PubMed] [Google Scholar]
- 105. Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat 2008; 4(3): 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Marí-Bauset S, Zazpe I, Mari-Sanchis A, et al. Food selectivity in autism spectrum disorders. J Child Neurol 2013; 29: 1554–1561. [DOI] [PubMed] [Google Scholar]
- 107. Raiten DJ, Massaro T. Perspectives on the nutritional ecology of autistic children. J Autism Dev Disord 1986; l6: 133–143. [DOI] [PubMed] [Google Scholar]
- 108. Schreck KA, Williams K, Smith AF. A comparison of eating behaviors between children with and without autism. J Autism Dev Disord 2004; 34: 433–438. [DOI] [PubMed] [Google Scholar]
- 109. Dominick KC, Davis NO, Lainhart J, et al. Atypical behaviors in children with autism and children with a history of language impairment. Res Dev Disabil 2007; 28: 145–162. [DOI] [PubMed] [Google Scholar]
- 110. Schmitt L, Heiss CJ, Campbell EE. A comparison of nutrient intake and eating behaviors of boys with and without autism. Topic Clin Nutr 2008; 23: 23–31. [Google Scholar]
- 111. Martins Y, Young RL, Robson DC. Feeding and eating behaviors in children with autism and typically developing children. J Autism Dev Disord 2008; 38: 1878–1887. [DOI] [PubMed] [Google Scholar]
- 112. Bandini LG, Anderson SE, Curtin C, et al. Food selectivity in children with autism spectrum disorders and typically developing children. J Pediatr 2010; 157(2): 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zimmer MH, Hart LC, Manning-Courtney P, et al. Food variety as a predictor of nutritional status among children with autism. J Autism Dev Disord 2011; 42: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Diolordi L, del Balzo V, Bernabei P, et al. Eating habits and dietary patterns in children with autism. Eat Weight Disord 2014; 19(3): 295–301. [DOI] [PubMed] [Google Scholar]
- 115. Marí-Bauset S, Llopis-González A, Zazpe-García I, et al. Nutritional status of children with autism spectrum disorders (ASDs): a case–control study. J Autism Dev Disord 2014; 45: 203–212. [DOI] [PubMed] [Google Scholar]
- 116. Hubbard KL, Anderson SE, Curtin C, et al. A comparison of food refusal related to characteristics of food in children with autism spectrum disorder and typically developing children. J Acad Nutr Diet 2014; 114(12): 1981–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Russell H, Oliver C. The assessment of food-related problems in individuals with Prader-Willi syndrome. Br J Clin Psychol 2003; 42: 379–392. [DOI] [PubMed] [Google Scholar]
- 118. Holland AJ, Treasure J, Coskeran P, et al. Measurement of excessive appetite and metabolic changes in Prader-Willi syndrome. Int J Obes Relat Metab Disord 1993; 17: 527–532. [PubMed] [Google Scholar]
- 119. Irizarry KA, Bain J, Butler MG, et al. Metabolic profiling in Prader-Willi syndrome and nonsyndromic obesity: sex differences and the role of growth hormone. Clin Endocrinol 2015; 83: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Cummings DE, Clement K, Purnell JQ, et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med 2002; 8: 643–644. [DOI] [PubMed] [Google Scholar]
- 121. Del Parigi A, Tschöp M, Heiman ML, et al. High circulating ghrelin: a potential cause for hyperphagia and obesity in Prader-Willi syndrome. J Clin Endocrinol Metab 2002; 87(12): 5461–5464. [DOI] [PubMed] [Google Scholar]
- 122. Kweh FA, Miller JL, Sulsona CR, et al. Hyperghrelinemia in Prader-Willi syndrome begins in early infancy long before the onset of hyperphagia. Am J Med Genet A 2014; 167(1): 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Bochukova EG, Lawler K, Croizier S, et al. A transcriptomic signature of the hypothalamic response to fasting and BDNF deficiency in Prader-Willi syndrome. Cell Rep 2018; 22(13): 3401–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Fountain MD, Aten E, Cho MT, et al. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet Med 2016; 19(1): 45127–45152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. McCarthy JM, McCann-Crosby BM, Rech ME, et al. Hormonal, metabolic and skeletal phenotype of Schaaf-Yang syndrome: a comparison to Prader-Willi syndrome. J Med Genet 2018; 55(5): 307–315. [DOI] [PubMed] [Google Scholar]
- 126. Bischof JM, Stewart CL, Wevrick R. Inactivation of the mouse MAGEL2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum Mol Genet 2007; 16: 2713–2719. [DOI] [PubMed] [Google Scholar]
- 127. Mercer RE, Michaelson SD, Chee MJS, et al. Magel2 is required for leptin-mediated depolarization of POMC neurons in the hypothalamic arcuate Nucleus in mice. PLoS Genet 2013; 9(1): e1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wijesuriya TM, De Ceuninck L, Masschaele D, et al. The Prader-Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum Mol Genet 2017; 26(21): 4215–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ding F, Li HH, Zhang S, et al. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 2008; 3(3): e1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Qi Y, Purtell L, Fu M, et al. Snord116 is critical in the regulation of food intake and body weight. Sci Rep 2016; 6(1): 18614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Polex-Wolf J, Lam BYH, Larder R, et al. Hypothalamic loss of Snord116 recapitulates the hyperphagia of Prader-Willi syndrome. J Clin Invest 2018; 128(3): 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Golding DM, Rees DJ, Davies JR, et al. Paradoxical leanness in the imprinting-centre deletion mouse model for Prader-Willi syndrome. J Endocrinol 2017; 232(1): 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Davies JR, Humby T, Dwyer DM, et al. Calorie seeking, but not hedonic response, contributes to hyperphagia in a mouse model for Prader-Willi syndrome. Eur J Neurosci 2015; 42(4): 2105–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cooper SJ, Al-Naser HA. Dopaminergic control of food choice: contrasting effects of SKF 38393 and quinpirole on high-palatability food preference in the rat. Neuropharmacology 2006; 50(8): 953–963. [DOI] [PubMed] [Google Scholar]
- 135. Tacer KF, Potts PR. Cellular and disease functions of Prader-Willi Syndrome gene MAGEL2. Biochem J 2017; 474: 2177–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. LaSalle JM, Reiter LT, Chamberlain SJ. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics 2015; 7: 1213–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wiater MF, Mukherjee S, Li A-J, et al. Circadian integration of sleep-wake and feeding requires NPY receptor-expressing neurons in the mediobasal hypothalamus. Am J Physiol Regul Integr Comp Physiol 2011; 301(5): R1569–R1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Willie JT, Chemelli RM, Sinton CM, et al. To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci 2001; 24(1): 429–458. [DOI] [PubMed] [Google Scholar]
- 139. Cheon CK. Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome. Ann Pediatr Endocrinol Metab 2016; 21(3): 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tan W-H, Bird LM, Thibert RL, et al. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A 2014; 164(4): 975–992. [DOI] [PubMed] [Google Scholar]
- 141. Sadeh A, Tikotzky L, Scher A. Parenting and infant sleep. Sleep Med Rev 2010; 14(2): 89–96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, PRISMA_2009_checklist_1 for Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes by Iiro Ilmari Salminen, Bernard J Crespi and Mikael Mokkonen in SAGE Open Medicine
Supplemental material, PRISMA_2009_flow_diagram_1 for Baby food and bedtime: Evidence for opposite phenotypes from different genetic and epigenetic alterations in Prader-Willi and Angelman syndromes by Iiro Ilmari Salminen, Bernard J Crespi and Mikael Mokkonen in SAGE Open Medicine