

Abstract
Oncolytic virotherapy is a promising approach for treating recurrent and/or drug-resistant ovarian cancer. However, its successful application in the clinic has been hampered by rapid immune-mediated clearance or neutralization of the virus, which reduces viral access to tumor foci. To overcome this barrier, patient-derived mesenchymal stem cells have been used to deliver virus to tumors, but variability associated with autologous cell isolations prevents this approach from being broadly clinically applicable. Here, we demonstrate the ability of an allogeneic, clonal neural stem cell (NSC) line (HB1.F3.CD21) to protect oncolytic viral cargo from neutralizing antibodies within patient ascites fluid and to deliver it to tumors within preclinical peritoneal ovarian metastases models. The viral payload used is a conditionally replication-competent adenovirus driven by the survivin promoter (CRAd-S-pk7). Because the protein survivin is highly expressed in ovarian cancer, but not in normal differentiated cells, viral replication should occur selectively in ovarian tumor cells. We found this viral agent was effective against cisplatin-resistant ovarian tumors and could be used as an adjunct treatment with cisplatin to decrease tumor burden without increasing toxicity. Collectively, our data suggest NSC-delivered CRAd-S-pk7 virotherapy holds promise for improving clinical outcome, reducing toxicities, and improving quality of life for patients with advanced ovarian cancer.
Keywords: oncolytic virotherapy, CRAd-S-pk7, ovarian cancer, neural stem cells, cellular therapy
Graphical Abstract
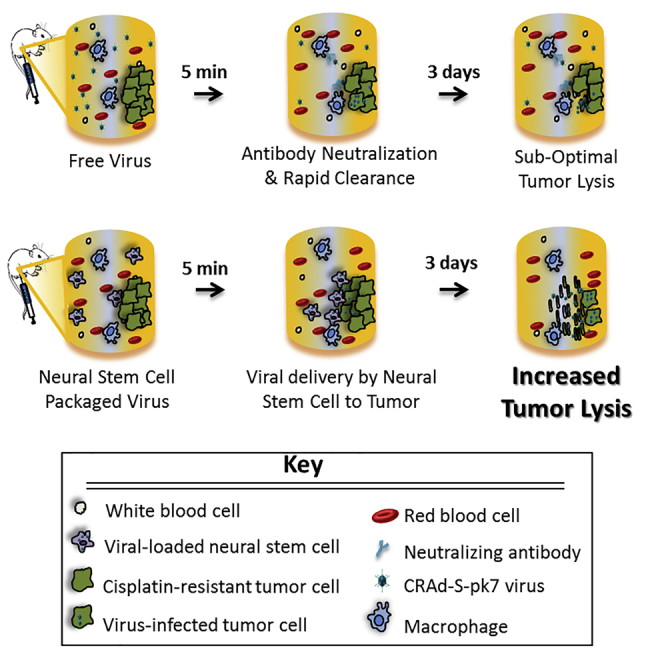
Introduction
Ovarian cancer is the most lethal gynecologic malignancy in the United States, because most patients already have abdominal metastases upon diagnosis. At this stage, their 5-year survival rate is only 34% following standard surgical debulking and chemotherapy.1 Thus, more effective and less toxic therapeutic approaches are urgently needed. Oncolytic virotherapy offers a new approach, whereby viruses are modified to preferentially replicate in tumor cells and destroy them in situ via direct lysis.2 The viral particles freed from lysed tumor cells continue to infect neighboring tumor cells, amplifying their anti-neoplastic effect until they reach normal tissue, at which point viral replication ceases.3 Oncolytic viruses can induce cancer cell death4 irrespective of chemoresistance5 and can stimulate immune-recognition of cancer cells because tumor antigens are exposed when the cancer cells lyse. To date, more than 11 oncolytic viruses have been tested in pre-clinical human ovarian cancer models, with 4 progressing to phase I/II clinical trials.2 Although these studies are still in early stages, all clinical trials so far have established the safety and non-toxicity of this approach.2 The challenge now is to achieve efficacy.
To date, adenovirus subtype 5 (Ad5)-based virotherapy agents have shown some of the best clinical results, as measured by the percentage of patients achieving stable disease and/or experiencing a partial response.6 Particularly effective are newer generation viruses with modified Ad5 capsids that enhance viral infection in situ and that are engineered to replicate only under the control of tumor-specific promoters.7 One such virus, CRAd-S-pk7, has been modified to replicate under the control of the survivin promoter.7 Survivin is a developmentally expressed protein that can suppress apoptosis and regulate cell division in a variety of drug-refractory cancers,8 including ovarian cancer.9, 10, 11 In addition, a poly-L-lysine (pk7) peptide was incorporated into the C terminus of the wild-type adenoviral fiber knob domain to enable more efficient loading into tumor cells.12
Although such transcriptional and transductional enhancements have improved oncoviral efficacy,7, 13, 14, 15 vector distribution remains a significant obstacle. Specifically, oncolytic viruses injected into the peritoneal space are subject to rapid clearance because of their small ∼100-nm size.16 The delivery hurdles for oncolytic adenoviruses are particularly high, because most of the population has pre-existing immunity since adenoviruses are a common human pathogen. Thus, the majority of administered CRAds do not exist as un-associated particles for longer than a few minutes,17 which limits their ability to infect tumors and reduces antitumor efficacy.
To overcome these barriers, there is increasing interest in developing tumor-tropic cell carriers for viral agents. The ideal cell carrier would be chromosomally normal and stable, support viral infection and amplification in situ, shield the loaded viruses from the host immune system,16 and most importantly, home to tumors so that viral penetration and distribution at tumor sites is improved.2, 7, 9 To this end, we have extensively characterized the immortalized HB1.F3.CD21 neural stem cell (NSC) line, showing it is tumor-tropic, chromosomally and functionally stable, non-tumorigenic, minimally immunogenic (human leukocyte antigen [HLA] class II negative18, 19), and clinically safe.20 We also established that these NSCs can improve tumor-localized production of the oncolytic adenovirus, CRAd-S-pk7, in glioma models.12, 21, 22, 23 The viral loading, release kinetics, and freeze-thaw standard operating procedures (SOPs) for HB1.F3.CD21 NSCs loaded with CRAd-S-pk7 (NSC.CRAd-S-pk7) have been established, and a good manufacturing practice (GMP) bank is currently in clinical trial as an adjunct oncolytic virotherapy for newly diagnosed glioma (ClinicalTrials.gov: NCT03072134). These established SOPs mean additional “off-the-shelf” allogeneic clinical banks can be generated, providing an accelerated route to clinical translation for ovarian cancer.
Here, we conduct in vitro and in vivo studies to assess the pre-clinical utility of NSC.CRAd-S-pk7 in the context of ovarian cancer metastases within the peritoneal cavity. Our studies show that NSC.CRAd-S-pk7 cells selectively target and penetrate tumor metastases, effectively delivering the CRAd-S-pk7 virus. The virus then replicates within tumor cells and lyses them. The resulting delay in tumor progression is as robust as that observed when treating with the commonly used chemotherapy, cisplatin, thus offering a potential strategy to minimize the toxicity of cisplatin treatments. We also found that NSC.CRAd-S-pk7 may have a synergistic therapeutic effect when combined with cisplatin, further reducing tumor burden without increasing toxicity.
Results
Survivin Expression in Ovarian Cancer
Because we planned to use the CRAd-S-pk7 virus, for which replication is under the control of the survivin promoter,24 we first assessed the frequency at which survivin expression is upregulated in ovarian cancers as compared with normal tissues to ensure our approach would be of practical utility for ovarian cancer. To do this, we analyzed survivin gene (BIRC5 gencode: ENSG00000089685.10) expression within the publically available GEO Affymetrix human U133A microarray dataset (GEO: GSE26712). This query dataset includes gene expression data for an extensive set of 185 samples from (90 optimally debulked/95 suboptimally debulked) primary ovarian tumors and 10 samples representing normal ovarian surface epithelium.11, 25 We found that 93.5% (173/185) of ovarian cancer patients represented in this dataset exhibited BIRC5 expression levels that exceeded those in the normal ovarian surface epithelium (Figure 1A). Furthermore, because we ultimately intend to deliver the NSC.CRAd-S-pk7 therapy intraperitoneally (i.p.), we needed to ensure that survivin expression is low, and viral replication thereby avoided, in healthy peritoneal organs. To do this, we analyzed surviving transcription expression levels within the Protein Atlas GTExPortal dataset, which revealed that although survivin is highly expressed in tumors, it is not highly expressed in normal adult organs within the peritoneal cavity (Figure 1B) or in organs outside the peritoneal cavity (Figure S1). It is noted that adult mesenchymal and intestinal progenitor cells express survivin,26, 27 and so utilizing the NSCs to distribute this virus solely to tumor locations should minimize any unintended off-target distribution.
Figure 1.

CRAd-S-pk7 NSCs for Ovarian Cancer
(A) GEO Accession Viewer data showing survivin (BIRC5 gene ID: 202095_s_at) expression in cisplatin-resistant patient tumors (185 samples) and 10 healthy tissues (10 samples). (B) Analysis of GTExPortal data available via Protein Atlas showing survivin expression in various peritoneal tissues. Expression values are shown in RPKM (reads per kilobase of transcript per million mapped reads), calculated from a gene model in which isoforms were collapsed to a single gene. No other normalization steps were applied. Boxplots show median and 25th and 75th percentiles; points are displayed as outliers if they are greater than or less than 1.5 times the interquartile range. (C) Cbioportal Oncoprint compact visualization of mRNA expression scores of three putative CRAd-S-pk7 entry receptors in the 100 patient samples included in The Cancer Genome Atlas serous ovarian cancer project that had survivin amplification (311 patients total). Red indicates amplification; blue indicates deep deletion. BIRC5, survivin; heparin sulfate proteoglycans: GPC1, glypican 1; HSPG2, perlecan; SDC1, Syndecan 1.
CRAd-S-pk7 Transduction in Ovarian Cancer
Based on our previous experience in the context of glioma, we expect that once CRAd-S-pk7-expressing NSCs localize to ovarian tumors, they will produce virus at the tumor sites for up to 3 days before achieving release of viral agent.23 Following this release, efficient infection of surrounding tumor cells will rely on rapid viral uptake by tumor cells expressing cell-surface receptors permissive for adenoviral entry. The poly-lysine modification to the fiber knob protein (pk7) contained within CRAd-S-pk7 permits the virus to bind to and enter tumor cells that express heparin sulfate proteoglycan family members such as perlecan (HSPG2), glypican-1 (GPC1), and syndecan-1 (SDC1).28, 29 To measure expression of these putative adenovirus entry receptors in ovarian cancer patients, we conducted Cbioportal Oncoprint analysis of samples from 100 patients with survivin amplification who were included in The Cancer Genome Atlas serous ovarian cancer project.24, 30 This analysis revealed variability in expression of these surface receptors, but 86% of ovarian cancer patients who overexpressed survivin also expressed at least one of these viral entry receptors (Figure 1C). This result implies that there is strong potential for enhanced viral infection to occur within patients, given the multiple routes available for viral entry.
CRAd-S-pk7 NSC Characterization
Our use of an immortalized cell line as the cell carrier enables the possibility of predictable, reproducible viral loading and release kinetics. Consistent with our previous reports,21 very efficient infection of NSCs by CRAd-S-pk7 was observed with 99.76% ± 0.20% of NSCs positive for the viral capsid protein, hexon, 1 day after transduction (Figures 2A and 2B). qPCR was used to confirm that the viral load per NSC (average = 644 viral particle [VP]/cell) was similar to the magnitude we previously reported21 (Figure 2C). Primer sequences are listed in Table 1. We previously described how the intracellular titer peaks 3 days after transduction, at which point the initial viral load amplifies by over a factor of 1 × 10.5, 21 As expected, the CRAD-S-pk7-infected NSCs began to undergo virus-induced cell lysis after 2–3 days in culture (Figures 2D and 2E).
Figure 2.

CRAd-S-pk7 NSC Characterization
(A) Flow cytometric quantification of hexon-positive NSCs after transduction (MOI = 50; 30 VP/ infectious unit [IFU]) with CRAd-S-pk7 virus. Anti-hexon samples: 99.76% ± 0.20% (+); isotype controls: 0.31% ± 0.06% (+); unstained controls: 0.25% ± 0.05% (+). (B) Fluorescent image showing infected NSCs stained with fluorescein isothiocyanate (FITC)-conjugated anti-hexon and counterstained with DAPI. Scale bar, 100 μm. (C) PCR quantification used to approximate viral load per NSC (644 VP/NSC). (D) Time-lapse bright-field images showing initial survival but eventual rounding and lysis due to viral burst within NSC.CRAd-S-pk7 cells (lower panels), but not parental NSCs (upper panels). (E) Software-automated quantification of phase-object confluency in each well demonstrating initial seeding and growth of NSC.CRAd-S-pk7 cells peaking at 2 days post-transduction prior to viral burst.
Table 1.
PCR Primer Sets
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| v-myc rxn 1 | Mari v-myc F: 5′-CCTTTGT TGATTTCGCCAAT-3′ | v-myc Inter R2: 5′-GCGAGCTTCTCCGACACCACC-3′ |
| Nested v-myc | GAG 1306 F: 5′-TCACAGCCAGATATCCAGCAGCTT-3′ | v-myc R1: 5′-AGTTCTCCTCCTCCTCCTCG-3′ |
| Hexon | Hex3 F: 5′-TTCCGCTTCACTGGACTCTT-3′ | Hex3 R: 5′-TGGACAGCGAGGAGAGAAG-3′ |
| GAPDH | Fwd: 5′-ATGTTCGTCATGGGTGTGAACCA-3′ | Rev: 5′-TGGCAGGTTTTTCTAGACGGCAG-3′ |
DNA purified from unmodified NSCs and NSC.CRAd-S-pk7 was used as a positive control for PCR amplification of the v-myc gene (replicon size 170 bp) and hexon (179 bp), respectively. Pure water was used as a negative control. PCR products were analyzed by agarose gel electrophoresis and staining of DNA bands with ethidium bromide.
NSC.CRAd-S-pk7 Tropism to Orthotopic Ovarian Cancer
To confirm that the viral agent did not interfere with the tumor-tropic properties of the NSCs toward ovarian cancer-derived cytokines, we conducted an in vitro Boyden chamber migration assay. The NSC.CRAd-S-pk7 effectively migrated to conditioned media from a range of ovarian cancer cell lines (Figure S2C) including the OVCAR8 and ID8 cell lines used for our xenograft and syngeneic mouse models (Figure 3A). In fact, NSC.CRAd-S-pk7 showed greater tropism to tumor-conditioned media than did the untransduced parental HB1.F3.CD NSCs.
Figure 3.

NSC-CRAd-S-pk7 Tropism to Ovarian Cancer
(A) Boyden migration assay comparing tropism of NSC-CRAd-S-pk7 and NSCs with ovarian cancer conditioned media versus U87 (+, “100”) and BSA (−) control media. (B and D) Organ plucks 3 weeks after tumor inoculation showing ovarian metastases (large white nodules) occupying omental tissue in both the immunodeficent (B) and immunocompetent (D) mouse models. (C and E) DiI-labeled NSCs (red) loaded with CRAd-S-pk7 (yellow) were injected i.p. into mice with established EGFP ovarian metastases (green). NSC.CRAd-S-pk7 distribution is shown in both (C) xenograft and (E) syngeneic metastatic ovarian cancer models. Scale bars: 50 μm (C, and applies to both image sets).
We have previously demonstrated that NSCs home to ovarian tumors after i.p. administration into mice bearing either SKOV3 or OVCAR8 i.p. xenografts.31 We chose to use the OVCAR8 cell line for our xenograft model given its ability to consistently engraft combined with its moderate genetic similarity to high-grade serous ovarian cancer tumor samples.32 To confirm that NSC.CRAd-S-pk7 could deliver the oncolytic viral agent to peritoneal metastases, we established EGFP-expressing OVCAR8 and ID8 disseminated peritoneal tumors in NOD scid gamma (NSG) or B6 mice, respectively (Figures 3B and 3D). Two weeks later, the mice were administered 1,1'-dioctadecyl-3,3,3',3'-tetramethylindocarbocyanine perchlorate (DiI)-labeled NSC.CRAd-S-pk7s. All peritoneal tissues were harvested 2 days after administration. To visualize NSC-mediated viral secretion, we performed anti-hexon viral staining on sectioned tumors to confirm viral distribution outside of the DiI-labeled NSCs but within the tumor (Figures 3C and 3E). Within both xenograft and syngeneic models, the yellow anti-hexon viral stain not only co-localized with the red DiI-labeled NSC.CRAd-S-pk7s, but also spread throughout the EGFP-positive tumor tissue (Figures 3C and 3E). This result suggests that at 2 days after administration, some of the virus is being released, whereas some is still contained within the NSCs, consistent with our in vitro viral burst kinetics. Although the yellow anti-hexon viral stain never extended beyond the green EGFP-positive tumor nodules into adjacent normal tissue, it cannot yet be determined whether the combined use of NSC carriers and survivin-driven viral replication effectively avoid off-target tissue distribution given that human adenoviruses do not replicate well within mouse tissue.
In Vitro Efficacy of NSC.CRAd-S-pk7
We next sought to determine the cytotoxicity of NSC.CRAd-S-pk7 against established ovarian cancer cells. We co-cultured NSC.CRAd-S-pk7 with three different human ovarian cancer cell lines (Skov3, OVCAR8, and Kuromochi using a 1:1 ratio of NSC.CRAd-S-pk7 to ovarian cancer cells) and observed efficient elimination of tumor cells from all three lines over the course of 5 days (Figure S2). In contrast, no cell killing was seen in co-cultures of parental NSCs and any of the three ovarian cancer cell lines. This indicates that the CRAd-S-pk7 virus could replicate within NSCs, infect neighboring tumor cells, and continue to amplify throughout the tumor culture.
In preparation for the in vivo studies within both xenograft and syngeneic models, we further explored the sensitivity of the human ovarian cancer line (OVCAR8) and a mouse ovarian cancer line (ID8) (Figure 4). NSC.CRAd-S-pk7 significantly reduced (>9-fold less than tumor-only control) the number of OVCAR8 cells by day 14 in culture, even when seeded at an initial ratio of 1:1,000 NSC.CRAd-S-pk7:OVCAR8 cells (Figures 4A and 4B). These data confirm the potent oncolytic potential of NSC.CRAd-S-pk7 to produce a considerable local bystander effect against the OVCAR8 cancer cell line. As expected, the virus was less potent against the faster replicating mouse ID8 line. Nonetheless, NSC.CRAd-S-pk7 significantly reduced (>40% less than tumor-only control) the number of ID8 cells by day 14 in culture, when seeded at an initial ratio of 1:10 NSC.CRAd-S-pk7:ID8 cells (Figures 4C and 4D). These data demonstrate that the ID8 line is semi-permissive for CRAd-S-pk7 replication, and thus useful for in vivo experiments within a B6 immunocompetent mouse model. Collectively, the in vitro cytotoxicity results confirm the potential for NSC.CRAd-S-pk7 to kill ovarian cancer cells. However, given that metastatic ovarian cancer spreads throughout the peritoneal cavity, we wanted to determine whether NSC.CRAd-S-pk7 would be effective in the peritoneal setting.
Figure 4.

Ovarian Cancer Lysis by CRAd-s-pk7 NSCs
NSC.CRAd-S-pk7 were co-cultured at the indicated seeding ratios with the human OVCAR8 (A and B) or mouse ID8 (C and D) ovarian tumor cell line and cultured for 14 days. (A and C) The fold increase in crystal violet absorption relative to day 1 (average ± SD) is shown for both OVCAR8 (A) and ID8 (C) cocultures. (B and D) Representative bright-field images of crystal violet-stained co-cultures are shown for both OVCAR8 (B) and ID8 (D) co-cultures. Scale bar, 50 μm.
NSCs Protect the CRAd-S-pk7 from Neutralizing Antibodies Present in Patient Ascites
Pre-existing anti-Ad5 antibodies (40%–69% of the adult population in the United States is seropositive to Ad533) present in ascites fluid within the peritoneal cavity can rapidly neutralize Ad5-based vectors, and thus can significantly hinder the clinical application of i.p.-administered CRAds for ovarian cancer. We sought to determine whether CRAd-S-pk7 was recognized by antibodies present in ascetic fluid. To do this, we applied cell-free ascetic fluid obtained from ovarian cancer patients during routine peritoneal drainage to a membrane onto which denatured, electrophoresed CRAd-S-pk7 samples were blotted (Figure 5A). The ascites fluid positively recognized the viral antigens hexon, penton base, and fiber.
Figure 5.

NSCs Enhance CRAd-s-pk7 Efficacy
(A and B) NSC protects CRAd-S-pk7 from adenovirus neutralization. (A) Blot analysis assessing recognition of adenovirus antigens hexon, penton base, and fiber by antibodies present in ascetic fluid. Dilutions of purified CRAd-S-pk7 virus were subjected to SDS-PAGE. Following semi-dry blotting and blocking in non-fat milk, membranes were incubated with ascites pooled from three patients with ovarian cancer. Bound antibodies were visualized using horseradish-peroxidase-conjugated anti-human IgG secondary antibodies. OVCAR8 human ovarian cancer cells served as a negative control. (B) Neutralization of adenovirus infectivity by ascites fluid. Two thousand OVCAR8.EGFP.ffluc cells/96-well plate were co-cultured with either 2,000 NSC.CRAd-S-pk7 (2.5 × 107 pfu) for 5 days or free CRAd-S-pk7 (2.5 × 107 pfu) for 3 days or NSC-CRAd-S-pk7 in the presence of serial 2-fold dilutions of ascetic fluid obtained from three different ovarian cancer patients. Ascitic fluid was replaced after 24 h with culture media. Data are presented as average raw luminescent signal ± SD. (C and D) NSC improves CRAd-S-pk7 delivery in vivo. (C) qPCR quantification of increased adenoviral E1A gene copy number in mouse tumors treated with NSC.CRAd-S-pk7 (black bars) in comparison with free virus (gray bars) 1 day after administration. (D) Tumor volume was determined by weighing the omentum (primary site of tumor formation) after three treatment rounds; each point indicates an individual mouse. Data for (C) and (D) represent mean ± SEM.
To investigate the possibility that NSCs could protect the CRAd-S-pk7 virus from pre-existing anti-adenovirus antibodies in ascetic fluid, we compared the adenovirus oncolytic potential of free CRAd-S-pk7 and NSC-CRAd-S-pk7 both with and without exposure to human ascites. Free CRAd-S-pk7 had reduced oncolytic ability upon 24-h exposure to ascites fluid, as evidenced by decreased ability to kill OVCAR8 tumor cells as compared with NSC.CRAd-S-pk7 and the tumor-only negative control (Figure 5B). In addition, the extent of viral inhibition increased at greater concentrations of the ascites fluid (Figure 5B). The NSCs seem to shield viruses from neutralizing antibodies within ascites, suggesting their potential to be effective viral carriers for i.p.-administered oncolytic therapies.
Treatment with NSC.CRAd-S-pk7 Slows Progression in Orthotopic Tumor Models
Next, we utilized our orthotopic mouse models to confirm that NSC-mediated CRAd-S-pk7 delivery improves in vivo efficacy relative to free virus administration. NSG mice (n = 7 per group) and B6 mice (n = 7 per group) were administered i.p. EGFP-expressing ovarian cancer (OVCAR8 and ID8, respectively). Two weeks after tumor cell inoculation, the mice were treated with either free CRAd-S-pk7 (3 weeks of 5 × 108 plaque-forming units [pfu]/day), NSC.CRAd-S-pk7 (3 weeks of 1 × 106 cells [5 × 108 pfu]/day), or PBS (control).
To assess the relative infectivity of CRAd-s-pk7 when delivered either as the free virus or using the NSC carrier, we used qPCR to quantify E1A levels within tumor mets harvested 1 day after the first treatment. As hypothesized, increased adenoviral E1A gene copy number (22.7-fold higher) was found in the tumors of mice treated with NSC.CRAd-s-pk7 instead of free virus (Figure 5C). To determine whether NSC-mediated improvements in viral load at the tumor would translate to improved treatment efficacy, mice were harvested 1 week after the third treatment round. The omentum, which is the primary site of tumor formation and growth, was collected from all mice and weighed. As expected, free virus administration resulted in a significantly reduced omental tumor burden as compared with mice that received only PBS injections (Figure 5D). Interestingly, even more substantial decreases in omental tumor burden were obtained from mice treated with NSC.CRAd-S-pk7 instead of free virus (Figure 5D). Together these data confirm the hypothesis that NSC carriers can improve the delivery and efficacy of CRAd-S-pk7 within the peritoneal setting.
Combination Treatment with NSC.CRAd-S-pk7 and Cisplatin In Vitro
The most active chemotherapeutic agent used to treat ovarian cancer is cisplatin, but even if patients initially respond, most ultimately die with platinum-resistant disease.33 Thus, it was important to determine whether NSC.CRAd-S-pk7 can help eliminate ovarian cancer cells that have become resistant to patient-tolerable doses of cisplatin. Co-culture of OVCAR8 tumor cells and NSC.CRAd-S-pk7 with increasing concentrations of cisplatin showed that the virus-loaded NSCs could kill ovarian tumor cells that were otherwise unaffected by low doses of cisplatin (<1 μM) (Figure 6A). Potential synergistic interactions between NSC.CRAd-S-pk7 and cisplatin were evaluated using the Chou-Talalay methodology, for which a combinatorial index (CI) of <0.9 is considered synergistic, a CI between 0.9 and 1.1 is considered additive, and a CI >1.1 is considered antagonistic.34 For the combination of cisplatin and NSC.CRAd-S-pk7, all CIs were <1 (Figure 6B), unless the concentration of cisplatin was extremely high (1,000 μM) or the NSC.CRAd-S-pk7:tumor ratio was very low (1:1,000) (data not shown). These data suggest the two agents may demonstrate synergistic efficacy at patient-tolerable doses.
Figure 6.

CRAd-S-pk7 NSCs Combination with Cisplatin (In Vitro and Flank Model)
(A) Log-dose versus response plot showing normalized OVCAR8.ffluc viability after co-culture with decreasing ratios of CRAd-S-pk7 NSCs while undergoing continuous cisplatin exposure. (B) Chou-Talalay plot showing log combinatorial index (CI) values: synergism (CI < 1), additive effect (CI = 1), and antagonism (CI > 1). (C) Pilot NSC.CRAd-S-pk7 efficacy study in flank model. Average tumor doubling times (±SD) for OVCAR8 xenograft-bearing mice that received the indicated treatments (n = 4 mice/group).
Combination Treatment with NSC.CRAd-S-pk7 and Cisplatin Slows Progression in a Flank Tumor Model
To determine whether the in vitro efficacy of NSC.CRAd-S-pk7 in combination with cisplatin also occurred in vivo, we performed a pilot study in which mice bearing subcutaneous flank OVCAR8.EGFP xenografts were treated with either i.p. cisplatin alone (4 mg/kg), intratumoral NSC.CRAd-S-pk7 alone (1 × 106 cells/5 × 108 pfu), the combination of intratumoral NSC.CRAd-S-pk7 and i.p. cisplatin (1 × 106 cells/5 × 108 pfu, 4 mg/kg), or PBS (control) (n = 4 mice per group). Mice began treatments approximately 2 weeks after injection of 5 million OVCAR8.EGFP cells, when the tumors were on average 0.5 cm in diameter (Table 2). The mice were treated for two cycles. Each cycle was 2 weeks long, consisting of 1 week on therapy and 1 week off. Tumor burden was evaluated via caliper measurements twice weekly over a 5-week period.
Table 2.
Flank OVCAR8 Tumor Model Treatment Regimen
| Schedule |
|||||||
|---|---|---|---|---|---|---|---|
| Tx Group | Agent (Dose) | Route | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
| 1 | PBS | i.p. | PBS | – | PBS | – | – |
| i.t. | PBS | PBS | PBS | PBS | PBS | ||
| 2 | cisplatin (4 mg/kg) | i.p. | cisplatin | – | cisplatin | – | – |
| i.t. | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | ||
| 3 | NSC-CRAd-S-pk7 (1e6 NSCs, 5e8 pfu) | i.p. | PBS | – | PBS | – | – |
| i.t. | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | ||
| 4 | cisplatin (4 mg/kg) + HSC-CRAd-5-pk7 (1e6 NSCs, 5e8 pfu) | i.p. | cisplatin | – | cisplatin | – | – |
| i.t. | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | ||
Tx, treatment; i.p., intraperitoneal; i.t., intratumoral.
Treatment with NSC.CRAd-S-pk7 slowed tumor progression both as a single agent and in combination with cisplatin as compared with control treatment with PBS (Figure 6C). As a single agent, NSC.CRAd-S-pk7 delayed the tumor volume doubling time from 7 to 14 days after the first treatment and from 15 to 26 days after the second treatment. When administered in combination with cisplatin, the tumor volume doubling time extended slightly to 15 days after the first treatment and 29 days after the second treatment (Figure 6C). In contrast, cisplatin alone delayed the tumor volume doubling time from 7 to 13 days after the first treatment, but appeared less effective after the second treatment, yielding a tumor volume doubling time that was not significantly different from that of the saline control (Figure 6C).
Combined Treatment with NSC.CRAd-S-pk7 and Cisplatin Slows Progression in Orthotopic Tumor Models
We next confirmed that co-administration of cisplatin did not interfere with NSC.CRAd-S-pk7 tropism within the peritoneal setting. Two weeks after tumor inoculation, NSC mice were administered either i.p. cisplatin alone (4 mg/kg), NSC.CRAd-S-pk7 alone (2 × 106 cells/1 × 109 pfu), or the combination of NSC.CRAd-S-pk7 and cisplatin (2 × 106 cells/1 × 109 pfu, 4 mg/kg) (n = 2 mice per group). Two days later, mice were euthanized and all peritoneal tissue harvested. To assess NSC.CRAd-S-pk7 and virus biodistribution, three tumors from each mouse were processed for PCR analysis. NSC.CRAd-S-pk7 were able to localize virus to peritoneal tumors in the presence of cisplatin, with hexon DNA detected in all six tumors obtained from mice treated with both agents (three of six were positive for v-myc DNA). Neither hexon nor v-myc DNA was detected in tumors obtained from mice treated with either cisplatin alone or PBS (Figure 7A; three representative tumors are shown per group).
Figure 7.

CRAd-S-pk7 NSCs Combination with Cisplatin (Orthotopic Model)
(A) PCR analysis for hexon (upper panel, indicates presence of virus) and v-myc (center panel, indicates presence of NSCs) in ovarian tumor metastases isolated 2 days post-NSC.CRAd-S-pk7 administration. GAPDH is shown as loading control. (B and C) NSC.CRAd-S-pk7 is effective against orthotopic ovarian cancer. (B) Bioluminescence images acquired on 11 days after tumor injection (post-treatment 1) to visualize tumor burden. (C) Quantitative representation of luminescent flux signal presented as the percent change in tumor flux signal from 5 days after tumor injection (pre-treatment) to 5 days after treatment round 1 (gray bars) and treatment round 2 (black bars). Error bars represent the SD. (D) Average percent change in mouse weight ± SEM for each treatment group. (E) Statistical analysis of clinical observation score pairwise comparisons acquired during treatment and 4 weeks afterward. Maximum daily score reflects the worst toxicity each mouse experienced after scoring for clinical symptoms, and early max score represents the earliest day each mouse reached its maximum daily score.
To assess the therapeutic efficacy of NSC.CRAd-S-pk7 as an adjunct to cisplatin therapy, NSG mice (n = 11 per group, combined from two independent experiments) were administered 2 million i.p. OVCAR8.EGFP.ffluc. One week after tumor cell injection, the mice were treated with three consecutive treatment cycles (Table 3). After each round of treatment, tumor burden was evaluated via bioluminescence imaging using the SPECTRAL Ami X imaging system (Figure 7B). In addition, mice were monitored daily using our clinical observation scoring system in which mice are numerically scored from 1 to 7 depending on the number of outward signs of toxicity they display. A higher daily score indicates more treatment-induced toxicity. Any mouse that received the maximum score of +7 or met any terminal criteria (see Materials and Methods) was euthanized immediately.
Table 3.
Orthotopic OVCAR8 Tumor Model Treatment Regimen
| Schedule |
|||||||
|---|---|---|---|---|---|---|---|
| Tx Group | Agent (Dose) | Route | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
| 1 | PBS | i.p. | PBS | PBS | PBS | PBS | PBS |
| 2 | cisplatin (4 mg/kg) | i.p. | cisplatin | PBS | cisplatin | PBS | PBS |
| 3 | NSC-CRAd-S-pk7 (1e6 NSCs, 5e8 pfu) | i.p. | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 | NSC-CRAd-S-pk7 |
| 4 | cisplatin (4 mg/kg) + NSC-CRAd-S-pk7 (1e6 NSCs, 5e8 pfu) | i.p. | cisplatin | PBS | cisplatin | PBS | PBS |
| i.p. | NSC-CRAd-S-pk7 | HSC-CRAd-S-pk7 | HSC-CRAd-S-pk7 | MSC-CRAd-S-pk7 | MSC-CRAd-S-pk7 | ||
Tx, treatment; i.p., intraperitoneal.
As expected, the average percent change in tumor flux was less in all treatment groups as compared with the PBS control group (Figure 7C). Furthermore, NSC.CRAd-S-pk7 treatment seemed as effective as cisplatin, because no significant differences in tumor flux were seen between the two treatment groups. In contrast with NSC.CRAd-S-pk7, cisplatin treatments were measurably more toxic than saline control treatments, with mice in this group experiencing accelerated weight loss (Figure 7D) and having both higher average maximum daily scores and earlier time to reach the maximum daily score (Figure 7E). These outward signs of toxicity were not apparent in the mice treated with NSC.CRAd-S-pk7. The combination of cisplatin and NSC.CRAd-S-pk7 resulted in lower average tumor flux compared with treating with cisplatin alone, but the results were not statistically significant. However, adding NSC.CRAd-S-pk7 did not significantly worsen the observed toxicity of cisplatin treatments, as measured by daily score (Figure 7E). This result implies potential for using NSC.CRAd-S-pk7 as an adjuvant treatment when administering cisplatin to achieve increased efficacy with no added toxicity.
Discussion
Identification of a well-characterized, scalable, off-the-shelf delivery system to improve the distribution of oncolytic viruses to ovarian cancer tumors will facilitate the clinical translation of novel virus-based therapies and may improve patient responses to currently available therapies. The virotherapy agent CRAd-S-pk7 is one virus, likely among many, that would benefit from a cell-based delivery vehicle given the high percentage of the human population that already has neutralizing antibodies against this virus family. Although other groups have used autologous cells for virus delivery (e.g., irradiated tumor cells, interleukin-2 [IL-2] expanded T cells,35 CD14+-derived monocytes,36 adipose tissue-derived mesenchymal stem cells [MSCs]2, 36, 37), we consider this an impractical strategy for broadly applied therapy given the potential heterogeneities among cell isolations in terms of infection efficiency, ex vivo virus loading capacities, chromosomal stabilities, and cell expansion potential without losing tropic potential. For example, Mader et al.37 recently demonstrated that it took 2 weeks after isolation to generate enough MSCs for patient treatment, 20% of which showed abnormal karyotypes. Therefore, we think an “off-the-shelf” allogeneic cell line is more practical for long-term scale-up and clinical translation.
The NSC.CRAd-S-pk7 cell line exhibited robust tropism toward and efficacy against several ovarian cancer lines in vitro. As previously observed in the context of glioma,38 NSC.CRAd-S-pk7 exhibit greater tropism to tumor-conditioned media as compared with uninfected NSCs. In this regard, previous reports have demonstrated that oncolytic viruses enhanced the expression of three chemoattractant receptors (CXCR4, c-met, VEGF-R2) on HB1.F3.CD NSCs, which may explain the increased tropism to tumor-conditioned media, although studies to clarify the mechanism for enhanced tropism are still needed.38 In the peritoneal cavity, however, tumor tropic cells may also rely heavily on adhesion molecules in addition to chemotaxis to localize to tumors.37 Therefore, further studies are needed to confirm whether this enhanced tropism is also observed in vivo.
Although we have previously demonstrated that the HB1.F3.CD cell line can improve adenovirus delivery within the brain, it was not yet clear whether this approach would work in the peritoneal setting, which is less immune-privileged than the brain. To test this approach, we used the clinical-equivalent research lot of NSC.CRAd-S-pk7 already developed for a glioma clinical trial (ClinicalTrials.gov: NCT03072134) and demonstrated that this well-characterized cell line can not only protect the CRAd-S-pk7 from pre-existing neutralizing antibodies present in patient ascites, but also improve CRAd-S-pk7 delivery and anti-tumor efficacy in orthotopic mouse models. This result is consistent with another study showing that MSCs carrying measles virus can home to ovarian tumor xenografts in passively immunized athymic mice and lead to a superior therapeutic outcome compared with virus alone.39 It is still unclear whether the increased therapeutic benefit is due solely to improvements in viral delivery, or whether in situ amplification of the virus contributes as well.
In both the flank and the orthotopic setting, we demonstrated that NSC.CRAd-S-pk7 is as effective at slowing tumor progression as 4 mg/kg/week cisplatin (equivalent to human clinical dose), but without the measurable toxicities associated with this chemotherapeutic, including dose-dependent renal tubule toxicity and neurotoxicity.40 Although the negligible toxicity due to NSC.CRAd-S-pk7 observed in our study may simply be because of poor infection of mouse tissues by human adenovirus, it is also consistent with clinical trial data reported to date. Of the 11 oncolytic viruses that have been tested in preclinical human ovarian cancer models, 4 have been tested in 9 different phase I/II clinical trials.2 Although these clinical trials are still in early stages, they have all established the safety and non-toxicity of oncolytic virus-based approaches.1 In direct contrast with trials testing traditional chemotherapies, not a single oncolytic virus trial has established a maximum tolerated dose because toxicities are so low.2, 41
One limitation of using NSC.CRAd-S-pk7 as a single agent is that its replication is conditional upon overexpression of survivin. Despite the redundancy of cell-death and cell-proliferation pathways, it has been suggested that survivin is required for cancer cell viability, an idea substantiated by cellular, molecular, and genetic approaches.37 It is worth noting, however, that many treatments that target a single gene fail because of the dynamic and heterogeneous nature of tumor genetics, so even though survivin appears necessary for cancer cells, combination treatment will likely be essential for sustained disease regression. Therefore, we explored in vitro the combination of survivin-targeted CRAd-S-pk7 with cisplatin.
In fact, we demonstrated significant synergy for the CRAd-S-pk7 and cisplatin combination in vitro, and confirmed the ability of this combination treatment to reduce tumor burden in both flank and orthotopic immunocompromised mice. This result is significant because including NSC.CRAd-S-pk7 as an adjuvant treatment could increase the therapeutic index of cisplatin. This finding is also consistent with other studies. For example, Adusumilli et al.42 reported that cisplatin selectively enhanced the tumor cell-killing effect of the oncolytic virus NV1066 in the context of mesothelioma. A similar adenoviral agent to ours (CRAd-S-RGD [arginine-lysine-aspartate]) was also investigated in combination with cisplatin for ovarian cancer. Their virus results in synergistically increased efficacy in a xenograft flank tumor model without a concomitant increase in toxicity.43 The RGD modification on the fiber knob domain of this virus enhances tumor cell infection via integrins instead of the polylysine-facilitated infection in our virus that utilizes proteoglycans. Finally, oncolytic herpes virus was found to enhance the efficacy of paclitaxel through various mechanisms, including microtubule acetylation, mitotic block, and apoptosis, in addition to its oncolytic effects.44
Certainly, further research into optimal dosing and schedules for combining cisplatin with NSC.CRAd-S-pk7 in ovarian cancer are still needed, in addition to determining the mechanism underlying this synergism. One possibility we explored was that tumor cells increase their expression of survivin when exposed to cisplatin in an effort to resist its cytotoxic effects. This effect has been previously observed in both human glioma and ovarian cancer cell lines, for which both irradiation and cisplatin, respectively, increase the activity of the survivin promoter.34 However, RT-PCR analysis of survivin expression in OVCAR8 and SKOV3 cell lines treated with cisplatin demonstrated that survivin expression did not significantly increase after up to 2 days of cisplatin exposure (data not shown). This result is consistent with bioinformatics analysis of GEO Accession Viewer data on patient tumors showing no reliable increase in survivin expression after patient tumors become cisplatin resistant (Figure S3). Thus, understanding how NSC.CRAd-S-pk7 enhances tumor killing beyond acting on cells that are not responsive or accessible to free cisplatin requires further study.
Collectively, our findings support further study of NSC.CRAd-S-pk7 within the peritoneal setting. Specifically, the CRAd-S-pk7 used in this study had no marker gene in an effort to study the effects of a clinical-equivalent research bank of the NSC.CRAd-S-pk7. Thus, we were limited in our ability to monitor the kinetics and biodistribution of the virus. Future studies should also include a time-course characterization of when and how efficiently NSCs reach the tumor, as well as a direct comparison of retention free-viral delivery versus NSC-mediated delivery, particularly after repeated administration rounds.45 Additionally, further assessment into the possibility that the CRAd-S-pk7 virus can stimulate anti-tumor immunogenicity, perhaps by exposing neoantigens during tumor cell lysis, would be beneficial. It may also prove fruitful to engineer the CRAd-S-pk7 virus to express additional therapeutic transgenes to enhance the host immune response against the cancer (e.g., granulocyte macrophage colony-stimulating growth factor).46
Overall, this study provides the first demonstration of the strong potential for oncoviral delivery using an off-the-shelf allogeneic cell line. Furthermore, we showed that the CRAd-S-pk7 oncolytic adenovirus has impressive anti-tumor activity against stage III ovarian cancer, on par with results observed using the gold standard treatment, cisplatin. We also demonstrated the potential of combining cisplatin with NSC.CRAd-S-pk7 to result in increased tumor killing than is possible with cisplatin alone. Upon further preclinical development using pre-immunized immunocompetent mouse models, this system has strong potential to improve the delivery of therapeutic oncolytic adenoviruses within the peritoneal cavity.
Materials and Methods
Microarray Analysis of Survivin and CRAd-S-pk7 Entry Receptor Expression in Patient Cohorts
Tumor versus normal Birc5 gene expression was calculated using 185 cisplatin-resistant patient tumors and 10 healthy tissue samples.11, 25 Leave-one-out cross-validation was applied to each tumor cohort and confirmed by a permutation test. External validation was conducted by applying the gene signature to a publicly available array database of expression profiles of advanced stage suboptimally debulked tumors. Data showing survivin expression in different peritoneal tissues were obtained from GTExPortal and modified to include only the peritoneal tissues of relevance to the present manuscript. Expression values are shown in RPKM (reads per kilobase of transcript per million mapped reads), calculated from a gene model with isoforms collapsed to a single gene. No other normalization steps have been applied. The mRNA expression scores for putative CRAd-S-pk7 viral entry receptors were obtained from The Cancer Genome Atlas serous ovarian cancer project24, 30 and modified to include only the 100 patients of 311 total patients who exhibited amplified survivin expression.
Human Tissue Procurement and Processing
Fresh tumors, non-malignant tissues, and ascites were obtained from patients who gave institutional review board (IRB)-approved informed consent (City of Hope [COH] IRB 15280) before tissue collection at the COH Medical Center. The fresh tumors and non-malignant tissues were either preserved as untreated controls or cut into ∼5-mm pieces that were then incubated in 24-well dishes with each well containing 0.5 mL of complete media and either 5e8 VP of free CRAd-S-pk7 or 5e5 NSC.CRAd-S-pk7. After a 6-h incubation, NSCs had plated and the tissue was floating, so Boyden chamber inserts were used to maintain contact between the NSCs and the tissue to facilitate viral transfer. Tissues were collected on days 1 and 3 when NSC lysis was observed. Tissues were washed, fixed, and processed for immunological staining or qPCR analysis of viral load. The fresh ascites was heat inactivated at 56°C for 60 min to eliminate complement proteins, but leave neutralizing antibodies intact as previously described.47 It was then frozen until used for western blot analysis and the viral neutralization assay.
Detection of CRAd5 Neutralizing Antibodies in Patient Ascites
Neutralizing antibodies against CRAd-S-pk7 were recognized in ascites samples by western blotting as previously described.48 In brief, CRAd-S-pk7 was diluted in PBS to concentrations of 125, 250, 500, and 1,000 ng protein/mL, then subjected to SDS-PAGE (10% NuPAGE Bis-Tris Gel; Thermo Fisher). Following electrophoresis, proteins were transferred by semi-dry electroblotting onto a nitrocellulose membrane (Bio-Rad), which was blocked with 5% non-fat dried milk/0.1% Tween in PBS (PBST) for 90 min at room temperature. Ascetic fluid diluted 1:1,000 in PBST was added to the membranes and incubated for 90 min at room temperature. Membranes were washed 3 × 10 min in PBST. Membranes were then incubated in a 1:10,000 solution of anti-human Ig (H+L), HRP conjugate (Promega) for 90 min at room temperature. After washing, membranes were developed with 3,3’, 5,5’-tetramethylbenzidine (TMB) stabilized substrate for horseradish peroxidase (Promega) and imaged.
Viral Neutralization Assay
A luminescent assay using the OVCAR8.EGFP.ffluc cell line was used to quantify viral neutralization after a 30-min exposure to heat-inactivated patient ascites fluid. A cell suspension was made of 105 OVCAR8.EGFP.ffluc cells/mL, and 100 μL was added to each well of a 96-well plate. The next day, heat-inactivated ascites was diluted using 5-serial doublings. Serum-negative controls were also included. To each dilution, 5 μL of free CRAd-S-pk7 virus (2.5e10 VP/mL) or 5e6 NSC.CRAd-S-pk7 cells were incubated for 30 min at 37°C. The ascites was then aliquoted into the 96-well plate containing OVCAR8.EGFP.ffluc tumor cells. After 8 h, the NSCs had adhered and media were replaced. Plates were incubated for another 4 days to allow oncolysis. On day 5, media were removed and replaced with media containing 100 μg/mL D-luciferin substrate. After 10 min, the resulting luciferase signal was read using a SpectraMax M3 microplate reader (Molecular Devices, CA, USA).
Cell Culture
NSC lines including the human, v-myc immortalized, HB1.F3.CD NSC line were obtained from Seung Kim (University of British Columbia, Canada).49 These were further modified by Dr. Maciej Lesniak (Northwestern University) to produce CRAd-S-pk7 as previously described.12 NSC lines were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Gemini Bio), 1% L-glutamine (Invitrogen), and 1% penicillin-streptomycin (Invitrogen) and maintained at 37°C in a humidified incubator (Thermo Electron Corporation) containing 6% CO2. Ovarian cancer cell lines were cultured in RPMI basal media with the same supplements. For all cell lines, when cells reached 80% confluency, they were passaged using a 0.25% trypsin and EDTA solution (Invitrogen); media were changed every 2–3 days. Ovarian cancer cell lines, Firefly luciferase-expressing OVCAR8 (OVCAR8.EGFP.ffluc), SKOV-3 (SKOV-3.ffluc), Kuramochi, OVCAR3, and CAOV3 cell lines, were generously provided by Dr. Carlotta Glackin. The ID8 murine glioma line was obtained from Dr. Katherine Roby (University of Kansas). A2780 and U87 human glioma cell lines were obtained from American Type Culture Collection. All tumor lines were used to generate tumor-conditioned media by replacing culture media with serum-free media when cells were 80%–100% confluent followed by a 48-h incubation period.
In Vitro Efficacy Co-culture Assay
Tumor cells were plated at 5 × 105 cells per well in 6-well plates. Select wells also received NSC.CRAd-S-pk7 at a 1:1, 1:10, 1:100, or 1:1,000 ratio. At select time points, wells were washed with PBS and stained with crystal violet or assessed for total DNA content (PicoGreen DNA quantification kit; Invitrogen). Wells were imaged using bright-field microscopy to obtain a visual representation of live cells per well. DNA content was quantified using a SpectraMax M3 microplate reader (Molecular Devices, CA, USA).
In Vitro Synergy Experiment: Chou-Talalay Analysis
OVCAR8.EGFP.ffluc cytotoxicity resulting from NSC.CRAd-S-pk7 cells, cisplatin chemotherapy, or both agents in combination was studied by quantifying ffluc expression remaining in culture after 3 days. Tumor cells were plated at 3 × 103 cells per well in 96-well plates. Select wells also received NSC.CRAd-S-pk7 at either a 1:1, 1:10, or 1:100 ratio. After overnight incubation, cisplatin was added in select wells at the indicated concentration. Serial dilutions of cisplatin were tested starting at a concentration of 10,000 μM. Cultures were incubated for 3 more days. Upon collection, media were replaced with that containing D-luciferin (200 μg/mL), and after a 10-min incubation, the resulting luminescent signal was quantified using a SpectraMax M3 microplate reader (Molecular Devices, CA, USA). Results are expressed as the percentage of surviving cells determined by comparing the luciferase signal of each sample relative to untreated control samples considered 100% viable. The interactions between the NSC.CRAd-S-pk7 viruses and cisplatin were evaluated by calculating Chou-Talalay combination indices (CI) using CompuSyn software (ComboSyn). Each condition was replicated in quadruplet, and the experiment was conducted twice.
In Vivo Subcutaneous Xenograft Ovarian Cancer Model
Female athymic nude mice 6–8 weeks of age (Charles River) were maintained under specific pathogen-free conditions at the COH Animal Resource Center, and all procedures were reviewed and approved by the COH Animal Care Committee. A subcutaneous flank xenograft model in nude mice was established using an EGFP and firefly luciferase-expressing ovarian cancer cell line, 2e6 OVCAR8.EGFP.ffluc. Approximately 2 weeks following tumor injections, when the tumors were on average 0.5 cm in diameter, the mice were divided into the four treatment groups: (1) PBS: 100 μL administered i.p. days 1–5; (2) cisplatin (4 mg/kg): 100 μL administered i.p. days 1 and 3; (3) NSC.CRAd-S-pk7 (1e6): 100 μL administered i.p. days 1–5; or (4) combination: NSC.CRAd-S-pk7 (1e6) administered i.p. days 1–5 and cisplatin (4 mg/kg) administered i.p. days 1 and 3. The mice were treated for three weekly cycles with 1 week off in between each cycle. The animals were observed for daily consumption of food and water, appearance, and body conditions. Tumor burden was evaluated via caliper measurements twice weekly over a 5-week period.
In Vitro Boyden Migration Assay
To evaluate cell migration, we used a classic Boyden chamber assay. In a 24-well tissue culture plate, 500 μL of target media (either containing only BSA as a negative control or derived from the culture of ovarian cancer cells) was added to each well. At a density of 1 × 105 cells/well, unmodified NSCs or NSC.CRAd-S-pk7 in DMEM and 5% w/v BSA were placed in transwell polycarbonate membrane cell culture inserts (Fisher) and incubated at 37°C for 4 h. After the incubation period, the transwell inserts were placed in a new 24-well tissue culture plate containing Accutase and incubated 10 min at 37°C. Detached cells were then transferred to a 96-well V-bottom plate, centrifuged at 1,500 rpm for 5 min, and resuspended in 1:1 PBS to Guava ViaCount Reagent (EMD Millipore). NSC migration to tumor-conditioned media was assessed using a Guava EasyCyte flow cytometer (EMD Millipore).
In Vivo NSC.CRAd-S-pk7 Tropism in Orthotopic Ovarian Cancer Model
Female NOD-SCID mice (Jackson Labs) that were 6–8 weeks old were inoculated with 2 million OVCAR8.EGFP.ffluc cells via i.p. injection. Alternatively, female B6 mice (National Cancer Institute [NCI]) that were 7–8 weeks old were inoculated with 5 million ID8.EGFP.ffluc cells via i.p. injection. After 3 weeks, mice (n = 3) were administered i.p. 2e6 DiI-labeled NSC.CRAd-S-pk7. Two days after NSC injection, tumors were harvested. Three tumors per mouse were digested using proteinase K, DNA was extracted (DNA Extraction from fixed tissues kit; Puragene), quantified using a NanoDrop, and then amplified by PCR using primers for v-myc and hexon to test for the presence of NSCs and viral particles, respectively. GAPDH was used as a loading control (351 bp). Primer sequences are listed. An additional three tumors per mouse were frozen in Tissue Tek OCT (Sakura Finetek USA) and sectioned on a Leica CM1510 S cryostat (Leica Biosystems). Sections (10 μm thick) were collected on positively charged slides (Thermo Fisher Scientific), immunostained for hexon (Goat Anti-Adenovirus FITC Conjugated Polyclonal Antibody, AB1056F; Millipore), counterstained with DAPI (1 μg/mL; Sigma), and then imaged using the Zeiss Axio Observer Z1fluorescence microscope (ZEISS Microscopy).
Assessment of In Vivo Efficacy Study in Orthotopic Ovarian Cancer Model
Female non-obese diabetic-severe combined immunodeficiency (NOD-SCID) mice (Jackson Labs) that were 6–8 weeks old were inoculated with 2 million OVCAR8.EGFP.ffluc cells via i.p. injection. Alternatively, female B6 mice (NCI) that were 7–8 weeks old were inoculated with 5 million ID8.EGFP.ffluc cells via i.p. injection. After 2 weeks, mice were divided into treatment groups: (1) PBS: 100 μL administered i.p. days 1–5; (2) cisplatin (4 mg/kg): 100 μL administered i.p. days 1 and 3; (3) NSC.CRAd-S-pk7 (1e6): 100 μL administered i.p. days 1–5; (4) combination: NSC.CRAd-S-pk7 (1e6) administered i.p. days 1–5 and cisplatin (4 mg/kg) administered i.p. days 1 and 3; or (5) matched-dose free CRAd-S-pk7 administered i.p. days 1–5. Both before and after treatment, tumor burden was evaluated weekly via bioluminescence using the SPECTRAL Ami X imaging system over a 5-week period. Firefly luciferase-expressing tumor cells were imaged in mice using a charge-coupled device camera (the SPECTRAL Ami X) coupled to the AmiX Image acquisition and analysis software. Mice received an i.p. injection of D-luciferin substrate suspended in PBS at 4.29 mg/mouse. Images were captured while the mice were anesthetized by isoflurane (1.5 L/oxygen, 4% isoflurane) and kept in an induction chamber. Light emission was measured over an integration time of 30 s at 9 min after injection of luciferin.
Clinical Observations
Treated mice were weighed weekly and scored daily Monday through Friday for general good health, i.e., food-water intake, urine and feces production, no signs of scruffy hair coat, emaciation, or hunched posture, or any debilitating terminal criteria secondary to tumor growth, including seizures, tremors, labored or difficult breathing, weight loss (>20% body weight), hypothermia or hyperthermia, impaired ambulation, obvious illness, or inability to remain upright. Any animal in distress was euthanized consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. Analysis of clinical observations was performed on symptoms following the start of treatment on study day 7 and continued through study day 60, when all mice were harvested. Least-squares analysis was used to compare the maximum score achieved by each mouse between each of the control and treatment groups. Mice were individually weighed on study days 1, 18, 24, 30, 35, 37, 39, 42, 46, 49, 51, 53, 56, 58, and 60. Mice weights were normalized with respect to their pre-treatment weight, then averaged for each time point.
Statistical Analysis
Data are presented as mean ± SEM unless otherwise stated. Statistical significance for percent change in tumor flux at each time point between two groups was examined using a two-sample t test with a two-sided alternative (*p < 0.05 deemed to be significant). The average max daily clinical observation score and early max score were also tested using a two-sample t test.
Author Contributions
Conceptualization, K.S.A., R.M., A.A.M.; Methodology, R.M., A.A.M., D.T.C., R.T., M.H.; Formal Analysis, R.M., X.L.; Investigation, R.M., A.A.M., J.B.-C., D.M., J.G.; Writing – Original Draft, R.M.; Writing – Review & Editing, R.M., J.B.-C., K.S.A., M.S.L., D.T.C.; Visualization: R.M., A.A.M., J.B.-C., K.S.A.; Funding Acquisition, R.M., A.A.M., K.S.A.; Resources, M.S.L., K.S.A.; Supervision, K.S.A., D.T.C.
Conflicts of Interest
K.S.A., D.T.C., and M.S.L. disclose patent holder and research funding. K.A. discloses employment, an advisory role, and stock ownership with CSO, TheraBiologics, Inc., and intellectual property with City of Hope and Harvard Patents. All other authors declare no competing interests.
Acknowledgments
This work was funded by STOP cancer, The Rosalinde and Arthur Gilbert Foundation, California Institute of Regenerative Medicine, the Alvarez Family Foundation, the Anthony F. & Susan M. Markel Foundation, the Daphna and Richard Ziman Family Foundation, the Ben and Catherine Ivy Foundation, the Accelerated Brain Cancer Cures Foundation, City of Hope, and National Cancer Institute grants R01CA198076, R01 FD004816, U01NS082328, R43CA86768, and R44CA8678. Research reported in this publication included work performed by X.L. in the City of Hope Biostatistics Core, which is supported by the National Cancer Institute of the NIH under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Materials Transfer Information is available from the City of Hope Office of Technology Licensing.
Footnotes
Supplemental Information includes three figures and can be found with this article online at https://doi.org/10.1016/j.omto.2018.12.003.
Contributor Information
Rachael Mooney, Email: rmooney@coh.org.
Karen S. Aboody, Email: kaboody@coh.org.
Supplemental Information
References
- 1.Cannistra S.A. Cancer of the ovary. N. Engl. J. Med. 2004;351:2519–2529. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 2.Li S., Tong J., Rahman M.M., Shepherd T.G., McFadden G. Oncolytic virotherapy for ovarian cancer. Oncolytic Virother. 2012;1:1–21. doi: 10.2147/OV.S31626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J., Hall R.R., Lesniak M.S., Ahmed A.U. Stem cell-based cell carrier for targeted oncolytic virotherapy: translational opportunity and open questions. Viruses. 2015;7:6200–6217. doi: 10.3390/v7122921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakamori M., Fu X., Meng F., Jin A., Tao L., Bast R.C., Jr., Zhang X. Effective therapy of metastatic ovarian cancer with an oncolytic herpes simplex virus incorporating two membrane fusion mechanisms. Clin. Cancer Res. 2003;9:2727–2733. [PubMed] [Google Scholar]
- 5.Ding J. Oncolytic virus as a cancer stem cell killer: progress and challenges. Stem Cell Investig. 2014;1:22. doi: 10.3978/j.issn.2306-9759.2014.12.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pesonen S., Kangasniemi L., Hemminki A. Oncolytic adenoviruses for the treatment of human cancer: focus on translational and clinical data. Mol. Pharm. 2011;8:12–28. doi: 10.1021/mp100219n. [DOI] [PubMed] [Google Scholar]
- 7.Ulasov I.V., Zhu Z.B., Tyler M.A., Han Y., Rivera A.A., Khramtsov A., Curiel D.T., Lesniak M.S. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum. Gene Ther. 2007;18:589–602. doi: 10.1089/hum.2007.002. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B., Pan J.S., Liu J.Y., Han S.P., Hu G., Wang B. Effects of chemotherapy and/or radiotherapy on survivin expression in ovarian cancer. Methods Find. Exp. Clin. Pharmacol. 2006;28:619–625. doi: 10.1358/mf.2006.28.9.1003581. [DOI] [PubMed] [Google Scholar]
- 9.Altieri D.C., Marchisio P.C. Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab. Invest. 1999;79:1327–1333. [PubMed] [Google Scholar]
- 10.Takai N., Miyazaki T., Nishida M., Nasu K., Miyakawa I. Expression of survivin is associated with malignant potential in epithelial ovarian carcinoma. Int. J. Mol. Med. 2002;10:211–216. [PubMed] [Google Scholar]
- 11.Bonome T., Levine D.A., Shih J., Randonovich M., Pise-Masison C.A., Bogomolniy F., Ozbun L., Brady J., Barrett J.C., Boyd J., Birrer M.J. A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 2008;68:5478–5486. doi: 10.1158/0008-5472.CAN-07-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmed A.U., Thaci B., Tobias A.L., Auffinger B., Zhang L., Cheng Y., Kim C.K., Yunis C., Han Y., Alexiades N.G. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J. Natl. Cancer Inst. 2013;105:968–977. doi: 10.1093/jnci/djt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borovjagin A.V., Krendelchtchikov A., Ramesh N., Yu D.C., Douglas J.T., Curiel D.T. Complex mosaicism is a novel approach to infectivity enhancement of adenovirus type 5-based vectors. Cancer Gene Ther. 2005;12:475–486. doi: 10.1038/sj.cgt.7700806. [DOI] [PubMed] [Google Scholar]
- 14.Tyler M.A., Ulasov I.V., Borovjagin A., Sonabend A.M., Khramtsov A., Han Y., Dent P., Fisher P.B., Curiel D.T., Lesniak M.S. Enhanced transduction of malignant glioma with a double targeted Ad5/3-RGD fiber-modified adenovirus. Mol. Cancer Ther. 2006;5:2408–2416. doi: 10.1158/1535-7163.MCT-06-0187. [DOI] [PubMed] [Google Scholar]
- 15.Ulasov I.V., Tyler M.A., Han Y., Glasgow J.N., Lesniak M.S. Novel recombinant adenoviral vector that targets the interleukin-13 receptor alpha2 chain permits effective gene transfer to malignant glioma. Hum. Gene Ther. 2007;18:118–129. doi: 10.1089/hum.2006.146. [DOI] [PubMed] [Google Scholar]
- 16.Mirahmadi N., Babaei M.H., Vali A.M., Dadashzadeh S. Effect of liposome size on peritoneal retention and organ distribution after intraperitoneal injection in mice. Int. J. Pharm. 2010;383:7–13. doi: 10.1016/j.ijpharm.2009.08.034. [DOI] [PubMed] [Google Scholar]
- 17.Fisher K. Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr. Opin. Mol. Ther. 2006;8:301–313. [PubMed] [Google Scholar]
- 18.Aboody K., Najbauer J., Metz M., D'Apuzzo M., Gutova M., Annala A., Synold T., Couture L., Blanchard S., Moats R. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci Transl Med. 2013;5:184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aboody K.S., Brown A., Rainov N.G., Bower K.A., Liu S., Yang W., Small J.E., Herrlinger U., Ourednik V., Black P.M. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Portnow J., Synold T.W., Badie B., Tirughana R., Lacey S.F., D’Apuzzo M., Metz M.Z., Najbauer J., Bedell V., Vo T. Neural stem cell-based anticancer gene therapy: a first-in-human study in recurrent high-grade glioma patients. Clin. Cancer Res. 2017;23:2951–2960. doi: 10.1158/1078-0432.CCR-16-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmed A.U., Thaci B., Alexiades N.G., Han Y., Qian S., Liu F., Balyasnikova I.V., Ulasov I.Y., Aboody K.S., Lesniak M.S. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol. Ther. 2011;19:1714–1726. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morshed R.A., Gutova M., Juliano J., Barish M.E., Hawkins-Daarud A., Oganesyan D., Vazgen K., Yang T., Annala A., Ahmed A.U. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther. 2015;22:55–61. doi: 10.1038/cgt.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thaci B., Ahmed A.U., Ulasov I.V., Tobias A.L., Han Y., Aboody K.S., Lesniak M.S. Pharmacokinetic study of neural stem cell-based cell carrier for oncolytic virotherapy: targeted delivery of the therapeutic payload in an orthotopic brain tumor model. Cancer Gene Ther. 2012;19:431–442. doi: 10.1038/cgt.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vathipadiekal V., Wang V., Wei W., Waldron L., Drapkin R., Gillette M., Skates S., Birrer M. Creation of a human secretome: a novel composite library of human secreted proteins: validation using ovarian cancer gene expression data and a virtual secretome array. Clin. Cancer Res. 2015;21:4960–4969. doi: 10.1158/1078-0432.CCR-14-3173. [DOI] [PubMed] [Google Scholar]
- 26.Martini E., Wittkopf N., Günther C., Leppkes M., Okada H., Watson A.J., Podstawa E., Backert I., Amann K., Neurath M.F., Becker C. Loss of survivin in intestinal epithelial progenitor cells leads to mitotic catastrophe and breakdown of gut immune homeostasis. Cell Rep. 2016;14:1062–1073. doi: 10.1016/j.celrep.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 27.O’Driscoll L., Linehan R., Clynes M. Survivin: role in normal cells and in pathological conditions. Curr. Cancer Drug Targets. 2003;3:131–152. doi: 10.2174/1568009033482038. [DOI] [PubMed] [Google Scholar]
- 28.Wickham T.J., Roelvink P.W., Brough D.E., Kovesdi I. Adenovirus targeted to heparan-containing receptors increases its gene delivery efficiency to multiple cell types. Nat. Biotechnol. 1996;14:1570–1573. doi: 10.1038/nbt1196-1570. [DOI] [PubMed] [Google Scholar]
- 29.Stoff A., Rivera A.A., Banerjee N.S., Mathis J.M., Espinosa-de-los-Monteros A., Le L.P., De la Torre J.I., Vasconez L.O., Broker T.R., Richter D.F. Strategies to enhance transductional efficiency of adenoviral-based gene transfer to primary human fibroblasts and keratinocytes as a platform in dermal wounds. Wound Repair Regen. 2006;14:608–617. doi: 10.1111/j.1743-6109.2006.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao P., Mooney R., Tirughana R., Abidi W., Aramburo S., Flores L., Gilchrist M., Nwokafor U., Haber T., Tiet P. Intraperitoneal administration of neural stem cell-nanoparticle conjugates targets chemotherapy to ovarian tumors. Bioconjug. Chem. 2017;28:1767–1776. doi: 10.1021/acs.bioconjchem.7b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Domcke S., Sinha R., Levine D.A., Sander C., Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013;4:2126. doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Helm C.W., States J.C. Enhancing the efficacy of cisplatin in ovarian cancer treatment—could arsenic have a role. J. Ovarian Res. 2009;2:2. doi: 10.1186/1757-2215-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chou T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 35.Ong H.T., Hasegawa K., Dietz A.B., Russell S.J., Peng K.W. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007;14:324–333. doi: 10.1038/sj.gt.3302880. [DOI] [PubMed] [Google Scholar]
- 36.Peng K.W., Dogan A., Vrana J., Liu C., Ong H.T., Kumar S., Dispenzieri A., Dietz A.B., Russell S.J. Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am. J. Hematol. 2009;84:401–407. doi: 10.1002/ajh.21444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mader E.K., Butler G., Dowdy S.C., Mariani A., Knutson K.L., Federspiel M.J., Russell S.J., Galanis E., Dietz A.B., Peng K.W. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J. Transl. Med. 2013;11:20. doi: 10.1186/1479-5876-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahmed A.U., Tyler M.A., Thaci B., Alexiades N.G., Han Y., Ulasov I.V., Lesniak M.S. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol. Pharm. 2011;8:1559–1572. doi: 10.1021/mp200161f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mader E.K., Maeyama Y., Lin Y., Butler G.W., Russell H.M., Galanis E., Russell S.J., Dietz A.B., Peng K.W. Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin. Cancer Res. 2009;15:7246–7255. doi: 10.1158/1078-0432.CCR-09-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozols R.F., Young R.C. High-dose cisplatin therapy in ovarian cancer. Semin. Oncol. 1985;12(4 Suppl 6):21–30. [PubMed] [Google Scholar]
- 41.Galanis E., Hartmann L.C., Cliby W.A., Long H.J., Peethambaram P.P., Barrette B.A., Kaur J.S., Haluska P.J., Jr., Aderca I., Zollman P.J. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adusumilli P.S., Chan M.K., Chun Y.S., Hezel M., Chou T.C., Rusch V.W., Fong Y. Cisplatin-induced GADD34 upregulation potentiates oncolytic viral therapy in the treatment of malignant pleural mesothelioma. Cancer Biol. Ther. 2006;5:48–53. doi: 10.4161/cbt.5.1.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B., Liu Y., Zhang P., Wei Y., Yin X., Zheng J. A novel CRAd in combination with cisplatin enhanced the antitumor efficacy in ovarian cancer. Int. J. Gynecol Cancer. 2011;21:1540–1546. doi: 10.1097/IGC.0b013e31823105ed. [DOI] [PubMed] [Google Scholar]
- 44.Lin S.-F., Gao S.P., Price D.L., Li S., Chou T.C., Singh P., Huang Y.Y., Fong Y., Wong R.J. Synergy of a herpes oncolytic virus and paclitaxel for anaplastic thyroid cancer. Clin. Cancer Res. 2008;14:1519–1528. doi: 10.1158/1078-0432.CCR-07-4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCoy K., Tatsis N., Korioth-Schmitz B., Lasaro M.O., Hensley S.E., Lin S.W., Li Y., Giles-Davis W., Cun A., Zhou D. Effect of preexisting immunity to adenovirus human serotype 5 antigens on the immune responses of nonhuman primates to vaccine regimens based on human- or chimpanzee-derived adenovirus vectors. J. Virol. 2007;81:6594–6604. doi: 10.1128/JVI.02497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grossardt C., Engeland C.E., Bossow S., Halama N., Zaoui K., Leber M.F., Springfeld C., Jaeger D., von Kalle C., Ungerechts G. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum. Gene Ther. 2013;24:644–654. doi: 10.1089/hum.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sprangers M.C., Lakhai W., Koudstaal W., Verhoeven M., Koel B.F., Vogels R., Goudsmit J., Havenga M.J., Kostense S. Quantifying adenovirus-neutralizing antibodies by luciferase transgene detection: addressing preexisting immunity to vaccine and gene therapy vectors. J. Clin. Microbiol. 2003;41:5046–5052. doi: 10.1128/JCM.41.11.5046-5052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stallwood Y., Fisher K.D., Gallimore P.H., Mautner V. Neutralisation of adenovirus infectivity by ascitic fluid from ovarian cancer patients. Gene Ther. 2000;7:637–643. doi: 10.1038/sj.gt.3301152. [DOI] [PubMed] [Google Scholar]
- 49.Kendall S.E., Najbauer J., Johnston H.F., Metz M.Z., Li S., Bowers M., Garcia E., Kim S.U., Barish M.E., Aboody K.S., Glackin C.A. Neural stem cell 7targeting of glioma is dependent on phosphoinositide 3-kinase signaling. Stem Cells. 2008;26:1575–1586. doi: 10.1634/stemcells.2007-0887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.