

Abstract
Two clinically distinct diseases, amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), have recently been classified as two extremes of the FTD/ALS spectrum. The neuropathological correlate of FTD is frontotemporal lobar degeneration (FTLD), characterized by tau-, TDP-43-, and FUS-immunoreactive neuronal inclusions. An earlier discovery that a hexanucleotide repeat expansion mutation in chromosome 9 open reading frame 72 (C9orf72) gene causes ALS and FTD established a special subtype of ALS and FTLD with TDP-43 pathology (C9FTD/ALS). Normal individuals carry 2–10 hexanucleotide GGGGCC repeats in the C9orf72 gene, while more than a few hundred repeats represent a risk for ALS and FTD. The proposed molecular mechanisms by which C9orf72 repeat expansions induce neurodegenerative changes are C9orf72 loss-of-function through haploinsufficiency, RNA toxic gain-of-function, and gain-of-function through the accumulation of toxic dipeptide repeat proteins. However, many more cellular processes are affected by pathological processes in C9FTD/ALS, including nucleocytoplasmic transport, RNA processing, normal function of nucleolus, formation of membraneless organelles, translation, ubiquitin proteasome system, Notch signalling pathway, granule transport, and normal function of TAR DNA-binding protein 43 (TDP-43). Although the exact molecular mechanisms through which C9orf72 repeat expansions account for neurodegeneration have not been elucidated, some potential therapeutics, such as antisense oligonucleotides targeting hexanucleotide GGGGCC repeats in mRNA, were successful in preclinical trials and are awaiting phase 1 clinical trials. In this review, we critically discuss each proposed mechanism and provide insight into the most recent studies aiming to elucidate the molecular underpinnings of C9FTD/ALS.
1. Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are two clinically distinct entities. ALS, also known as motor neuron disease (MND), Lou Gehrig's disease, and Charcot disease, affects both upper and lower motor neurons leading to hyperreflexia, spasticity, fasciculations, and muscular atrophy [1]. The disease onset is mostly after 60 years of age with a prevalence of around 5 cases per 100,000 [2]. ALS patients can be divided into subgroups according to neuropsychological deficits: ALS with cognitive impairment (ALS-ci), ALS with behavioral impairment (ALS-bi), and ALS with combined cognitive and behavioral impairment (ALS-cbi) [3]. In FTD, neurodegeneration affects the frontal and temporal lobes causing frontotemporal lobar degeneration (FTLD) and is associated with changes in behavior and personality, deficits in frontal executive functions, and language impairment [3]. FTD is considered to be one of the most common forms of dementia in the population under 65 years of age with the prevalence around 10 cases per 100,000 [4]. FTD patients are divided in three clinical syndromes according to their symptomatology: two language variants (progressive nonfluent aphasia (PNFA) and semantic dementia (SD)), and behavioral variant of frontotemporal dementia (bvFTD) [3]. Based on the clinical, genetic, and neuropathological overlap between ALS and FTD, these two diseases are now considered as two extremes of the FTD/ALS spectrum (Figure 1) [1]. About 15% of FTD patients show symptoms of ALS, and up to 50% of ALS patients have symptoms of FTD [5].
Figure 1.

Influence of different genes on FTD/ALS clinical spectrum.
2. Main Features of ALS and FTLD-TDP Neuropathology
The vast majority of ALS cases (ALS-TDP) and the most common FTD pathological subtype (FTLD-TDP) show TDP-43 (TAR DNA-binding protein 43) immunoreactive aggregates forming characteristic inclusions (for overview see [6, 7]). The hallmarks of ALS with TDP-43 pathology (ALS-TDP) are skein-like, granular, and compact inclusions in motor neurons, whereas in FTLD-TDP, there are four distinct morphological types (A–D) with characteristic distribution and morphology of dystrophic neurites, cytoplasmic, and intranuclear inclusions. There is a good correlation with both clinical phenotype and genetic alterations [8]. For example, cases with hexanucleotide repeat expansions in C9orf72 are typically associated with type A and type B pathology, whereas mutations in the progranulin gene GRN and valosin-containing protein gene (VCP) are associated with type A and type D, respectively. Regarding the correlation with clinical phenotypes, for example, PNFA is associated with type A, SD with type C, and inclusion body myopathy with frontotemporal dementia (IBMFD) with type D. In both ALS and FTLD, there is glial pathology mainly affecting oligodendroglia, with TDP-43-immunoreactive cytoplasmic inclusions [6, 7]. While most of the ALS cases are sporadic, about half of all FTD patients show a familial pattern of inheritance linked to mutations in several different genes [6, 9–11].
3. Hexanucleotide Repeat Expansion in C9orf72 Gene
A mutation in chromosome 9 open reading frame 72 (C9orf72) has been identified as a common genetic cause of both ALS and FTD. The link of ALS and FTD to chromosome 9 was first reported in 2006 in two independent studies [12, 13]. It was later shown that the mutation is an expansion of GGGGCC (G4C2) hexanucleotide repeat in a noncoding region of the C9orf72 gene [14–16], which is the most common genetic cause of ALS and FTD (so called C9FTD/ALS) in certain populations.
In humans, three transcript variants of C9orf72 have been identified. Transcript variants 2 and 3 give rise to identical protein isoforms of 481 amino acids, while the 3′ site of transcript variant 1 is truncated and encodes an isoform of 222 amino acids. The hexanucleotide repeat is located within the promoter region for transcript variant 2, which corresponds to the position of the first intron in transcript variants 1 and 3 depending on the transcription start site used [17, 18].
While the number of G4C2 repeat units in the DNA of healthy individuals is up to 25, the number of repeats in the DNA of ALS and FTD patients is usually 400 to several thousand [14–16, 19–21]. A small percentage of patients have shorter expansions, from 45–80 repeats [19], and an even shorter expansion, around 30 repeats, has been associated with the disease [15]. Notably, there is an apparent gap between short pathogenic repeat sizes of 45 to 80 and long expansions from 400 to several thousand units. This is likely due to high genomic instability of the intermediate long repeats, which may have a tendency to either expand or contract [19]. Interestingly, longer expansions have been recently correlated with an earlier onset of disease [19]. In contrast, other studies detected either positive correlation [22–25] or no association [22, 25] between disease severity and expansion size. Additionally, the results of these studies were variable depending on the tissue in which expansion size was measured [22, 25].
4. C9orf72 Protein
The function of C9orf72 protein is still unclear. Bioinformatics predictions suggest that C9orf72 belongs to the family of DENN (differentially expressed in normal and neoplastic cells) proteins, which function as RabGEF (guanine exchange factor) regulators of membrane trafficking by activating RabGTPases [26, 27]. RabGEF facilitates a release of GDP from Rab and exchanges it for GTP. To support the role of C9orf72 as RabGEF, a study conducted on neuronal cell lines and human spinal cord tissue revealed that it was colocalized and coprecipitated with Rab proteins [28]. Therefore, C9orf72 could be involved in Rab-mediated cellular trafficking and protein degradation. Additionally, C9orf72 protein was detected at presynaptic sites, and as it interacts with Rab proteins, it is believed that C9orf72 could regulate synaptic vesicles as RabGEF for RAB3 proteins [29]. Xiao et al. detected interactions of C9orf72 with different components of the nuclear pore complex (NPC) and nuclear receptors in human brain tissue, indicating a possible role of C9orf72 in nucleocytoplasmic trafficking [30]. Nuclear transport is compromised in different C9FTD/ALS models (such as Drosophila, induced pluripotent stem cells (iPSC) derived neurons, human brain tissue, yeast cells, mouse primary neurons, and primary human dermal fibroblasts) [31–33]. As knockdown of C9orf72 leads to disruption in endosomal trafficking and formation of autophagosome [28], it is proposed that C9orf72 may be also involved in the regulation of endosomal trafficking and autophagy. Additional studies established the involvement of C9orf72 in the regulation of autophagy [34–36]. In studies conducted on cell lines, it was also shown that C9orf72 could be involved in the regulation of lysosomal function [37, 38] and in the formation of stress granules [39]. In mice, C9orf72 is required for the normal macrophage and microglial function [40]. It was shown that decreased levels of C9orf72 cause dysfunctional microglia that are related to neurodegeneration [40].
5. Neuropathological Features of C9FTD/ALS
Cases with hexanucleotide repeat expansion in C9orf72 are typically associated with type A and B FTLD-TDP pathology, as previously mentioned. The neuropathology of C9FTD/ALS shows pathognomonic ubiquitin- and p62-positive and, rarely, TDP-43-containing inclusions in the cerebellum (Purkinje cells and granular cells) and hippocampus [41, 42]. A unique feature is the presence of aggregating dipeptide repeat proteins within a proportion of inclusion bodies, some of which may not show phosphorylated TDP-43 (pTDP-43) immunoreactivity [43, 44]. Ultrastructurally, the characteristic inclusions in FTLD and ALS with C9orf72 mutation are granular and filamentous [45].
6. Potential Mechanisms of C9orf72 Hexanucleotide Repeat Expansion-Mediated Neurodegeneration
Three mechanisms have been proposed for G4C2 hexanucleotide repeat expansion (HRE) in C9orf72 to induce neurodegenerative changes: (1) loss of C9orf72 function through haploinsufficiency, (2) toxic gain-of-function due to the generation of aberrant HRE-containing RNA, and (3) toxic gain-of-function through the accumulation of dipeptide repeat proteins (DPR) translated from hexanucleotide repeat RNA. Potential mechanisms by which C9orf72 repeat expansions result in neurodegeneration are summarized in Figure 2. Microphotograph with characteristic histopathological changes is given in Figure 3. Studies investigating these mechanisms are summarized in Table 1.
Figure 2.

Potential mechanisms of C9orf72 hexanucleotide repeat expansion (HRE)-mediated neurodegeneration. Pathology due to repeats in C9orf72 gene may emerge from C9orf72 haploinsufficiency, RNA toxicity, and DPR accumulation. HRE in the noncoding region of the C9orf72 gene (1) form G-quadruplex structures (2). RNA transcribed from HRE DNA region can form different structures including G-quadruplexes (3) and RNA hairpins (4). HRE-containing RNA form RNA foci (5), which bind RNA-binding proteins. The last possible mechanism underlying pathology in C9FTD/ALS is through the repeat-associated non-ATG (RAN) translation, in which five different dipeptide repeat proteins can be formed—poly-GA, poly-GP, and poly-GR from the sense strand and poly-GP, poly-PA, and poly-PR from the antisense strand (6).
Figure 3.
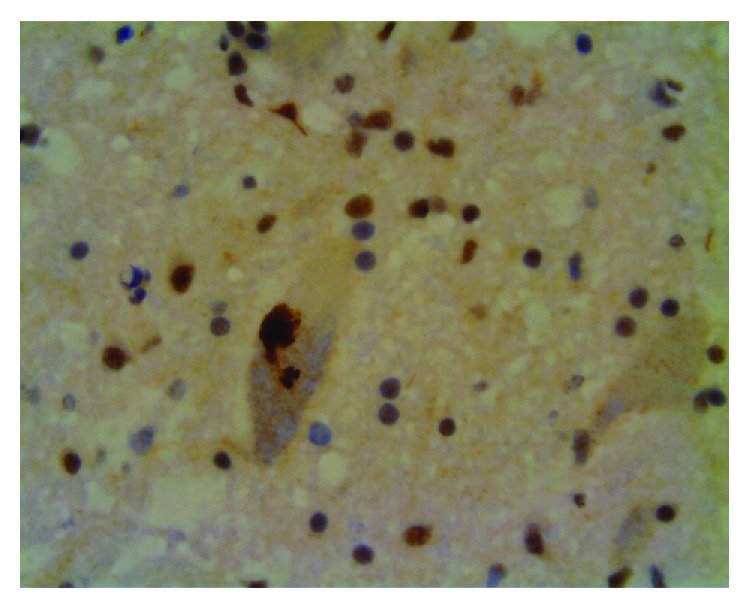
TDP-43-immunoreactive cytoplasmic inclusions, finely granular aggregates, and lack of nuclear labelling in a spinal cord motoneuron of a patient with ALS caused by C9orf72 hexanucleotide repeat expansion.
Table 1.
Summary of studies on the mechanisms by which C9orf72 repeat expansions cause neurodegeneration.
| Reference | Molecular mechanism supported by the study | Experimental model | ||
|---|---|---|---|---|
| Loss-of-function | Gain-of-function | |||
| RNA-mediated gain-of-function | Protein-mediated gain-of-function | |||
| Renton et al. [15] | + | Human DNA Human brain tissue Cell culture |
||
|
| ||||
| DeJesus-Hernandez et al. [14] | + | + | Human DNA Human brain tissue Cell culture |
|
|
| ||||
| Gijselinck et al. [16] | + | Human brain tissue Human DNA |
||
|
| ||||
| Fratta et al. [58] | + | + | Human DNA | |
|
| ||||
| Therrien et al. [52] | + | Caenorhabditis elegans | ||
|
| ||||
| Belzil et al. [135] | + | Human brain tissue Cell culture |
||
|
| ||||
| Gendron et al. [67] | + | + | Human DNA Human brain tissue Cell lines |
|
|
| ||||
| Zu et al. [93] | + | + | Human brain tissue Human RNA Cell culture |
|
|
| ||||
| Mizielinska et al. [68] | + | Human brain tissue | ||
|
| ||||
| Lee et al. [70] | + | Human brain tissue Cell culture Zebrafish embryos Rat brain |
||
|
| ||||
| Sareen et al. [71] | + | iPSC-derived neurons | ||
|
| ||||
| Xu et al. [72] | + | Cell culture Drosophila |
||
|
| ||||
| Lagier-Tourenne et al. [54] | + | Human brain tissue Cell culture |
||
|
| ||||
| Donnelly et al. [69] | + | Human brain tissue Cell culture iPSC-derived neurons |
||
|
| ||||
| Ciura et al. [53] | + | Zebrafish Human brain tissue Cell culture |
||
|
| ||||
| Wen et al. [94] | + | + | Cell culture Drosophila |
|
|
| ||||
| Liu et al. [50] | + | + | Human autopsy tissue Human DNA Cell culture |
|
|
| ||||
| May et al. [138] | + | Human brain samples Cell culture |
||
|
| ||||
| Zhang et al. [102] | + | Human brain samples Cell culture |
||
|
| ||||
| Cooper-Knock et al. [73] | + | Human brain samples Cell culture |
||
|
| ||||
| Su et al. [134] | + | + | Cell culture | |
|
| ||||
| Kwon et al. [95] | + | Cell culture | ||
|
| ||||
| Haeusler et al [51] | + | Human brain tissue iPSC-derived neurons Cell culture |
||
|
| ||||
| Mizielinska et al. [87] | + | + | Drosophila | |
|
| ||||
| Russ et al. [139] | + | + | Human DNA | |
|
| ||||
| Prudencio et al. [113] | + | + | Human brain tissue | |
|
| ||||
| Chew et al. [140] | + | + | Mouse model expressing 66 G4C2 repeats | |
|
| ||||
| Freibaum et al. [32] | + | + | Drosophila | |
|
| ||||
| Yang et al. [101] | + |
Drosophila
Human brain samples Cell culture iPSC-derived neurons |
||
|
| ||||
| Jovičić et al. [33] | + | Yeast cells Cell culture |
||
|
| ||||
| Yamakawa et al. [99] | + | Cell culture Mice brains |
||
|
| ||||
| Hu et al. [141] | + | Cell culture | ||
|
| ||||
| Gendron et al. [110] | + | Human brain samples | ||
|
| ||||
| Tao et al. [98] | + | Cell culture | ||
|
| ||||
| Tran et al. [86] | + |
Drosophila
iPSC-derived neurons Human brain tissue |
||
|
| ||||
| Koppers et al. [55] | + | + | Conditional C9orf72 knockout mouse model | |
|
| ||||
| Cooper-Knock et al. [83] | + | Human brain tissue | ||
|
| ||||
| Zhang et al. [31] | + | + |
Drosophila
iPSC-derived neurons Human brain tissue |
|
|
| ||||
| Rossi et al. [84] | + | Cell culture | ||
|
| ||||
| Cooper-Knock et al. [116] | + | Cell culture | ||
|
| ||||
| Schweizer Burguete et al. [85] | + | Cell culture Drosophila iPSC-derived neurons |
||
|
| ||||
| Liu et al. [92] | + | + | BAC mouse model of C9FTD/ALS | |
|
| ||||
| Kanekura et al. [96] | + | Human brain samples Cell culture |
||
|
| ||||
| Boeynaems et al. [142] | + | Drosophila | ||
|
| ||||
| Chang et al. [103] | + | Cell culture | ||
|
| ||||
| Flores et al. [143] | + | Cell culture | ||
|
| ||||
| Lin et al. [126] | + | Cell culture | ||
|
| ||||
| Lopez-Gonzalez et al. [144] | + | iPSC-derived neurons | ||
|
| ||||
| Liu et al. [145] | + | Human DNA and RNA | ||
|
| ||||
| Dodd et al. [146] | + | |||
|
| ||||
| Zhang et al. [147] | + | Mice that exhibit poly(GA) pathology | ||
|
| ||||
| Westergard et al. [148] | + | iPSC-derived neurons Cell culture |
||
|
| ||||
| Sellier et al. [36] | + | Cell culture Zebrafish |
||
|
| ||||
| Mori et al. [149] | + | + | Human brain samples Cell culture |
|
|
| ||||
| Gijselinck et al. [19] | + | Human DNA | ||
|
| ||||
| Ugolino et al. [150] | + | C9orf72 knockout mice C9orf72 knockdown cell models |
||
|
| ||||
| Lee et al. [125] | + |
Drosophila
Cell culture |
||
|
| ||||
| O'Rourke et al. [40] | + | + | C9orf72 +/− and C9orf72 −/− mice | |
|
| ||||
| Atanasio et al. [56] | + | + | C9orf72-deficient mouse | |
|
| ||||
| Jiang et al. [91] | + | + | C9orf72 +/− and C9orf72 −/− mice Mice expressing BAC with the human expanded C9orf72 gene |
|
|
| ||||
| Webster et al. [34] | + | Cell culture iNeurons (obtained by differentiation of iNPCs) |
||
|
| ||||
| Ji et al. [151] | + | C9orf72 knockout mouse model Cell culture |
||
|
| ||||
| Ohki et al. [152] | + | Zebrafish | ||
|
| ||||
| Liu et al. [153] | + | Cell culture | ||
|
| ||||
| Hu et al. [154] | + | Cell culture | ||
|
| ||||
| Green et al. [155] | + | Cell culture | ||
|
| ||||
| Schludi et al. [156] | + | Transgenic mice expressing codon-modified (GA)149 | ||
|
| ||||
| Gupta et al. [157] | + | Cell culture | ||
|
| ||||
| Khosravi et al. [158] | + | Cell culture | ||
|
| ||||
| Shi et al. [159] | + | Cell culture | ||
|
| ||||
| Saberi et al. [160] | + | Human brain samples | ||
|
| ||||
| Kramer et al. [105] | + | Cell culture | ||
|
| ||||
| Herranz-Martin et al. [161] | + | + | Mice that overexpress 10 or 102 interrupted G4C2 repeats | |
|
| ||||
| Zhou et al. [162] | + | Cell culture Human brain samples |
||
|
| ||||
| Lehmer et al. [163] | + | Human CSF | ||
|
| ||||
| Hautbergue et al. [82] | + | + |
Drosophila
Cell culture |
|
|
| ||||
| Boeynaems et al. [100] | + | Cell culture | ||
|
| ||||
| Maharjan et al. [39] | + | + | + | Cell culture |
|
| ||||
| Moens et al. [88] | + | + | Drosophila | |
|
| ||||
| Cheng et al. [164] | + | Cell culture | ||
|
| ||||
| Swinnen et al. [165] | + | Zebrafish | ||
|
| ||||
| Shi et al. [59] | + | + | Cell culture iPSC-derived neurons Human brain samples |
|
|
| ||||
| Simone et al. [137] | + | + | iPSC-derived neurons Drosophila |
|
|
| ||||
| Tabet et al. [166] | + | Human brain sections Cell culture |
||
|
| ||||
| Corrionero and Horvitz [167] | + | Caenorhabditis elegans | ||
|
| ||||
| Zamiri et al. [168] | + | CD spectroscopy UV melting Gel electrophoresis |
||
|
| ||||
| Swaminathan et al. [104] | + | Zebrafish | ||
|
| ||||
| Yeh et al. [169] | + | Zebrafish | ||
|
| ||||
| Meeter et al. [170] | + | Human brain samples | ||
| Nonaka et al. [171] | + | Cell culture | ||
|
| ||||
| Frick et al. [29] | + | Human brain samples Cell culture C9orf72 knockout mice |
||
BAC: bacterial artificial chromosome; CSF: cerebrospinal fluid; iNPCs: induced neural progenitor cells; iPSC: induced pluripotent stem cells. Studies investigating pathological mechanisms of C9orf72 HRE but not clearly supporting any of the three proposed disease mechanisms are not included in this table. Search for these studies was completed on May 20, 2018. The literature search was performed in Google Scholar using the keywords: “C9orf72”, “mechanism”, “pathological”, “loss-of-function”, “gain-of-function”, “haploinsufficiency”, “RNA foci”, and “DPR”. The literature search was conducted by two independent researchers.
6.1. Loss of C9orf72 Function through Haploinsufficiency
Carriers of C9orf72 HRE have decreased levels of C9orf72 transcripts [14, 16], which are also reflected in the decreased C9orf72 protein levels in the frontal and temporal cortex [20], presumably due to the loss of transcription from the mutant allele carrying the HRE. A systematic study of C9orf72 levels in patients with HRE revealed decreased levels of C9orf72 transcripts in comparison to non-HRE patients and controls [46]. Interestingly, the levels of the long C9orf72 protein isoform were decreased in the brain, while levels of the short C9orf72 protein isoform were increased (as detected by Western blot). Immunohistochemical analysis of spinal motor neurons showed decreased expression of short C9orf72 protein isoform on nuclear membrane in C9ALS cases compared to controls, while subcellular localization of long C9orf72 protein isoform was unchanged [30]. Another study detected 80% reduction of long C9orf72 protein isoform in the cerebellum of C9orf72 HRE carriers in comparison to controls [29]. C9orf72 hexanucleotide repeat expansion might induce DNA hypermethylation and consequently lead to decreased C9orf72 transcription [47]. HRE is methylated when the number of repeats is larger than 90 [48]. Moreover, the DNA from a fraction of C9orf72 HRE carriers is also methylated in the 5′ CpG island [47, 49, 50]. The repeat expansion-induced DNA methylation in the C9orf72 promoter region, which results in downregulated C9orf72 transcription, therefore provides a likely explanation for the association between the size of repeat expansion and the age of onset of the disease [19]. More precisely, the authors tested C9orf72 promoter activity in human kidney and neuroblastoma cell lines and observed reduced C9orf72 transcription in cells with larger repeats and increased methylation. Thus, they proposed that higher methylation of C9orf72 promoter may be an explanation of how repeat expansion could lead to loss-of-function, without excluding the possibility that repeat expansion could also lead to toxic gain-of-function [19]. There is also a possibility that higher-order DNA structures formed on G4C2 repeats could lead to abortive transcription of C9orf72 and therefore be the cause for decreased C9orf72 transcription [51].
C9orf72 loss-of-function in C. elegans and zebrafish models results in motor neuron degeneration, indicating a possible disease mechanism [52, 53]. However, human C9orf72 gene has only partial homology with its ortholog in C. elegans and zebrafish. Thus, loss-of-function mechanisms were assessed in C9orf72 knockout mice as the mouse ortholog of C9orf72 gene is more similar to the human C9orf72 gene [18]. Various knockout and knockdown models have been developed, yet none of the mouse models showed phenotypes characteristic for ALS or FTD [35, 40, 54–57], indicating that C9orf72 loss-of-function is not the true cause of disease. The observation that patients homozygous for C9orf72 repeat expansion do not have more severe symptoms of disease compared to heterozygotes further supports these findings [58, 59] (Table 2).
Table 2.
Evaluation of potential mechanisms underlying pathology in C9FTD/ALS.
| Molecular mechanism underlying pathology in C9FTD/ALS | Pros | Cons |
|---|---|---|
| Loss-of-function | C9orf72 loss-of-function models in C. elegans and zebrafish result in motor neuron degeneration | C9orf72 loss-of-function mouse models do not show phenotype characteristic for ALS and FTD |
| Carriers of C9orf72 HRE have decreased levels of C9orf72 mRNA and proteins in the brain | Patients homozygous for C9orf72 repeat expansion do not have more severe symptoms of disease | |
|
| ||
| RNA-mediated gain-of-function | HRE-containing RNA transcripts accumulate and form nuclear aggregates, or RNA foci, in the brain of patients with mutated C9orf72 | Drosophila models of RNA toxic gain-of-function fail to produce neurodegeneration |
| Sequestration of RNA-binding proteins into RNA foci can disrupt RNA processing, translation, nucleocytoplasmic transport, and granule transport and lead to nucleolar stress | The results on RNA toxic gain-of-function mouse models are conflicting and need to be further investigated | |
| Higher abundance of RNA foci in patients carrying C9FTD/ALS HRE is associated with earlier disease onset | ||
|
| ||
| Protein-mediated gain-of-function | Drosophila model of protein-mediated gain-of-function develops neurodegeneration | Amounts of DPR in the brain do not correlate with clinical phenotype, severity of diseases, and neurodegeneration |
| DPR disrupt nucleocytoplasmic transport, RNA processing, translation, ubiquitin proteasome system, formation of stress granule, and Notch signalling pathway and can lead to nucleolar stress | Abundance of DPR is low in the brain regions most affected by ALS and FTD | |
ALS: amyotrophic lateral sclerosis; C9FTD/ALS: hexanucleotide repeat expansion in C9orf72 causing ALS and FTD; DPR proteins: dipeptide repeat proteins; FTD: frontotemporal dementia; HRE: hexanucleotide repeat expansion.
6.2. Toxic Gain-of-Function due to Aberrant HRE-Containing RNA
6.2.1. Higher-Order Structures of DNA and RNA Formed by C9orf72 HRE Sequence
Due to the uniformity of the G4C2 sequence and the abundance of guanine nucleotides, expanded hexanucleotide repeats in the C9orf72 gene form higher-order DNA structures called G-quadruplexes [51, 60]. G-quadruplexes formed in the DNA can adopt both parallel- and antiparallel-stranded conformations, and increasing the length of the repeats creates a heterogeneous mixture of these structures. Both C-rich sense and antisense strands can assemble as i-motifs and hairpin structures [61]. G-quadruplex structures formed in the C9orf72 HRE region cause generation of truncated RNA transcripts that are aborted in the hexanucleotide repeat region [51]. These aberrant RNA transcripts containing repetitive hexanucleotide sequence can also form G-quadruplexes and RNA hairpin structures. Additionally, HRE-containing RNA can form hybrids with HRE-containing DNA called R-loops [51, 62–64]. Together, these higher-order structures of DNA and RNA are thought to act as promoters and regulatory elements affecting replication, transcription, and translation of the surrounding region [65, 66], and to exert deleterious effect on cells by causing nucleolar stress and impeding RNA processing [51], as discussed below.
6.2.2. Sequestration of RNA-Binding Proteins into Nuclear Aggregates by the C9orf72 HRE-RNA
HRE-containing RNA transcripts accumulate and form nuclear aggregates, or RNA foci, in the brain of patients with C9orf72 HRE [14]. As the transcription of C9orf72 HRE DNA region can also occur in the antisense direction, antisense RNA is also found within foci in the brains of patients with C9orf72 HRE [67, 68]. Several proteins specifically bind to C9orf72 HRE-containing RNA, including ADARB2, ALYREF, hnRNP H, hnRNP A1, nucleolin, Pur α, and SRSF2 [51, 69–73]. A number of RNA-binding proteins (RBP) are implicated in the HRE-induced neurodegeneration.
(1) Nucleolin. One of the main constituents of the nucleolus, nucleolin, has a high affinity for C9orf72 RNA containing G-quadruplex structures of G4C2 repeat [51]. Nucleolin bound to RNA G-quadruplexes dislocates from the nucleoli and disperses through the nucleus. This results in impaired rRNA processing, followed by decreased maturation of ribosomes, and finally, accumulation of untranslated mRNA in the neuronal cytoplasm [51].
(2) Pur α. Another protein that has been identified as a HRE-RNA binding is Pur α [72]. Involvement of Pur α in the pathogenesis of ALS/FTD is supported by experiments showing that Pur α overexpression alleviates HRE-mediated toxicity. In other words, Pur α overexpression prevents cell death both in mammalian cells and in Drosophila [72]. Pur α is a component of the RNA-transport granules—particles that carry mRNAs to the nerve fibers where translation of those mRNAs into proteins occurs [74, 75]—and is involved in the regulation of the cell cycle [76] and in cell differentiation [77, 78]. Sequestration of Pur α by the C9orf72 hexanucleotide RNA repeats could impair neuronal mRNA transport thus leading to neurodegeneration [79].
(3) hnRNPs. The largest group of proteins sequestered by the C9orf72 HRE-containing RNA are heterogeneous nuclear ribonucleoproteins (hnRNPs) [44, 51]. It was shown that C9orf72 interacts with hnRNP-U, hnRNP-F, hnRNP-K [51], hnRNP-A2/B1, and hnRNP-A1 [28], while hnRNP A1 and hnRNP H colocalize with RNA foci [70, 71]. HnRNP H is involved in the regulation of RNA processing [80]; thus, hnRNP H sequestration in RNA cause aberrant RNA processing and may enhance neurodegeneration [70].
(4) ADARB2. An RNA-editing enzyme ADARB2 also interacts with the HRE-containing RNAs and colocalizes with RNA foci in C9ALS cases [69]. Although the mechanism by which ADARB2 sequestration in RNA foci could contribute to HRE-mediated toxicity is unclear, it was proposed that this protein may be important for RNA foci formation because ADARBP knockdown results in the reduction of neurons that contain RNA foci [69].
(5) SRSF1 and SRSF2. It was shown that serine-arginine-rich splicing factor 1 (SRSF1) and SRSF2 colocalize with RNA foci [70]. Given that SRSF2 is a marker of nuclear speckles, nuclear regions important for storage of splicing factors [81], its accumulation in RNA foci could disrupt the function of these speckles and lead to aberrant RNA processing [73]. Additionally, binding of the nuclear export adaptor SRSF1 to C9orf72 repeats promotes export of C9orf72 repeats from the nucleus. As it was demonstrated that SRSF1 knockdown in C9FTD/ALS Drosophila model blocks neurodegeneration, SRSF1 could be a potential therapeutic target [82].
(6) ALYREF. Because aberrant nuclear transport is observed in different C9FTD/ALS models [31–33], the observation that ALYREF (Aly/REF export factor) also colocalizes with RNA foci in C9FTD/ALS cases [70, 73, 83] is of interest. However, although knockdown of nuclear export adaptor SRSF1 blocks neurodegeneration in Drosophila, only a modest decrease in neurodegeneration was observed after knockdown of ALYREF [82].
In conclusion, toxic gain-of-function due to aberrant HRE-containing RNA can lead to dysfunctional RNA processing [73], aberrant translation [84], nucleolar stress [51], disrupted nucleocytoplasmic transport [31], and dysfunction in granule transport [85]. Additionally, it was observed that higher abundance of RNA foci in the frontal cortex of C9FTD patients is associated with earlier disease onset [68]. However, several experimental studies failed to detect neurodegeneration in in vivo C9FTD/ALS models displaying RNA foci (Table 2). Neurodegeneration was not observed in Drosophila models carrying 160 G4C2 repeats [86], 288 G4C2 repeats [87], and 1000 G4C2 repeats [88]. More precisely, Drosophila models carrying “RNA-only” repeats that formed RNA foci but not DPRs failed to produce neurodegeneration, while Drosophila models carrying “pure repeats” that could form both RNA foci and DPRs displayed neurodegeneration [87, 88]. However, two transgenic C9FTD/ALS mouse models with both RNA and protein toxic gain-of-function failed to produce neurodegeneration [89, 90], while two other transgenic mouse models showed signs of neurodegeneration [91, 92]. Regarding the latter two C9FTD/ALS mice models, whether neurodegeneration was caused by toxic HRE-containing RNAs or DPRs translated from hexanucleotide repeat RNA remains to be assessed [91, 92].
6.3. Toxic Gain-of-Function through Accumulation of Dipeptide Repeat Proteins Translated from C9orf72 HRE-Containing RNA
RNA transcripts of C9orf72 HRE region can undergo repeat-associated non-ATG (RAN) translation in which different DPR proteins are synthesized [43, 44, 93]. Poly-Gly-Ala, poly-Gly-Pro, and poly-Gly-Arg are translated from different open reading fragments on the sense transcript and poly-Gly-Pro, poly-Pro-Arg, and poly-Pro-Ala from the antisense transcript. Toxicity of DPR proteins seems to be mainly dependent on arginine-containing DPR proteins, particularly poly-Pro-Arg [33, 87, 94, 95]. Arginine-rich DPRs appear to disrupt primarily nucleocytoplasmic transport [33] and RNA processing [33, 95], which can cause dysregulation of translation [96, 97], nucleolar stress [98], disturb ubiquitin proteasome system [99], affect the formation of stress granules [100], and influence the Notch signalling pathway [101]. Additionally, it was shown that poly-Gly-Ala DPR proteins can disturb the ubiquitin proteasome system and cause endoplasmic reticulum stress [102], and enhance the formation of toxic amyloid fibrils [103].
Although there is molecular evidence of DPR proteins' toxicity in in vitro and in vivo C9FTD/ALS models [104, 105], post mortem analyses of human brain revealed inconsistent results (Table 2). No correlation of DPR proteins with clinical phenotype, severity of disease, and neurodegeneration was observed, and the abundance of DPR proteins was low in the brain regions most affected in ALS and FTD [106–109]. This lack of correlation may signal that neurons carrying toxic DPR proteins are possibly dead at the time of autopsy [18]. Additionally, there were discrepancies in the outcome of the studies comparing the distribution of DPR proteins between ALS and FTD cases [107, 110, 111].
7. Cellular Processes Affected in C9FTD/ALS
Although many cellular processes are affected by C9orf72 repeat expansion, including translation [96], ubiquitin proteasome system [99], Notch signalling pathway [101], granule transport [85], and normal function of TDP-43 (for review see [17]), here we discuss the cellular processes affected by C9orf72 repeat expansion for which sufficient information is available.
7.1. Effects of DPR Proteins and HRE-Containing RNAs on Nucleocytoplasmic Transport
A hallmark morphological feature of TDP-43 proteinopathies is the cytoplasmic accumulation and aggregate formation of TDP-43. We have demonstrated earlier that impaired nucleocytoplasmic transport plays a major role in this process [112]. Importantly, C9orf72 repeat expansion compromises nucleocytoplasmic transport of proteins and RNA through the nuclear pores. A study in yeast expressing arginine-rich DPR constructs demonstrated that one of the main targets of toxic DPR proteins is nucleocytoplasmic transport [33]. In support of these findings, genetic screens in Drosophila designed to identify genes linked to HRE-induced toxicity also identified components of the nuclear pore complex and nucleocytoplasmic transport machinery [31, 32]. Furthermore, RCC1, which is a human protein required for nucleocytoplasmic transport, is mislocalized in cells derived from patients with the C9orf72 HRE, providing preliminary evidence that C9FTD/ALS HRE might affect nucleocytoplasmic transport in human cells as well [33]. Taken together, the studies in yeast and Drosophila provided the first evidence that C9orf72 repeat expansion causes degeneration by impairing nucleocytoplasmic transport of proteins and RNA [31–33]; however, the effect of C9FTD/ALS HRE on nucleocytoplasmic transport in human cells needs to be further investigated. These findings open a possibility of targeting nucleocytoplasmic transport as a potential new therapeutic target in ALS and FTD [31, 32]. Additionally, as mRNA export factor ALYREF [70, 73, 83] and Ran GTPase-activating protein (RanGAP, regulator of nucleocytoplasmic transport) [31] colocalize with RNA foci (observed in C9FTD/ALS brains) and their function is disrupted, it seems that not only DPRs can lead to aberration of nucleocytoplasmic transport but also HRE-containing RNAs.
7.2. C9orf72 Hexanucleotide Repeat Expansion and RNA Processing
Several studies reported different transcriptional profiles between C9FTD/ALS patients and controls [54, 69, 71, 113–115]. Additionally, Cooper-Knock et al. observed an enrichment in RNA splicing factors in C9FTD/ALS patients [116], further supporting the observation of aberrant RNA processing caused by pathological processes in C9FTD/ALS. The main mechanism that leads to disturbed RNA processing in C9FTD/ALS patients is through sequestration of RBP in RNA foci [14, 73], but it could be also caused by DPR proteins as they can bind to RPB too [33, 95].
7.3. HRE-Containing RNAs and Dipeptide Repeat Proteins Can Lead to Nucleolar Stress
The way by which nucleolin binding to RNA foci disrupts the normal function of the nucleolus [51] was discussed above. It was also observed that DPR proteins can cause nucleolar stress in cell lines [98]. Enlargement of the nucleolus was observed in in vitro [94, 98] and in vivo [101] C9FTD/ALS models expressing arginine-rich DPR proteins. Enlargement of the nucleolus in these models leads to its fragmentation and decreased maturation of rRNA [98].
7.4. The Effect of DPRs on Formation and Function of Membraneless Organelles
Cells possess several RNA and protein-containing membraneless organelles, collectively referred to as ribonucleoprotein (RNP) granules, which separate from the cytoplasm or nucleoplasm into a distinct liquid phase-like state that is typically slightly denser than the surrounding [117]. Examples of such organelles are nucleoli and Cajal bodies in the nucleus and processing bodies (P-bodies), stress granules, and transport granules in the cytoplasm [117–119]. Formation of such compartments is triggered by intrinsically disordered low complexity polypeptide sequences present within RBP, which have the ability to undergo phase transitions [120–122]. Furthermore, it has been shown that nuclear membraneless suborganelles, such as Cajal bodies and nuclear speckles, can be nucleated by several coding and noncoding RNAs through the recruitment of proteins residing in these nuclear bodies [123]. Stress induces the formation of many membraneless compartments [124]. Lee et al. reported that arginine-rich DPRs bind low complexity polypeptide sequences of RBP that are components of membraneless organelles. Arginine-rich DPRs alter phase separation of those proteins and, in that way, disturb the function of membraneless organelles [125]. Liquid-liquid phase separation of RBP is very important for the normal formation of membraneless organelles. Other authors also showed that arginine-rich DPRs disturb the function and dynamics of membraneless organelles [126], more precisely, stress granules [98, 100]. Because it has been proposed that stress granules could be seeding points where aggregation of pathological proteins in FTD and ALS begins [127, 128], understanding the influence of DPR proteins on dynamics of membraneless organelles is highly relevant in this context.
7.5. C9orf72 HRE Affects Autophagy and Apoptosis
Adulterations in the autophagic pathway can alter protein homeostasis, causing detrimental effects on neurons that have been implicated in neurodegenerative diseases like Parkinson's disease and tauopathies. Autophagy prevents proteotoxic cell death by removing damaged, misfolded, and unwanted proteins [129]. Hindrance of C9orf72 function leads to a consequential decrease in Rab GTPase activity, a protein involved in membrane fusion, vesicle formation, and vesicle trafficking, which impairs the endocytosis process and autophagy. This causes an increased amount of p62/SQSTM1 and TDP-43, known markers of ALS-FTD [36]. Using C9orf72-deficient mice and cell lines, Sullivan et al. demonstrated that C9orf72 forms a binding complex with SMCR8 and WDR41, which allow it to interact with FIP200/Ulk1/ATG13/ATG101 complex and initiate autophagy [35]. Additionally, C9orf72 HRE have been implicated in increased endoplasmic reticulum stress by causing dysregulation of its calcium channels that leads to neuron apoptosis. This has been quantified by observing a decrease in antiapoptotic genes Bcl-2 and BcL-XL, while causing an increased expression of proapoptotic gene BAK [130].
7.6. C9orf72 HRE and Neuroinflammation
Experimental studies investigating C9orf72 loss-of-function in C9orf72-deficient mice revealed an increase in proinflammatory cytokines, lymphadenopathy, splenomegaly, alterations in myeloid cells from spleen, and in some cases of autoimmunity [40, 56, 57]. Additionally, a human autopsy study showed that C9ALS cases had more severe microglial pathology in the medulla and motor cortex than ALS cases without C9orf72 HRE [131]. These findings raised questions about increased neuroinflammation in C9FTD/ALS patients and possible involvement of activated microglia in disease pathogenesis. It was proposed that microglia could represent a link between three potential pathological mechanisms of C9orf72 HRE, whereby microglia is being most affected by loss of C9orf72 function, while neurons are most affected by a gain-of-function mechanism. Altogether, increased neuroinflammation, accumulation of RNA foci, and DPRs could lead to neuronal death (reviewed in [132]).
8. Potential Therapeutic Approaches
Potential therapeutics most commonly target HRE-containing RNA, because its degradation abolishes RNA toxicity and the formation of DPR proteins by RAN translation. The following approaches have been considered so far.
8.1. Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) targeting HRE-containing RNAs were tested in different studies [54, 69, 91]. After treatment of C9FTD/ALS cells in in vitro [54, 69, 71] and in vivo models with ASOs (mice expressing bacterial artificial chromosome (BAC) with the human expanded C9orf72 gene [91] and adeno-associated virus (AAV) (G4C2)66 mice [133]), a reduction in RNA foci was observed. Additionally, treatment with ASOs also led to a decrease in DPRs [91, 133]. Although ASOs used in these studies did not affect the levels of normal C9orf72 protein [54, 71, 91], the possibility of total silencing C9orf72 cannot be confirmed, so these approaches need to be considered with caution. A clinical trial for ASO targeting HRE-containing RNAs (WVE-3972-01; https://adisinsight.springer.com/trials/700291284) is expected in the fourth quarter of 2018.
8.2. Small Molecules
The advantage of using small molecules as therapeutics for C9FTD/ALS is that these compounds could not only target HRE-containing RNAs or DPR proteins but also cellular processes affected by pathology underlying C9FTD/ALS. Su et al. developed three small molecules that target hexanucleotide repeat region of RNAs and could stop RAN translation in (G4C2)66-expressing COS7 cells [134]. As epigenetic alterations were observed in C9FTD/ALS cases, epigenetic changes in the C9orf72 gene were also targeted. Usage of G-quadruplex-binding small molecules yielded increased expression of C9orf72 protein [135–137].
9. The Mechanisms of C9orf72 HRE-Mediated Neurodegeneration Are Not Mutually Exclusive
It is possible that the proposed mechanisms of C9FTD/ALS coexist and act in a combined manner. Maharjan et al. suggested that HRE in C9orf72 gene affects normal expression of C9orf72, diminishes levels of C9orf72 protein, and consequently impairs the formation of stress granules during the cellular stress (caused by formation of RNA foci and DPRs). In other words, C9orf72 loss-of-function made cells more sensitive to toxicity caused by gain-of-function mechanisms [39]. Additionally, Lall and Baloh [132] proposed a model that unifies all three pathological mechanisms mentioned above. Other authors stressed the importance of generation of rodent experimental models in which all three mechanisms coexist, for example, by crossing C9orf72 knockout mouse and mouse carrying BAC with human expanded C9orf72 gene [18]. Although it was shown that ASO targeting HRE-containing RNAs do not affect the levels of normal C9orf72 protein [54, 71, 91], the possibility of silencing C9orf72 totally should not be overlooked. As ASO targeting HRE-containing RNAs moves into phase 1 of clinical trials, the development of novel cellular and animal experimental models that exhibit all three pathological mechanisms is highly relevant.
10. Conclusions
Seven years after the discovery of hexanucleotide repeat expansion in C9orf72 gene, a lot of progress has been made in the clarification of molecular mechanisms through which C9orf72 repeat expansions cause neurodegeneration. Three possible, not mutually exclusive, mechanisms could together contribute to the pathogenesis of disease [39]. The majority of the studies investigating the molecular mechanisms of pathological processes in C9FTD/ALS support either toxic HRE-RNA or DPR-dependent gain-of-function, with many studies supporting both mechanisms (Table 1). Hence, to better identify potential therapeutic targets, further studies are needed to fully understand molecular events underlying pathological processes in C9FTD/ALS.
Acknowledgments
We thank Mirta Boban for her help in improving the initial version of the manuscript. This work was supported by the Croatian Science Foundation grant IP-2014-09-9730 to GŠ, by the Scientific Centre of Excellence for Basic, Clinical and Translational Neuroscience CoreNeuro of the Croatian Institute for Brain Research, University of Zagreb Medical School, grant KK.01.1.1.01.0007, and in part by NIH grant P50 AG005138 to PRH.
Abbreviations
- ALS:
Amyotrophic lateral sclerosis
- ALS-bi:
ALS with behavioral impairment
- ALS-ci:
ALS with cognitive impairment
- ALS-cbi:
ALS with combined cognitive and behavioral impairment
- ALYREF:
Aly/REF export factor
- ASOs:
Antisense oligonucleotides
- BAC:
Bacterial artificial chromosome
- bvFTD:
Behavioral variant of frontotemporal dementia
- C9orf72:
Chromosome 9 open reading frame 72
- C9FTD/ALS:
Hexanucleotide repeat expansion in C9orf72 causing ALS and FTD
- CSF:
Cerebrospinal fluid
- DENN proteins:
Differentially expressed in normal and neoplasia proteins
- DPR proteins:
Dipeptide repeat proteins
- FTD:
Frontotemporal dementia
- FTLD:
Frontotemporal lobar degeneration
- FUS:
Fused in sarcoma
- G4C2:
GGGGCC
- GEF:
Guanine exchange factor
- hnRNPs:
Heterogeneous nuclear ribonucleoproteins
- HRE:
Hexanucleotide repeat expansion
- IBMFD:
Inclusion body myopathy with frontotemporal dementia
- iNPCs:
Induced neural progenitor cells
- iPSC:
Induced pluripotent stem cells
- MND:
Motor neuron disease
- NCL:
Nucleolin
- NPC:
Nuclear pore complex
- P-bodies:
Processing bodies
- PNFA:
Progressive nonfluent aphasia
- pTDP-43:
Phosphorylated TDP-43
- RAN translation:
Repeat-associated non-ATG translation
- RanGAP:
Ran GTPase-activating protein
- RBP:
RNA binding proteins
- RNP:
Ribonucleoprotein
- SD:
Semantic dementia
- SRSF:
Serine-arginine-rich splicing factor
- TDP-43:
TAR DNA-binding protein 43
- VCP:
Valosin-containing protein.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.van Langenhove T., van der Zee J., van Broeckhoven C. The molecular basis of the frontotemporal lobar degeneration–amyotrophic lateral sclerosis spectrum. Annals of Medicine. 2012;44(8):817–828. doi: 10.3109/07853890.2012.665471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Worms P. M. The epidemiology of motor neuron diseases: a review of recent studies. Journal of the Neurological Sciences. 2001;191(1-2):3–9. doi: 10.1016/s0022-510x(01)00630-x. [DOI] [PubMed] [Google Scholar]
- 3.Strong M. J., Abrahams S., Goldstein L. H., et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2017;18(3-4):153–174. doi: 10.1080/21678421.2016.1267768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ratnavalli E., Brayne C., Dawson K., Hodges J. R. The prevalence of frontotemporal dementia. Neurology. 2002;58(11):1615–1621. doi: 10.1212/WNL.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 5.Ng A. S. L., Rademakers R., Miller B. L. Frontotemporal dementia: a bridge between dementia and neuromuscular disease. Annals of the New York Academy of Sciences. 2015;1338(1):71–93. doi: 10.1111/nyas.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hortobágyi T., Cairns N. Amyotrophic lateral sclerosis and frontotemporal dementia. In: Kovacs G., editor. Neuropathology of Neurodegenerative Diseases: A Practical Guide. Cambridge University Press; 2015. pp. 209–248. [Google Scholar]
- 7.Hortobágyi T., Cairns N. J. Chapter 26 - Amyotrophic lateral sclerosis and non-tau frontotemporal lobar degeneration. Handbook of clinical neurology. 2017;145:369–381. doi: 10.1016/B978-0-12-802395-2.00026-2. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie I. R. A., Neumann M., Baborie A., et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathologica. 2011;122(1):111–113. doi: 10.1007/s00401-011-0845-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chow T. W., Miller B. L., Hayashi V. N., Geschwind D. H. Inheritance of frontotemporal dementia. Archives of Neurology. 1999;56(7):817–822. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosso S. M., Kaat L. D., Baks T., et al. Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126(9):2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 11.Strong M. J., Hortobágyi T., Okamoto K., Kato S. Amyotrophic lateral sclerosis, primary lateral sclerosis and spinal muscular atrophy. In: Dickson D., Weller R., editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Wiley-Blackwell; 2011. pp. 418–433. [DOI] [Google Scholar]
- 12.Vance C., al-Chalabi A., Ruddy D., et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain. 2006;129(4):868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 13.Morita M., al-Chalabi A., Andersen P. M., et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66(6):839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- 14.DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Renton A. E., Majounie E., Waite A., et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gijselinck I., van Langenhove T., van der Zee J., et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. The Lancet Neurology. 2012;11(1):54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 17.Todd T. W., Petrucelli L. Insights into the pathogenic mechanisms of chromosome 9 open reading frame 72 (C9orf72) repeat expansions. Journal of Neurochemistry. 2016;138:145–162. doi: 10.1111/jnc.13623. [DOI] [PubMed] [Google Scholar]
- 18.Wen X., Westergard T., Pasinelli P., Trotti D. Pathogenic determinants and mechanisms of ALS/FTD linked to hexanucleotide repeat expansions in the C9orf72 gene. Neuroscience Letters. 2017;636:16–26. doi: 10.1016/j.neulet.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gijselinck I., on behalf of the BELNEU CONSORTIUM13, Van Mossevelde S., et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Molecular Psychiatry. 2016;21(8):1112–1124. doi: 10.1038/mp.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waite A. J., Bäumer D., East S., et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiology of Aging. 2014;35(7):1779.e5–1779.e13. doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dols-Icardo O., Garcia-Redondo A., Rojas-Garcia R., et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Human Molecular Genetics. 2014;23(3):749–754. doi: 10.1093/hmg/ddt460. [DOI] [PubMed] [Google Scholar]
- 22.van Blitterswijk M., DeJesus-Hernandez M., Niemantsverdriet E., et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. The Lancet Neurology. 2013;12(10):978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck J., Poulter M., Hensman D., et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. The American Journal of Human Genetics. 2013;92(3):345–353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hübers A., Marroquin N., Schmoll B., et al. Polymerase chain reaction and Southern blot-based analysis of the C9orf72 hexanucleotide repeat in different motor neuron diseases. Neurobiology of Aging. 2014;35(5):1214.e1–1214.e6. doi: 10.1016/j.neurobiolaging.2013.11.034. [DOI] [PubMed] [Google Scholar]
- 25.Nordin A., Akimoto C., Wuolikainen A., et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Human Molecular Genetics. 2015;24(11):3133–3142. doi: 10.1093/hmg/ddv064. [DOI] [PubMed] [Google Scholar]
- 26.Zhang D., Iyer L. M., He F., Aravind L. Discovery of novel DENN proteins: implications for the evolution of eukaryotic intracellular membrane structures and human disease. Frontiers in Genetics. 2012;3:p. 283. doi: 10.3389/fgene.2012.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Levine T. P., Daniels R. D., Gatta A. T., Wong L. H., Hayes M. J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29(4):499–503. doi: 10.1093/bioinformatics/bts725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farg M. A., Sundaramoorthy V., Sultana J. M., et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Human Molecular Genetics. 2014;23(13):3579–3595. doi: 10.1093/hmg/ddu068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frick P., Sellier C., Mackenzie I. R. A., et al. Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathologica Communications. 2018;6(1):p. 72. doi: 10.1186/s40478-018-0579-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao S., MacNair L., McGoldrick P., et al. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Annals of Neurology. 2015;78(4):568–583. doi: 10.1002/ana.24469. [DOI] [PubMed] [Google Scholar]
- 31.Zhang K., Donnelly C. J., Haeusler A. R., et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freibaum B. D., Lu Y., Lopez-Gonzalez R., et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jovičić A., Mertens J., Boeynaems S., et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nature Neuroscience. 2015;18(9):1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Webster C. P., Smith E. F., Bauer C. S., et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. The EMBO Journal. 2016;35(15):1656–1676. doi: 10.15252/embj.201694401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sullivan P. M., Zhou X., Robins A. M., et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathologica Communications. 2016;4(1):p. 51. doi: 10.1186/s40478-016-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sellier C., Campanari M. L., Julie Corbier C., et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ ataxin-2 to induce motor neuron dysfunction and cell death. The EMBO Journal. 2016;35(12):1276–1297. doi: 10.15252/embj.201593350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amick J., Roczniak-Ferguson A., Ferguson S. M. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Molecular Biology of the Cell. 2016;27(20):3040–3051. doi: 10.1091/mbc.e16-01-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amick J., Ferguson S. M. C9orf72: at the intersection of lysosome cell biology and neurodegenerative disease. Traffic. 2017;18(5):267–276. doi: 10.1111/tra.12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maharjan N., Künzli C., Buthey K., Saxena S. C9ORF72 regulates stress granule formation and its deficiency impairs stress granule assembly, hypersensitizing cells to stress. Molecular Neurobiology. 2017;54(4):3062–3077. doi: 10.1007/s12035-016-9850-1. [DOI] [PubMed] [Google Scholar]
- 40.O'Rourke J. G., Bogdanik L., Yanez A., et al. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351(6279):1324–1329. doi: 10.1126/science.aaf1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Sarraj S., King A., Troakes C., et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathologica. 2011;122(6):691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 42.Murray M. E., DeJesus-Hernandez M., Rutherford N. J., et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathologica. 2011;122(6):673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ash P. E. A., Bieniek K. F., Gendron T. F., et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mori K., Weng S. M., Arzberger T., et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 45.Troakes C., Maekawa S., Wijesekera L., et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2012;32(5):505–514. doi: 10.1111/j.1440-1789.2011.01286.x. [DOI] [PubMed] [Google Scholar]
- 46.van Blitterswijk M., Gendron T. F., Baker M. C., et al. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathologica. 2015;130(6):863–876. doi: 10.1007/s00401-015-1480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xi Z., Zinman L., Moreno D., et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. American Journal of Human Genetics. 2013;92(6):981–989. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xi Z., Zhang M., Bruni A. C., et al. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathologica. 2015;129(5):715–727. doi: 10.1007/s00401-015-1401-8. [DOI] [PubMed] [Google Scholar]
- 49.Xi Z., Rainero I., Rubino E., et al. Hypermethylation of the CpG-island near the C9orf72 G4C2-repeat expansion in FTLD patients. Human Molecular Genetics. 2014;23(21):5630–5637. doi: 10.1093/hmg/ddu279. [DOI] [PubMed] [Google Scholar]
- 50.Liu E. Y., Russ J., Wu K., et al. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathologica. 2014;128(4):525–541. doi: 10.1007/s00401-014-1286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haeusler A. R., Donnelly C. J., Periz G., et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507(7491):195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Therrien M., Rouleau G. A., Dion P. A., Parker J. A. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS One. 2013;8(12, article e83450) doi: 10.1371/journal.pone.0083450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ciura S., Lattante S., le Ber I., et al. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Annals of Neurology. 2013;74(2):180–187. doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- 54.Lagier-Tourenne C., Baughn M., Rigo F., et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(47):E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koppers M., Blokhuis A. M., Westeneng H. J., et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Annals of Neurology. 2015;78(3):426–438. doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atanasio A., Decman V., White D., et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production and glomerulonephropathy in mice. Scientific Reports. 2016;6(1):p. 23204. doi: 10.1038/srep23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sudria-Lopez E., Koppers M., de Wit M., et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathologica. 2016;132(1):145–147. doi: 10.1007/s00401-016-1581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fratta P., Poulter M., Lashley T., et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathologica. 2013;126(3):401–409. doi: 10.1007/s00401-013-1147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi Y., Lin S., Staats K. A., et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nature Medicine. 2018;24(3):313–325. doi: 10.1038/nm.4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Šket P., Pohleven J., Kovanda A., et al. Characterization of DNA G-quadruplex species forming from C9ORF72 G4C2-expanded repeats associated with amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Neurobiology of Aging. 2015;36(2):1091–1096. doi: 10.1016/j.neurobiolaging.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 61.Kovanda A., Zalar M., Šket P., Plavec J., Rogelj B. Anti-sense DNA d(GGCCCC)n expansions in C9ORF72 form i-motifs and protonated hairpins. Scientific Reports. 2015;5(1, article 17944) doi: 10.1038/srep17944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fratta P., Mizielinska S., Nicoll A. J., et al. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Scientific Reports. 2012;2(1):p. 1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reddy K., Zamiri B., Stanley S. Y. R., Macgregor R. B., Jr., Pearson C. E. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. Journal of Biological Chemistry. 2013;288(14):9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reddy K., Schmidt M. H. M., Geist J. M., et al. Processing of double-R-loops in (CAG)·(CTG) and C9orf72 (GGGGCC)·(GGCCCC) repeats causes instability. Nucleic Acids Research. 2014;42(16):10473–10487. doi: 10.1093/nar/gku658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kendrick S., Hurley L. H. The role of G-quadruplex/i-motif secondary structures as cis-acting regulatory elements. Pure and Applied Chemistry. 2010;82(8):1609–1621. doi: 10.1351/PAC-CON-09-09-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brooks T. A., Kendrick S., Hurley L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. The FEBS Journal. 2010;277(17):3459–3469. doi: 10.1111/j.1742-4658.2010.07759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gendron T. F., Bieniek K. F., Zhang Y. J., et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathologica. 2013;126(6):829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mizielinska S., Lashley T., Norona F. E., et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathologica. 2013;126(6):845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Donnelly C. J., Zhang P. W., Pham J. T., et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee Y.-B., Chen H. J., Peres J. N., et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Reports. 2013;5(5):1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sareen D., O'Rourke J. G., Meera P., et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Science Translational Medicine. 2013;5(208, article 208ra149) doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu Z., Poidevin M., Li X., et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(19):7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cooper-Knock J., Walsh M. J., Higginbottom A., et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014;137(7):2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kiebler M. A., Bassell G. J. Neuronal RNA granules: movers and makers. Neuron. 2006;51(6):685–690. doi: 10.1016/j.neuron.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 75.Kanai Y., Dohmae N., Hirokawa N. Kinesin transports RNA. Neuron. 2004;43(4):513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 76.White M. K., Johnson E. M., Khalili K. Multiple roles for Puralpha in cellular and viral regulation. Cell Cycle. 2009;8(3):1–7. doi: 10.4161/cc.8.3.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Khalili K., del Valle L., Muralidharan V., et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Molecular and Cellular Biology. 2003;23(19):6857–6875. doi: 10.1128/MCB.23.19.6857-6875.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mishra M., Del Valle L., Otte J., Darbinian N., Gordon J. Pur-alpha regulates RhoA developmental expression and downstream signaling. Journal of Cellular Physiology. 2013;228(1):65–72. doi: 10.1002/jcp.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolozin B., Apicco D. RNA binding proteins and the genesis of neurodegenerative diseases. Advances in Experimental Medicine and Biology. 2015;822:11–15. doi: 10.1007/978-3-319-08927-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chaudhury A., Chander P., Howe P. H. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: focus on hnRNP E1’s multifunctional regulatory roles. RNA. 2010;16(8):1449–1462. doi: 10.1261/rna.2254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spector D. L., Lamond A. I. Nuclear speckles. Cold Spring Harbor Perspectives in Biology. 2011;3(2, article a000646) doi: 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hautbergue G. M., Castelli L. M., Ferraiuolo L., et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nature Communications. 2017;8, article 16063 doi: 10.1038/ncomms16063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cooper-Knock J., Higginbottom A., Stopford M. J., et al. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathologica. 2015;130(1):63–75. doi: 10.1007/s00401-015-1429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rossi S., Serrano A., Gerbino V., et al. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. Journal of Cell Science. 2015;128(9):1787–1799. doi: 10.1242/jcs.165332. [DOI] [PubMed] [Google Scholar]
- 85.Burguete A. S., Almeida S., Gao F.-B., Kalb R., Akins M. R., Bonini N. M. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. eLife. 2015;4, article e08881 doi: 10.7554/eLife.08881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tran H., Almeida S., Moore J., et al. Differential toxicity of nuclear RNA foci versus dipeptide repeat proteins in a Drosophila model of C9ORF72 FTD/ALS. Neuron. 2015;87(6):1207–1214. doi: 10.1016/j.neuron.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mizielinska S., Gronke S., Niccoli T., et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moens T. G., Mizielinska S., Niccoli T., et al. Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathologica. 2018;135(3):445–457. doi: 10.1007/s00401-017-1798-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.O’Rourke J. G., Bogdanik L., Muhammad A. K. M. G., et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901. doi: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peters O. M., Cabrera G. T., Tran H., et al. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88(5):902–909. doi: 10.1016/j.neuron.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang J., Zhu Q., Gendron T. F., et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liu Y., Pattamatta A., Zu T., et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90(3):521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 93.Zu T., Liu Y., Banez-Coronel M., et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(51):E4968–E4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wen X., Tan W., Westergard T., et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84(6):1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kwon I., Xiang S., Kato M., et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345(6201):1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kanekura K., Yagi T., Cammack A. J., et al. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Human Molecular Genetics. 2016;25(9):1803–1813. doi: 10.1093/hmg/ddw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hartmann H., Hornburg D., Czuppa M., et al. Proteomics and C9orf72 neuropathology identify ribosomes as poly-GR/PR interactors driving toxicity. Life Science Alliance. 2018;1(2, article e201800070) doi: 10.26508/lsa.201800070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tao Z., Wang H., Xia Q., et al. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Human Molecular Genetics. 2015;24(9):2426–2441. doi: 10.1093/hmg/ddv005. [DOI] [PubMed] [Google Scholar]
- 99.Yamakawa M., Ito D., Honda T., et al. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Human Molecular Genetics. 2015;24(6):1630–1645. doi: 10.1093/hmg/ddu576. [DOI] [PubMed] [Google Scholar]
- 100.Boeynaems S., Bogaert E., Kovacs D., et al. Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Molecular Cell. 2017;65(6):1044–1055.e5. doi: 10.1016/j.molcel.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang D., Abdallah A., Li Z., Lu Y., Almeida S., Gao F.-B. FTD/ALS-associated poly(GR) protein impairs the notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathologica. 2015;130(4):525–535. doi: 10.1007/s00401-015-1448-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Y.-J., Jansen-West K., Xu Y. F., et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathologica. 2014;128(4):505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chang Y.-J., Jeng U.-S., Chiang Y.-L., Hwang I.-S., Chen Y.-R. The glycine-alanine dipeptide repeat from C9orf72 Hexanucleotide expansions forms toxic amyloids possessing cell-to-cell transmission properties. Journal of Biological Chemistry. 2016;291(10):4903–4911. doi: 10.1074/jbc.M115.694273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Swaminathan A., Bouffard M., Liao M., et al. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Human Molecular Genetics. 2018;27(10):1754–1762. doi: 10.1093/hmg/ddy083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kramer N. J., Haney M. S., Morgens D. W., et al. CRISPR–Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nature Genetics. 2018;50(4):603–612. doi: 10.1038/s41588-018-0070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mackenzie I. R., Arzberger T., Kremmer E., et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathologica. 2013;126(6):859–879. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- 107.Davidson Y. S., Barker H., Robinson A. C., et al. Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathologica Communications. 2014;2(1):p. 70. doi: 10.1186/2051-5960-2-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mackenzie I. R. A., Frick P., Grässer F. A., et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathologica. 2015;130(6):845–861. doi: 10.1007/s00401-015-1476-2. [DOI] [PubMed] [Google Scholar]
- 109.Gomez-Deza J., Lee Y. B., Troakes C., et al. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathologica Communications. 2015;3(1):p. 38. doi: 10.1186/s40478-015-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gendron T. F., van Blitterswijk M., Bieniek K. F., et al. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathologica. 2015;130(4):559–573. doi: 10.1007/s00401-015-1474-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.German Consortium for Frontotemporal Lobar Degeneration, Bavarian Brain Banking Alliance, Schludi M. H., et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathologica. 2015;130(4):537–555. doi: 10.1007/s00401-015-1450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nishimura A. L., Župunski V., Troakes C., et al. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133(6):1763–1771. doi: 10.1093/brain/awq111. [DOI] [PubMed] [Google Scholar]
- 113.Prudencio M., Belzil V. V., Batra R., et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nature Neuroscience. 2015;18(8):1175–1182. doi: 10.1038/nn.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Barker H. V., Niblock M., Lee Y. B., Shaw C. E., Gallo J. M. RNA misprocessing in C9orf72-linked neurodegeneration. Frontiers in Cellular Neuroscience. 2017;11:p. 195. doi: 10.3389/fncel.2017.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kumar V., Hasan G. M., Hassan M. I. Unraveling the role of RNA mediated toxicity of C9orf72 repeats in C9-FTD/ALS. Frontiers in Neuroscience. 2017;11:p. 711. doi: 10.3389/fnins.2017.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cooper-Knock J., Bury J. J., Heath P. R., et al. C9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS One. 2015;10(5, article e0127376) doi: 10.1371/journal.pone.0127376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brangwynne C. P. Phase transitions and size scaling of membrane-less organelles. The Journal of Cell Biology. 2013;203(6):875–881. doi: 10.1083/jcb.201308087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Berry J., Weber S. C., Vaidya N., Haataja M., Brangwynne C. P. RNA transcription modulates phase transition-driven nuclear body assembly. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(38):E5237–E5245. doi: 10.1073/pnas.1509317112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Aguilera-Gomez A., Rabouille C. Membrane-bound organelles versus membrane-less compartments and their control of anabolic pathways in Drosophila. Developmental Biology. 2017;428(2):310–317. doi: 10.1016/j.ydbio.2017.03.029. [DOI] [PubMed] [Google Scholar]
- 120.Han T. W., Kato M., Xie S., et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149(4):768–779. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 121.Kato M., Han T. W., Xie S., et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149(4):753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin Y., Protter D. S. W., Rosen M. K., Parker R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Molecular Cell. 2015;60(2):208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shevtsov S. P., Dundr M. Nucleation of nuclear bodies by RNA. Nature Cell Biology. 2011;13(2):167–173. doi: 10.1038/ncb2157. [DOI] [PubMed] [Google Scholar]
- 124.Rabouille C., Alberti S. Cell adaptation upon stress: the emerging role of membrane-less compartments. Current Opinion in Cell Biology. 2017;47:34–42. doi: 10.1016/j.ceb.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 125.Lee K.-H., Zhang P., Kim H. J., et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167(3):774–788.e17. doi: 10.1016/j.cell.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lin Y., Mori E., Kato M., et al. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell. 2016;167(3):789–802.e12. doi: 10.1016/j.cell.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.King O. D., Gitler A. D., Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Research. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ramaswami M., Taylor J. P., Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154(4):727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Loos B., Klionsky D. J., Wong E. Augmenting brain metabolism to increase macro- and chaperone-mediated autophagy for decreasing neuronal proteotoxicity and aging. Progress in Neurobiology. 2017;156:90–106. doi: 10.1016/j.pneurobio.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 130.Dafinca R., Scaber J., Ababneh N., et al. C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells. 2016;34(8):2063–2078. doi: 10.1002/stem.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brettschneider J., Toledo J. B., van Deerlin V. M., et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012;7(6, article e39216) doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lall D., Baloh R. H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. The Journal of Clinical Investigation. 2017;127(9):3250–3258. doi: 10.1172/JCI90607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gendron T. F., Chew J., Stankowski J. N., et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Science Translational Medicine. 2017;9(383, article eaai7866) doi: 10.1126/scitranslmed.aai7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Su Z., Zhang Y., Gendron T. F., et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83(5):1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Belzil V. V., Bauer P. O., Prudencio M., et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathologica. 2013;126(6):895–905. doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zeier Z., Esanov R., Belle K. C., et al. Bromodomain inhibitors regulate the C9ORF72 locus in ALS. Experimental Neurology. 2015;271:241–250. doi: 10.1016/j.expneurol.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Simone R., Balendra R., Moens T. G., et al. G‐quadruplex‐binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Molecular Medicine. 2018;10(1):22–31. doi: 10.15252/emmm.201707850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.May S., Hornburg D., Schludi M. H., et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathologica. 2014;128(4):485–503. doi: 10.1007/s00401-014-1329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Russ J., Liu E. Y., Wu K., et al. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathologica. 2015;129(1):39–52. doi: 10.1007/s00401-014-1365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chew J., Gendron T. F., Prudencio M., et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348(6239):1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hu J., Liu J., Li L., Gagnon K. T., Corey D. R. Engineering duplex RNAs for challenging targets: recognition of GGGGCC/CCCCGG repeats at the ALS/FTD C9orf72 locus. Chemistry & Biology. 2015;22(11):1505–1511. doi: 10.1016/j.chembiol.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Boeynaems S., Bogaert E., Michiels E., et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Scientific Reports. 2016;6(1):p. 20877. doi: 10.1038/srep20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Flores B. N., Dulchavsky M. E., Krans A., et al. Distinct C9orf72-associated dipeptide repeat structures correlate with neuronal toxicity. PLoS One. 2016;11(10, article e0165084) doi: 10.1371/journal.pone.0165084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lopez-Gonzalez R., Lu Y., Gendron T. F., et al. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron. 2016;92(2):383–391. doi: 10.1016/j.neuron.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu F., Liu Q., Lu C. X., et al. Identification of a novel loss-of-function C9orf72 splice site mutation in a patient with amyotrophic lateral sclerosis. Neurobiology of Aging. 2016;47:219.e1–219.e5. doi: 10.1016/j.neurobiolaging.2016.07.027. [DOI] [PubMed] [Google Scholar]
- 146.Dodd D. W., Tomchick D. R., Corey D. R., Gagnon K. T. Pathogenic C9ORF72 antisense repeat RNA forms a double helix with tandem C:C mismatches. Biochemistry. 2016;55(9):1283–1286. doi: 10.1021/acs.biochem.6b00136. [DOI] [PubMed] [Google Scholar]
- 147.Zhang Y.-J., Gendron T. F., Grima J. C., et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nature Neuroscience. 2016;19(5):668–677. doi: 10.1038/nn.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Westergard T., Jensen B. K., Wen X., et al. Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Reports. 2016;17(3):645–652. doi: 10.1016/j.celrep.2016.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mori K., Nihei Y., Arzberger T., et al. Reduced hnRNPA3 increases C9orf72 repeat RNA levels and dipeptide-repeat protein deposition. EMBO Reports. 2016;17(9):1314–1325. doi: 10.15252/embr.201541724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ugolino J., Ji Y. J., Conchina K., et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genetics. 2016;12(11, article e1006443) doi: 10.1371/journal.pgen.1006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ji Y. J., Ugolino J., Brady N. R., Hamacher-Brady A., Wang J. Systemic deregulation of autophagy upon loss of ALS- and FTD-linked C9orf72. Autophagy. 2017;13(7):1254–1255. doi: 10.1080/15548627.2017.1299312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ohki Y., Wenninger-Weinzierl A., Hruscha A., et al. Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Molecular Neurodegeneration. 2017;12(1):p. 6. doi: 10.1186/s13024-016-0146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu J., Hu J., Ludlow A. T., et al. C9orf72 disease-related foci are each composed of one mutant expanded repeat RNA. Cell Chemical Biology. 2017;24(2):141–148. doi: 10.1016/j.chembiol.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hu J., Rigo F., Prakash T. P., Corey D. R. Recognition of c9orf72 mutant RNA by single-stranded silencing RNAs. Nucleic Acid Therapeutics. 2017;27(2):87–94. doi: 10.1089/nat.2016.0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Green K. M., Glineburg M. R., Kearse M. G., et al. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nature Communications. 2017;8(1):p. 2005. doi: 10.1038/s41467-017-02200-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schludi M. H., Becker L., Garrett L., et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathologica. 2017;134(2):241–254. doi: 10.1007/s00401-017-1711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gupta R., Lan M., Mojsilovic-Petrovic J., et al. The proline/arginine dipeptide from hexanucleotide repeat expanded C9ORF72 inhibits the proteasome. eNeuro. 2017;4(1):ENEURO.0249–ENEU16.2017. doi: 10.1523/ENEURO.0249-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Khosravi B., Hartmann H., May S., et al. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Human Molecular Genetics. 2017;26(4):790–800. doi: 10.1093/hmg/ddw432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shi K. Y., Mori E., Nizami Z. F., et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(7):E1111–E1117. doi: 10.1073/pnas.1620293114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Saberi S., Stauffer J. E., Jiang J., et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathologica. 2018;135(3):459–474. doi: 10.1007/s00401-017-1793-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Herranz-Martin S., Chandran J., Lewis K., et al. Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Disease Models & Mechanisms. 2017;10(7):859–868. doi: 10.1242/dmm.029892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhou Q., Lehmer C., Michaelsen M., et al. Antibodies inhibit transmission and aggregation of C9orf72 poly‐GA dipeptide repeat proteins. EMBO Molecular Medicine. 2017;9(5):687–702. doi: 10.15252/emmm.201607054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lehmer C., Oeckl P., Weishaupt J. H., et al. Poly‐GP in cerebrospinal fluid links C9orf72‐associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Molecular Medicine. 2017;9(7):859–868. doi: 10.15252/emmm.201607486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cheng W., Wang S., Mestre A. A., et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2α phosphorylation. Nature Communications. 2018;9(1):p. 51. doi: 10.1038/s41467-017-02495-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Swinnen B., Bento-Abreu A., Gendron T. F., et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathologica. 2018;135(3):427–443. doi: 10.1007/s00401-017-1796-5. [DOI] [PubMed] [Google Scholar]
- 166.Tabet R., Schaeffer L., Freyermuth F., et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nature Communications. 2018;9(1):p. 152. doi: 10.1038/s41467-017-02643-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Corrionero A., Horvitz H. R. A C9orf72 ALS/FTD ortholog acts in endolysosomal degradation and lysosomal homeostasis. Current Biology. 2018;28(10):1522–1535.e5. doi: 10.1016/j.cub.2018.03.063. [DOI] [PubMed] [Google Scholar]
- 168.Zamiri B., Mirceta M., Abu-Ghazalah R., Wold M. S., Pearson C. E., Macgregor R. B., Jr Stress-induced acidification may contribute to formation of unusual structures in C9orf72-repeats. Biochimica et Biophysica Acta (BBA) - General Subjects. 2018;1862(6):1482–1491. doi: 10.1016/j.bbagen.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 169.Yeh T.-H., Liu H. F., Li Y. W., et al. C9orf72 is essential for neurodevelopment and motility mediated by cyclin G1. Experimental Neurology. 2018;304:114–124. doi: 10.1016/j.expneurol.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 170.Meeter L. H. H., Gendron T. F., Sias A. C., et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Annals of Clinical Translational Neurology. 2018;5(5):583–597. doi: 10.1002/acn3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nonaka T., Masuda-Suzukake M., Hosokawa M., et al. C9ORF72 dipeptide repeat poly-GA inclusions promote intracellular aggregation of phosphorylated TDP-43. Human Molecular Genetics. 2018;27(15):2658–2670. doi: 10.1093/hmg/ddy174. [DOI] [PubMed] [Google Scholar]