

ABSTRACT
Host responses to tumor cells include tumor suppressing or promoting mechanisms. We sought to detail the effect of Hedgehog (Hh) pathway inhibition on the composition of the mammary tumor immune portfolio. We hypothesized that Hh signaling mediates a crosstalk between breast cancer cells and macrophages that dictates alternative polarization of macrophages and consequently supports a tumor-promoting microenvironment. We used an immunocompetent, syngeneic mouse mammary cancer model to inhibit Hh signaling with the pharmacological inhibitor, Vismodegib. Using molecular and functional assays, we identified that Hedgehog (Hh) signaling mediates a molecular crosstalk between mammary cancer cells and macrophages that culminates in alternative polarization of macrophages. We carried out an unbiased kinomics and genomics assessment to unravel changes in global kinomic and gene signatures impacted by Hh signaling. Our investigations reveal that in an immunocompetent mammary cancer model, the administration of Vismodegib led to changes in the portfolio of tumor-infiltrating immune cells. This was characterized by a marked reduction in immune-suppressive innate and adaptive cells concomitant with an enrichment of cytotoxic immune cells. Breast cancer cells induce M2 polarization of macrophages via a crosstalk mediated by Hh ligands that alters critical kinomic and genomic signatures. Macrophage depletion improved the benefit of Hedgehog inhibition on eliciting an immunogenic, pro-inflammatory profile. We define a novel role for Hh signaling in disabling anti-tumor immunity. Inhibition of Hh signaling presents with dual advantages of tumor cell-targeting as well as re-educating a dysfunctional tumor microenvironment.
Keywords: breast cancer, Hedgehog signaling, tumor microenvironment, tumor associated macrophages, metastasis
Introduction
The tumor microenvironment (TME) consists of multiple cell populations that participate in crosstalk with tumor cells,1,2 thus playing a major role in regulating tumor growth and progression. A subset of myeloid cells circulate through the blood as monocytes, and in response to homing signals,3 travel to reach a resting destination where they differentiate into macrophages. Macrophages are abundantly found in the tumor milieu and can comprise up to 50% of the tumor mass in solid tumors.4 Tumor-associated macrophages (TAMs) display a high degree of plasticity as they regulate a range of different and opposing functions depending on the tumor-derived cytokine stimulus they receive.5 This versatility in function and phenotype is co-opted by tumor cells resulting in enhanced cancer progression and metastasis.6,7 Tumor-associated macrophages are simplistically classified into two major phenotypes: M1 – classically activated and M2 – alternatively activated macrophages; emulating helper T cells type I (Th I) and type II (Th II) respectively.8,9 Classically activated M1 macrophages are polarized by pro-inflammatory cytokines such as TNF-α,10 LPS, and IFN-γ, which activate target genes responsible for mounting an anti-tumorigenic immune response.11 Alternatively activated M2 macrophages are stimulated by many cytokines, the most prominent being IL-4 and IL-13,12,13 which activate a STAT6 signaling cascade that promotes the transcription of genes such as IL-10 and TGF-β which mediate an anti-inflammatory and reparative response.14 STAT6 target genes suppress immune system function, allow tissue remodeling, and induce angiogenesis.15,16 Collectively, M2 macrophages permit the sustenance and proliferation of tumor cells, extravasation, and establishment of a suitable metastatic niche. It is believed that tumor cells secrete cytokines that influence TAMs to express an M2 program, a characteristic often associated with poor clinical outcome in multiple cancer types.17–19 Hypoxia in the tumor milieu recruits TAMs and sustains tumor progression through inducing the expression of Sema3A in tumors.20 The phenotype of the recruited TAMs is fine-tuned to M2-like MHC-IIlo TAM that express greater levels of hypoxia-regulated genes and prototypical M2 markers.21
Hedgehog (Hh) signaling is an essential pathway for normal mammalian development.22 The pathway is initiated by the binding of one of three ligands: Sonic hedgehog (SHH), Desert hedgehog (DHH), or Indian hedgehog (IHH) to the Patched1 (PTCH1) receptor. Upon ligand binding, PTCH1 relieves its inhibitory action on Smoothened (SMO), triggering a signaling cascade culminating in the translocation of the glioma associated oncogene homolog (GLI) transcription factors to the nucleus where they activate the transcription of genes that impact cell proliferation, differentiation, epithelial-mesenchymal transition, and stem cell maintenance.23,24 Although Hh signaling is tightly regulated and is minimally activated in adult tissues, its deregulation is a feature of multiple cancers. Previous reports from our lab and others have established the role of Hh signaling in mediating breast cancer progression through upregulating drug resistance25 and enhancing metastasis.26,27
In this study, we investigated the role of Hh signaling in impacting anti-tumor immune functions. Our investigations reveal that Hh inhibition elicits an altered portfolio of tumor-infiltrating immune cells, particularly characterized by a reduction in immune-suppressive cells concomitant with an enrichment of cytotoxic immune cells that cumulatively results in reduced metastases. Mechanistically we identified that Hh signaling alters a critical kinomic signature that enables macrophages to assume an alternative M2 phenotype. Importantly, depletion of macrophages ameliorates the effects of Hh inhibition. Our investigations provide insight into a novel role for Hh signaling in sculpting anti-tumor immunity.
Results
Inhibiting hedgehog signaling in vivo blunts the mammary tumor-associated inhibitory immune portfolio and elicits an inflammatory immune response
We sought to determine the effects of inhibiting Hh signaling on the tumor microenvironment of mammary tumors. We administered the FDA-approved, orally available pharmacological SMO/Hh inhibitor, Vismodegib thrice weekly for 4 weeks to female BALB/c mice bearing orthotopic mammary 4T1 tumors (Figure 1(a)). There were no notable differences in primary tumor growth between DMSO and Vismodegib-treated groups (Supplementary Figure 1A) until day 28 when the Vismodegib-treated tumors seemed to slow their growth. Vismodegib-treated tumors demonstrated a statistically significant increase in TUNEL-positive apoptotic cells (Figure 1(b)), Annexin V-stained sorted tumor cells (Figure 1(c); Supplementary Figure 1B), and reduced numbers of epithelial cells (CD24-positive) (Supplementary Figure 1C), cumulatively suggesting elevated apoptosis. Mice were euthanized four weeks after surgical resection of the primary tumor to enable visible enumeration of metastases. Vismodegib-treated mice exhibited significantly decreased pulmonary metastases compared to vehicle-treated mice (Figure 1(d), Supplementary Figure 1D).
Figure 1.
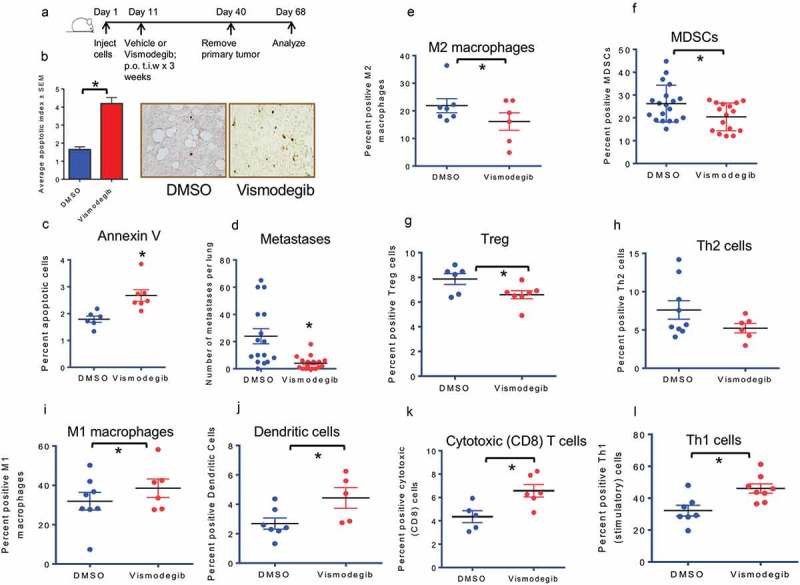
Inhibiting Hedgehog signaling in vivo blunts the inhibitory immune response and elicits an inflammatory immune response(a) Schematic of Hh inhibition strategy used in vivo. (b) Tumor sections from DMSO and Vismodegib-treated mice were assessed for apoptosis by TUNEL staining. Tumors from Vismodegib-treated mice show significantly greater levels of apoptotic cells relative to vehicle-treated mice (p < 0.0001). (c)Tumors from Vismodegib-treated mice show significantly greater levels of late apoptotic cells relative to vehicle-treated mice (p = 0.0055). Tumor cells from DMSO and Vismodegib-treated mice were analyzed by flow cytometry analysis for late apoptotic cells as indicated by PI and Annexin V following the exclusion of lineage positive immune cells (CD45, CD31, and Ter119) and inclusion of CD24+ ve epithelial cells. (d) Vismodegib-treated mice show significantly decreased pulmonary metastasis. Lung metastasis in mice was assessed by microscopic counting of lungs fixed and stained in Bouin’s solution post tumor resection (p = 0.0009). (E-L) Following resection and processing of the primary tumor, non-viable cells were excluded and immune cell infiltrates were assayed based on the following markers: (e) M2 macrophages: CD11b, Arg1, and CD206 positive cells (p = 0.04). (f) Myeloid-derived suppressor cells: CD11b, LY6G and LY6C double positive cells (p = 0.02). (g) Regulatory T cells: CD3, CD4, FOXP3, and CD25 positive cells (p = 0.03). (h) Type II helper T cells: CD3, CD4, and GATA3 positive cells (p = 0.1). (i) M1 macrophages: CD11b, F4/80 and CD80 positive cells (p = 0.042). (j) Dendritic cells: CD11c and MHC II positive cells (p = 0.04). (k) Cytotoxic CD8 T cells: CD3, CD8, and Granzyme B positive cells (p = 0.015). (l) Activated Type I helper T cells: CD3, CD4, and IFN-γ positive cells (p = 0.0078). *p< statistically significant difference relative to DMSO-treated mice.
In order to characterize overall changes in the immune portfolio of the primary tumor, we conducted a comprehensive flow cytometric analysis of the tumor-infiltrating immune population (Supplementary Figure 2). This investigation revealed a significant reduction of suppressive myeloid cells, including M2 macrophages (Figure 1(e)) and MDSCs (Figure 1(f)) in Vismodegib-treated animals. In addition, regulatory T cells (Figure 1(g)) were also significantly reduced in Vismodegib-treated mice. There was a decrease in type II helper T cells (Figure 1(h)) in Vismodegib-treated mice, albeit not statistically significant. Furthermore, Vismodegib-treated mice showed decreased numbers of T cells expressing inhibitory CTLA4 (Supplementary Figure 3A) and PD-1 (Supplementary Figure 3B), albeit not statistically significant. Overall, inhibiting Hh signaling notably decreased the population of immunosuppressive cells of the innate and adaptive immune system. The results indicate that Hh signaling enables an immunosuppressive, pro-tumorigenic microenvironment for breast cancer cells.
Further characterization of tumor immune cell infiltrate revealed a notable shift from an immune suppressive microenvironment to a pro-inflammatory, immunogenic milieu. Hh inhibition significantly upregulated infiltration of antigen-presenting, innate immune M1 macrophages (Figure 1(i)) and dendritic cells (Figure 1(j)). Vismodegib also elicited a significant infiltration of cytotoxic CD8 T cells (Figure 1(k)) and activated CD4 T cells (Figure 1(l)). No significant changes were observed in infiltrating natural killer cells (Supplementary Figure 3C). These data suggest that systemic inhibition of Hh signaling in tumor-bearing mice alters the phenotypic landscape of tumor-immune cell infiltrates. These infiltrates are enriched in innate immune cells (M1 macrophages and dendritic cells) that are efficient in presenting tumor antigens. The overall reduction in the immunosuppressive framework concomitant with an increase in tumor-reactive cells of the adaptive immune system (activated Granzyme B-positive cytotoxic T cells) complements the enhanced tumor cell apoptosis and reduced metastasis seen in Vismodegib-treated animals.
There was a notable shift in the ratios of the tumor-infiltrating effector T cells (Teffs):Tregs (Supplementary Figure 4A) and functional CD8-GzmB-positive T cells:Tregs (Supplementary Figure 4B) in the Vismodegib-treated mice indicating an overall increase in the immune reactive T cells relative to immunosuppressive Treg cells. The trends in cellular proportions of Teffs:MDSCs (Supplementary Figure 4C) and CD8-GzmB-positive Tcells:MDSCs (Supplementary Figure 4D) were decidedly increased in Vismodegib-treated tumors, albeit not statistically significant. As such, Hh inhibition shifts the tumor immune microenvironment from suppressive to inflammatory. In concordance with this observation, F4/80-positive macrophages from the primary tumor of Vismodegib treated mice demonstrated a significant decrease in urea production indicative of an overall reduced arginase activity in tumor-associated macrophages (Supplementary Figure 4E). These data collectively suggest a notable role for Hh signaling in creating a favorable, immunosuppressive environment that enables progression and metastasis.
Hedgehog signaling enables alternative activation of macrophages
Given the functional plasticity of macrophages and their relevance to breast cancer progression, we queried the role of Hh signaling in influencing their activation state. The process of acquiring an alternatively activated M2 phenotype [RAW 264.7 cells (Supplementary Figure 5A-C) and BMDMs (Supplementary Figure 5D-F)] is kinetically characterized by the upregulation of Arg1 and Cd206 simultaneous with elevated levels of the Hh transcription factor Gli1, a bonafide Hh activation marker. This is accompanied by elevated levels of the Ihh ligand in the macrophages (Supplementary Figure 5G) and functional activation of Hh signaling, as evidenced by upregulation of a Gli1 reporter plasmid (Supplementary Figure 5H). Our data is in concordance with the finding that Hh ligand-producing macrophages are involved in fibrogenic and angiogenic responses.28 Overall, these results indicate that the acquisition of an M2 phenotype of macrophages upregulates Hh ligand expression and engages transcriptional activation of Hh signaling.
In order to establish the functional relevance of activation of Hh signaling, we incorporated recombinant SHH protein in polarization conditions. Exogenous SHH protein further potentiated the expression of M2 markers Arg1 (Figure 2(a); Supplementary Figure 6A) and Cd206 (Figure 2(b)), while successfully increasing the expression of Gli1 (Figure 2(c)). In contrast, the small molecule Gli inhibitor GANT61 attenuated the gene expression of Arg1 (Figure 2(d)) and Cd206 (Figure 2(e); Supplementary Figure 6B). In addition to pharmacological inhibition, we validated the effects of Hh blockade by stably knocking down Gli1 in RAW 264.7 cells using shRNA. Abrogating endogenous Gli1 expression reduced the ability of macrophages to launch an M2 polarization program (Figure 2(f)). Targeting of SMO, the regulatory molecule of the Hh pathway, with the SMO inhibitor BMS-833923, attenuated Arg1 (Figure 2(g)) and Cd206 (Figure 2(h)) in M2 polarized macrophages. To assess the functional outcome of Hh inhibition on the phagocytic capacity of macrophages, we enumerated the fluorescently labeled bacterial particles that were phagocytosed by the M2 polarized macrophages. Inhibiting Hh signaling with the BMS compound or Vismodegib, significantly enhanced the phagocytic capacity of the alternatively polarized macrophages (Figure 2(i)). As such, inhibiting Hh signaling in macrophages using two distinct approaches, inhibiting Gli and Smo, inhibited alternative polarization of macrophages. This suggests that Hh signaling significantly inactivates the phagocytic functions of macrophages. Cumulatively, our data establishes the critical importance of Hh signaling in enabling alternative activation of macrophages.
Figure 2.
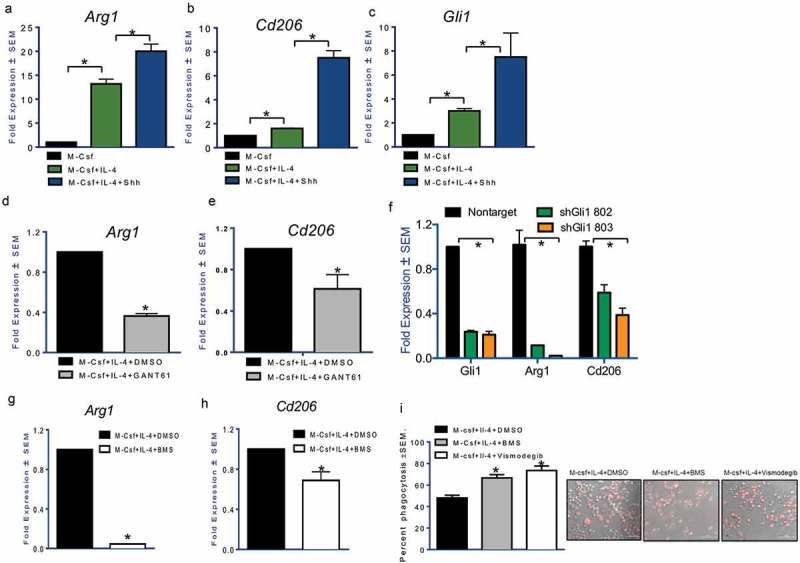
Hh signaling potentiates alternative activation of macrophages.The addition of recombinant Sonic Hedgehog ligand (100 nM) for 24 hours notably upregulates (a) Arg1 (p < 0.0001 and p = 0.0008) and (b) Cd206 (p < 0.0001 and p = 0.001). Shh ligand treatment was done in serum-free conditions. (c) Gli1 expression (p = 0.0007 and p < 0.0001) is an indication of activated Hh signaling. Gli inhibitor, GANT61 (10 μM for 24 hours) reduces the expression of (d) Arg1 (p < 0.0001) and (e) Cd206 (p = 0.0082). Transcript levels of Arg1 (p = 0.0023 for sh802 and p = 0.0032 for sh803) and Cd206 (p = 0.0087 for sh802 and p = 0.0015 for sh803) are significantly decreased in polarized RAW 264.7 cells transduced with an RFP expressing short hairpin RNA targeting Gli1 (p < 0.0001 for both shRNAs) (f). The expression of (g) Arg1 (p < 0.0001) and (h) Cd206 (p = 0.016) was significantly decreased in M2 polarized macrophages treated with the SMO inhibitor, BMS-833923 (2.5 μM). (i) Phagocytosis of bacterial particles was assessed using fluorescence microscopy in M2 stimulated macrophages inhibited for Hh signaling with BMS-833923 (2.5 μM) (p < 0.0001) or Vismodegib (20 μM) (p = 0.0006). Inhibition of Hh signaling significantly upregulates phagocytic activity. *statistically significant difference.
Inhibition of Hedgehog signaling facilitates classical activation of macrophages
Having demonstrated that alternative polarization of macrophages elicits Hh activation, we undertook investigations to evaluate the role of this signaling in classically activated macrophages. Polarization of macrophages towards an inflammatory type was characterized by significant upregulation of the expression of inflammatory genes iNos, Il-12, and Tnf-α (Figure 3(a-c)). Concomitantly, Gli1 expression was significantly downregulated (Figure 3(d)). When classically polarized macrophages were treated with GANT61, there was a significant upregulation in iNos expression (Figure 3(e)); Il-12 did not demonstrate a change but Tnf-α increased significantly (Figure 3(f-g)). The decrease in Gli1 expression in the presence of GANT61 substantiates inhibition of Hh signaling (Figure 3(h)). Thus, Hh signaling is decreased during classical polarization of macrophages and its inhibition potentiates expression of inflammatory cytokines.
Figure 3.
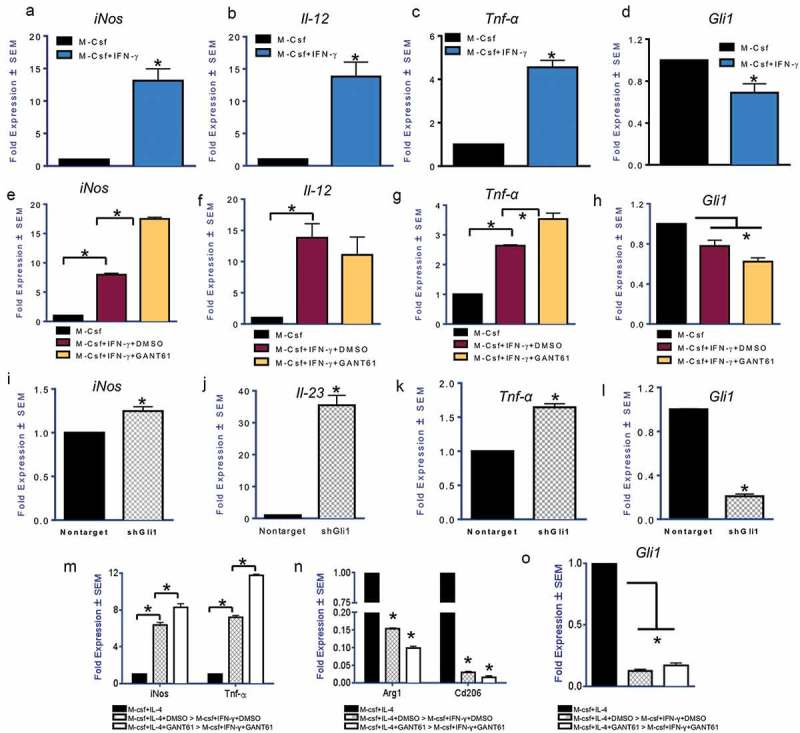
Inhibition of Hedgehog signaling facilitates classical activation of macrophages (a-d) Transcript levels of iNos (p = 0.029), Il-12 (p = 0.022), and Tnf-α (p = 0.0085), in macrophages treated with IFN-γ (50 ng/ml for 24 hours) are significantly increased with a concomitant reduction in the levels of Gli1 (p = 0.048). (e-h) Treatment of inflammatory macrophages with GANT61 (10 μM) causes an upregulation in the expression of iNos (p = 0.0002) and Tnf-α (p = 0.008). Il-12 expression did not change significantly. Gli1 transcript levels are reduced (p = 0.03) indicative of reduced potency of Hh signaling. (i-l) Stable silencing of Gli1 (p = 0.0008) in RAW 264.7 cells is consistent with an increase in the expression of iNos (p = 0.009), Il-23 (p = 0.0089), and Tnf-α (p = 0.0020) when assayed in M2 polarizing conditions. (m-o) Macrophages initially polarized to be immunosuppressive can be reversed to assume an inflammatory phenotype when treated with IFN-γ (50 ng/ml). Inhibition of Gli with GANT61 (p = 0.004) permits a further increase in expression of iNos (p = 0.007 and 0.023) and TNF-α (p = 0.008 and 0.005) with a concomitant decrease in Arg1 (p = 0.002 and 0.01) and Cd206 (p = 0.0007 and 0.002). *statistically significant difference.
We hypothesized that inhibition of Hh signaling in alternatively polarized macrophages may pivot their expression of inflammatory cytokines. While inhibition of Hh signaling in M2 polarized macrophages dampened their alternative polarization (Figure 2), GANT61 enhanced the expression of iNos and Il-23 in alternatively polarized macrophages (Supplementary Figure 6C). In agreement with this, we registered a significant potentiation in inflammatory markers iNos (Figure 3(i)), Il-23 (Figure 3(j)), and Tnf-α (Figure 3(k); Supplementary Figure 6D) in macrophages stably silenced for Gli1 (Figure 3(l)). Tnf-α cytokine levels were also elevated in macrophages treated with GANT61 or the SMO inhibitor BMS-833923 (Supplementary Figure 6E). BMS-833923 also significantly increased the levels of Tnf-α, Il-23, and iNOS (Supplementary Figures 6F-H). This was substantiated by notably increased iNOS activity and iNOS protein levels (Supplementary Figure 6H) and accompanied by decreased expression of arginase (Supplementary Figure 6I). We then investigated whether inhibiting Hh signaling in M2 macrophages primes them for reversal towards an inflammatory type. We treated M2 polarized macrophages with GANT61, then changed the medium to provide an inflammatory stimulus (IFN-γ). As depicted in Figure 3(m), GANT61 primed M2 macrophages to acquire an inflammatory phenotype when stimulated with IFN-γ as indicated by the upregulation of iNos and Tnf-α and concomitant reduction of M2 signature markers Arg1 and Cd206 (Figure 3(n)). Thus, inhibiting Hh signaling (Figure 3(o)) in alternatively polarized, immunosuppressive M2 macrophages enables their plasticity and conversion to an inflammatory macrophage.
Mammary-tumor derived Hh ligands promote alternative polarization of macrophages
Breast tumor cells are known to promote alternative polarization of macrophages.29 In order to investigate whether the crosstalk between breast cancer cells and macrophages elicits activation of Hh signaling, we cultured macrophages with conditioned medium (CM) derived from two invasive and metastatic mouse mammary carcinoma cells, 4T07 and 4T1.30 Conditioned medium from both cell types potentiated the expression of Arg1 and Cd206 (Figure 4(a-d)). Inhibiting Hh signaling in macrophages with Gli-targeting GANT61 or Smo inhibitor KAAD-cyclopamine attenuated the effects of conditioned medium (Figure 4(a-d)) simultaneous with a reduction in the expression of bonafide Hh/Gli target genes, Ptch and Gli1 (Supplementary Figure 7A). Both mammary carcinoma cells expressed elevated Shh and Dhh (Ihh was not detectable) (Supplementary 7B) relative to normal mammary gland-derived NMuMG cells. In order to assign a role for Hh ligands produced by mammary carcinoma cells, we used the 5E1 antibody to neutralize Hh ligands from the medium.31 Squelching Hh ligands from the 4T1 tumor cell CM decreased M2 associated makers Arg1, Cd206, and Il-10 (Figure 4(e-g)). Similarly, squelching Hh ligands from 4T07-derived CM blunted its effects on macrophage polarization (Figure 4(h-j); Supplementary Figure 7C). Notably, Hh/Gli signaling is also important in the dialog between human macrophages and breast cancer cells. The M2 polarization of THP-1 human monocytic cells was characterized by a significant increase in GLI1 expression (Supplementary Figure 8A). Inhibiting Hh/GLI signaling led to a decrease in M2 signature genes Fibronectin 1, CCL22, and MRC1 (CD206) with a concomitant increase in TNF-α (Supplementary Figure 8B). Squelching Hh ligands from the conditioned medium of two triple-negative breast cancer cells, SUM1315 and SUM159, decreased expression of CCL22 and CD206 with a simultaneous increase in TNF-α (Supplementary Figure 8C). As such, inhibiting Hh signaling at all levels – Hh ligand availability, SMO activity, and Gli activity – decreased the ability of macrophages to become alternatively polarized. Collectively, the data demonstrate that tumor-derived Hh ligands promote alternative polarization of macrophages.
Figure 4.
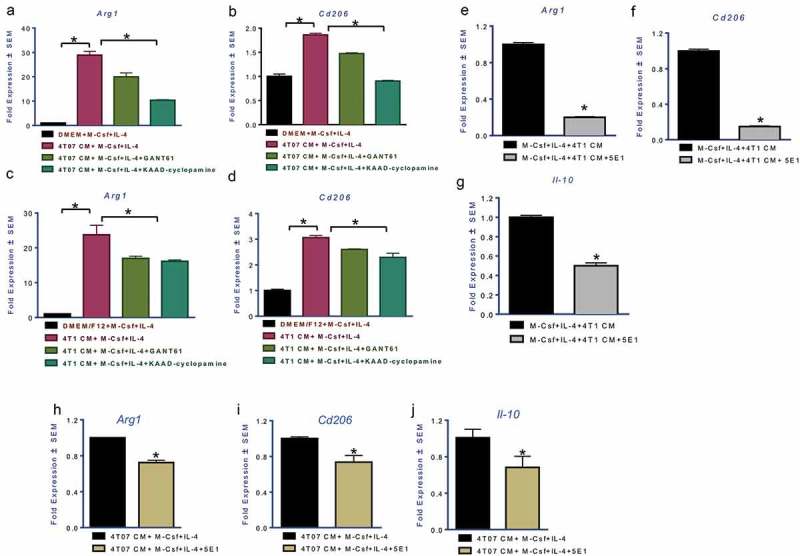
Mammary carcinoma cells upregulate alternative polarization of macrophages by eliciting activation of Hedgehog signaling Transcript levels of Arg1 (a (p = 0.0008), c (p = 0.01)) and Cd206 (b (p = 0.002), d (p = 0.002)) are significantly upregulated in presence of conditioned medium (CM) from 4T07 and 4T1 invasive mammary carcinoma cells. Treatment with GANT61 (10 μM) or KAAD-Cyclopamine (20 μM) significantly decreased the immunosuppressive polarization induced by the CM (Arg1 in 4T07 cells: p = 0.0034; Arg1 in 4T1 cells: p = 0.022; Cd206 in 4T07 cells: p = 0.0013; Cd206 in 4T1 cells: p = 0.006). The addition of Hh ligand squelching antibody, 5E1 (2.5 μg/ml), significantly decreases the effect of the 4T1 and 4T07 CM on its ability to induce a potent M2 phenotype marked by Arg1 (e, h), Cd206 (f, i), and Il-10 (g, j) (p < 0.0001 for all comparisons). *statistically significant difference.
Hedgehog signaling alters molecular mechanisms that dictate alternative macrophage polarization
To understand the role of Hh signaling in M2 activation of macrophages, we adopted an unbiased kinomics approach. This technique analyzes phospho-peptide alterations in the perspective of known kinase signatures to identify changes in the cellular phospho-signaling network (Supplementary Figures 9A, B). Kinomic analysis revealed significant changes in a network encompassing STAT6, p38 MAPK, and JAKs (Figure 5(a)). Phosphorylation of p38 follows activation of Il-4ra and precedes STAT6 activation.32 GANT61 decreased the levels of phospho-p38 and also reduced phosphorylation of upstream signal transducers, JAK1 and JAK3 (Figure 5(b); Supplementary Figure 9C). Phosphorylation of STAT6 on the tyrosine 641 residue is essential for the dimerization and translocation to the nucleus where it promotes transcription of M2 target genes. This signature phosphorylation of STAT6 was notably reduced in M2 macrophages treated with GANT61 or macrophages silenced for Gli1 expression (Figure 5(b); Supplementary Figure 9D).
Figure 5.
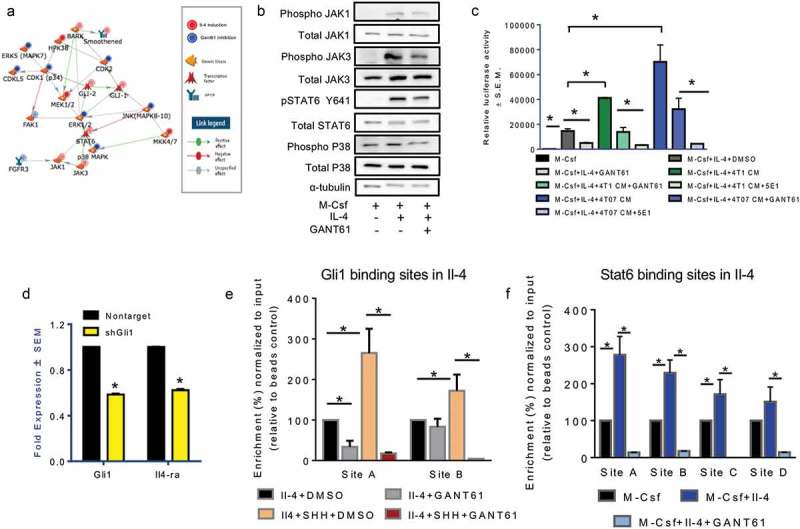
Hedgehog signaling mediates molecular mechanisms that dictate alternative macrophage polarization and its blockade promotes a pro-inflammatory, immunogenic profile. (a) Network model generated from kinomics assessment of M2 polarized macrophages treated with DMSO (vehicle control) or GANT61 (10 μM). Phosphorylated tyrosine and serine/threonine residues were assessed using PamGene PamStation Kinomic Array platform. (b) Inhibition of Hh signaling decreases phosphorylation of JAK1, JAK3, STAT6, and p38 kinases. Lysates from M2-polarized RAW 264.7 cells treated with DMSO (vehicle control) or GANT61 (10 μM) were assessed for total JAK1, JAK3, STAT6, p38 and pJAK1, pJAK3, pSTAT6, and p-p38 expression using western blot analysis. α-tubulin was used as a loading control. (c) Inhibiting Hh signaling significantly decreases the activity of the STAT6 luciferase reporter (p = 0.0007). CM from 4T1 (p = 0.0002) and 4T07 cells (p = 0.0022) potently activates STAT6 activity; squelching of Hh ligands with 5E1 antibody (4T1CM: p < 0.0001; 4T07 CM: p = 0.001) or inhibition with GANT61 (4T1CM: p = 0.02; 4T07 CM: p = 0.0015) significantly decreased the ability of the CM to activate STAT6. RAW 264.7 cells were transfected with the p4X STAT6 plasmid, and cultured to induce M2 polarization in presence or absence of vehicle control, DMSO, or GANT61 (10 μM) or CM from 4T1 and 4T07 cells with or without 5E1 (2.5 μg/ml) or GANT61 (10 μM). *p < 0.05. (d) The transcript levels of Il-4Ra (p < 0.00010) are significantly decreased in macrophages silenced for Gli1 (p < 0.0001) and polarized toward the M2 phenotype. (e) ChIP was performed in macrophages treated with M-Csf and Il-4 and DMSO or GANT61 (10 μM) in the presence and absence of SHH (100 nM) using an anti-GLI1 antibody followed by qPCR with primers specific for two putative Gli1 binding sites on the IL-4 promoter. (f) Stat6 was ChIP-ed in macrophages treated with M-Csf and Il-4 and DMSO or GANT61 (10 μM) followed by qPCR with primers specific for four putative Stat6 binding sites on the IL-4 promoter. *statistically significant difference.
In order to dissect the impact of Hh signaling on molecular mechanisms underlying alternative macrophage polarization, we evaluated the IL-4/STAT6 signaling axis, a major signaling conduit that regulates the expression of genes characteristic of M2 macrophages. While STAT6 activity was upregulated with IL-4, GANT61 significantly downregulated STAT6-mediated transcription activity (Figure 5(c)). Tumor cell-derived conditioned medium further potentiated STAT6 activity. Squelching Hh ligands from tumor cell-derived medium with 5E1 or inhibiting GLI proteins in macrophages with GANT61 significantly reduced STAT6 mediated transcription activity (Figure 5(c)). One of the main transcriptionally activated targets of STAT6 activity is Il-4ra.33 Thus, we hypothesized that Hh signaling enables the activation of a feed-forward loop of STAT6-Il-4ra. We evaluated Il-4ra levels in GANT61 treated or Gli1-silenced macrophages. M2 polarized macrophages generated from these cells showed significantly reduced transcript levels of Il-4ra (Figure 5(d); Supplementary Figure 9E). This is in agreement with decreased P-STAT6 in Gli1-silenced macrophages treated with Il-4 (Supplementary Figure 9D). In order to determine the molecular nature of this regulation, we assessed by chromatin immunoprecipitation if Il-4ra and Il-4 are direct transcriptional targets of STAT6 and Gli1. The promoters of both genes bear putative binding sites for STAT6 and Gli1 (Supplementary Figure 10A). Binding of Gli1and STAT6 to the respective sites in the promoter of Il-4 is diminished in presence of GANT61 (Figure 5(e-f)). SHH enriched the occupancy of Gli1 at the Il-4 promoter that was significantly decreased with GANT61 (Figure 5(e)). STAT6 and Gli1 also appear to directly regulate ll-4ra (Supplementary Figure 10B-C. The primer sequences used for these analyses are listed in Supplementary Figure 10D). Through these studies we have identified that Gli1 directly impacts the activation state of M2 macrophages.
In order to identify changes in the overall transcript profile of M2 macrophages, we analyzed changes in global gene expression profile by RNAseq analysis. Inhibiting Hh signaling in M2 polarized macrophages has distinct effects on the gene expression profile (Figure 6(a)). The gene signature of GANT61-treated immunosuppressive macrophages was similar to that of macrophages from genetically engineered STAT6 knockout BALB/c mice (Figure 6(b)). Gene set enrichment analysis of M2 macrophages demonstrates an immunosuppressive signature while macrophages treated with GANT61 showed an overlap with enhanced MHC processing signature of dendritic cells as well as upregulated inflammatory responses in bacterial infections (Figure 6(c); Supplementary Table 1) indicating that the inhibition of Hh signaling in M2 macrophages dampens their anti-inflammatory functions and enhances their antigen presentation abilities and immune system activation properties. Together, this data shows that Hh inhibition in M2 macrophages mechanistically alters their polarization and promotes a reversal of their anti-immunogenic functions.
Figure 6.
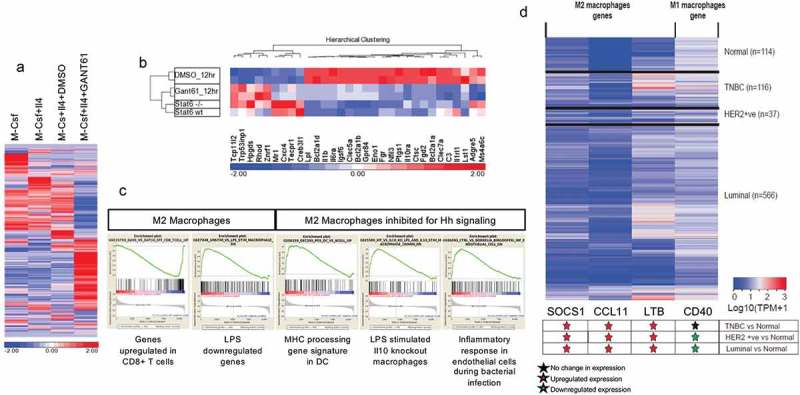
Inhibiting Hedgehog signaling upregulates an inflammatory gene expression signature (a) Heat map of the gene signature of macrophages derived from RAW 264.7 cells treated with M-Csf (M0 state), M-Csf and IL-4 (M2 macrophages), M-Csf, IL-4, and DMSO (M2 and vehicle control), and M-Csf, IL-4, and 20 μM GANT61 (M2 inhibited for Hh signaling). GANT61-treated macrophages demonstrate clear differences from vehicle controls. Log2-transformed RPKM-normalized intensities were used for Z-normalization with color indicating above (in red) or below (in blue) average. (b) GANT61-treated macrophages demonstrate similarity to Stat6 (-/-) macrophages, although Stat6 (-/-) and Stat6 (WT) show fewer differences among these genes. (c) Gene set enrichment analysis of RNAseq data depicts that the molecular gene signature of M2 macrophages is characteristically immunosuppressive. Inhibition of Hh/Gli signaling reveals an overlap with inflammatory signature in bacterial infections and antigen presentation by dendritic cells. (d) Heatmap showing expression level of M1 and M2 macrophage genes in normal (n = 114), HER2 Positive (n = 37), Luminal (n = 566), and triple negative breast cancer [TNBC] (n = 116) samples from TCGA breast invasive carcinoma [BRCA] dataset. Transcript per million (TPM) value for each sample is obtained by multiplying RSEM scaled_estimate by 106.The significance of differential expression is estimated via student’s T-test.
In order to ascertain the clinical significance of M2 macrophages in breast cancer, we queried the TCGA breast cancer data for STAT6 target genes that critically impact alternative polarization of macrophages. STAT6 target genes (SOCS1, CCL11 and LTB) were all significantly upregulated across all breast cancer subtypes (Figure 6(d)). The inflammatory macrophage marker CD40 was downregulated in HER2-positive and luminal breast cancer subtypes. As such, breast cancers show attributes indicative of enrichment of immunosuppressive macrophages.
Macrophage depletion improves the benefit of inhibiting Hedgehog signaling on eliciting a pro-inflammatory, immunogenic profile
Thus far our data provides evidence for Hh/Gli signaling in eliciting an immune suppressive tumor milieu and in mechanistically influencing an alternative M2 activation state in macrophages. In order to investigate the possibility of eliminating macrophages to ameliorate the activity of Hh inhibition, we depleted the animals of macrophages using liposomal clodronate followed by Vismodegib administration (Figure 7(a); Supplementary Figure 11). While Vismodegib in combination with control liposomes elicited apoptosis, the overall extent of apoptosis was significantly increased when Vismodegib was co-administered with liposomal clodronate (Figure 7(b)). We then analyzed the primary tumor mass for its portfolio of immune-suppressive and immune-stimulatory cells. The combination of liposomal clodronate with Vismodegib yielded a significantly reduced population of M2 polarized macrophages, MDSCs, Th2 cells and Treg cells (Figure 7(c-f)). Simultaneous with this, we registered a significant increase in the inflammatory M1 macrophages, dendritic cells, cytotoxic T cells, and Th1 cells (Figure 7(g-j)). Cumulatively, the data clearly demonstrates that clodronate treatment improves the effectiveness of Hh inhibition on re-configuring the immune cell portfolio of the mammary tumor to a more potent immune-stimulatory type.
Figure 7.
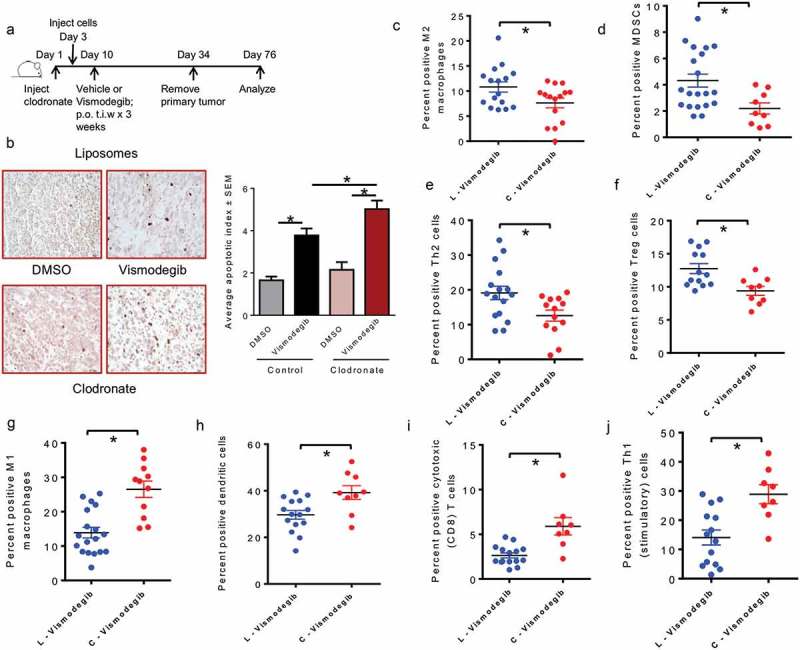
Macrophage depletion improves the benefit of inhibiting Hedgehog signaling on eliciting a pro-inflammatory, immunogenic response (a) Schematic of macrophages depletion using liposomal clodronate followed by Hh inhibition strategy used in vivo. (b) Tumor sections from DMSO and Vismodegib-treated mice were assessed for apoptosis by TUNEL staining. Tumors from Vismodegib-treated mice in the control liposomes (L) group show significantly greater levels of apoptotic cells relative to vehicle-treated mice (p < 0.0001). Tumors from Vismodegib-treated mice depleted for macrophages with clodronate (C) display a significantly higher apoptotic index than their vehicle treated counterparts (p < 0.0001). Following resection and processing of the primary tumor, non-viable cells were excluded and immune cell infiltrates were assayed based on the following markers: (c) M2 macrophages: CD11b, LY6C-ve, F4/80 + ve, Arg1, and CD206 positive cells (p = 0.0151). (d) Myeloid-derived suppressor cells: CD11b, LY6G and LY6C double positive cells (p = 0.0133). (e) Type II helper T cells: CD3, CD4, and GATA3 positive cells (p = 0.0184). (f) Regulatory T cells: CD3, CD4, and FOXP3, and CD25 positive cells (p = 0.0056). (g) M1 macrophages: F4/80, CD80 and CD86 positive cells (p < 0.0001). (h) Dendritic cells: CD11c, MHC II and CD86 positive cells (p = 0.0085). (i) Cytotoxic CD8 T cells: CD3, CD8, and Granzyme B positive cells (0.0005). (j) Activated Type I helper T cells: CD3, CD4, and IFN-γ positive cells (p = 0.0021). *p< statistically significant difference relative to DMSO-treated mice.
Discussion
Since the pathway was first identified, years of research have cemented the tumorigenic role of Hh signaling in multiple tumor models. The autonomous activation of Hh signaling in tumor cells is vital for carcinogenesis, tumor progression, and metastasis. Studies in pancreatic ductal adenocarcinoma (PDAC) specifically,34,35 have characterized paracrine activation of Hh signaling where tumor-derived ligands activate Hh signaling in stromal cells and facilitate the establishment of a metastatic niche. Crosstalk between tumors and surrounding stroma leads to microenvironmental changes that favor immune evasion of tumor cells, leading to tumor progression and metastasis. However, the influence of Hh signaling on tumor associated immune cells remains largely undiscovered. Our investigations have elucidated a functional link between aberrant Hh signaling in breast cancer cells and alternative macrophage polarization, thereby characterizing a novel mechanism by which Hh signaling dictates immune suppression.
Recent studies have highlighted the significance of macrophages in breast cancer progression. In solid tumors, TAMs comprise approximately 5−50% of the tumor mass.29 IL-4 produced by CD4 + T cells in mammary tumors polarizes macrophages toward an immunosuppressive TAM phenotype expressing Arg1 and TGF-ß. The upregulation of HIF-1α and HIF-2α in macrophages encountering a hypoxic tumor milieu enables metabolic adaptation to an oxygen limiting environment and further facilitates immunosuppressive functions.7 Murray et al have proposed a set of standards encompassing three guiding principles for macrophage nomenclature – the source of macrophages, definition of the activators, and a consensus collection of markers to describe macrophage activation.36 Macrophage polarization is a continuum that spans two extremes from the classically activated M1 macrophages to the alternatively activated M2 macrophages. The tumor microenvironment principally pivots the tumor-killing M1 macrophages to tumor-promoting M2 macrophages. While M1-dominant macrophages stimulate naïve T cells to launch a Th1/cytotoxic response, M2-dominant macrophages stimulate a Th2-type response associated with antibody production.37 Linde et al., have reported a causal role for macrophages in early dissemination of breast cancer cells that affects long-term metastasis.38 The enrichment of infiltrating macrophages in the breast tumor microenvironment correlates with higher tumor grade and lower relapse-free survival in patients.4,7 High numbers of CD163+ M2-macrophages were strongly associated with fast proliferation, poor differentiation, estrogen receptor negativity, and histological ductal type.29 Thus, targeting macrophages presents as a novel therapeutic strategy for breast cancer treatment. Indeed, in vivo investigations have employed different agents to block several processes involved in macrophage recruitment and maturation including CCL2-CCR239 and CSF1/CSF1R blockade,40 both of which have been launched into clinical trials in solid tumors. Though these approaches are promising, they systemically abrogate the recruitment and activation of all macrophages, including the anti-tumorigenic M1 phenotype. Thus, more appealing approaches have emerged to particularly target and reverse the polarization of pro-tumorigenic M2 macrophages and reprogram them to the immune stimulating, anti-tumorigenic M1 macrophages. Several groups have employed this strategy in vivo to stimulate immunogenic macrophages by administering CD40 agonists41 and stimulating STAT1 function.42 However, these previously mentioned approaches exclusively target macrophages, which are significant players in immune tolerance, but not necessarily the primary drivers for breast cancer tumorigenesis. Through our investigations, we report for the first time, that Hh signaling regulates a transcriptional program and underlying molecular mechanisms that dictate alternative macrophage polarization towards the M2 phenotype. Importantly we report that breast cancer cells polarize macrophages towards the immunosuppressive M2 type by a paracrine crosstalk that is mediated by Hh ligands produced by the tumor cells. Interfering with the crosstalk significantly attenuated the M2 phenotype. Inhibiting Hh signaling even primed the M2 macrophages to respond more potently to inflammatory stimuli and set the macrophages on a path towards reverting to M1 macrophages. In alignment with the priming of macrophages towards an M1 type, we were able to identify molecular signatures associated with enhanced pro-inflammatory as well as antigen processing and presentation attributes.
We uncovered a molecular network that identified that Hh signaling interfaces with the IL-4/STAT6 signaling cascade mediated by the involvement of p38 MAPK. p38 activation precedes STAT6 activation downstream of IL-4 binding to its receptors; inhibition of p38 blocks STAT6 activation and reduces the expression of M2 target genes.32 Our investigations reveal that inhibiting Hh signaling disengages the feed-forward loop that amplifies IL-4ra. Hh signaling has been reported to stimulate the differentiation of type II helper T cells through Gli2 induction of IL-4 expression.43 Through this investigation we have identified Gli1 occupancy on the promoters of Il-4 and Il4ra; this is significantly decreased with GANT61. Thus, we have established that the feed-forward loop involving Il-4 and Il-4r is disrupted when Hh signaling is inhibited. This consequently decreases STAT6 phosphorylation when Hh is inhibited. Our findings cumulatively underscore the immune-inhibitory effects of Hh signaling in macrophages.
These mechanistic findings were recapitulated in the mammary tumor microenvironment that was characterized by remarkable changes in the portfolio of tumor infiltrating immune populations when the animals were treated with the orally available SMO inhibitor, Vismodegib. Overall, Vismodegib increased the population of immune cells with anti-tumor activity. The most striking changes we observed were in dendritic cell infiltrates and cytotoxic CD8-positive T cell numbers; both cell types were upregulated in Vismodegib-treated mice. Concomitantly, the pro-tumorigenic M2 polarized macrophages and MDSCs were characteristically reduced. Interestingly, the numbers of CTLA4-expressing and PD-1 expressing inhibitory cells were also reduced. Therapeutic blocking antibodies against CTLA4 and PD-1 co-inhibitory receptors have reached routine clinical use. Suppression of the CTLA4 and PD-1 pathway enables the expansion of tumor-specific T cells.44 Thus, the decrease in the CTLA4-expressing and PD-1 expressing T-cells represents a reduction in the population of exhausted T-cells and corroborates with an increase in the population of granzyme B expressing CD 8-positive T-cells. These altered T-lymphocyte profiles may contribute to an elevated apoptotic tumor cell population in the Vismodegib-treated tumors.
In agreement with the mechanistic changes identified, Vismodegib-treated animals showed overall reduced arginase activity of the tumor-associated macrophages. As such, inhibition of Hh signaling reduced the functional potency of the immunosuppressive macrophages with simultaneous upregulation of the antigen presenting cells and adaptive cytotoxic cells. These findings may explain the increased tumor cell apoptosis in Vismodegib-treated animals. The cumulative changes in the tumor were reflected in overall decreased pulmonary metastases, indicating that the changes in the tumor-associated immune portfolio had determinative effects on the metastatic potential of the tumor cells. This combination of immune cell changes establishes a role for Hh inhibition in causatively promoting the anti-tumor immunogenic response. Given the prominent role of infiltration of TAM in immune suppression, angiogenesis, and tumor aggressiveness, we tested the potential benefit of depleting macrophages on the benefits of the Hh inhibitor. We employed the liposome mediated macrophage “suicide” approach to eliminate macrophages.45 The combination of macrophage depletion and Hh inhibitor generated a robust antitumor immune portfolio, thereby offering additional insights into the interaction between Hh signaling and immune functions and offer a novel therapeutic approach for interfering in tumor progression through re-configuring the immune microenvironment.
With regards to its role in influencing the immune system, Hh signals for differentiation, survival and proliferation in the early stages of T cell development, before T cell receptor (TCR) gene rearrangement. In more mature T lineage cells, Hh signaling also modulates TCR signal strength46–48 and can impact CD4(+) T cell effector function.49 As such, the studies pertaining to the involvement of Hh signaling have principally investigated its role in T cell development and effector functions. Our investigation provides evidence for Hh signaling in influencing the ratio of tumor-associated Teffs:Tregs and Teffs:MDSCs. Specifically, inhibition of Hh with Vismodegib increased these cellular proportions, clearly signifying a shift from an immunosuppressive infiltrating configuration to an immune reactive type.
We provide compelling evidence that demonstrates the determinative effects of the Hh signaling pathway on programmatically altering molecular mechanisms of M2 macrophages. Additionally, this work offers a multi-faceted basis for using Hh inhibitors to target breast cancer progression, not just through effects on tumor cells, but also the tumor immune compartment. Our findings demonstrate that Hh signaling inhibition not only diminishes pro-tumorigenic myeloid populations of M2 macrophages and MDSCs, but it also sculpts macrophages to assume an anti-tumor phenotype and propels the activation of tumor-associated cytotoxic T cells. Overall, we establish a novel role for the Hh signaling pathway in functionally editing the innate immune system in breast cancer and eliciting the inflammatory response of the adaptive immune cells. Thus, inhibiting Hh signaling can re-program the dysfunctional tumor immune microenvironment and mitigate breast cancer metastasis.
Materials and methods
Cell culture
4T1 murine tumor cell line was cultured in Dulbecco’s modified minimum essential medium/F12 nutrient mixture supplemented with 2.5% heat-inactivated fetal bovine serum. RAW 264.7 and 4T07 murine cells were cultured in Dulbecco’s modified Eagle’s medium (Life Technologies, Carlsbad, CA) supplemented with 5% fetal bovine serum (Gibco Life Technologies). Cell lines were maintained at 37°C in a humidified 5 % CO2 incubator.
RAW 264.7 cells were stably silenced for endogenous Gli1 using SMARTvector lentiviral shRNA cloned into a murine CMV-RFP plasmid (GE Dharmacon, Lafayette, CO). Cells were transfected with Lipofectamine 2000 (Life Technologies) and RFP-expressing cells were selected with puromycin. A non-targeting vector plasmid was utilized as a control. 4T1 and RAW264.7 cells were obtained from ATCC approximately 8–10 years ago; 4T07 cells were gifted to us in 2014 by Dr. Yibin Kang, Princeton University50 and originally referenced in Aslakson and Miller.51 Cells were frozen in early passages, while the culture was actively growing. Cells were replaced from frozen stocks after a maximum of twelve passages or three months continuous culture. Cell lines were periodically (once every six months) confirmed negative for mycoplasma contamination using PCR assays.
BMDM prep
Tibias and femurs were resected from 6–8 week old mice, bone marrow was flushed from bones in αMEM medium supplemented with 10% fetal bovine serum and 1x Penicillin-Streptomycin using a 26G needle. Bone marrow was incubated overnight at 37°C. The following day, the bone marrow was centrifuged at 1400 RPM, and then ACK lysis buffer (Lonza, Basel, Switzerland) was added to cell pellet and incubated at 37°C for 5 minutes to eliminate red blood cells. The cells were centrifuged at 1400 RPM for 5 minutes, then M-CSF (25 ng/ml) in αMEM medium supplemented with 10% fetal bovine serum and 1x Penicillin-Streptomycin was added to cells and incubated for 7 days to differentiate bone marrow monocytes into macrophages, with one media change on day 4 post plating.
Quantitative real-time polymerase chain reaction
RNA from cells was harvested using Qiagen RNeasy Mini Kit (Valencia, CA) according to protocol. cDNA was synthesized from total RNA using High Capacity cDNA Reverse Transcription Kit (Life Technologies) under the following protocol: 25°C for 10 minutes, 37°C for 120 minutes, 85°C for 5 seconds. Real time PCR was performed using TaqMan Fast Advanced Master Mix (Applied Biosystems) and Taqman gene expression assay probes for Il-10, Arginase 1, CD206, iNos, Il-23, Il-12, Tnf-α, Gli1, Gli2, and Ptch1 (Life Technologies, Carlsbad, CA). Experiments were performed in triplicate and GAPDH served as an endorse control gene.
Western blotting
Cells (RAW 264.7) were treated with M-Csf (20 ng/ml) overnight followed by DMSO or GANT61 (10 μM) four hours prior to treatment with IL-4 (10 ng/ml) for 15 minutes. Whole cell lysates were collected in NP-40 lysis buffer and immunoblotted for total JAK1 (#3332S), phospho JAK1 Y1022/1023 (#3331S), total JAK3 (#8863S), phospho JAK3 Y980/981 (#5031S), total p38 (#9212S), phospho p38 Thr180/Tyr182 (#9211S), total STAT6 (#9362S), and phospho Tyr641 STAT6 (#9361S; Cell Signaling, Danvers, MA). To assess protein levels of Arginase 1and iNOS, RAW 264.7 cells (stably transfected with a non-target shRNA, Gli1shRNA, or treated with GANT61 (10 μM) or BMS-833923 (5 μM)) were treated with M-csf (20 ng/ml) and IL-4 (10 ng/ml) for 24 hours. Whole cell lysates were collected in NP-40 lysis buffer and immunoblotted for Arginase 1 (#93668S, Cell Signaling) or iNOS (#2982, Cell Signaling).
Kinomic profiling
Cells (RAW 264.7) were treated with M-Csf (20 ng/ml) overnight followed by DMSO or GANT61 (10 μM) four hours prior to treatment with IL-4 (10 ng/ml) for 15 minutes. Whole cell lysates were collected in M-PER Mammalian Protein Extraction Reagent (Thermofisher, Rockford, IL) supplemented with Halt protease and phosphatase inhibitors (1:100; Thermofisher). Lysates were loaded onto PamChip microarray tyrosine or serine/threonine chips imprinted with 144 tyrosine or 144 serine/threonine kinase substrates, respectively. Changes in individual peptide phosphorylation were imaged with FITC conjugated phospho-specific antibodies, and signal was quantified using BioNavigator. Lists of altered peptides were exported and analyzed for probable upstream kinases with tools such as Kinexus Phosphonet, as well as advanced Pathway Analysis and network modeling using GeneGo MetaCore.
Reporter assays
RAW 264.7 were transfected with reporter plasmids (p4X STA6 or pGreenFire-Gli) using Lipofectamine 2000 (Life Technologies). P4X STAT6 consists of 4 STAT6 binding sites upstream of a luciferase gene (Addgene, Cambridge, MA). pGreenFire-Gli encodes four Gli transcription response element sites upstream of GFP and luciferase genes (System Biosciences, Palo Alto, CA). Cells were treated with 20 ng/ml M-Csf overnight and with DMSO or 10 μM GANT61 four hours prior to treatment with 10 ng/ml IL-4 for 12 hours. Cells were lysed and luminescence was measured using GloMax Luminometer (Promega, Madison, WI). The readings were normalized to total protein concentration.
Next-generation sequencing
mRNA-sequencing was performed on the Illumina HiSeq2500. Briefly, the quality of total RNA was assessed using the Agilent 2100 Bioanalyzer. Total RNA (RIN of ≥ 7.0) was used for the RNA library prep. The SureSelect Stranded mRNA library generation kit was used as per the manufacturer’s instructions (Agilent, Santa Clara, CA) to generate the sequence ready libraries. mRNA-Sequencing was done on the Illumina HiSeq2500 with onboard clustering by loading 11pM of the pooled libraries and paired end 50bp sequencing by standard techniques (Illumina, Inc., San Diego CA). Approximately 30 million reads were generated per sample.
Gene expression data analysis
Gene expression analysis was performed using UAB Cheaha HPC cluster, Galaxy platform and software packages of Partek Flow and Genomics Suite (PGS, Partek, St Louis, MO). Briefly, sequence reads in fastq format were trimmed at each end of reads using read quality score,52 vendor sequencing adaptors removed, and reads aligned against human genome hg19 using STAR algorithm.53 Resulting bam files were used for gene expression quantification after removing PCR duplicates using samtools (http://www.htslib.org). Reads per kilobase of transcript per million mapped reads (RPKM) normalization54 was performed before any statistical analysis. For GEO dataset, gene level quantification and statistics were performed using Partek PGS. For hierarchical clustering, gene list of significant changes of various comparisons were used to generate heatmap. Log2-transformed RPKM-normalized intensities were used for Z-normalization. Gene Set Enrichment Analysis (GSEA) including leading edge analyses were performed using software from Broad Institute.55,56
Chromatin immunoprecipitation (ChIP)
Cells were pretreated with 10 µM GANT61 for 4 hours and then treated with 20 ng/ml M-Csf with or without 20 ng/mL Il-4 for 1 hr in combination with GANT61 or DMSO and/or SHH (100 nM). After one hour cells were processed using the Simple Chip Plus Enzymatic kit (Cell Signaling) as per manufacturer’s protocol. 10 µg of cross-linked chromatin was immunoprecipitated with either 2 µg of anti-Stat6 (Cell Signaling) or anti-Gli1 (Novus). Chromatin was eluted from the IP and cross-links were reversed followed by column purification of DNA. Purified DNA from ChIP and input was subjected to real-time quantitative PCR to quantitate the amount of DNA associated with Gli or Stat6 in the Il4ra or Il4 promoter sequence. PCR was done using 2X Maxima SYBR Green Master Mix (Thermo Scientific) with primer pairs to amplify various regions of the respective promoters. Primer pairs and associated schematic are detailed in Supplementary Figure 9. CT values of input DNA was used to calculate percent input of immunoprecipitation utilizing the following calculation: Percent Input = 2% x 2(C[T] 2% Input Sample-C[T] IP Sample) and percent enrichment as compared to corresponding controls is depicted. Each reaction was done in triplicate using an Applied Biosystems Step One Plus.
TUNEL staining
Tumors were fixed in neutral-buffered formalin and embedded in paraffin. Sections were cut and stained using the Click-iT TUNEL Colorimetric IHC Detection Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Nuclei were counted in ten fields for each sample. The apoptotic index was determined using the following formula: number of TUNEL positive nuclei/total number of nuclei in a given field x 100.
TNFα sandwich ELISA
TNFα cytokine levels in RAW 264.7 supernatants were assayed using a mouse specific sandwich DuoSet ELISA kit (DY410, R&D systems) following manufacturer’s directions. Absorbance readings were acquired at 450 nm and 540 nm to account for wavelength correction.
Nitric oxide production assay
NOS production was measured in samples using Nitric Oxide Synthase Activity Assay Kit (Abcam, Cambridge, UK, ab211083). Briefly, RAW264.7 cells were treated with M-csf (20 ng/ml) and IL-4 (10 ng/ml) for 24 hours followed by IFN-γ stimulation (50 ng/m) for an additional 24 hours. Cell lysates were generated using assay buffer supplemented with Halt protease and phosphatase inhibitors (1:100; Thermofisher) and quantified using the Eon Microplate spectrophotometer (BioTek, Winooski, VT). The assay was done per the manufacturer’s instructions. The optical density was measured in wells at 540 nm using the Eon Microplate spectrophotometer.
Animals
Luciferase-expressing 4T1 cells (5 x 105) suspended in HBSS were injected into the inguinal mammary fat pad of eight week old female BALB/c mice. Tumor progression was documented by palpation three times weekly and bioluminescent imaging (BLI) using the IVIS Imaging System (Xenogen Corp., Alameda, CA) once weekly. Briefly, mice were injected intraperitoneally with D-luciferin (150 mg/kg body weight) and anesthetized using isoflurane gas. Ten minutes later, photographic images were obtained using Living Image Software. Beginning ten days post injection of tumor cells, mice were orally gavaged with 100 μl (2 mg/mouse) of Vismodegib (Selleck Chemicals, Houston, TX) or DMSO as a vehicle control thrice weekly for 3.5 weeks. Tumors were harvested at an approximate mean tumor diameter of 5–6 mm in order to avoid confounding effects of necrosis. Lung metastases were enumerated using a Nikon StereoZoom microscope. The animal studies have been conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) of The University of Alabama at Birmingham. We have used Vismodegib at a concentration of 20 μM for in vitro studies and administered 2 mg/mouse in animal studies. This dose is consistent with that used in patients and corresponds to 100 mg/kg dose. The pharmacokinetics (PK) and pharmacodynamics of Vismodegib have been worked out; at 100 mg/kg dose, the Area Under the Curve (AUC) for the PK of Vismodegib is 413 μM/hour.57 Thus, the doses used in our studies are well below the PK of Vismodegib.
Clodronate treatment
Two days prior to the injection of luciferase-expressing 4T1 tumor cells mice were intraperitoneally injected with control liposomes or liposomal clodronates (Encapsula Nanosciences, Nashville, TN; 200 μl/mouse) and once weekly thereafter throughout the duration of the study. Tumor progression was documented by palpation three times weekly and BLI. Beginning ten days post injection of tumor cells, mice were orally gavaged with 100 μl (2 mg/mouse) of Vismodegib or DMSO as a vehicle control thrice weekly for 3.5 weeks.
Flow cytometry
Tumors were surgically excised from animals and incubated in dissociation solution using the Mouse Tumor Dissociation Kit (Milteni Biotec, San Diego, CA) for 45 minutes on a shaker at 37°C. Cells were passed through a 100 μm cell strainer (Thermo Fisher Scientific, Waltham, MA) to ensure a single cell suspension. Cells recovered were strained through a 70 μm cell strainer (Thermo Fisher Scientific). Tumor and organ-derived cells were treated with ACK buffer (Lonza, Basel, Switzerland) to lyse red blood cells. Post processing, cells were counted and incubated with an FC receptor blocker anti-mouse CD16/32 antibody (BioLegend, San Diego, CA). Cells were then stained with fixable viability dye eFluor 450 (eBioscience, now Thermo Fisher) in PBS, incubated with primary conjugated antibody cocktail specific to cell type (Biolegend), and analyzed using a BD LSRII Analyzer. In all cases, we employed FMO controls to guide our gating strategy. Analysis of the acquired files was done using FlowJo v 10 software. For samples requiring nuclear stain, True-Nuclear Transcription factor buffer set (BioLegend) was used. Sample preparation for samples requiring intracellular stains was done using a separate Fixation buffer and Intracellular Staining Permeabilization Wash Buffer (Biolegend). The gating strategies used are detailed in Supplementary Figures 12–16.
Urea production assay
Dissociated cells were stained with brilliant violet 650-conjugated F4/80 antibody. Post staining, cells were washed and F4/80 positive cells were sorted using BD FACS Aria. Cells (5 x 104) were plated in duplicate in a 96-well plate in RPMI supplemented with 1% FBS. Twenty-four hours later, urea in the supernatant from each well was assessed using QuantiChrom™ Urea Assay Kit (DIUR-100, BioAssay Systems, Hayward, CA) according to manufacturer’s instructions. Absorbance was read at 520 nm using the Eon Microplate spectrophotometer (BioTeck, Winooski, VT).
Annexin V apoptosis assay
Following tumor dissociation as described above, tumor cells were stained with Pe-Cy7 conjugated anti-mouse CD31, Ter119, and CD45 to exclude endothelial, erythroid, and lymphocyte cells respectively. Epithelial cells were stained with anti-mouse PE conjugated CD24. Annexin V staining was done according to manufacturer’s protocol (BD, Franklin Lakes, NJ).
Phagocytosis assay
Cells (RAW 264.7) were plated in a black walled 96-well plate and polarized with M-Csf+ LPS or IL-4, with or without simultaneous treatment with Hh inhibitors. Twenty-four hours post treatment, media was aspirated and 100 μl of reconstituted pHrodo™ Red E. coli BioParticles (ThermoFisher) were added to each well. Two hours later, cells were washed with PBS and red fluorescently-labeled cells were digitally documented and quantified using the 40X objective of Nikon Eclipse Ti-U microscope (Nikon, Tokyo, Japan).
Statistical analysis
For statistical analysis we used the student’s t-test or ANOVA analysis, as appropriate, and plotted using GraphPad Prism 7 software (La Jolla, CA). Statistical significance was determined for p ≤ 0.05. Specific p-values are listed in the corresponding figure legend.
Funding Statement
This work was supported by the National Institutes of Health [CA194048];National Institutes of Health [CA169202];National Institutes of Health [UL1TR001417];U.S. Department of Defense [W81XWH-14-1-0516];U.S. Department of Defense [W81XWH-18-1-0036];U.S. Department of Veterans Affairs [BX003374];
Acknowledgments
We acknowledge funding from the Department of Defense (W81XWH-14-1-0516 and W81XWH-18-1-0036), NCI R01CA169202, The Breast Cancer Research Foundation of Alabama (BCRFA), and The George G. and Amelia G. Tapper Foundation to L.A.S.; NCI R01CA194048 and BX003374 to R.S.S.; UAB Center for Clinical and Translational Science Grant Number UL1TR001417 by NCATS, NIH to D.C. The authors would like to thank: Dr. Shamik Das, Dr. Hawley C. Pruitt, Mateus Mota, Tshering Lama-Sherpa, and Dominique Hinshaw for their technical assistance with experimental procedures throughout this study; Dr. Selvarangan Ponnazhagan, Dr. Anandi Sawant, Dr. Jonathan Hensel, Dr. Tika Benveniste, and Dr. Xu Feng, D for their scientific advice and guidance throughout this study; UAB Kinomics Core under the supervision of Dr. Christopher Willey and Dr. Joshua Anderson for the kinomic study analysis; Heflin Center Genomics Core and the UAB Comprehensive Cancer Center with grant P30CA013148; The UAB Comprehensive Flow Cytometry Core supported by NIH Grants P30 AR048311 and P30 AI027667, and UAB Comprehensive Cancer Center’s Preclinical Imaging Shared Facility with technical assistance from Ms. Sharon Samuel (Center Core Support Grant P30 CA013148).
Disclosure of Potential Conflicts of Interest
None of the authors have a competing financial interest to report for this work.
Supplementary material
Supplementary material for this article can be accessed here.
Abbreviations
- CD
Cluster of differentiation
- CM
Conditioned medium
- DC
Dendritic cells
- DHH
Desert hedgehog
- GANT61
Gli antagonist
- Gli
Glioma-associated oncogene homolog
- Hh
Hedgehog
- HIF
Hypoxia-inducible factor
- IFN-γ
Interferon gamma
- IHH
Indian hedgehog
- IL
Interleukin
- JAK
Janus kinase
- M1
Classically activated macrophages
- M2
Alternatively activated macrophages
- MAPK
Mitogen-activated protein kinases
- M-csf
Macrophage colony stimulating factor
- MDSC
Myeloid-derived suppressor cells
- NK
Natural killer cells
- PDAC
Pancreatic ductal adenocarcinoma
- PTCH1
Patched 1
- SHH
Sonic hedgehog
- SMO
Smoothened
- SOCS
Suppressor of cytokine signaling
- STAT
Signal transducer and activator of transcription
- TAM
Tumor-associated macrophages
- TME
Tumor microenvironment
- TNF-α
Tumor necrosis factor alpha
- Th1
Type I helper T cells
- Th2
Type II helper T cells
- Treg
Regulatory T cells
References
- 1.Mao Y, Keller ET, Garfield DH, Shen K, Wang J.. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013;32(1–2):303–315. DOI: 10.1007/s10555-012-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turley SJ, Cremasco V, Astarita JL.. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15(11):669–682. DOI: 10.1038/nri3902. [DOI] [PubMed] [Google Scholar]
- 3.Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. 2015;33:643–675. DOI: 10.1146/annurev-immunol-032414-112220. [DOI] [PubMed] [Google Scholar]
- 4.Obeid E, Nanda R, Fu YX, Olopade OI. The role of tumor-associated macrophages in breast cancer progression (review). Int J Oncol. 2013;43(1):5–12. DOI: 10.3892/ijo.2013.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, Roy S. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. 2015;185(10):2596–2606. DOI: 10.1016/j.ajpath.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, Tsai MF, Chen CH, Yang PC. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol. 2005;23(5):953–964. DOI: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 7.Williams CB, Yeh ES, Soloff AC. Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer. 2016;2DOI: 10.1038/npjbcancer.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. DOI: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13 DOI: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kroner A, Greenhalgh AD, Zarruk JG. Dos Santos RP, Gaestel M, David S. TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron. 2014;83(5):1098–1116. DOI: 10.1016/j.neuron.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 11.Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer. 2015;15:577 DOI: 10.1186/s12885-015-1546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Veremeyko T, Siddiqui S, Sotnikov I, Yung A, Ponomarev ED. IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS ONE. 2013;8(12):e81774 DOI: 10.1371/journal.pone.0081774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem. 2011;286(3):1786–1794. DOI: 10.1074/jbc.M110.169367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–462. DOI: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lesterhuis WJ, Punt CJ, Hato SV, Eleveld-Trancikova D, Jansen BJ, Nierkens S, Schreibelt G, de Boer A, Van Herpen CM, Kaanders JH, et al. Platinum-based drugs disrupt STAT6-mediated suppression of immune responses against cancer in humans and mice. J Clin Invest. 2011;121(8):3100–3108. DOI: 10.1172/JCI43656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukushi J, Ono M, Morikawa W, Iwamoto Y, Kuwano M. The activity of soluble VCAM-1 in angiogenesis stimulated by IL-4 and IL-13. J Immunol. 2000;165(5):2818–2823. [DOI] [PubMed] [Google Scholar]
- 17.Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, Zhao YW, Wei YQ. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS ONE. 2012;7(12):e50946 DOI: 10.1371/journal.pone.0050946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niino D, Komohara Y, Murayama T, Aoki R, Kimura Y, Hashikawa K, Kiyasu J, Takeuchi M, Suefuji N, Sugita Y, et al. Ratio of M2 macrophage expression is closely associated with poor prognosis for Angioimmunoblastic T-cell lymphoma (AITL). Pathol Int. 2010;60(4):278–283. DOI: 10.1111/j.1440-1827.2010.02514.x. [DOI] [PubMed] [Google Scholar]
- 19.Medrek C, Ponten F, Jirstrom K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer. 2012;12:306 DOI: 10.1186/1471-2407-12-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, Deschoemaeker S, Van Ginderachter JA, Tamagnone L, Mazzone M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24(6):695–709. DOI: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, Movahedi K, Houbracken I, Schouppe E, Elkrim Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74(1):24–30. DOI: 10.1158/0008-5472.CAN-13-1196. [DOI] [PubMed] [Google Scholar]
- 22.Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15(6):801–812. DOI: 10.1016/j.devcel.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris LG, Samant RS, Shevde LA. Hedgehog signaling: networking to nurture a promalignant tumor microenvironment. Mol Cancer Res. 2011;9(9):1165–1174. DOI: 10.1158/1541-7786.MCR-11-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer. 2016;15:24 DOI: 10.1186/s12943-016-0509-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das S, Samant RS, Shevde LA. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to smoothened-targeting Hedgehog inhibition. J Biol Chem. 2013;288(17):11824–11833. DOI: 10.1074/jbc.M112.432302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, Kim JS, Oh SC. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71(22):7061–7070. DOI: 10.1158/0008-5472.CAN-11-1338. [DOI] [PubMed] [Google Scholar]
- 27.Das S, Tucker JA, Khullar S, Samant RS, Shevde LA. Hedgehog signaling in tumor cells facilitates osteoblast-enhanced osteolytic metastases. PLoS ONE. 2012;7(3):e34374 DOI: 10.1371/journal.pone.0034374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira TA, Xie G, Choi SS, Syn WK, Voieta I, Lu J, Chan IS, Swiderska M, Amaral KB, Antunes CM, et al. Macrophage-derived Hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni. Liver Int. 2013;33(1):149–161. DOI: 10.1111/liv.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Monkkonen J, Kellokumpu-Lehtinen PL, Lauttia S, Tynninen O, Joensuu H, et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015;17:101 DOI: 10.1186/s13058-015-0621-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heppner GH, Miller FR, Shekhar PM. Nontransgenic models of breast cancer. Breast Cancer Res. 2000;2(5):331–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maun HR, Wen X, Lingel A, de Sauvage FJ, Lazarus RA, Scales SJ, Hymowitz SG. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J Biol Chem. 2010;285(34):26570–26580. DOI: 10.1074/jbc.M110.112284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jimenez-Garcia L, Herranz S, Luque A, Hortelano S. Critical role of p38 MAPK in IL-4-induced alternative activation of peritoneal macrophages. Eur J Immunol. 2015;45(1):273–286. DOI: 10.1002/eji.201444806. [DOI] [PubMed] [Google Scholar]
- 33.Khaled WT, Read EK, Nicholson SE, Baxter FO, Brennan AJ, Came PJ, Sprigg N, McKenzie AN, Watson CJ. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development. 2007;134(15):2739–2750. DOI: 10.1242/dev.003194. [DOI] [PubMed] [Google Scholar]
- 34.Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, Hanahan D. GLI1 is regulated through smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36. DOI: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Damhofer H, Medema JP, Veenstra VL, Badea L, Popescu I, Roelink H, Bijlsma MF. Assessment of the stromal contribution to Sonic Hedgehog-dependent pancreatic adenocarcinoma. Mol Oncol. 2013;7(6):1031–1042. DOI: 10.1016/j.molonc.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. DOI: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–6173. [DOI] [PubMed] [Google Scholar]
- 38.Linde N, Casanova-Acebes M, Sosa MS, Mortha A, Rahman A, Farias E, Harper K, Tardio E, Reyes Torres I, Jones J, et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun. 2018;9(1):21 DOI: 10.1038/s41467-017-02481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7(19):28697–28710. DOI: 10.18632/oncotarget.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Gillam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–5069. DOI: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–1043. DOI: 10.1158/1078-0432.CCR-12-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front Immunol. 2017;8:828 DOI: 10.3389/fimmu.2017.00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furmanski AL, Barbarulo A, Solanki A, Lau CI, Sahni H, Saldana JI, D’Acquisto F, Crompton T. The transcriptional activator Gli2 modulates T-cell receptor signalling through attenuation of AP-1 and NFkappaB activity. J Cell Sci. 2015;128(11):2085–2095. DOI: 10.1242/jcs.165803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U.S.A. 2010;107(9):4275–4280. DOI: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Rooijen N, Hendrikx E. Liposomes for specific depletion of macrophages from organs and tissues. Methods Mol Biol. 2010;605:189–203. DOI: 10.1007/978-1-60327-360-2_13. [DOI] [PubMed] [Google Scholar]
- 46.Rowbotham NJ, Hager-Theodorides AL, Furmanski AL, Crompton T. A novel role for Hedgehog in T-cell receptor signaling: implications for development and immunity. Cell Cycle. 2007;6(17):2138–2142. DOI: 10.4161/cc.6.17.4644. [DOI] [PubMed] [Google Scholar]
- 47.Rowbotham NJ, Hager-Theodorides AL, Furmanski AL, Ross SE, Outram SV, Dessens JT, Crompton T. Sonic hedgehog negatively regulates pre-TCR-induced differentiation by a Gli2-dependent mechanism. Blood. 2009;113(21):5144–5156. DOI: 10.1182/blood-2008-10-185751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Outram SV, Hager-Theodorides AL, Shah DK, Rowbotham NJ, Drakopoulou E, Ross SE, Lanske B, Dessens JT, Crompton T. Indian hedgehog (Ihh) both promotes and restricts thymocyte differentiation. Blood. 2009;113(10):2217–2228. DOI: 10.1182/blood-2008-03-144840. [DOI] [PubMed] [Google Scholar]
- 49.Stewart GA, Lowrey JA, Wakelin SJ, Fitch PM, Lindey S, Dallman MJ, Lamb JR, Howie SE. Sonic hedgehog signaling modulates activation of and cytokine production by human peripheral CD4+ T cells. J Immunol. 2002;169(10):5451–5457. [DOI] [PubMed] [Google Scholar]
- 50.Sethi N, Dai X, Winter CG, Kang Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell. 2011;19(2):192–205. DOI: 10.1016/j.ccr.2010.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52(6):1399–1405. [PubMed] [Google Scholar]
- 52.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. DOI: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. DOI: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94 DOI: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U.S.A. 2005;102(43):15545–15550. DOI: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. DOI: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 57.Wong HAB, West KA, Pacheco P, La H, Januario T, Yauch RL, de Sauvage FJ, Gould SE. Pharmacokinetic–pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent Hedgehog pathway activation. Clin Cancer Res. 2011;17(14):4682–4692. DOI: 10.1158/1078-0432.CCR-11-0975. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.