

ABSTRACT
We explored whether engineering of T cell specificity and effector function improves immunotherapy of solid tumors. Although IL-12 can enhance cancer immunity, a strategy of safe IL-12 delivery without toxicity is currently lacking. We engineered T cells to express IL-12 controlled by the NFAT promoter responsive to TCR stimulation, or by the Tet-On promoter responsive to doxycycline. In vivo, NFAT-engineered T cells caused lethal toxicity, while Tet-engineered T cells were safe in the absence of doxycycline. Combining gene transfer of the melanoma-specific TRP2-TCR with Tet-IL-12 engineering revealed that temporal induction of IL-12 was essential to inhibit the growth of B16F10 melanoma tumors. Induced IL-12 increased the number of tumor-infiltrating T cells and also prevented the down-modulation of the TRP2-TCR and the associated up-regulation of the PD1 marker that was observed in the absence of IL-12. In addition, temporal induction of IL-12 expression also increased the number of plasmacytoid DC in the tumor micro-environment. We show that repeated induction of IL-12 can be used to enhance control of tumor growth without encountering systemic toxicity. The observation that TCR engineering combined with Tet-regulated IL-12 expression can achieve tumor immunity without the side effects that are usually associated with the in vivo use of IL-12 warrants translation of this concept into the clinic.
Keywords: Immunotherapy, TCR, IL-12, T cells, adoptive, engineering, NFAT, Tet
Introduction
The genetic engineering T cell specificity with chimeric antigen receptors (CARs) or T cell receptors (TCRs) has provided a robust platform for directing T cell immunity towards defined cancer antigens.1–3 Dramatic clinical benefits have been seen with CAR-engineered T cells specific for CD19, a cancer-associated antigen present on most B-lineage leukemias.4,5 In contrast to leukemias, solid cancers establish a complex tumor stromal environment consisting of new blood vessels, mesenchymal cells and hematopoietic cells including myeloid derived suppressor cells. The establishment of an immune suppressive micro-environment is one of the mechanisms promoting escape of developing tumours from natural immune surveillance.6,7
IL-12 is a potent immune stimulatory cytokine capable of inducing pro-inflammatory Th1 and Th17 immune responses.8 Murine models have revealed that IL-12 delivery to sites of tumor growth can convert myeloid suppressor cells into immune-stimulatory cells and also activate local macrophages and NK cells to contribute to tumor immunity.9,10 While adoptive transfer of tumor-specific T cells engineered to express IL-12 constitutively resulted in improved tumor protection, this approach also carried a high risk of IL-12 mediated toxicity, as was previously seen in patients treated by administration of recombinant IL-12.11 Hence, several groups have explored antigen-specific T cell delivery of IL-12 to tumors using the DNA binding sites of the nuclear factor of activated T cells (NFAT) combined with a minimal IL-2 promoter.9,12,13 Upon stimulation of G-protein-coupled cell surface receptors or tyrosine-coupled receptors, such as TCRs, the calcium-regulated NFAT proteins translocate to the nucleus where, in combination with other transcription factors, they activate expression of genes containing NFAT binding sites.14 Although murine models have demonstrated that NFAT-regulated IL-12 expression in tumor-specific T cells resulted in enhanced tumor immunity and reduced toxicity, recent clinical trials in melanoma showed that patient T cells engineered with the same NFAT-IL-12 construct caused unacceptable toxicity that required termination of the trial.15
The purpose of this study was to explore whether the Tet-On system could be used to generate antigen-specific T cells with tightly controlled IL-12 expression.16 We demonstrate that adoptive transfer of T cells with Tet-regulated IL-12 is safe in the absence of doxycycline induction, while ‘leaked’ expression by T cells with NFAT-regulated IL-12 expression caused lethal toxicity. Transient induction of IL-12 was sufficient to reprogram the tumor micro-environment, boost the numbers of tumor-infiltrating T cells with reduced expression of PD1, which resulted in improved tumor protection.
Results
Genetic engineering using TCR and IL-12 gene constructs
In order to test the most effective way of combining TCR gene transfer with regulated IL-12 expression, we generated several retroviral constructs containing: i) TCR alpha/beta chains with truncated CD19 as marker for transduction; ii) constitutively expressed IL-12 with GFP as marker; iii) NFAT-regulated IL-12 with GFP as marker; iv) and IL-12 regulated by the Tet transactivator that required the presence of doxycycline (Dox) to activate expression from the tet-O7 promoter (Figure 1(a)). In this vector the Q8 marker (truncated CD34) served to identify transduced T cells and GFP served as marker for induced IL-12 expression.
Figure 1.
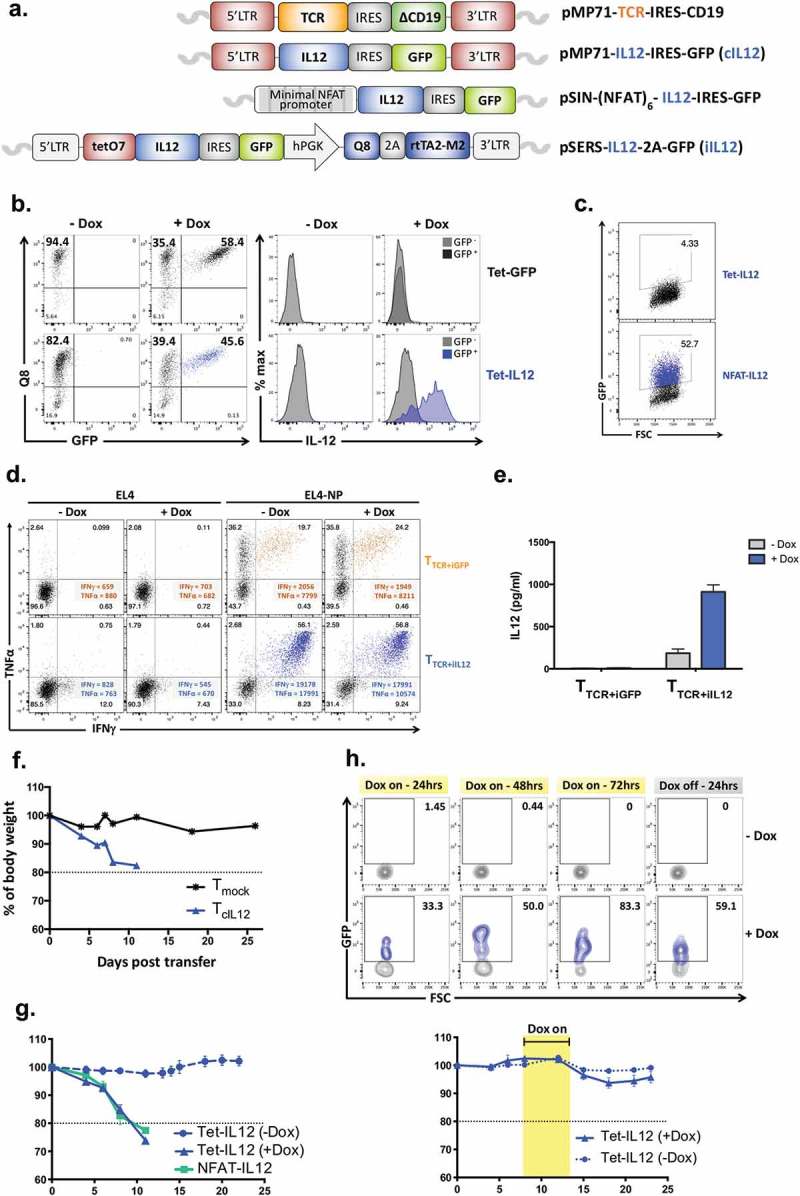
Design of IL-12 and TCR vectors and in vitro validation.
(a) Diagrams representing the molecular structure of the retroviral vectors. IRES; internal ribosome entry site, LTR; long terminal repeat, GFP; green fluorescent protein, NFAT; nuclear factor activated T cells. (b) Anti-CD3/CD28-activated splenocytes were transduced with the Tet-IL-12 construct (Tet-IL-12) or vector control (VC) construct containing GFP only (Tet-GFP), and treated with Dox (1 µg/ml) overnight or left untreated. Representative flow cytometry plots showing the expression of Q8 and GFP in transduced T cells demonstrating the transduction efficacy and the level of induction in the presence and absence of Dox. Representative histogram overlay showing intracellular IL-12 staining in GFP-positive (induced) and GFP-negative (non-induced) cells after 4hrs treatment with BFA. The experiments were done at least 3 times with similar results. (c) Anti-CD3/CD28-activated splenocytes were transduced with NFAT-IL-12 construct or mock-transduced and analyzed by flow cytometry for GFP expression the following day. (d) Representative flow cytometry plots depicting intracellular IFNγ and TNFα staining of T cells transduced with NP-specific F5-TCR and either Tet-IL-12 (TTCR+iIL-12) or Tet-GFP vector control (TTCR+iGFP), and stimulated with EL4 (control) or EL4-NP tumor cells expressing the cognate antigen for 4hrs in the presence and absence of Dox. Dot plots show live-gated TCR-expressing cells (CD19+). Data shown represents at least 3 independent experiments. (e) Measurements of IL-12 secretion in culture supernatant of transduced T cells by enzyme-linked immunosorbent assay (ELISA). Graph shows mean ± SEM of duplicate values from two experiments. (f) Mean of body weight measurements over time post transfer of 5 × 105 TcIL-12 or Tmock transduced cells into sublethally irradiated (4Gy) recipient mice; baseline is 100%. n = 3 mice per group. (g)Mean of body weight measurements over time of mice receiving 5 × 105 Tet-IL-12 or NFAT-IL-12 transduced T cells. Mice received Tet-IL-12 transduced T cells were split into two cohorts: one received Dox (2mg/ml) in drinking water (+ Dox) and the other cohort left untreated (-Dox). n = 5 mice per group. (h) Kinetics of transient IL-12 induction in vivo. C57BL/6 mice (Thy1.2+) were sublethally irradiated (4Gy) and injected intravenously with 5 × 105 Tet-IL-12 transduced T cells (Thy1.1+). On day 4 post T cell transfer, mice were split into two groups, one group received Dox-containing water (2mg/ml) for 3 consecutive days and the other group left untreated. Blood samples were obtained at 24hrs, 48hrs and 72hrs following Dox administration, and 24hrs following Dox withdrawal. Representative flow cytometry plots showing the levels of GFP expression. Cells were pre-gated on PI- singlet Thy1.1+ lymphocytes. n = 4 mice (-Dox); n = 6 mice (+ Dox) (top). Mean of body weight measurements over time showing lack of toxicity with temporal Dox induction post transfer of 5 × 105 Tet-IL-12 transduced cells into sublethally irradiated (4Gy) recipient mice. Mice received Tet-IL-12 transduced T cells were split into two cohorts: one received Dox (2mg/ml) in drinking water (+ Dox) for 3 days and the other cohort left untreated (-Dox). Graph shows mean ± SEM. n = 5 mice per group (bottom).
In vitro analysis of engineered T cells
In a first set of validation experiments primary mouse T cells were transduced with the Tet-IL-12 construct, or with an identical GFP vector control (VC) construct in which IL-12 was deleted. In the absence of Dox, staining with anti-CD34 (Q8) antibodies revealed that both vectors transduced more than 80% of T cells (Figure 1(b)). When Dox was added to the transduced primary T cells, most but not all Q8-positive cells started to express high levels of GFP. Intracellular IL-12 staining was used to demonstrate that all GFP-positive cells transduced with the Tet-IL-12 vector also expressed IL-12, while all GFP-negative cells were negative for IL-12. This indicated that GFP was a reliable marker to identify IL-12 producing cells. The control of expression by Dox was effective as no intracellular IL-12 was detectable when transduced cells were not exposed to Dox (Figure 1(b)). The transduction of primary mouse T cells with the NFAT-IL-12-GFP construct (Figure 1(a)) revealed that a large proportion of transduced cells expressed GFP in the absence of TCR stimulation (Figure 1(c)). As expected, the GFP-positive cells also expressed IL-12 as determined by intracellular cytokine staining (not shown). Together the data indicated that freshly transduced mouse T cells displayed robust control of GFP/IL-12 expression using the Tet regulation system, but not the NFAT system.
In the next set of experiments, we tested how Dox-induced IL-12 expression affected the antigen-specific response of TCR transduced primary T cells. C57BL/6 T cells were transduced with the F5-TCR specific for an H2-Db-presented peptide of the influenza nucleoprotein (NP). The F5-TCR vector was transferred into T cells together with the vector containing Tet-IL-12 (TTCR+iIL-12) or the control vector lacking IL-12 (TTCR+iGFP). Transduced T cells were then stimulated in the absence or presence of Dox with EL4 control cells or EL4-NP expressing the TCR-recognized target antigen. Figure 1(d) shows that more than 35% of T cells transduced with the F5-TCR and the vector control construct produced TNFα when stimulated with EL4-NP, and approximately 20% of T cells produced TNFα as well as IFNγ. As expected, the pattern of cytokine production was very similar in the absence and presence of Dox. However, the pattern of cytokine production was markedly different for T cells transduced with the F5-TCR and the Tet-IL-12 vector. All T cells responding to the NP antigen produced high levels of IFNγ and TNFα. The Tet-IL-12 vector increased the level of antigen-specific IFNγ production (measured by MFI) by nearly 10-fold compared to T cells transduced with TCR and GFP control vector (Figure 1(d)). This is consistent with the Th1-polarising effects of IL-12.17 However, in the absence of Dox we observed a similar enhancement of IFNγ production, suggesting leaked IL-12 expression despite the lack of detectable IL-12 by intracellular staining (Figure 1(b)). We therefore tested culture supernatant of transduced primary T cells for detachable IL-12 secreted over a 24 h period, which revealed low level of IL-12 in the absence of Dox which was strongly increase in the presence of Dox (Figure 1(e)).
In vivo safety assessment
The next set of experiments explored the in vivo safety profile of engineered T cells expressing IL-12. Primary T cells from C57BL/6 mice were transduced with retroviral constructs expressing IL-12 constitutively or controlled by the Tet-On or by NFAT regulatory elements. In order to mimic an adoptive cell therapy setting, non-myeloablative conditioning was used prior to adoptive transfer of 5 × 105 transduced T cells into recipient mice. T cells expressing IL-12 constitutively caused acute weight loss of more than 20%, at which point the animals had to be sacrificed according to UK Home Office regulations (Figure 1(f)). The adoptive transfer of 5 × 105 T cells containing the Tet-IL-12 construct caused similar acute weight loss in animals receiving Dox, but not in control animals that were not treated with Dox (Figure 1(g)). This indicated that the ‘leaked’ IL-12 expression detected in vitro (Figure 1(e)) did not cause IL-12 driven weight loss in vivo. In contrast, adoptive transfer of 5 × 105 T cells containing the NFAT-IL-12 construct caused acute weight loss in the absence of TCR stimulation (Figure 1(g)). Finally, we tested whether short-term IL-12 induction was associated with in vivo toxicity. On day 4 after adoptive transfer of Tet-IL-12 T cells animals received Dox for 3 consecutive days, and the analysis of blood samples indicated that at day 3 after Dox administration 83% of transferred Thy1.1 T cells expressed GFP, followed by a rapid decline 24 h after Dox discontinuation (Figure 1(h)). No GFP expression was detectable in transferred Thy1.1 T cells from mice that did not receive Dox (Figure 1(h), top panels). Importantly, temporal IL-12 expression did not result in detectable weight loss (Figure 1(h), bottom). Together, these experiments indicated that Tet-regulated IL-12 expression was safe in vivo, and that transient administration of Dox efficiently induced IL-12 expression in the majority of engineered T cells but was insufficient to cause toxicity.
Analysis of tumor infiltrating T cells
To explore the role of IL-12 in tumor immunity, C57BL/6 mice with established B16F10 tumors were conditioned with 4Gy total body irradiation followed by adoptive T cell transfer. T cells were engineered to express the TRP2-TCR (specific for tyrosinase-related-protein) or Tet-IL-12 or TRP2-TCR as well as Tet-IL-12. Due to the observed fatal toxicity, vectors expressing constitutive IL-12 and NFAT-IL-12 were not tested in the tumor setting. The “non-toxic” 3-day Dox administration was used to assess the effect of temporal IL-12 induction on tumor immunity (Figure 2(a)). Five days after adoptive cell therapy the numbers and phenotype of T cells and myeloid cells in the B16F10 tumors were analyzed.
Figure 2.
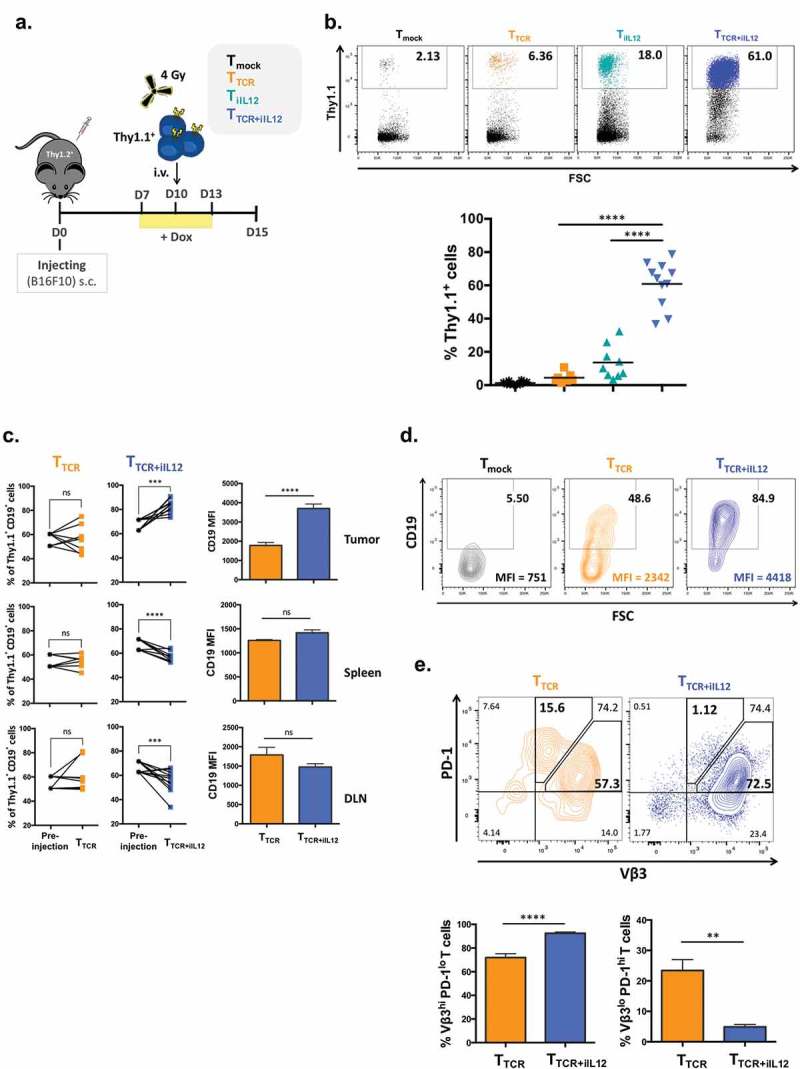
Temporal induction of IL-12 in TCR-engineered T cells increases the numbers of T cells in the tumor and prevents PD-1 upregulation.
(a) Experimental setup. C57BL/6 female mice (Thy1.2+) were inoculated subcutaneously with 5 × 105 B16 melanoma cells. 10 days later, mice were sublethally irradiated with 4Gy TBI 3-4hrs prior receiving intravenous injection of 2 × 106 T cells (Thy1.1+) that were transduced with TRP2-TCR (TTCR), Tet-IL-12 (TiIL-12), TRP2-TCR + Tet-IL-12 (TTCR+iIL-12), or mock-transduced T cells (Tmock). All mice received Dox (2mg/ml) in drinking water 2–3 days prior receiving T cell infusion and kept on Dox water for another 3 days. Tumors, spleens and lymph nodes were harvested on day 5 after the adoptive transfer and analyzed by flow cytometry. (b) Representative dot plots showing the percentage of transferred cells (Thy1.1+) (top) and pooled summary data (bottom). Cells were pre-gated on live-singlet lymphocytes. Symbols represent individual mice and bars indicate group averages. P values: TTCR versus TTCR+iIL-12 transduced cells and TiIL-12 versus TTCR+iIL-12 transduced cells; p < 0.0001. (c) Relative accumulation of TCR-expressing cells (CD19+) in tumor, spleen and draining lymph nodes (DLN) 5 days post T cell transfer. Cells were pre-gated on live-singlet transferred Thy1.1+ T cells. Data shown are cumulative results from at least two independent experiments. P values for the frequency of TCR+ cells: TTCR versus pre-injection in the tumor, spleen and DLN; p > 0.05, TTCR+iIL-12 versus pre-injection in the tumor; p = 0.0010, spleen; p = 0.0020 and DLN; p = 0.0049. P values for CD19 MFI: TTCR versus TTCR+iIL-12 in the tumor; p < 0.0001, spleen and DLN; p > 0.05. Symbols represent individual mice (TTCR, n = 8 and TTCR+iIL-12, n = 11). (d) Representative flow cytometry plots depicting the percentage of TCR-expressing cells (CD19+) and mean fluorescence intensity (MFI) of CD19 within the adoptively transferred Thy1.1+ T cells. Cells were pre-gated on live-singlet lymphocytes. (e) Representative flow cytometry plots depicting PD-1 staining profile of TCR-expressing cells (Vβ3+) in the tumor which divided them into two populations: Vβ3hi PD-1lo and Vβ3lo PD-1hi T cells (top) and bar charts representing summary data pooled from two independent experiments (bottom). P values: percentage of Vβ3hi PD-1lo T cells in TTCR versus TTCR+iIL-12; p < 0.0001, percentage of Vβ3lo PD-1hi T cells in TTCR versus TTCR+iIL-12; p = 0.0011.
The TRP2-TCR was inserted into our retroviral vector that also contained the truncated CD19 as a marker of transduction efficacy (Figure 1(a)). When primary mouse T cells were transduced with this construct, the level of TRP2-TCR expression correlated with the level of CD19 expression (suppl Fig 1A). Double transduction with the TCR and the Tet-IL-12 vectors generated a large number of T cells expressing both CD19/TCR and the Q8/CD34 marker (suppl Fig 1B). For the in vivo experiments 2 × 106 T cells expressing the TRP2-TCR only, or 2 × 106 cells expressing both TCR plus the Q8/CD34 were adoptively transferred into B16F10 tumor bearing mice. T cells engineered with only the TRP2-TCR showed modest enrichment in the tumor compared to mock transduced T cells (Figure 2(b)). However, dramatically increased tumor infiltration was observed after adoptive transfer of T cells engineered with the TRP2-TCR and Tet-IL-12. While Tet-IL-12 alone also increased T cell frequency, the combination of melanoma-specificity and induced IL-12 expression was most potent in boosting the frequency of adoptively transferred T cells in the tumor (Figure 2(b)). Figure 2(c) demonstrated that IL-12 promoted the selective accumulation of transduced CD19-positive T cells in the tumor, which was associated with a reduction in the frequency of the CD19-positive T cells in spleen and lymph nodes. This suggested that selective recruitment of TRP2-specific T cells from the periphery into the tumor was one factor by which IL-12 increased the frequency of antigen-specific T cells in the tumor microenvironment. In addition to increasing T cell numbers, IL-12 also mediated enhanced expression level of CD19/TCR in tumor-resident T cells compared to T cells found in the spleen and lymph nodes (Figure 2(c,d)). In contrast, TRP2-TCR engineered T cells that were unable to produce IL-12 displayed a phenotype that was characterized by reduced TCR levels and elevated expression of the PD1 marker (Figure 2(e)). The frequency of TCR-low/PD1-high T cells was high when T cells were unable to produce IL-12, and was substantially reduced by the temporal induction of IL-12 (Figure 2(e)). Correspondingly, IL-12 increased the frequency of TCR-high/PD1-low T cells in the tumor micro-environment. Together, these data indicated that transient expression of IL-12 in the first 3 days after adoptive cell therapy increased the accumulation of T cells expressing high levels of TCR and low levels of PD1 in the tumor.
Analysis of tumor infiltrating myeloid cells
Tumors of mice treated as described above with T cells transduced with TCR or Tet-IL-12 or TCR plus Tet-IL-12 were analyzed for the presence of dendritic cells, macrophages and CD11bpos/Gr1low myeloid suppressor cells using the staining panels shown in Figure 3(a). As described previously, IL-12 reduced the relative frequency and absolute numbers of CD11b+/Gr1low myeloid suppressor cells (Figure 3(b)). However, we also observed an IL-12 dependent increase in CD11b−/Gr1+ cells. Further analysis of this population of cells showed B220 expression combined with low/intermediate levels of CD11c and MHC class II (I-Ab), a phenotype that is characteristic of murine plasmacytoid DC (Figure 3(c)). The frequency of these plasmacytoid DC in the tumor was substantially increased when T cells were engineered to express the TRP2-TCR and Tet-IL-12 (Figure 3(c)).
Figure 3.
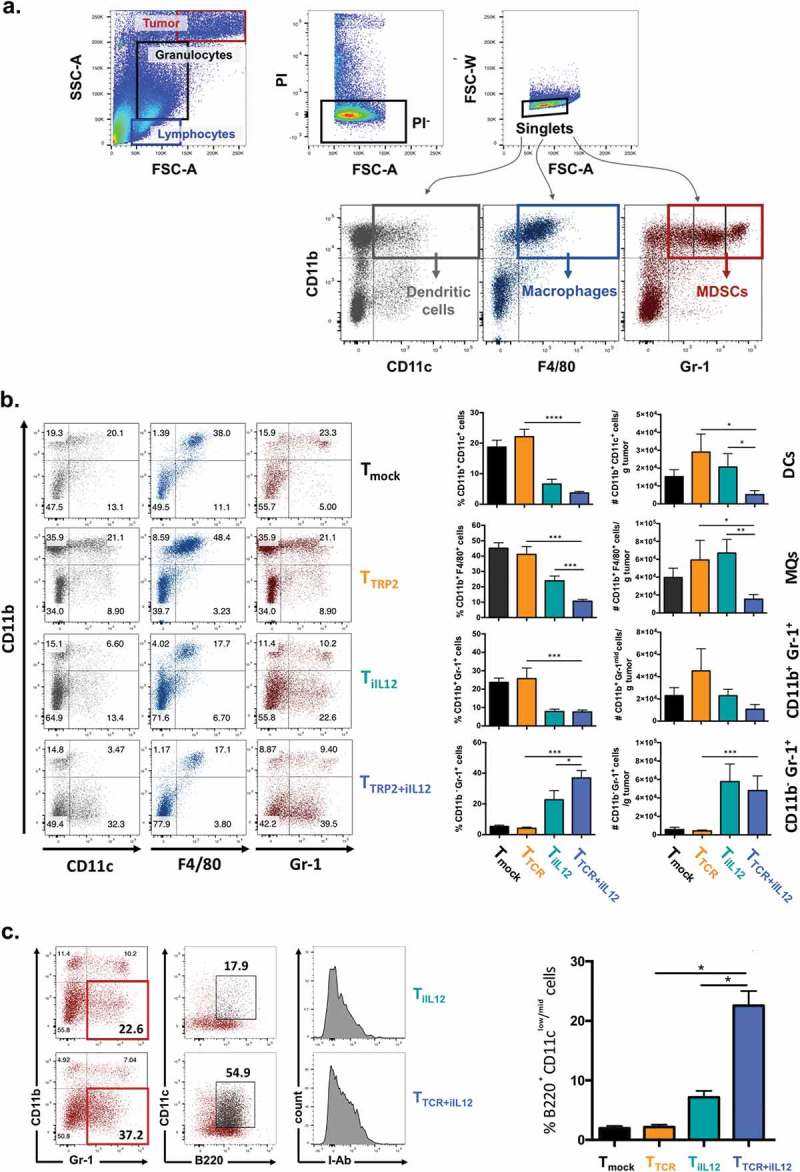
Transient IL-12 induction changes the myeloid cell composition of the tumor microenvironment.
C57BL/6 female mice bearing subcutaneous melanoma for 10 days were sublethally irradiated with 4Gy TBI prior to adoptive T cell transfer of Tmock, TTCR, TiIL-12 or TTCR+iIL-12 transduced T cells. All mice received Dox-containing water (2mg/ml) 2–3 days prior receiving T cell infusion and kept on Dox water for another 3 days. Tumor infiltrating cells were harvested on day 5 post T cell transfer and analyzed by flow cytometry. (a) Gating strategy. Using forward scatter (FSC) and side scatter (SSC), tumor cells (FSC/SSC high), granulocytes (SSC intermediate) and lymphocytes (FSC/SSC low) were identified. PI− singlet granulocytes were assessed for their surface expression of CD11b, CD11c, F4/80 and Gr-1 to identify different myeloid-derived cell populations including: CD11b+CD11c+; dendritic cells (DCs), CD11b+F4/80+; macrophages (MQs) and CD11b+Gr-1+ myeloid-derived suppressor cells (MDSCs), including monocytic MDSCs (Gr-1mid; MDSC-M) and granulocytic MDSCs (Gr-1hi; MDSC-G). (b) Representative dot plots showing tumor infiltrating myeloid-derived cells 5 days after the adoptive T cell transfer (left) and pooled summary data (right). Cells were pre-gated on live-singlet granulocytes. Graphs are showing mean ± SEM. P values: frequency of DCs in mice treated with TTCR versus TTCR+iIL-12; p = 0.0001, frequency of MQs in mice treated with TTCR versus TTCR+iIL-12; p = 0.0001, TTCR+iIL-12 versus TiIL-12; p = 0.0008, frequency of MDSCs in mice treated with TTCR versus TTCR+iIL-12; p = 0.0003, frequency of CD11b−Gr-1+ in mice treated with TTCR versus TTCR+iIL-12; p = 0.0001, TTCR+iIL-12 versus TiIL-12; p = 0.0432, total number of DCs in mice treated with TTCR versus TTCR+iIL-12; p = 0.0195, TTCR+iIL-12 versus TiIL-12; p = 0.0249, total number of MQs in mice treated with TTCR versus TTCR+iIL-12; p = 0.0441, TTCR+iIL-12 versus TiIL-12; p = 0.0091, total number of CD11b−Gr-1+ in mice treated with TTCR versus TTCR+iIL-12; p = 0.0031. Data shown are cumulative results of two independent experiments. Number of mice per group: Tmock, n = 10; TTCR, n = 7; TiIL-12, n = 8; TTCR+iIL-12, n = 11. (c) Identification of pDC subset infiltrating the tumor at day 5 following T cell transfer. CD11b−Gr-1+ population were analyzed for the surface expression of CD11c, B220 and MHC class II molecules (I-Ab). Representative plots showing tumor-infiltrating pDCs in mice treated with TiIL-12 and TTCR+iIL-12 cells (left) and pooled summary data for the frequency of B220+CD11clo/mid cells pre-gated on live-singlet CD11b−Gr-1+ granulocytes in mice treated with Tmock, TTCR, TiIL-12 or TTCR+iIL-12 transduced T cells (right). Graphs are showing mean ± SEM. P values: frequency of pDCs in mice treated with TTCR versus TTCR+iIL-12 and TiIL-12 versus TTCR+iIL-12; p = 0.0159, total number of pDCs in mice treated with TTCR versus TTCR+iIL-12; p = 0.015.
Tumor inhibition by IL-12
Mice with establish B16F10 tumors were conditioned and then treated by adoptive transfer of T cells expressing TRP2-TCR or TRP2-TCR plus Tet-IL-12. T cells engineered with only Tet-IL-12 were used to reveal non-antigen-specific effects of IL-12. Dox-induction of IL-12 was either done once in the first 3 days, or twice in the first 3 days and again on day 10–13 after T cell transfer (Figure 4(a)). Figure 4(b) shows that T cells expressing TCR were unable to inhibit the growth of B16F10 tumors. T cells engineered with Tet-IL-12 mediated a small reduction in tumor growth, but the most pronounced inhibition was seen with T cells expressing TCR plus Tet-IL-12. The more effective growth inhibition correlated with the improved survival of mice treated with TCR+ Tet-IL-12 T cells compared to T cell expressing only TCR or only Tet-IL-12 (Figure 4(c)). However, despite improved protection, the T cell mediated immunity was insufficient to prevent tumor progression. We tested whether the B16F10 tumors that grew in mice treated with TCR+ Tet-IL-12 T cells were escape variants that had lost the TCR-recognized target antigen. To address this, tumors of T cell treated mice were reisolated and tested for their ability to stimulate effector function of TRP-TCR transduced T cells. Figure 4(d) demonstrates that all reisolated tumors were not antigen-escape variants and stimulated TCR transduced T cells as effectively as B16F10 cells that were cultured in vitro for the same time period as tumors that grew in vivo.
Figure 4.
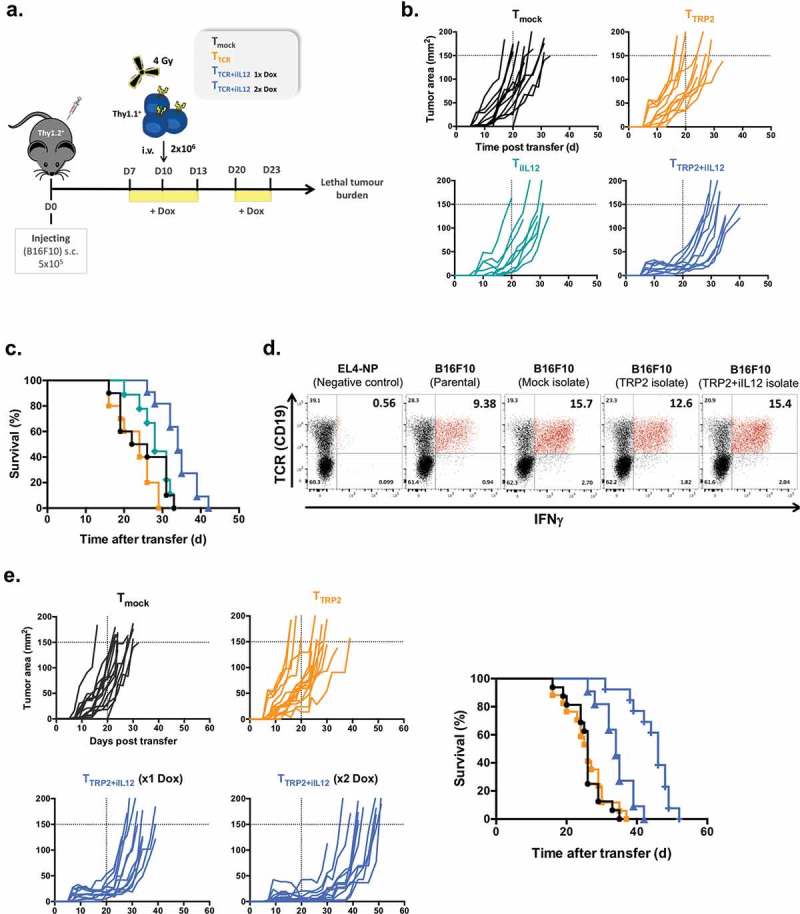
IL-12 overexpression in TCR-engineered T cells delay the development of lethal tumor burden in B16 melanoma-bearing mice.
(a) Experimental setup. C57BL/6 (Thy1.2+) female mice bearing 10 days B16F10 melanoma were sublethally irradiated and injected intravenously with 2 × 106 transduced T cells (Thy1.1+) including TTCR, TiIL-12, TTCR+iIL-12 or Tmock cells. All mice received Dox (2mg/ml) in drinking water 2–3 days before T cell transfer and kept on Dox for another 3 days. In another set of experiments, mice that were treated with TTCR+iIL-12 cells were split into two cohorts: one group had single induction of IL-12 (at d10) and the other group had double induction of IL-12 (at d10 and d20 post tumor injection). (b) Tumor size over time post T cell transfer until mice reached lethal tumor burden. (c) Kaplan-Meyer survival plots of mice treated with TTCR, TiIL-12, TTCR+iIL-12 or Tmock cells. P values: Tmock versus TTCR or TiIL-12; p > 0.05 (ns), TTCR versus TTCR+iIL-12; p < 0.0001. Data shown in B and C are cumulative results of at least two independent experiments. Number of mice per group: Tmock, n = 10; TTCR, n = 10; TiIL-12, n = 9; TTCR+iIL-12, n = 11. (d) B16F10 tumors were re-isolated from mice treated with TTCR, TTCR+iIL-12 or Tmock cells at the time point when mice reached lethal tumor burden. The tumor cells were pre-treated overnight with IFNγ-containing medium and co-cultured with TRP2-TCR-transdcued T cells (CD19; TCR reporter marker) for 5hrs in the presence of BFA. Representative dot plots showing antigen-specific production of IFNγ by TCR-positive cells. Parental B16F10 melanoma cells and EL4-NP tumor cells were used as a positive and negative controls, respectively. Cells were pre-gated on live-singlet lymphocytes. (e) B16F10 tumor growth kinetics until mice reached lethal tumor burden (left) and Kaplan-Meyer survival plot of mice received TTCR, TTCR+iIL-12 (1x Dox; single induction), TTCR+iIL-12 (2x Dox; double induction) or Tmock transduced T cells. P values: TTCR versus TTCR+iIL-12 (1x Dox); p = 0.0002, TTCR versus TTCR+iIL-12 (2x Dox); p < 0.0001, TTCR+iIL-12 (1x Dox) versus TTCR+iIL-12 (2x Dox); p = 0.0004 (right). Data shown represent pooled data from three independent experiments. Number of mice per group: Tmock, n = 15; TTCR, n = 17; TTCR+iIL-12 (1x Dox), n = 12; TTCR+iIL-12 (2x Dox), n = 13.
In a final set of experiments, we tested whether repeated IL-12 induction could further enhance the level of tumor protection. Figure 4(e) demonstrates that repeated IL-12 induction at day 10–13 after adoptive cell transfer was more effective in inhibiting tumor growth than a single cycle of IL-12 induction during the initial 3 days following T cell transfer. Similarly, two cycles of IL-12 induction resulted in substantial survival benefits by nearly doubling the survival time compared with mice treated with engineered T cells expressing only the TRP2-TCR (Figure 4(e)). At the time when mice reached lethal tumor burden, we analyzed the number and phenotype of adoptively transferred T cells. This revealed that the T cell frequency in the tumors, spleen and bone marrow was similar in mice treated with TCR-only, or TCR+ Tet-IL-12 T cells (suppl. Fig 2), although the average time from adoptive transfer to lethal tumor burden was 25 days for the TCR-only group, and 35 or 45 days for the TCR+ Tet-IL-12 group (Figure 4(c)). Furthermore, the CD62L, CD127, PD-1 and annexinV expression profiles of tumor infiltrating T cells were similar in all groups (suppl. Fig 3C and E). In contrast, proliferation of tumor infiltrating T cells, as assessed by Ki67 staining, decreased over time from 40% at day 25 to 20% at day 45 (suppl. Fig 3D). Together, this data suggested that tumor infiltrating T cells developed over time a partial hypo-responsiveness that was not prevented by the temporal induction of IL-12.
Discussion
Although IL-12 is a potent immune stimulatory cytokine, its clinical application to boost immunity in cancer patients has been limited by severe side effects including the death of two patients in early clinical trials.18 Subsequent trials in patients with head and neck cancer involved the injection of recombinant IL-12 at the tumor site, which was associated with high level of systemic IFNγ production and substantial toxicity necessitating the termination of the clinical trial.11 Similarly, toxicity was seen in melanoma patients treated with autologous tumor infiltrating lymphocytes (TIL) that were genetically engineered to express IL-12 under the control of NFAT regulatory elements.15 In this melanoma trial, T cell doses that mediated tumor regression also induced severe toxicity in 5 out of 16 treated patients, possibly caused by high level systemic IL-12 and IFNγ that was detectable in the serum of patients. This trial experience has indicated that NFAT-regulated IL-12 expression is not sufficiently safe to achieve tumor immunity without substantial toxicity in patients.
This was somewhat surprising, as previous studies in murine systems did not report systemic toxicity with NFAT regulated IL-12 expression. In one study, human T cells engineered to express NFAT-regulated human IL-12 and a CAR specific for human carcinco-embryonic antigen (CEA) were tested in immune-deficient mice bearing human CEA-positive tumors.9 The lack of detectable toxicity in these studies may be related to the relative poor expansion and survival of human T cells in immune-deficient mice, and to a species barrier impairing the biological activity of human IL-12 in mice. However, similar to our work reported here, Zhang and colleagues have used the B16 melanoma model for adoptive therapy with TCR-transgenic T cells engineered to express IL-12 in an NFAT-regulated fashion.12 In these studies, the adoptive transfer of up to 3 million TCR transgenic T cells specific for the pmel melanoma antigen provided protection against B16 tumors in the absence of detectable toxicity. In contrast, we observed lethal toxicity when 2 million T cells containing the NFAT IL-12 construct were transferred into conditioned mice. Experimental differences might contribute to the observed differences in the outcome. In our studies 2 million T cells that were successfully transduced with the NFAT IL-12 construct were adoptively transferred into mice, compared to a total dose of 3 million pmel T cells where the transduction efficiency with NFAT IL-12 was not reported. It is therefore possible that the total number of NFAT IL-12 engineered T cells in our study was greater than the maximal dose reported by Zhang et al. In addition, it is conceivable that the ‘natural’ TCR repertoire of bulk T cells used in our studies may contain self-reactive T cells that are absent in the repertoire of pmel-TCR transgenic mice. In this case, the recognition of self-antigens might trigger increased IL-12 release by NFAT regulated bulk T cells but not by pmel-TCR transgenic T cells. Irrespective of the differences between the two murine studies, the clinical experience with NFAT IL-12 transduced TIL in melanoma patients is in line with the substantial toxicity risk observed in this report.15 Similarly a recent study in HLA-A0201 transgenic mice bearing HLA-transfected B16 tumors showed that adoptive transfer of T cells with NFAT regulated IL-12 expression resulted in severe toxicity in recipient mice.19
We found that Dox is efficient in regulating IL-12 expression in vivo. Although the in vitro studies revealed low IL-12 leakage in the absence of Dox, this did not cause toxicity in vivo, which was strictly dependent on the continued administration of Dox after T cell transfer. Adding Dox to the drinking water efficiently induced IL-12 production within 24 hours and returned to baseline 24–48 hours after removal of Dox from the water. Surprisingly, administration of Dox in the first 3 days after T cell transfer was sufficient to increase T cell numbers in the tumors, reduce expression of the PD1 exhaustion marker and change the myeloid cell composition of the tumor microenvironment, including an increase in the numbers of plasmacytoid DCs. We did not see an IL-12 mediated increase in macrophages in the tumor, which was seen when NFAT IL-12 human T cells were tested in immune-deficient mice.9 In line with the report by Kerkar et al. using murine T cells expressing IL-12 constitutively, we also found a reduction in the number of tumor resident macrophages and myeloid derived suppressor cells after transient IL-12 induction in the first 3 days after T cell transfer.10 The transient expression of IL-12 may not only reduce the risk of toxicity but also avoid the negative effects of IL-12 on T cells. For example, studies of human T cells have shown that continued exposure to high level IL-12 can induce exhaustion markers and reduce T cell effector function.20 Similarly, in vivo murine experiments indicated that low dose recombinant IL-12 was able to enhance viral immunity, while high doses were ineffective.21 In the recent clinical trial with NFAT IL-12 engineered TIL, the authors observed poor persistence of the transferred T cells and speculated that this might be due to the inhibition of T cell proliferation by high level IL-12 production. It is therefore possible that transient IL-12 expression may avoid some of the negative effects that are associated with continued exposure of T cells to IL-12. In our experiments we found that transient IL-12 induction in engineered T cells increased the numbers of T cells in the tumor and also prevented the up-regulation of the PD1 exhaustion marker. We do not fully understand the mechanisms by with IL-12 prevented/reversed the expression of PD1. A recent human study has shown that IL-12 can function as 3rd signal to restore functional activity of exhausted human T cells.22 In this study, improved T cell function was associated with IL-12 mediated down-modulation of PD1, which is similar to the IL-12 effect observed in our study.
Our data suggest that the IL-12 driven accumulation of T cells in the tumor mirco-environment was partly due to selective recruitment of TCR-positive T cells from spleen and lymph nodes into the tumor (Figure 2(c)), and partly to an increase in proliferation of tumor resident T cells as determined by increased staining for Ki67 (data not shown). It is likely that the increased numbers of plasmacytoid DC (Figure 3(c)) improved T cell stimulation by cross presentation of B16 melanoma antigens to TRP2-postive T cells, although it is difficult to experimentally dissect the precise role of this DC subset in our model.
The potential immunogenicity of the bacterially-derived Tet trans-activator protein could limit the survival of engineered T cells in vivo. A previous study has identified epitopes in the trans-activator protein that can be recognized by cytotoxic T cells.23 In this study vectors were injected into muscle tissue to achieve expression of the trans-activator protein. The induction of cytotoxic T cell responses against the identified epitopes was dependent upon the extent of local inflammation associated with different vector types, and was not seen when ‘weakly immunogenic’ AAV vectors were used.23 In our study no vectors were used in vivo, and it is possible that the expression of the trans-activator protein in ex-vivo engineered autologous T cells may not be sufficiently immunogenic to stimulate cytotoxic T cell responses in vivo.
The Dox-regulated system described here offers the option to repeat IL-12 expression in vivo at defined time intervals. We found that two cycles of IL-12 induction were superior to one cycle in providing tumor protection against B16 melanoma. A further advantage of the system described here is that doxycycline and other tetracylins antibiotics have an extensive safety track record in patients, facilitating their clinical use in trials to assess the feasibility, safety and efficacy of the Dox-regulated IL-12 expression in cancer patients.
Materials and methods
Mice
Female C57BL/6 and C57BL/6 (Thy1.1+) mice used in this study were bred in-house in the animal facility, a specific pathogen-free facility, at University College London-based at the Royal Free Hospital or obtained from Charles Rivers Laboratories. All animal experiments were performed in accordance with the United Kingdom Home office regulations.
Cell lines
Phoenix Ecotropic (ECO), an adherent packaging cell lines (Nolan Laboratory, Stanford, CS, USA), were used to produce viral supernatants containing retrovirus particles for transduction of mouse T cells. The ECO cells were maintained in IMDM supplemented with 10% heat-inactivated fetal calf serum, 1% of 2 mM L-glutamine and 1% of 100U/ml Penicillin/Streptomycin.
EL4 is a murine lymphoma cell line (H-2b) and EL4-NP is a variant stably expressing the influenza A derived nucleoprotein (NP) peptide (a kind gift from Dr B. Stockinger, National Institute for Medical Research, London). B16F10 is a murine melanoma cell line derived from a C57BL/6 mouse developed melanoma. All tumor cell lines were maintained in RPMI supplemented with 10% heat-inactivated fetal calf serum, 1% of 2 mM L-glutamine and 1% of 100U/ml Penicillin/streptomycin.
Retroviral constructs
The retroviral F5-TCR and TRP2-TCR vectors (pMP71-TCRα-2A-TCRβ-IRES-CD19), which encodes the TCR α/β chains linked by 2A sequences, followed by an internal ribosome entry site (IRES) and truncated CD19 (ΔCD19) as reporter marker, were modified from pMX-TCRα-IRES-TCRβ (a kind gift from Prof T. Schumacher, Netherland Cancer Institute, Amsterdam). These vectors were modified to encode a truncated CD19 (ΔCD19) that includes the transmembrane and extracellular domains of mouse CD19 downstream of an internal ribosome entry site (IRES) sequence. The F5-TCR recognizes the influenza A virus nucleoprotein (NP366-379) peptide in the context of murine MHC class I (H-2Db). The TRP2-TCR recognizes the Tyrosinase related protein-2 (TRP2181-188) in the context of murine MHC class I (H-2Kb). Both TCRs were codon optimized and have an additional cysteine residue in the constant region. The retroviral vector expressing constitutive IL-12 (cIL-12), pMP71-IL-12-IRES-GFP, was generated by cloning mouse single chain IL-12 containing the p40 and p35 subunits of IL-12 linked by (Gly4Ser)3 flexible linker, followed by IRES and green fluorescent protein (GFP) into the pMP71 vector backbone. The IL-12p40-(Gly4Ser)3-IL-12p35 was obtained from the self-inactivating retroviral vector pSIN-(NFAT)6-IL-12-IRES-GFP under the control of nuclear factor activated T cells (NFAT)-responsive promoter containing six NFAT-binding motifs combined with a minimal IL-2 promoter (a kind gift from Dr T. Schumacher, The Netherlands Cancer Institute and from Dr H. Abken, University of Cologne).
The pSERS retroviral vectors, all-in-one Tet-On inducible system, used to regulate the transgene expression16 was modified to encode mouse single chain IL-12 linked to GFP via a ribosomal skipping P2A sequence (Tet-IL-12) or GFP alone (Tet-GFP), which is under the control of tetracyclin-responsive promoter containing repeats of tet operator sequences fused to a minimal promoter (Tet-O7). This vector also encodes the reverse tet-responsive transactivator (rtTA2-M2) which is fused to a minimal epitope called Q8 (a truncated human CD34 linked to a CD8 stalk) via a ribosomal skipping foot-and-mouth disease 2A sequence, and is constitutively expressed by the human phosphoglycerate kinase (hPGK) promoter.
Retroviral transduction of T cells
For retroviral production, the ECO packaging cells were transiently transfected with the retroviral vectors using FuGENE® HD transfection reagent (Promega), and viral supernatants were harvested 48 hours following transfection. For retroviral transduction, splenocytes from C57BL/6 female mice were harvested and activated with CD3/CD28 antibody coated beads (Dynabeads-Mouse T cell activator, Gibco). 24 hours later, activated splenocytes were transduced with the retroviral supernatants containing the indicated vectors or mock-transduced using RetroNectin (Takara-Bio-Otsu, Japan)-coated plates. After spinning the plate at 712g for 90 minutes at 32 C, cells were cultured in the presence of 20 U/ml IL-2 (Roche).
Flow cytometry
The following monoclonal antibodies (mAbs) were used: anti-murine Thy1.1 (H1S51), CD19 (ID3), CD11b (M1/70), F4/80 (BM8) supplied by eBiosciences; anti-murine Vβ3 (KJ25), PD-1 ‘CD279ʹ (J43), IL-12 (C15.6), IFNγ (XMG1.2), TNFα (MP6-XT22), CD11c (HL3), Gr-1 ‘Ly-6G and Ly-6C’ (RB6-8C5), B220 (RA3-6B2) and I-Ab (MHCII) (AF6-120.1) supplied by BD Pharmigen; and anti-human CD34 (QBEND/10) supplied by AbD Serotec. Single cell suspensions prepared from mouse tissues were initially FC-blocked with anti-mouse CD16/32 (eBiosciences). For intracellular cytokine staining, the BD Cytofix/Cytoperm™ kit (BD Biosciences) was used. To select for viable cells, propidium iodide (BD Pharmigen) was used, and alternatively, Fixable Viability Dye was used when cells were fixed for intracellular staining. Data were acquired using BD™ LSRII or BD LSRFortessa™ flow cytometers (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Cytokine release assay
Engineered T cells were stimulated and assessed 4–5 days following transduction. Transduced T cells were co-cultured with tumor cells either EL4 or EL4-NP (in a responder-to-target ratio of 1:2), and incubated in supplemented RPMI in a 24 well tissue culture plate. T cells were also stimulated (nonspecifically) with PMA (50ng/ml) and Ionomycin (1μg/ml) as a positive control for intracellular cytokine staining. Stimulated T cells were treated with 10μg/ml BFA (Sigma) at the beginning of the stimulation to block cytokine secretion and were incubated for 4-5hrs under standard tissue culture conditions. Following the incubation period, cells were washed and stained with mAbs.
Adoptive transfer and tumor challenge
C57BL/6 (Thy1.2+) female recipient mice at 8–12 weeks of age were injected subcutaneously (s.c.) with 5 × 105 B16 melanoma cells. After 10 days, mice were sub-lethally irradiated with 4 Gy total body irradiation (TBI) and received intravenous (i.v.) injection of 2 × 106 transduced T cells. Tumors were measured overtime at different time intervals using a digital caliper; tumor size was calculated using the following formula (a x b x 3/4, where a is the horizontal diameter and b is the vertical diameter of the tumor.
Statistical analysis
All statistical analysis was performed using GraphPad Prism software version 6.0 (GraphPad Software, USA). To calculate p values and test for significant differences as specified in the figure legends, a two-tailed Mann-Whitney test or two-tailed Wilcoxon matched-pairs signed rank test were utilized. The log-rank (Mantel Cox) test was utilized for comparison of survival curve. A significant difference between groups was indicated by a p value of ≤ 0.05
Acknowledgments
AA was supported by a grant from the King Abdulaziz University, Jeddah, Saudi Arabia. This work was also supported by the Bloodwise Program Grant 13004 to HJS.
Conflict-of-interest disclosure
HJS is scientific advisor and shareholder of Cell Medica. GB is an employee of and recipient of equity from Kite Pharma EU B.V.
Author contributions
AH, PV, and BS provided reagents and scientific advise. GB helped with the design of experiments and data analysis. AA designed and performed experiments, analysed data and wrote the paper. HJS designed experiments, analysed data and wrote the paper.
Supplemental data
Supplemental data for this article can be accessed here.
References
- 1.Barrett DM, Grupp SA, June CH.. Chimeric antigen receptor- and TCR-modified T cells enter main street and wall street. J Immunol. 2015;195:755–761. doi: 10.4049/jimmunol.1500751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morris EC, Stauss HJ. Optimizing T-cell receptor gene therapy for hematologic malignancies. Blood. 2016;127:3305–3311. doi: 10.1182/blood-2015-11-629071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riviere I, Sadelain M. Chimeric antigen receptors: a cell and gene therapy perspective. Mol Ther. 2017;25:1117–1124. doi: 10.1016/j.ymthe.2017.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in children and young adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, Sauter C, Wang Y, Santomasso B, Mead E, et al. Long-term follow-up of CD19 CAR therapy in acute Lymphoblastic Leukemia. N Engl J Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 9.Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 10.Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, Leonardi AJ, Morgan RA, Wang E, Marincola FM, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. 2011;121:4746–4757. doi: 10.1172/JCI58814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Herpen CM, van der Laak JA, de Vries IJ, van Krieken JH, de Wilde PC, Balvers MG, Adema GJ, De Mulder PHM. Intratumoral recombinant human interleukin-12 administration in head and neck squamous cell carcinoma patients modifies locoregional lymph node architecture and induces natural killer cell infiltration in the primary tumor. Clin Cancer Res. 2005;11:1899–1909. doi: 10.1158/1078-0432.CCR-04-1524. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, Rosenberg SA, Morgan RA. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19:751–759. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257:83–90. doi: 10.1111/imr.12125. [DOI] [PubMed] [Google Scholar]
- 14.Hooijberg E, Bakker AQ, Ruizendaal JJ, Spits H. NFAT-controlled expression of GFP permits visualization and isolation of antigen-stimulated primary human T cells. Blood. 2000;96:459–466. [PubMed] [Google Scholar]
- 15.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21:2278–2288. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinz N, Schambach A, Galla M, Maetzig T, Baum C, Loew R, Schiedlmeier B. Retroviral and transposon-based tet-regulated all-in-one vectors with reduced background expression and improved dynamic range. Hum Gene Ther. 2011;22:166–176. doi: 10.1089/hum.2010.099. [DOI] [PubMed] [Google Scholar]
- 17.Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13:722–728. doi: 10.1038/ni.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen J. IL-12 deaths: explanation and a puzzle. Science. 1995;270:908. [DOI] [PubMed] [Google Scholar]
- 19.Kunert A, Chmielewski M, Wijers R, Berrevoets C, Abken H, Debets R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology. 2017;7:e1378842. doi: 10.1080/2162402X.2017.1378842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, Witzig TE, Ansell SM. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest. 2012;122:1271–1282. doi: 10.1172/JCI59806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orange JS, Wolf SF, Biron CA. Effects of IL-12 on the response and susceptibility to experimental viral infections. J Immunol. 1994;152:1253–1264. [PubMed] [Google Scholar]
- 22.Schurich A, Lj P, Lubowiecki M, Singh HD, Gill US, Kennedy PT, Nastouli E, Tanwar S, Rosenberg W, Maini MK, et al. The third signal cytokine IL-12 rescues the anti-viral function of exhausted HBV-specific CD8 T cells. PLoS Pathog. 2013;9:e1003208. doi: 10.1371/journal.ppat.1003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ginhoux F, Turbant S, Gross DA, Poupiot J, Marais T, Lone Y, Lemonnier FA, Firat H, Perez N, Danos O, et al. HLA-A*0201-restricted cytolytic responses to the rtTA transactivator dominant and cryptic epitopes compromise transgene expression induced by the tetracycline on system. Mol Ther. 2004;10:279–289. doi: 10.1016/j.ymthe.2004.05.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.