

ABSTRACT
This study aimed to establish the number of expression-based molecular subclasses in cutaneous melanoma, identify their dominant biological pathways and evaluate their clinical relevance. To this end, consensus clustering was performed separately on two independent datasets (n = 405 and n = 473) composed of publicly available cutaneous melanoma expression profiles from previous studies. Four expression-based molecular subclasses were identified and labelled ‘Oxidative phosphorylation’, ‘Oestrogen response/p53-pathway’, ‘Immune’ and ‘Cell cycle’, based on their dominantly expressed biological pathways determined by gene set enrichment analysis. Multivariate survival analysis revealed shorter overall survival in the ‘Oxidative phosphorylation’ subclass compared to the other subclasses. This was validated in a third independent dataset (n = 214). Finally, in a pooled cohort of 76 patients treated with anti-PD-1 therapy a trend towards a difference in response rates between subclasses was observed (‘Immune’ subclass: 65% responders, ‘Oxidative Phosphorylation’ subclass: 60% responders, other subclasses: <50% responders). These findings support the stratification of cutaneous melanoma in four expression-based molecular subclasses.
Keywords: Cutaneous melanoma, gene expression, consensus clustering, pooled analysis, molecular classification, anti-PD-1 therapy
Introduction
Despite the introduction of new systemic therapies, around 40% of patients with advanced cutaneous melanoma still die within the first year after initiation of systemic therapy.1 Therefore, a need for novel treatment strategies remains. A promising approach to guide the development of novel therapeutic strategies is the stratification of tumours into gene expression-based subclasses.2 Jönssen et al. identified four subclasses that were associated with overall survival (OS) in 57 stage IV melanoma patients.3,4 In 333 mRNA-sequencing (mRNA-seq) samples from The Cancer Genome Atlas (TCGA), three subclasses were identified. These subclasses were univariately associated with post accession survival, defined as time from date of sample collection until death.5 Finally, single-cell RNA-seq of 1,246 melanoma cells obtained from 14 patients identified two subclasses.6 Generally, subclasses have been assigned a biological annotation based on a limited subset of genes and their prognostic value has been assessed in limited numbers of patients or without adjustment for validated prognostic factors.
A previous attempt by Lauss et al. to determine the similarity between subclasses showed only a limited overlap in genes (n = 34) between the genes used to define the molecular subclasses in TCGA (n = 1465) and Lund (n = 452).7 Sample classification between the TCGA and Lund schemes applied in four datasets showed a limited overlap. As described above, research in melanoma has provided conflicting results concerning the number and biological nature of such subclasses. Finally, the association between subclass assignment and response to anti-PD-1 therapy has not previously been assessed.
Our hypothesis was that with a large pooled analysis of multiple datasets the number of subclasses in cutaneous melanoma could be identified with more certainty. In addition, we hypothesized that this refined sub classification of melanomas would be related to tumour biology and that the subclasses would have potential prognostic and predictive value for the individual patient. Therefore, the aim of this study was to refine expression-based molecular subclasses in cutaneous melanoma and to identify their dominant biological pathways. To this end, we applied the same consensus clustering procedure from the ground up in two independent datasets. Next, we extensively characterized the identified subclasses at the individual gene and biological pathway level. Furthermore, we assessed their prognostic value, which we validated in a third independent dataset, and evaluated their association to response to anti-PD-1 therapy.
Results
Data acquisition
We extracted 405 microarray gene expression profiles (Affymetrix HG-U133A or HG-U133 Plus 2.0) and available clinical data of patients with cutaneous melanoma from the Gene Expression Omnibus (GEO) and labelled these as the GEO-dataset. Additionally, the mRNA-seq and clinical data from the cutaneous melanoma set from TCGA were collected, containing 473 samples representing 469 patients. This dataset was labelled the TCGA-dataset. A third independent dataset containing 214 samples (Illumina HumanHT-12) was obtained from GEO accession number GSE65904 and labelled the Cirenajwis-dataset. Finally, processed expression data from pre-treatment melanomas obtained from patients undergoing anti-PD-1, anti-CTLA-4 or anti-PD-1/anti-CTLA-4 combination therapy was obtained from GEO accession numbers GSE78220 (Hugo-dataset), GSE91061 (Riaz-dataset) and GSE115821 (Auslander-dataset).
The GEO-dataset contained more stage III/IV samples (~90%) compared to the TCGA-dataset (~41%) and fewer stage I/II samples (~5%) compared to the TCGA-dataset (~50%) (Table 1). In the GEO-dataset 14% of samples (n = 55) were obtained from primary tumours, 27% (n = 108) from (sub)cutaneous or in transit metastases and 47% (n = 191) from metastases in distant or regional lymph nodes, viscera, or an unknown location. In 13% of samples (n = 51) it was unknown whether the biopsy was taken from a primary tumour or metastatic lesion. In the TCGA-dataset 22% of samples (n = 102) were obtained from primary tumours, 47% (n = 222) from regional lymph node metastases, 21% (n = 99) from (sub)cutaneous or in-transit metastases and 9% (n = 43) from distant metastases in soft tissue, distant lymph nodes, viscera, bones, CNS or an unknown location. Of the biopsies taken from primary tumours, 69% (n = 70) were staged as stage I/II disease and of the biopsies taken from regional lymph node metastases 42% (n = 94) were staged as stage III disease. Of all samples in the TCGA-dataset, 28% (n = 130) were obtained within 30 days and 39% (n = 182) within 90 days after the time of diagnosis. However, it should be noted that 59% (n = 277) of the tumour biopsies in the TCGA-dataset were taken more than 90 days later than the moment of initial diagnosis and staging, and therefore not always optimally reflect the tumour biology at time of initial diagnosis. According to the available data, 3% of patients (n = 12) in the TCGA-dataset were treated with BRAF/MEK-inhibitors, 4% were treated with immune checkpoint inhibitors (n = 19) and 9% were treated with chemotherapy (dacarbazine or other; n = 44). Of all patients 15% (n = 69) were treated with other systemic therapies such as interferon and interleukin-2 or with vaccination therapy. In 69% of patients (n = 325) no data on systemic therapy was available. Of these patients, 48% (n = 156) had stage I/II disease and 36% (n = 117) had stage III disease. Therefore, it can be assumed that these patients were treated with local excision of the tumour, with or without lymph node dissection and/or radiotherapy and did not receive systemic therapy. The remaining patients were either staged as stage IV disease (n = 20) or had an unknown tumour stage (n = 26). It was unknown if these patients received systemic therapy.
Table 1.
Summary of the baseline characteristics of patients in the GEO-, TCGA- and Cirenajwis-dataset.
| Patient & tumour characteristics | GEO-dataset n (%) | TCGA-dataset n (%) | Cirenajwis-dataset n (%) | |||
|---|---|---|---|---|---|---|
| Total number of patients | 405 | 469 | 214 | |||
| Sex | ||||||
| Male | 129 (31.9) | 289 (61.6) | 124 (57.9) | |||
| Female | 101 (24.9) | 180 (38.4) | 89 (41.6) | |||
| NA | 175 (43.2) | 0 (0) | 1 (0.5) | |||
| Age (yrs) | ||||||
| Median (IQR) | 66 (51–78) | 58 (48–71) | 64 (54–74) | |||
| Tumour stage (AJCC 7th ed.) | ||||||
| Tumour in situ (stage 0) | 0 (0) | 7 (1.5) | 0 (0) | |||
| Low stage (stage I or II) | 22 (5.4) | 235 (50.1) | 0 (0) | |||
| High stage (stage III or IV) | 363 (89.6) | 191 (40.7) | 214 (100) | |||
| NA | 20 (4.9) | 36 (7.7) | 0 (0) | |||
| Sample obtained from | ||||||
| Primary tumour | 56 (13.8) | 102 (21.7) | 16 (7.5) | |||
| Lymph node metastasis | 15 (3.7) | 229 (48.9) | 128 (59.8) | |||
| Regional | Unknown | 222 (47.3) | 127 (59.3) | |||
| Distant | Unknown | 7 (1.5) | 1 (0.5) | |||
| (Sub)cutaneous or in transit metastasis | 108 (26.7) | 99 (21.1) | 41 (19.2) | |||
| Visceral metastasis | 20 (2.5) | 26 (5.5) | 10 (4,7) | |||
| Bone metastasis | 0 (0) | 6 (1.3) | 0 (0) | |||
| CNS metastasis | 0 (0) | 2 (0.4) | 0 (0) | |||
| Unknown location of distant metastasis | 155 (38.3) | 2 (0.4) | 19 (8.9) | |||
| Unknown primary or metastatic lesion | 51 (12.6) | 3 (0.6) | 0 (0) | |||
Abbreviations: CNS: Central nervous system, ed.: edition, NA: Not Available, IQR: Interquartile range, yrs: years.
The Cirenajwis-dataset consisted solely of samples obtained from patients with stage III/IV disease. Treatment data in the Cirenajwis-dataset was not available. The Hugo-dataset consisted of 27 pre-treatment tumour samples from patients treated with anti-PD-1 therapy. In the Riaz-dataset, 49 pre-treatment tumour samples were available obtained from patients treated with anti-PD-1 therapy and in the Auslander-dataset six pre-treatment samples were available obtained from three patients treated with anti-PD-1 therapy. Relevant sample, patient and treatment characteristics for all datasets are provided in Supplementary Table 1.
Consensus clustering
We identified four consensus clusters in the GEO-dataset, with clustering stability increasing up to four clusters (Figure 1(a)Supplementary Fig. S1A-C). In the TCGA-dataset we observed a more gradual levelling of clustering stability (Figure 1(b), Supplementary Figure 1D-F). When evaluating five clusters we observed four clusters of substantial size (range n = 35 to n = 169) representing ~99% of samples in the TCGA-dataset. Cluster assignments in both datasets are provided in Supplementary Table 2.
Figure 1.
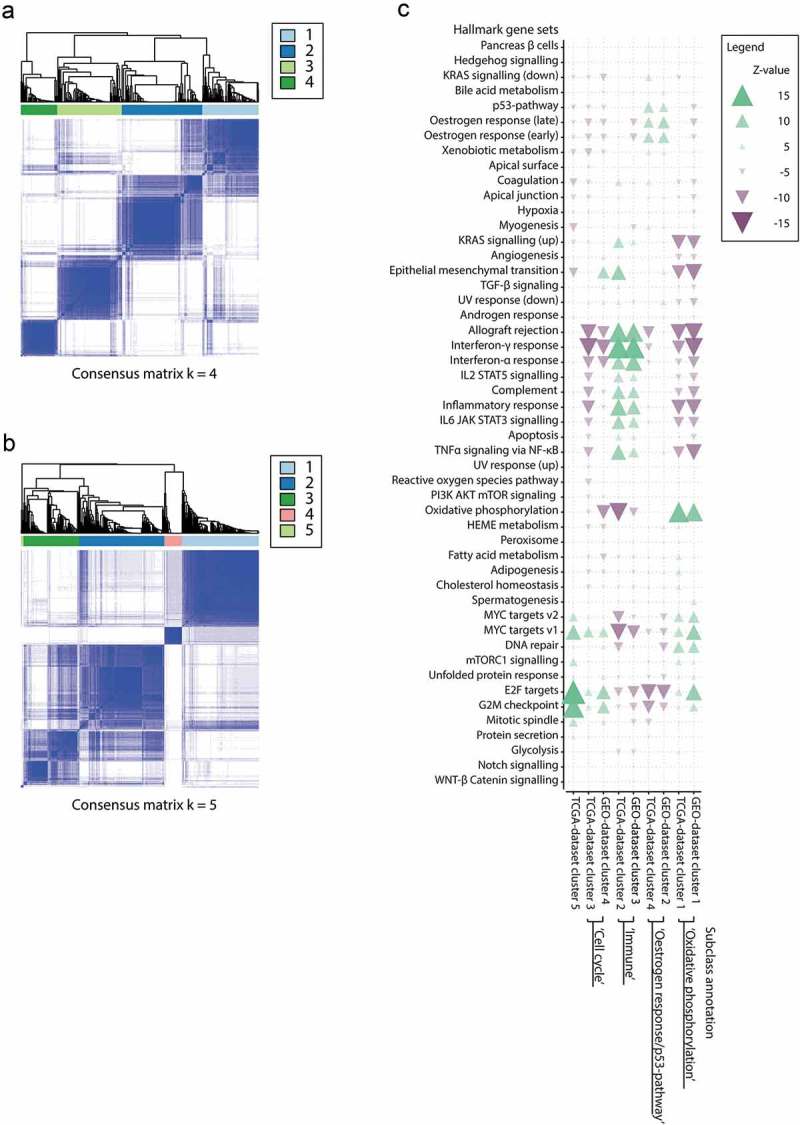
(a) Consensus clustering for the GEO-dataset showing four clusters of substantial size in the GEO-dataset at k = 4. (b) Consensus clustering for the TCGA-dataset showing four large and one small cluster (n = 5) at k = 5. (c) Enrichment overview of Hallmark gene sets for all paired GEO- and TCGA-dataset consensus clusters. Green arrows indicate enrichment of a Hallmark gene set in upregulated genes, purple arrows indicate enrichment of Hallmark gene sets in downregulated genes. Transparency and arrow width represent the significance level of enrichment.
A high concordance with a clear one-to-one mapping between clusters identified independently in the GEO- and TCGA-dataset was observed at the individual gene and biological pathway level across all gene set databases from the Molecular Signature Database (Supplementary Figure 2).
Biological characterization
GEO-dataset cluster 1 showed the strongest one-to-one mapping (R = 0.68) with TCGA-dataset cluster 1. These clusters were assigned the subclass label ‘Oxidative phosphorylation’ because the Hallmark gene set oxidative phosphorylation was the most significantly enriched pathway in both clusters. In addition, we observed high expression of the genes microphthalmia-associated transcription factor (MITF) and peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PPARGC1A, also known as PGC1α) (Figure 1(c)).
We observed high similarity (R = 0.47) between GEO-dataset cluster 2, and TCGA-dataset cluster 4 and these were assigned the subclass label ‘Oestrogen response/p53-pathway’ because in both the top-3 enriched Hallmark gene sets were: oestrogen response early, oestrogen response late and p53-pathway.
GEO-dataset cluster 3 showed the strongest one-to-one mapping (R = 0.61) with TCGA-dataset cluster 2. In both clusters, the most enriched Hallmark gene sets were immune related, such as the interferon-γ response gene set. We therefore labelled these consensus clusters ‘Immune’.
GEO-dataset cluster 4, and TCGA-dataset cluster 3, showed high similarity (R = 0.60) and were assigned the label ‘Cell cycle’ as in both the top three enriched gene sets included the Hallmark gene sets E2F targets and G2M checkpoint.
Detailed gene set enrichment results are provided as Supplementary Data.
Comparison with previously published molecular subclasses
We compared our subclasses with those defined by Jönsson et al.3 and found the strongest concordance between our ‘Oxidative phosphorylation’ and their ‘Pigmentation’ subclass (R = 0.64), our ‘Immune’ and their ‘Immune high’ subclass (R = 0.79), our ‘Cell cycle’ and their ‘Proliferative’ subclass (R = 0.60), and between our ‘Oestrogen response/p53-pathway’ and their ‘Normal-like’ subclass (R = 0.78).
Comparison of our subclasses with those defined by the TCGA5 showed high enrichment of the gene set representing their ’Immune’-gene set in our ‘Immune’ subclass, high enrichment of the their ‘MITF-low’-gene set in our ‘Cell cycle’ subclass and high enrichment of the ‘Keratin’-gene set in our ‘Oestrogen response/p53-pathway’ subclass. None of the gene sets representing the subclasses identified by TCGA Network showed strong enrichment within our ‘Oxidative phosphorylation’ subclass. However, the gene set belonging to the ‘MITF-high’ subclass identified by Tirosh et al.6 did show strong enrichment within our ‘Oxidative phosphorylation’ subclass. The gene set belonging to the ‘AXL-high’ subclass as defined by Tirosh et al. was not enriched in a specific subclass identified in our study.
A detailed overview of the concordance between our subclasses and previously defined subclasses is shown in Figure 2 and Supplementary Table 3.
Figure 2.
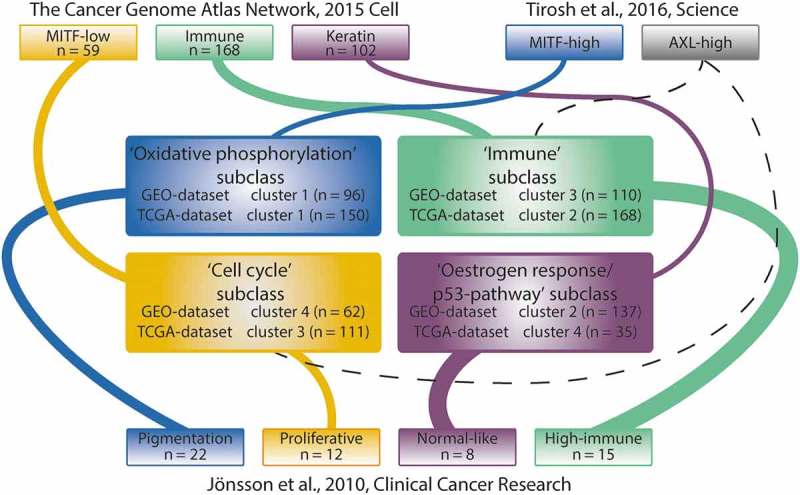
Concordance with previously described subclasses. Concordance between our subclasses and those described by TCGA Network and Tirosh et al. was determined by performing GSEA using the representative set of genes for each of the previously described subclasses.5,6 Concordance with the subclasses defined by Jönsson et al. was assessed by matching the gene expression-based centroid for each subclass with the Z-transformed p-values resulting from the pairwise class comparisons.4 Subsequently, Spearman’s rank correlations were calculated between the centroids and the ranked lists with the Z-transformed two-sided p-values. The thickness of the lines between subclasses indicates the significance level of concordance.
Association with clinicopathological variables
No difference in Breslow thickness, presence of tumour ulceration or frequency of BRAF, NRAS, KIT or NF1 mutations was observed between subclasses in the TCGA-dataset. KRAS mutations were only observed within the ‘Oxidative phosphorylation’ subclass, but the prevalence was low (n = 5 out of 150). However, it should be noted that the association between subclass assignment and baseline tumour characteristics is preferably assessed in samples obtained from the primary melanoma lesion, since metastatic lesions may not share their phenotype with the primary lesion. All associations between subclasses and clinicopathological variables in the TCGA-dataset are provided in Supplementary Table S4.
In the GEO-dataset, estimated fractions of CD8+ T-cells, CD4+ T-cells, M1 macrophages, plasma cells and naïve B-cells were higher in the ‘Immune’ subclass compared to the other subclasses (Figure 3 and Supplementary Figure 3). In the ‘Oestrogen response/p53-pathway’ subclass, a higher fraction of resting mast cells and resting and activated dendritic cells was observed. The highest fraction of activated mast cells was observed in the ‘Cell cycle’ subclass.
Figure 3.
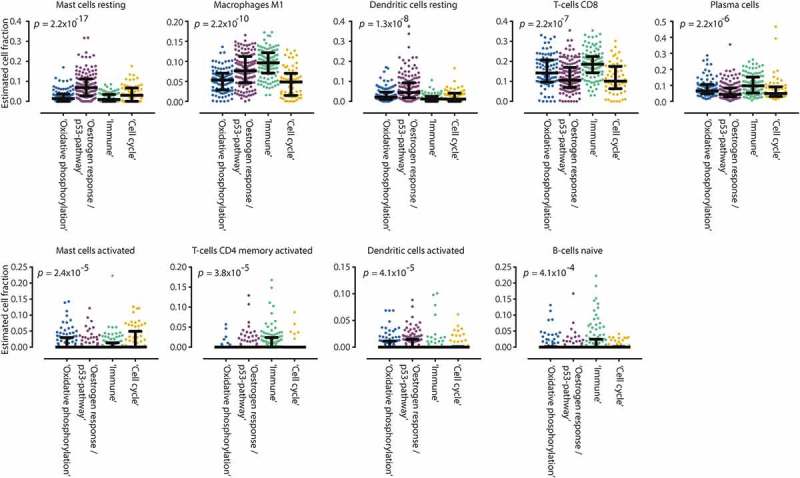
Boxplots of estimated immune cell fractions in each GEO-dataset consensus cluster. We applied the recently developed CIBERSORT method to estimate the fraction of 22 immune cell types. In the ‘Oestrogen response/p53-pathway’ subclass, higher estimated fractions of resting mast cells were observed as compared to the other clusters. In the ‘Immune’ subclass, estimated fractions of M1 macrophages, activated CD4 + memory and CD8 + T-cells, naïve B-cells and plasma cells were higher as compared to other subclasses.
Survival analysis
In the TCGA-dataset subclass assignment was associated with OS independently of age, gender and stage in a multivariate Cox regression analysis (Table 2, number of samples and events in each dataset are listed in table footnote). Longer OS was observed in the ‘Immune’ subclass compared to the ‘Oxidative phosphorylation’ subclass (Hazard-ratio (HR): 2.01, p-value: 0.004) and compared to the ‘Cell cycle’ subclass (HR: 1.51, p-value: 0.04) (Figure 4(a)). Survival analysis in the Cirenajwis-dataset showed similar results (‘Immune’ vs. ‘Oxidative phosphorylation’ subclass HR: 2.55, p-value: 0.0003; ‘Immune’ vs. ‘Cell cycle’ subclass HR: 2.19, p-value: 0.008) (Figure 4(b)). Post accession survival, defined as time from sample procurement to death or lost to follow-up, was shorter in the ‘Immune’ subclass compared to both the ‘Oxidative phosphorylation’ subclass (HR: 1.68, p-value: 0.008) and the ‘Cell cycle’ subclass (HR: 1.69, p-value: 0.009) (Figure 4(c)). It should be taken into account that these plots do not visualize the effects of other factors besides subclass assignment that influence OS such as age, gender and tumour stage. Similarly, the Log Rank (Mantel-Cox) test does not take into account these factors. Additionally, the same trend in association of OS with subclass assignment was observed in the GEO-dataset for both the ‘Immune’ and ‘Oxidative phosphorylation’ subclass, although limited data was available (Supplementary Figure 4).
Table 2.
Results of the multivariate Cox-Regression analysis in the TCGA-dataset for endpoint overall survival (OS) and post accession survival (PAS) and in the Cirenajwis-dataset for endpoint OS.
| Dataset | Endpoint | Covariates | Variables | HR (95%-CI) | Wald-test p-value |
|---|---|---|---|---|---|
| TCGAa | OS | Subclass | Immune | 1.00 | 3.00 x 10−06 |
| Oxidative phosphorylation | 2.01 (1.37–2.96) | ||||
| Cell cycle | 1.51 (1.02–2.56) | ||||
| Oestrogen response/p53-pathway | 1.44 (0.52–4.05) | ||||
| Gender | Female | 1.00 | |||
| Male | 1.02 (0.73–1.42) | ||||
| Age | Continuous | 1.02 (1.01–1.03) | |||
| Tumour stage | Stage 0 (Melanoma in situ) | 1.00 | |||
| Stage I/II | 0.75 (0.30–1.87) | ||||
| Stage III | 1.22 (0.48–3.09) | ||||
| Stage IV | 1.98 (0.64–6.19) | ||||
| Unknown | 0.57 (0.20–1.62) | ||||
| TCGAa | PAS | Subclass | Immune | 1.00 | 0.008 |
| Oxidative phosphorylation | 1.68 (1.15–2.47) | ||||
| Cell cycle | 1.69 (1.14–2.51) | ||||
| Oestrogen response/p53-pathway | 0.62 (0.22–1.73) | ||||
| Gender | Female | 1.00 | |||
| Male | 1.11 (0.80–1.55) | ||||
| Age | Continuous | 1.01 (1.00–1.02) | |||
| Tumour stage | Stage 0 (Melanoma in situ) | 1.00 | |||
| Stage I/II | 0.31 (0.12–0.76) | ||||
| Stage III | 0.32 (0.13–0.80) | ||||
| Stage IV | 0.46 (0.15–1.40) | ||||
| Unknown | 0.46 (0.17–1.26) | ||||
| Cirenajwisb | OS | Subclass | Immune | 1.00 | 3,00 x 10−05 |
| Oxidative phosphorylation | 2.55 (1.54–4.21) | ||||
| Cell cycle | 2.19 (1.22–3.92) | ||||
| Oestrogen response/p53-pathway | 3.16 (1.14–8.79) | ||||
| Gender | Female | 1.00 | |||
| Male | 1.39 (0.92–2.12) | ||||
| Age | Continuous | 1.00 (0.99–1.02) | |||
| Tumour stage | General (metastasized) | 1.00 | |||
| In-transit | 0.43 (0.18–1.00) | ||||
| Local | 0.19 (0.06–0.63) | ||||
| Primary | 0.03 (0.00–0.23) | ||||
| Regional | 0.29 (0.17–0.52) |
a Number of patients in the TCGA-dataset available for survival analysis (both OS and PAS): 455. Number of events in the TCGA-dataset: 162.
b Number of samples in the Cirenajwis-dataset available for survival analysis: 203. Number of events in the Cirenajwis-dataset: 99. Abbreviations: 95%-CI: 95%-confidence interval, HR: Hazard-ratio, OS: Overall survival, PAS: Post accession survival.
Figure 4.
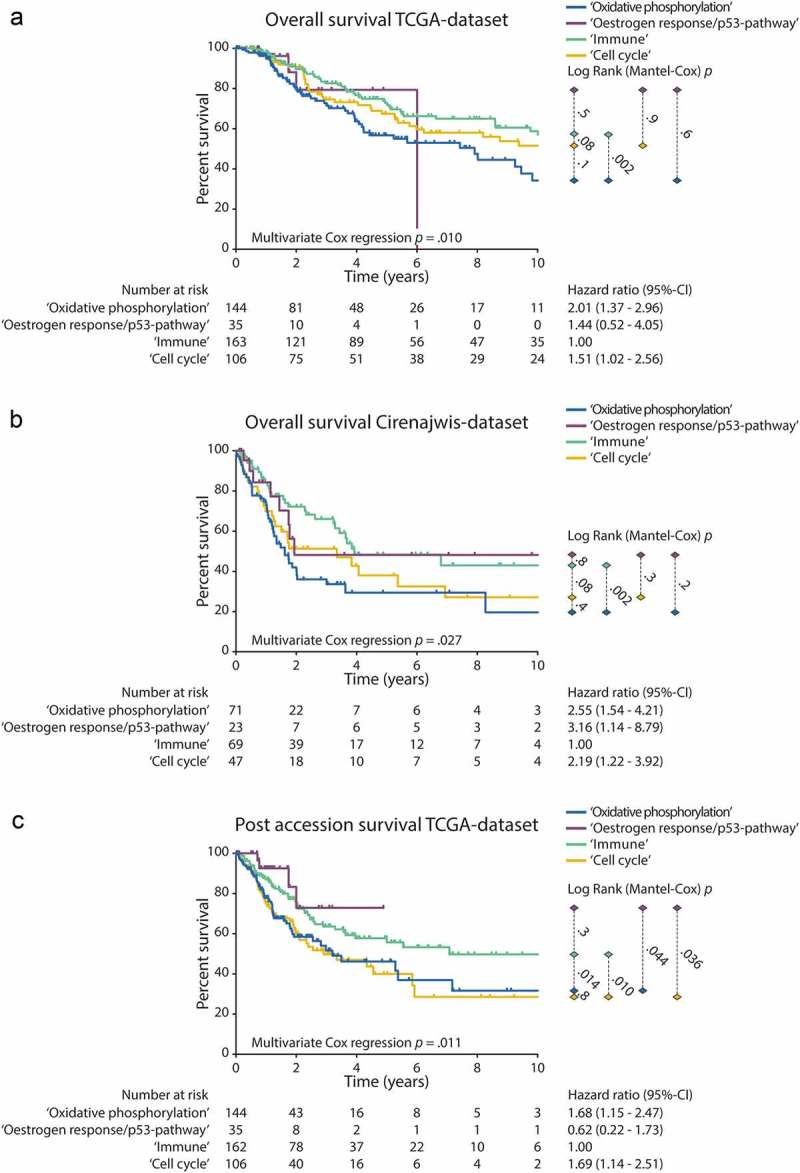
Survival analysis. Kaplan Meier curves are shown for each subclass. The p-values right from the curves illustrate the difference between individual curves, as calculated with the Log Rank (Mantel-Cox)-test. The hazard-ratios and 95%-confidence intervals of the multivariate cox-regression analysis for the association between subclass assignment with survival are shown right from the number at risk table. In each analysis, the ‘Immune’ subclass was selected as the reference group. Covariates in the multivariate cox regression analysis were: Gender, Age and Tumour stage. (a) OS in the TCGA-dataset. (b) OS in the Cirenajwis-dataset. (c) Post accession survival in the TCGA-dataset.
Response to anti-PD-1 therapy
In the Hugo-dataset, ten patients were assigned to the ‘Oxidative phosphorylation’ subclass, six patients to the ‘Immune’ subclass, four patients to the ‘Oestrogen response/p53-pathway’ subclass and seven patients to the ‘Cell cycle’ subclass. In the Riaz-dataset, 20 patients were assigned to ‘Oxidative phosphorylation’ subclass, 11 patients to the ‘Immune’ subclass, ten patients to the ‘Oestrogen response/p53-pathway’ subclass and eight to the ‘Cell cycle’ subclass. When pooling the data, we observed a difference in the distribution of responders between the subclasses, with 18 responders (60%) in the ‘Oxidative phosphorylation’, eleven responders (65%) in the ‘Immune’ subclass, six (43%) responders in the ‘Oestrogen response/p53-pathway’ subclass and six responders (40%) in the ‘Cell cycle’ subclass (Table 3). The subclass annotation for each sample in the Hugo-, Riaz- and Auslander-datasets are provided in Supplementary Table 5.
Table 3.
Number of responders and non-responders in the pooled data of the Hugo- and Riaz-dataset.
| Subclass | Responders, n (%) | Non-responders, n (%) |
|---|---|---|
| Cell cycle | 6 (40) | 9 (60) |
| Immune | 11 (65) | 6 (35) |
| Oestrogen response/p53-pathway | 6 (43) | 8 (57) |
| Oxidative phosphorylation | 18 (60) | 12 (40) |
Discussion
In this largest pooled analysis of expression profiles of cutaneous melanoma samples thus far, we identified four gene expression-based molecular subclasses that showed an association with survival. Additionally, we observed a trend towards differential response rates to anti-PD-1 therapy between these subclasses.
In contrast to the four subclasses based on complex tissue biopsies identified in this study, single-cell RNA-seq identified only two subclasses.6 Expression profiling of complex tissue biopsies measures the average expression pattern of all cells present in the sample. These may include tumour cells as well as other cells present in the tumor-microenvironment.8 The samples in the GEO- and TCGA-datasets are all obtained from such complex tissue biopsies. Therefore, it may be deemed likely that the additional subclasses we identified capture all cellular components within a tumour lesion. Since the tumour-microenvironment (TME) is related to melanoma development and progression, these components are of interest from a biological point of view.9 None of the subclasses we identified showed high similarity to the ‘AXL-high’ subclass detected using single-cell RNA-seq. This may be due to intra-tumour heterogeneity causing the ‘AXL-high’ subclass population in a tumour to be overshadowed by more prominent subpopulations present within a complex biopsy.8
We observed the shortest OS in the ‘Oxidative phosphorylation’ subclass. In this subclass, we also observed high expression of MITF, a known biomarker for poor prognosis in melanoma.10 MITF is critical in reprogramming metabolism through direct regulation of PGC1α expression, a transcriptional co-activator that enhances mitochondrial biogenesis and oxidative phosphorylation.10,11 These processes are required for ATP and biomass production and thus support cancer cell proliferation.12–14 Drugs targeting oxidative phosphorylation, such as mitochondrial complex 1 inhibitors and glutamine uptake inhibitors, reduce melanoma cell growth in vitro and in xenograft models in vivo.15,16 During oxidative phosphorylation reactive oxygen species (ROS) are generated.17 In a mouse model it has been shown that the deacetylase inhibitor vorinostat can lead to a lethal ROS level in melanoma cells with increased ROS levels and can induce responses in patients with BRAF resistant melanoma.18 Further investigation of vorinostat is of interest in the subclass of melanoma with enhanced oxidative phosphorylation. We also observed a trend towards a higher tumour response rate (60%) to anti-PD-1 therapy in patients with melanomas within the ‘Oxidative phosphorylation’ subclass. In many cancer types, including melanoma, glucose uptake is high.19 Subsequent high glycolytic rates provide ATP and glycolytic intermediates to support biomass production and results in excretion of metabolites such as lactate acid and CO2.20 These excreted metabolites are a major cause of acidification of the tumour-microenvironment. Low pH-levels impair NK- and T-cell effector function.21 Enhanced utilization of oxidative phosphorylation might reduce lactate acid production, thereby providing an immune permissive tumour-microenvironment leading to a more potent response to anti-PD-1 therapy. Modulating tumour metabolism, either by targeting enhanced oxidative phosphorylation or by reducing tumour lactate production in combination with checkpoint inhibition may be interesting treatment strategy.
Alternatively, the high expression of genes related to oxidative phosphorylation could reflect the expression pattern of other cells in the TME, such as T-cells or macrophages. In T-cells, metabolic rewiring towards aerobic glycolysis upon T-cell activation is essential to support the differentiation and function of T-effector cells.22,23 PD-1 ligation of T-cells rewires T-cell metabolism towards an increased rate of fatty acid oxidation and increased utilization of endogenous lipids for β-oxidation to sustain their survival.22 This phenotype is associated with differentiation towards memory T-cells.24 In addition, PD-1 ligation of T-cells results in a significant decrease in levels of the cellular antioxidant glutathione, indicating a more oxidative environment. It could be hypothesized that the relative high expression of genes related to oxidative phosphorylation in the ‘Oxidative phosphorylation’ subclass could reflect a phenotype that has resulted from PD-1 ligation of T-cells in the TME. This hypothesis implies an important role for PD-1/PD-L1 pathway activation for successful immune evasion by the tumour in this subclass. In theory, blocking the PD-1/PD-L1 pathway to reinvigorate the anti-cancer immune response would be especially effective in this subclass.
Additionally, in patients with coronary artery disease, enhanced influx of pyruvate into the mitochondria of macrophages leads to enhanced expression of PD-L1 by macrophages through activation of the p-SMAD1/5/IRF1 signalling cascade by BMP4.25 Assuming that the same mechanism can occur in macrophages in the TME in melanoma, it could also be hypothesized that the high expression of genes related to oxidative phosphorylation in the ‘Oxidative phosphorylation’ subclass reflect the expression pattern of macrophages utilizing enhanced oxidative phosphorylation. Subsequent enhanced expression of PD-L1 by these macrophages could then lead to suppression of the anti-tumour immune response. Again, this hypothesis implies a central role for the PD-1/PD-L1 pathway to counteract the anti-tumour immune response, providing a theoretical basis for anti-PD-1 therapy in this subclass. Our observation of a trend towards a higher response rate to anti-PD-1 therapy in the ‘Oxidative phosphorylation’ subclass is in line with these hypotheses.
The ‘Immune’ subclass was associated with longer OS compared to other subclasses. In this subclass, we observed relatively high estimated fractions of M1 macrophages, CD8+ T-cells and high enrichment of the interferon-γ response pathway. Both high CD8+ T-cell density in the tumour-microenvironment and the presence of a strong interferon-γ cytolytic T-cell signature have been related to a favourable response to immune checkpoint inhibitors.26 Recently, it was shown that in breast cancer patients a higher fraction of M1 macrophages in the tumour is independently associated with a higher rate of pathological complete response and prolonged OS.27 The association of the M1 macrophage subpopulation with disease outcome in breast cancer and the high M1 macrophage fraction in the ‘Immune’ subclass in melanoma is of particular interest considering the development of therapeutic interventions targeting macrophages.28
The ‘Cell cycle’ subclass was characterized by high expression of E2F-target genes. E2F is activated by cyclin-dependent kinases (CDK) 4 and 6 through the CDK4-pathway.29 Pre-clinical studies have shown effectiveness of CDK4-pathway inhibition in melanoma cell lines, especially when combined with BRAF- and MEK-inhibitors.30 Moreover, a phase I trial in 26 melanoma patients treated with a selective CDK4/6 inhibitor showed a partial response in one and stable disease in six patients.31 Notably, the patient with partial response was shown to have an NRAS mutation and copy-number loss at the Inhibitors of CDK4 (INK4) locus, inducing aberrant CDK4 and CDK6 activity. In light of these publications a possible E2F-target driven melanoma subclass is of particular interest.
Due to the low number of samples in the ‘Oestrogen response/p53-pathway’ subclass, the association with OS could not be ascertained. However, the expression of oestrogen response pathway genes may have clinical implications. Hormone therapy, such as the selective oestrogen receptor modifier tamoxifen, has shown to have varying effects on OS in cutaneous melanoma in various studies.32 Enhanced expression of oestrogen receptor signalling pathways conferred resistance to MAPK-inhibiting therapies in melanoma cell lines.33 The identification of a subclass with high oestrogen response pathways might justify renewed interest in research towards hormonal therapy in cutaneous melanoma in this subclass.
A large proportion of samples included in the TCGA-dataset was not collected at initial diagnosis but during the course of disease. Expression profiles obtained from these samples might not optimally reflect the molecular state of the disease at initial diagnosis and therefore are not preferably used to assess OS.9 However, the clear association between subclass assignment and OS in both the TCGA-dataset and the Cirenajwis-dataset and the clear association between subclass assignment and post accession survival in the TCGA-dataset underlines the clinical relevance. Additionally, the relevance of the subclasses in relation to contemporary therapies such as immune checkpoint inhibitors could only be assessed in a very small set of samples. These limitations emphasize the need to validate these subclasses in a homogeneous set of primary, untreated cutaneous melanoma samples. In addition, the clinical relevance of the subclasses should be validated in a prospective observational cohort of patients with untreated metastasized melanoma eligible for anti-PD-1, anti-CTLA-4 or combination therapy, with inclusion of a tumour biopsy prior to the start of systemic therapy. However, despite the different profiling methods that were used to generate the GEO- and TCGA-datasets, and despite the differences in patient and tumour characteristics, there was a high degree of reproducibility between the four identified subclasses within the datasets. This reflects the solidity of the subclasses identified in this study.
Our analysis provides, for the first-time, compelling cross-platform evidence for the existence of four molecular subclasses in cutaneous melanoma. Additionally, we show that these subclasses are characterised by distinct biological pathways and have a significant association with disease outcome. Finally, we show for the first time a trend towards differential response rates to anti-PD-1 therapy between these subclasses. These results can guide clinicians and drug developers in developing new treatment strategies in cutaneous melanoma.
Methods
Consensus clustering and biological characterization
Consensus clustering (ConsensusClusterPlus, version 1.24.0) and biological characterization with gene set enrichment analysis (GSEA) was performed independently in the GEO and TCGA-dataset. To identify differentially expressed genes in a cluster, we performed a class comparison between the set of samples assigned to the cluster and the set of samples assigned to any of the other clusters. Subsequently, GSEA – utilizing a large panel of gene set databases available at the Molecular Signature Database – was performed on the ranked list of differentially expressed genes per cluster.
To assess the concordance between the clusters identified in the GEO- and TCGA-dataset, we determined Spearman’s rank correlations between the ranked lists of differentially expressed genes and performed average linkage hierarchical clustering on the gene set enrichment vectors of individual consensus clusters.
Concordance with previously reported subclasses
Concordance between subclasses identified in this study and the subclasses described by TCGA Network and Tirosh et al. was determined by performing GSEA with the set of representative genes for each of the previously described subclasses.5,6 Jönsson et al. reported a gene expression-based centroid per subclass.3 Spearman’s rank correlations were calculated between these centroids and our ranked lists of differentially expressed genes.
Determination of in silico immune cell type fractions
CIBERSORT was used to estimate the fractions of 22 immune cell types for each sample in the GEO-dataset.34 Associations between consensus clusters and immune cell type fractions were determined with the Kruskal-Wallis test.
Survival analysis
Multivariate Cox regression analysis was performed to assess the association of subclass assignment with OS and post accession survival in the TCGA-dataset, with covariates age, gender and stage for OS and age, gender and biopsy location for post accession survival. Association of subclass assignment with OS was validated in the Cirenajwis-dataset in the same manner.
A detailed description of all methods, including the method for subclass assignment in the Cirenajwis-dataset, Hugo-dataset, Riaz-dataset and Auslander-dataset, is provided in the Supplementary Methods.
Abbreviations
95%-CI 95%-confidence interval
CDK cyclin-dependent kinases
CNS Central nervous system
ed. edition
GEO Gene Expression Omnibus
HR Hazard-ratio
INK4 Inhibitors of CDK4
IQR Interquartile range
MITF microphthalmia-associated transcription factor
mRNA-seq mRNA-sequencing
NA Not Available
OS Overall survival
PAS Post accession survival
PFS Progression free survival
PGC1α peroxisome proliferator-activated receptor gamma co-activator 1 alpha
ROS reactive oxygen species
TCGA The Cancer Genome Atlas
TME Tumour-microenvironment
yrs years
Funding Statement
This work was supported by the Dutch Cancer Society [RUG 2016–10034] to E.G.E. de Vries; a NWO-VENI grant [916–16025] to R.S.N. Fehrmann; the Bas Mulder Award [RUG 2013–5960] to R.S.N. Fehrmann; and a Mandema Stipendium [no grant number] to R.S.N. Fehrmann.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplymentary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Ugurel S, Rohmel J, Ascierto PA, Flaherty KT, Grob JJ, Hauschild A, Larkin J, Long GV, Lorigan P, McArthur GA, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies update 2017. Eur J Cancer. 2017;83:247–257. PMID: 28756137. doi: 10.1016/j.ejca.2017.06.028. [DOI] [PubMed] [Google Scholar]
- 2.Sanoudou D, Mountzios G, Arvanitis DA, Pectasides D.. Array-based pharmacogenomics of molecular-targeted therapies in oncology. The Pharmacogenomics J. 2012;12(3):185–196. PMID: 22249357. doi: 10.1038/tpj.2011.53. [DOI] [PubMed] [Google Scholar]
- 3.Jonsson G, Busch C, Knappskog S, Geisler J, Miletic H, Ringer M, Lillehaug JR, Borg A, Lonning PE.. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin Cancer Res. 2010;16(13):3356–3367. PMID: 20460471. doi: 10.1158/1078-0432.CCR-09-2509. [DOI] [PubMed] [Google Scholar]
- 4.Cirenajwis H, Ekedahl H, Lauss M, Harbst K, Carneiro A, Enoksson J, Rosengren F, Werner-Hartman L, Törngren T, Kvist A, et al. Molecular stratification of metastatic melanoma using gene expression profiling: prediction of survival outcome and benefit from molecular targeted therapy. Oncotarget. 2015;6(14):12297–12309. PMID: 25909218. doi: 10.18632/oncotarget.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cancer Genome Atlas Network Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681–1696. PMID: 26091043. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Lian C, Murphy G, Fallahi-Sichani M, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352(6282):189–196. PMID: 27124452. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauss M, Nsengimana J, Staaf J, Netwon-Bishop J, Jönsson G. Consensus of melanoma gene expression subtypes converges on biological entities. J Invest Dermatol. 2016;136:2502–2505. PMID: 27345472. doi: 10.1016/j.jid.2016.05.119. [DOI] [PubMed] [Google Scholar]
- 8.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Naheb BV, Curry WT, Martuza RL, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. PMID: 24925914. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandner JM, Haass NK. Melanoma’s connections to the tumour microenvironment. Pathology. 2013;45(5):443–452. PMID: 23851614. doi: 10.1097/PAT.0b013e328363b3bd. [DOI] [PubMed] [Google Scholar]
- 10.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23(3):302–315. PMID: 23477830. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23(3):287–301. PMID: 23416000. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for KRAS-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107(19):8788–8793. PMID: 20421486. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2001;35(8):427–433. PMID: 20570523. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lakhter AJ, Hamilton J, Konger RL, Brustovetsky N, Broxmeyer HE, Naidu SR. Glucose-independent acetate metabolism promotes melanoma cell survival and tumor growth. J Biol Chem. 2016;291(42):21869–21879. PMID: 27539851. doi: 10.1074/jbc.M115.712166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schöckel L, Glasauer A, Basit F, Bitschar K, Truong H, Erdmann G, Algire C, Hägebarth A, Willems PH, Kopitz C, et al. Targeting mitochondrial complex I using BAY 87–2243 reduces melanoma tumor growth. Cancer Metab. 2015;3(11). PMID: 26500770. doi: 10.1186/s40170-015-0138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Q, Beaumont KA, Otte NJ, Font J, Bailey CG, van Geldermalsen M, Sharp DM, Tiffen JC, Ryan RM, Jormakka M, et al. Targeting glutamine transport to suppress melanoma cell growth. Int J Cancer. 2014;135(5):1060–1071. PMID: 24531984. doi: 10.1002/ijc.28749. [DOI] [PubMed] [Google Scholar]
- 17.Emanuelle S, D’Anneo A, Calvaruso G, Cernigliaro C, Giuliano M, Lauricella M. The double-edged sword profile of redox-signalling: oxidative events as molecular switches in the balance between cell physiology and cancer. Chem Res Toxicol. 2018;31(4):201–210. PMID: 29513521. doi: 10.1021/acs.chemrestox.7b00311. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, Bosma A, Song JY, Zevenhoven J, Los-de Vries GT, et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell. 2018;173(6):1413–1425. PMID: 29754815. doi: 10.1016/j.cell.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 19.Böhme I, Bosserhoff AK. Acidic tumor microenvironment in human melanoma. Pigment Cell Melanoma Res. 2016;29(5):508–523. PMID: 27233233. doi: 10.1111/pcmr.12495. [DOI] [PubMed] [Google Scholar]
- 20.Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, Kreutz M. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol. 2017;8:248 PMID: 28337200. doi: 10.3389/fimmu.2017.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher K, Hoffman P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. PMID: 17255361. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 22.Frauwirth KA, Thompson CB. Regulation of T lymphocyte metabolism. J Immunol. 2004;172(8):4661–4665. PMID: 15067038. doi: 10.4049/jimmunol.172.8.4661. [DOI] [PubMed] [Google Scholar]
- 23.Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000;6(3):683–692. PMID: 11030347. doi: 10.1016/S1097-2765(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 24.Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. PD-1 alters T0cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692 PMID: 25809635. doi: 10.1038/ncomms7692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe R, Shirai T, Namkoong H, Zhang H, Berry GJ, Wallis BB, Schaefgen B, Harrison DG, Tremmel JA, Giacomini JC, et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J Clin Invest. 2017;127(7):2725–2738. PMID: 28604383. doi: 10.1172/JCI92167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hegde PS, Karanika V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clinical Cancer Res. 2016;22(8):1865–1874. PMID: 17255361. doi: 10.1158/1078-0432.CCR-15-1507. [DOI] [PubMed] [Google Scholar]
- 27.Bense RD, Sotiriou C, Piccart-Gebhart MJ, Haanen JB, van Vugt MA, de Vries EG, Schröder CP, Fehrmann RSN. Relevance of tumor-infiltrating immune cell composition and functionality for disease outcome in breast cancer. J Natl Cancer Inst. 2016;109(1):1–9. PMID: 27737921. doi: 10.1093/jnci/djw192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–662. PMID: 27055731. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheppard KE, McArthur GA. The Cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res. 2013;19(19):5320–5328. PMID: 24089445. doi: 10.1158/1078-0432.CCR-13-0259. [DOI] [PubMed] [Google Scholar]
- 30.Martin CA, Cullinane C, Kirby L, Abuhammad S, Lelliott EJ, Waldeck K, Young RJ, Brajanovski N, Cameron DP, Walker R, et al. Palbociclib synergizes with BRAF and MEK inhibitors in treatment naïve melanoma but not after the development of BRAF inhibitor resistance. Int J Cancer. 2017;142(10):2139–2152. PMID: 29243224. doi: 10.1002/ijc.31220. [DOI] [PubMed] [Google Scholar]
- 31.Patnaik A, Rosen LS, Tolaney SM, Tolcher AW, Goldman JW, Gandhi L, Papadopoulos KP, Beeram M, Rasco DW, Hilton JF, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6(7):740–753. PMID: 27217383. doi: 10.1158/2159-8290.CD-16-0095. [DOI] [PubMed] [Google Scholar]
- 32.Ribeiro MP, Santos AE, Custódio JB. Rethinking tamoxifen in the management of melanoma: new answers for an old question. Eur J Pharmacol. 2015;764:372–378. PMID: 26165763. doi: 10.1016/j.ejphar.2015.07.023. [DOI] [PubMed] [Google Scholar]
- 33.Martz CA, Ottina KA, Singleton KR, Jasper JS, Wardell SE, Peraza-Penton A, Anderson GR, Winters PS, Wang T, Alley HM, et al. Systemic identification of signalling pathways with potential to confer anticancer drug resistance. Science Signal. 2014;7(357):1–22. PMID: 25538079. doi: 10.1126/scisignal.aaa1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. PMID: 25822800. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.