

 Schematic overview of the varied strategies for the discovery of monoamine oxidase inhibitors.
Schematic overview of the varied strategies for the discovery of monoamine oxidase inhibitors.
Abstract
Neuropsychiatric disorders, such as Alzheimer's disease (AD), Parkinson's disease (PD) and depression, have seriously inconvenienced the lives of patients. Growing evidence indicates that these diseases are closely related to the monoamine oxidase (MAO) enzyme, making it an attractive target for the exploitation of potent MAO inhibitors (MAOIs) with high selectivity and low side effects. Although various MAOIs have been discovered, the discovery of an ideal MAOI is not an easy task. In this review, we discuss the currently available rational design strategies for obtaining ideal MAOIs, including ligand-based and receptor-based design strategies, and these strategies were further illustrated with the aid of specific examples from the recent literature. To better understanding the biological activity of MAO, we also highlight the binding modes of typical inhibitors against MAO. Besides, advanced strategies for finding upcoming potent MAOIs were prospected.
1. Introduction
Nowadays, neuropsychiatric disorders, such as Alzheimer's disease (AD), Parkinson's disease (PD) and depression have become a serious social problem attracting worldwide attention. So far, however, there is still no effective treatment or alternative options to cure these diseases, largely owing to an insufficient understanding of the complex pathogenesis.1,2 Among the well-studied etiologies, the abnormal expression of mitochondrial enzyme monoamine oxidases (MAO, EC 1.4.3.4) has been recognized as a major cause. This is based on the fact that a neurodegenerative disorder is the result of excessive monoamine metabolites produced by the over-expression of MAO.3–5 All these physiological and pathological characteristics of MAO make it a promising biotarget for the development of MAO inhibitors (MAOIs) with therapeutic effects for the neuropsychiatric disorders.
Monoamine oxidases6 (MAO, EC 1.4.3.4) are flavin-binding (FAD) proteases that catalyze the oxidation of structurally diverse monoamines, including the neurotransmitters dopamine, norepinephrine, serotonin (5-HT), tyramine, 2-phenylethylamine (PEA), and exogenous amines—including the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).7,8 MAO is an outer membrane mitochondrial enzyme existing in two isoforms, MAO-A and MAO-B, which are both encoded in the genes on the X chromosome (Xp11.23–11.4)9,10 and have substrate specificities.11 In the human body, MAO-A is the main isoform in the intestine, placenta and heart, while MAO-B is mainly distributed in the platelets, brain glial cells and liver cells.12 These two isoforms have substrate specificities such that MAO-A catalyzes the oxidative deamination of serotonin (5-HT), adrenaline and noradrenaline (NA), whereas MAO-B positively deaminates β-phenethylamine and benzylamine.11,13 The normal degradation of these molecules ensures the proper functioning of synaptic neurotransmission, which regulates the emotional behaviors and other brain functions. As a consequence, the monoaminergic signaling is considered to be the key mechanism that regulates mood and controls motor, sensory and cognitive functions. The degradation process (Fig. 1) involves the MAO-assisted catalysis of monoamines into aldehydes, followed by the oxidation to the corresponding acids by aldehyde dehydrogenase (ALDH) or the conversion into alcohols or glycols by aldehyde reductase (ALR). However, this process brings about the production of chemical species such as hydrogen peroxide, ammonia and aldehydes. Particularly, hydrogen peroxide can trigger the production of reactive oxygen species (ROS).7 Moreover, it is intriguing that the increased expression of MAO in human brain has a strong correlation with age. In addition, the expression level of MAO-B is approximately 4 times more than that of MAO-A. Excessive expression of MAO-B in the elderly results in the production of a large amount of hydrogen peroxide (H2O2) that triggers the production of reactive oxygen species (ROS). The high levels of ROS cause mitochondrial damage, leading to neuronal cell death, which is tightly implicated in the pathogenesis of various neurodegenerative disorders, especially AD and PD.5,14 It has also been demonstrated that there is a correlation between severe depression and the increased MAO-A concentration in the cerebral cortex.15,16 Due to such key pharmacological effects, MAO-A and MAO-B are regarded as important drug targets for the treatment of neuropsychiatric disorders.17
Fig. 1. MAO catalyzes the oxidative deamination of monoamines. NADH is a key cofactor for the latter reaction.

Over the past few years, efforts have been put into the development of promising compounds that target MAO receptor, leading to the generation of a spectrum of new chemical entities with desirable properties. Broadly, MAOIs are classified into non-selective and selective, and reversible or irreversible inhibitors. A detailed classification of MAOIs is given in Table 1 and Fig. 2.18 As shown in Table 1 and Fig. 2, although MAO-A inhibitors are used for depression19 and MAO-B inhibitors are used in PD treatment in association with L-DOPA and/or DA agonists20 and for treating AD,21 many side effects were observed due to the lack of affinity and selectivity towards the specific isoforms. Moreover, owing to their irreversible binding property, they exhibit typical long-lasting enzyme inhibition, which will cause some toxicity, such as orthostatic hypotension, hepatotoxicity and hypertensive crisis (these conditions are termed as the cheese effect or cheese reaction).22,23
Table 1. Classification of MAOIs.
| Group | Class | Compound | MAO selectivity | Application |
| First generation MAOIs (non-selective) | Hydrazines | Iproniazid 1 | A and B | Antidepressant |
| Hydrazines | Isoniazid 2 | A and B | Antidepressant | |
| Hydrazines | Octamoxin 3 | A and B | Antidepressant | |
| Nitrofuran | Furazolidone 4 | A and B | Antidepressant | |
| Curcuminoids | Curcumin 5 | A and B | Antidepressant | |
| Flavonoid glycosides | Myricetin 6 | A and B | Anti-Alzheimer | |
| Quinolinamine | Echinopsidine 7 | A and B | Antidepressant | |
| Second generation MAOIs (selective-irreversible) | Propargylamines | Selegiline 8 | B | Anti-Parkinson |
| Propargylamines | Rasagiline 9 | B | Anti-Parkinson | |
| Propargylamines | Clorgyline 10 | A | Antidepressant | |
| Third generation MAOIs (selective-reversible) | Piperidylbenzofurans | Brofaromine 11 | A | Antidepressant |
| Morpholinobenzamide | Moclobemide 12 | A | Antidepressant | |
| Indole derivative | Prilindole 13 | A | Antidepressant |
Fig. 2. Known MAO inhibitors.

As stated above, it is pivotal to develop a novel class of potent, selective and reversible MAO inhibitors. Thus, great attention must be focused on the rational design strategies for developing desirable MAOIs. In this review, we provide an overview of the currently available design strategies for obtaining potent MAOIs (Fig. 3), with illustrative examples. In addition, we also describe how each strategy can be used to design MAOIs and give a brief glimpse into the SAR investigation of the representative compounds. Also, other advanced strategies for discovering upcoming relative MAOIs are also sought out.
Fig. 3. Schematic overview of the varied strategies for the discovery of monoamine oxidase inhibitors.

2. Structures of MAO-A and MAO-B
Primary structures deduced from cDNA clones suggest that the two MAO isoforms (MAO-A and MAO-B) are respectively composed of 527 and 520 amino acid identities with molecular weights of 59 700 and 58 800, respectively, and have a 70% amino acid identity.24–26 The secondary structures of MAO-A and MAO-B were studied by Fourier transform attenuated total reflection spectroscopy (FTIR-ATR). The FTIR-ATR experimental results showed that the two MAO isoforms have significant folding and molecular difference,27 while identifying four extremely conserved regions in the MAO isozymes, namely, an ADP-binding unit (residues 6–43); a putative substrate binding domain (residues 178–221); a site for the covalent attachment of FAD (residues 350–458); a C-terminus region (residues 491–511). The C-terminus region was predicted to form a transmembrane associated R-helix.28
The X-ray crystal structures of human MAO-B and human MAO-A were reported in 2002 and 2005, respectively,29,30 which showed that the human MAO-B crystals are dimers, while the human MAO-A crystals are monomeric units. Although, in both the isoforms, their cavities are usually hydrophobic, details of the active site architectures show disparities in their structural properties.31 The structural comparison of human MAO-A and MAO-B32,33 showed a significant conformational distinction in the cavity-shaping loops of MAO-A (from residues 210–216) and MAO-B (residues 201–207), which resulted in the MAO-A isoform accommodating bulkier ligands, while the MAO-B isoform preferring smaller ones due to its small entrance cavity. The volumes of human MAO-A and MAO-B cavities are ∼400 Å3 and ∼700 Å3, respectively. In addition, the MAO-B cavity consists of the active site cavity (∼400 Å3) for substrate binding and the entrance cavity (∼300 Å3),34 which are separated by Ile199 and Tyr326 residues. For clarity, the 3D crystallographic structures of MAO ‘enzyme-ligand’ complexes (A: clorgyline complexed with MAO-A, PDB code: ; 2Z5X;35 B: deprenyl complexed with MAO-B, PDB code: ; 2BYB 36) are exemplified in Fig. 4. We have also drawn the cavity of MAO-A (∼400 Å3) and the entrance cavity of MAO-B (∼300 Å3) using the red frames in Fig. 4a and b, respectively. Particularly, Ile199 in MAO-B acts as a structural determinant of substrate and inhibitor recognition. The Ile199 residue conformation determines the plasticity of the catalytic site of the MAO-B protein. The orientation of this “gated” Ile199 depends on the property of the binding ligand, adopting two different conformations (“closed” and “open”), which determines the catalytic domain of MAO-B as a bipartite cavity and a large single cavity, respectively.37,38 Evidently, we also see that the MAO-A isoform can accommodate bulkier ligands, while the MAO-B isoform prefers smaller ones due to its small entrance cavity, from Fig. 4a and b, respectively. In contrast, in human MAO-A, the corresponding cavity entrance residue is Phe208, but Phe208 does not function as a gating residue. On the other hand, Tyr326 in hMAO-B did not directly partition the two cavities, but produced a restriction that is more pronounced than that in hMAO-A, where Ile335 occupied the equivalent position.18 Related studies have also revealed that a single mutation in MAOs, in which the Phe-208 residue in MAO-A was replaced by the corresponding Ile residue of MAO-B, was sufficient to convert the type A substrate selectivity to type B and vice versa.39 The active site residues, i.e. Tyr (Tyr407 and Tyr 444 in MAO-A; Tyr 398 in MAO-B) and Lys (Lys305 in MAO-A and Lys296 in MAO-B), remain conserved in both the enzymes. Other non-conserved active site residues (which do not significantly affect the shapes of the cavities) are Asn181 and Ile180 in MAO-A and Cys172 and Leu171 in MAO-B. Hence, the Phe208 and Ile335 residues of MAO-A and the Ile199 and Tyr326 residues of MAO-B are the key determinants for the differential substrate and inhibitor specificities of the two isoforms.
Fig. 4. (a) The co-crystallized structure of clorgyline with hMAO-A (PDB ID: 2Z5X). (b) The co-crystallized structure of deprenyl with hMAO-B (dimeric form) (PDB ID: ; 2BYB).

In order to make a rational use of design strategies, we need to understand where and how the substrates (or the inhibitors) enter the MAO-A and MAO-B active sites. Structural data suggested that the MAO-A and MAO-B ligands chase the same pathways for binding with the target protein.40 Furthermore, it was also revealed that the cavity-shaping loop (residues 210–216 in MAO-A) assumed a helical conformation, which was conserved in all MAO-A structures, except in hMAO-A complexed with clorgyline. In general, the cavity of MAO-B is hydrophobic in nature, however, the MAO-B isoform contains a small, highly conserved hydrophilic area in the entrance cavity.41 The cavity can hold a small inhibitor (e.g. isatin) or a cavity-filling ligand (e.g. rasagiline42) depending on the conformation of the gating residue Ile199. It can hold a small inhibitor as the active site is restricted to a smaller cavity separated from an entrance cavity space. By contrast, binding of inhibitors, such as rasagiline or deprenyl41 can induce a mid-span type of cavity because these ligands are large enough to push the gating residue Ile199 into the open conformation. It is worth noting that this cavity plasticity in MAO-B is probably determined by the subtle conformational changes.
3. Medicinal chemistry strategies in the ligand-based design for discovering MAO inhibitors
The typical drawbacks of MAO inhibitors, for instance, irreversibility and low selectivity, necessitate an ongoing search for novel MAO inhibitors with alternative and rational design strategies. In this part of the paper, we will describe the ligand-based design strategy for MAO inhibitors with case studies.
3.1. Using available 3D-QSAR or SAR results to guide the design of compounds
The ligand-based design strategy uses only ligand information for predicting its activity, which depends on its similarity/dissimilarity to the previously known active ligands, including pharmacophore modelling,43 3D-quantitative structure–activity relationship (3D-QSAR) approach,44 matched molecular pairs (MMP) search45 and so on. In this part, we will mainly describe the approach of 3D-QSAR and SAR. The use of available 3D-QSAR or SAR results has been validated to be not only a fast and simple but also an effective method for detecting pharmacophores or active fragments that can be rapidly used to develop MAO inhibitors. Such moieties play a decisive role in improving drug properties and avoiding deficiencies when used either as a scaffold core or as a peripheral replacement.
When establishing a 3D-QSAR model, the following steps are generally followed:46 (1) selecting a set of compounds that are biologically active for MAO-A and MAO-B; (2) determining the pharmacodynamic structures of selected bio-active molecules and superimposing with the test molecules; (3) computing the space parameters that consist of R2 (leave-n-out cross validation for the training set), Q2 (leave-n-out cross validation for the test set); (4) regression analysis of the spatial parameters of the test molecule and its corresponding biological activity to obtain 3D-QSAR; (5) testing the predictive ability of the 3D-QSAR model (pharmacological evaluation). Bautista-Aguiler et al.47 applied a 3D-QSAR approach to determine the specific molecular determinants of MAO-A or MAO-B and created a 3D-QSAR model for assessing the MAO-A or MAO-B inhibition activity of a designed molecule, aiming to design potent inhibitors with the optimal selectivity profile for MAO. As a result, they identified compound 14 (Fig. 5) as a potent inhibitor of hMAO-A (IC50 = 5.5 ± 1.4 nM) in the nanomolar concentration range and a moderately potent inhibitor of hMAO-B (IC50 = 150 ± 31 nM). Using the same method, Pisani et al.48 designed and developed a series of novel coumarin-based MAO inhibitors in 2015. The biological evaluation of these novel coumarin derivatives demonstrated the reliability of their models. In particular, the coumarin derivatives 15, 16 and 17 (Fig. 5) were endowed with high MAO-B inhibitory potency (pIC50 = 8.13, 7.89 and 7.82, respectively) and good selectivity over MAO-A.
Fig. 5. Discovery of MAOIs via QSAR or SAR results.

Furthermore, Castagnoli and Gnerre49,50 proposed the ligand-based design and created 3D-QSAR models for the specific recognition of MAO-B inhibitors. According to their 3D-QSAR models, they found that the heterocyclic structure is a vital pharmacophore for MAO-B inhibitor designing. Thus, more and more heterocyclic structures have been reported for their potent inhibition activity towards MAO-B. For instance, oxadiazole and pyrroles derivatives51,52 have been suggested as MAO-B inhibitors. It is interesting that the transition metal complexes based on these heterocyclic compounds can also be used as MAO-B inhibitors. In 2017, Yang et al.53 designed and synthesized piperazine derivatives as ligands, coordinated with transition metals, with the aim of obtaining novel complexes. Interestingly, these complexes are found to have potential activity for the inhibition of MAO-B. For instance, compound 18 (rat MAO-B IC50 = 1.85 ± 0.31 μM) (Fig. 5) was a potent inhibitor of MAO-B. Analogously, dual inhibition of A2A (adenosine A2A receptor,54 a target receptor related to the neuropsychiatric disorders) and MAO-B is an effective treatment in neuropsychiatric disorders. However, it is difficult to balance the effectiveness of a test compound against A2A and MAO-B at the same time. However, Bhayye et al. also used a 3D-quantitative structure–activity relationship (3D-QSAR)-based approach to address balance activity and have made corresponding progress.55 In addition, it is also very promising to use the SAR results to guide the design of compounds. In the case of finding MAO inhibitors based on a novel chalcone, Mathew et al.56 reported the MAO inhibitory activity of these novel chalcone thorough a SAR study. Guided by this SAR study, they designed and synthesized a series of chlorinated thienyl chalcone derivatives. Among them, compound 19 (Fig. 5) was a selective hMAO-A inhibitor (Ki = 0.49 ± 0.02 μM), while compound 20 (Fig. 5) was found to have the best inhibitory activity and higher selectivity towards hMAO-B with Ki and SI values of 0.31 ± 0.02 μM and 16.84, respectively. As described above, it is very promising to use the available QSAR or SAR results to guide the design of prospective compounds.
3.2. Molecular hybridization
Molecular hybridization is regarded as a perfect strategy for identifying bioactive compounds through the rational assembling of essential pharmacophoric elements or different bioactive fragments to generate a new hybrid entity that was endowed with significantly improved affinity and potency towards MAOs compared to the parent compound.57 Moreover, the novel hybrid entity can not only act on the monoamine oxidase but also on other targets, such as acetylcholinesterase (AChE) or butyrylcholinesterase (BChE).
In order to find the ideal compounds with different biological activities that simultaneously target both MAO-B and AChE, Farina et al.58 built a molecular framework of multitargeting ligands following a hybridization strategy. Starting from the known ligands of the two enzymes, simple pharmacophore motifs were selected and joined in a unique molecular entity. In detail, the 2H-chromen-2-one ring59 was chosen as the moiety that can efficiently fit the MAO-B enzymatic cleft, and a protonatable basic moiety was chosen that might bind with AChE effectively (Fig. 6). Ultimately, the reliability of this method was also confirmed by a bioactivity evaluation. Among these compounds, 21, 22, 23 and 24 (Fig. 6) showed excellent activity profiles with nanomolar inhibitory potency on hMAO-B, high MAO-B over MAO-A selectivity and submicromolar potency on hAChE.
Fig. 6. Hybridization process and representative MAOIs and hAChE inhibitors.

It was reported that pyridoxine (vitamin B6, Fig. 7) is an effective antioxidant60 because it can inhibit the production of free radicals and can be used as a quencher for singlet oxygen.61 Besides, resveratrol (Fig. 7), as a natural product, has a wide range of biological activities such as antioxidant and neuroprotective properties.62 Moreover, resveratrol has the ability to inhibit monoamine oxidase and its stilbene structure is important for related biological activities.63 Besides, it has been reported that molecules bearing the phenolic Mannich base moieties may display potent antioxidant and AChE inhibitory activities.64,65 Therefore, in an extension of this method, Yang et al.66 designed, synthesized and evaluated the biological activities of pyridoxine–resveratrol hybrids Mannich base derivatives. The results showed that most of the synthesized compounds could selectively inhibit acetylcholinesterase (AChE) and MAO-B. Among them, compound 25 (Fig. 7) exhibited the highest MAO-B inhibition with an IC50 value of 2.68 ± 0.01 μM. In addition, the binding mode of compound 25 complexed with MAO-B (Fig. 7c) shows H-bonding between the oxygen of phenolic hydroxyl group on the benzene ring of compound 25 and the hydrogen on the Ile198 nitrogen of MOA-B, and between the oxygen on the morpholine and the hydrogen on the Ser59 nitrogen of MOA-B. Compared with the complex with MOA-B in Fig. 7c, compound 25 forms only one H-bonding with MAO-A in Fig. 7b. It manifested that the two H-bonding interactions with MAO-B in Fig. 7c are beneficial for the enhancement of binding affinity from the point of activity data. This also explains why compound 25 was more active against MAO-B than MAO-A.
Fig. 7. (a) Hybridization process and representative MAO and AChE inhibitors. (b) The binding mode of compound 25 complexed with MAO-A (PDB code: ; 2Z5X). (c) The binding mode of compound 25 complexed with MAO-B (PDB code: ; 2V60).

Hu et al.67 used the active fragments of the well-known MAO-B inhibitors (compounds 26, 27, 28 (ref. 68)) (Fig. 8) to design a series of compounds. Among them, compound 29 displayed significant MAO-B inhibition activity and selectivity. Notably, the styrol–formamido group at position-3′ in compound 29 might have promoted the activity and selectivity of 8-phenyl-xanthine analogues. Although 29 is not more active than compounds 26–28, it has excellent selectivity. In addition, the introduction of F atoms can increase its biological activity and improve metabolic stability.
Fig. 8. Discovery of MAO-B inhibitors via an active fragment combination approach.

Thus, these cases described here provide a proof-of-concept for the utility of this methodology, namely molecular hybridization strategy, in the discovery of novel molecules from substructures of pre-existing MAO inhibitors. Thus, this method should be potentially applicable for the efficient construction of various compound collections of advantageous structures in the future.
3.3. Others
In addition to the above strategies, in this section we also describe other strategies, including the open or close ring strategy and the optimization of parent compounds.
3.3.1. Open or close ring strategy
It is well known that a fascinating area for the further development of small-molecular drugs is to synthesize ring-opened or ring-closed analogues. These derivatives often exhibit potency similar to that of the parent compound and may have superior pharmacokinetic properties.
For the ring-opened analogues approach, interestingly, when the heterocycles of hetero-benzofused derivatives are opened, the compounds still exhibit potent MAO inhibitory activity, for instance, the evolution of compounds from compounds 30 (ref. 69) to 31 (ref. 70) (Fig. 9). In 2017, Lan et al.71 used this method to design and synthesize a new series of small molecules that bear a benzyloxy substituent. After the evaluation of their activity, Lan et al. found that most of the as-synthesized compounds were potent and selective MAO-B inhibitors and were weak inhibitors of MAO-A. In particular, compounds 32 (IC50 = 0.35 μM) and 34 (IC50 = 0.19 μM) were the most potent MAO-B inhibitors, exhibiting the highest selectivity for MAO-B (32, SI > 285.7-fold and 34, SI = 146.8-fold). Furthermore, among the new compounds, those with para-substitution of the benzyloxy phenyl ring were more potent for MAO-B inhibition than those with meta-substitution (Fig. 9).
Fig. 9. Discovery of MAO inhibitors via a ring-opened strategy.

The ring-closed analogues approach, namely conformational restriction, was adopted in 2015 by Badavath et al.72 to design a chalcone that is similar to the aryl-α, β-unsaturated carbonyl moiety of curcumin. However, by reversing the position of the double bond and the carbonyl group, the chalcone was then cyclized to provide a series of new pyrazoline derivatives (Fig. 10). Among these compounds, compound 36 (hMAO-A, Ki = 0.14 μM, SI = 9 × 10–7) exhibited pivotal hMAO-A inhibition activity. Furthermore, in an extension of this research, in 2016, Badavath et al.73 also used the same approach to design a series of new 2-methoxy-4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl) phenol derivatives. These compounds were synthesized and tested for their human MAO inhibitory activity, and compound 37 (Fig. 10) was found to be a potent inhibitor of hMAO-A with Ki = 0.06 ± 0.003 μM (selectivity index: SI = 1.02 × 10–5). Moreover, it was found to be better than the standard drug, moclobemide (hMAO-A with Ki = 0.11 ± 0.01 μM) with selectivity index SI = 0.049. The binding mode of compound 37 complexed with MAO-A (Fig. 10b) is H-bonding between the oxygen atom on the sulfonamide of compound 37 and the hydrogen on the Tyr69 nitrogen of MAO-A, and between the hydrogen of phenolic hydroxyl group on the benzene ring of compound 37 and the oxygen atom on the carbonyl group of Arg51 of MAO-A. Compared with MAO-A in Fig. 10b, it is fortunate that compound 37 did not produce any H-bonding with MAO-B in Fig. 10c, which is consistent with the activity data.
Fig. 10. (a) Discovery of MAO-A inhibitors via a ring-closed strategy. (b) The binding mode of compound 37 complexed with MAO-A (PDB code: ; 2BXR). (c) The binding mode of compound 37 complexed with MAO-B (PDB code: ; 2BYB).

In addition, in 2016, Pisani et al.74 built a new molecular framework using the method of ring-closed strategy (Fig. 11), which can also improve the solubility of compounds. Subsequently, the designed compounds were tested in vitro to evaluate their ability to inhibit both AChEs and MAOs. The researchers found a promising hit compound 38, exhibiting a high hMAO-B inhibitory activity (IC50 = 30 nM) and good MAO B/A selectivity (selectivity index, SI = 94) along with a micromolar level AChE inhibition (IC50 = 1.03 μM). The binding mode of compound 38 complexed with MAO-B (Fig. 11c) shows two important H-bonds between the oxygen atom on the carbonyl group of compound 38 with the hydrogens on Tyr60 and Ser59 of MAO-B. Compared with Fig. 11c, the oxygen atom on the carbonyl group of compound 38 formed only one H-bonding with Tyr197 in MAO-A, as shown in Fig. 11b. It also indicates that compound 38 could bind better with MAO-B, which is also consistent with the activity data.
Fig. 11. (a) Discovery of MAO-B and AChE inhibitors via a ring-closed strategy. (b) The binding mode of compound 38 complexed with MAO-A (PDB code: ; 2Z5X). (c) The binding mode of compound 38 complexed with MAO-B (PDB code: ; 2V60).

As described above, these ring-opened or ring-closed derivatives seemed to be promising starting points for further investigation into new MAO inhibitors.
3.3.2. Optimization of the parent compounds
In order to find the ideal MAOIs, many parent compounds have also been explored though structural optimizations. In this section, several representative favoured scaffolds, including caffeines, chalcones and isatins are discussed.
3.3.2.1. Caffeines
Caffeine (Fig. 12) has been used as a privileged scaffold for the design of monoamine oxidase (MAO) A and B enzyme inhibitors. It has been demonstrated that the substitution at the C8-position of caffeine produces structures with high MAO inhibitory potency. In general, all the substituted product structures are high-potency MAO-B inhibitors.75 Especially, C8 substituents with styryl and benzyloxy moieties have been shown to be particularly effective.76 For instance, compounds 39 (ref. 77) and 40 (ref. 78) are potent inhibitors of MAO-B, with IC50 values 0.128 μM and 0.068 μM, respectively.
Fig. 12. Caffeine derivatives as h-MAO-B inhibitors.

3.3.2.2. Chalcones
Chalcone (1,3-diphenyl-2-propen-1-one) is an open-chain flavonoid, which is widely found in edible plants, and has a broad range of biological activities, such as anti-inflammatory and neuroprotective effects.79,80 Therefore, using chalcone as the core skeleton, different substituents are introduced, which have good inhibitory activity against MAO-B. Some substitutions include the introduction of an electron-withdrawing group (CF3), e.g. compound 42 (hMAO-B IC50 = 0.55 ± 0.07 μM);81 an electron-donating group (–OCH3, OH), e.g. compound 43 (hMAO-B IC50 = 0.0044 ± 0.00027 μM);82 other aromatic rings, e.g. furans, including compound 44 (hMAO-B IC50 = 0.174 ± 0.033 μM)83 (Fig. 13).
Fig. 13. Chalcone derivatives as hMAO-B inhibitors.

3.3.2.3. Isatin congeners
Isatin (2,3-dioxindole, Fig. 14) has been reported as a well-known pharmacological agent that has a broad biological activity, and is a potent inhibitor of MAO, particularly of MAO-B.84 Thus, isatin is a valuable starting point in the design of potent compounds that target MAO. Fig. 14a illustrates a few examples of isatin derivatives that possess potent activity against MAO (compound 45,85 compound 46 (ref. 86) and compound 47 (ref. 87)). The binding mode of compound 47 complexed with MAO-B (Fig. 14c) shows that compound 47 forms two important H-bonding with Tyr435. Compared with Fig. 14c, the oxygen atom on the carbonyl group of compound 47 formed only one H-bonding with Asn181 in MAO-A in Fig. 14b. This result shows that compound 47 could bind better with MAO-B, which is also consistent with the activity data.
Fig. 14. (a) Isatin congeners derivatives as hMAO inhibitors. (b) The binding mode of compound 47 complexed with MAO-A (PDB code: ; 2Z5X). (c) The binding mode of compound 47 complexed with MAO-B (PDB code: ; 2V5Z).

As we all know, many natural products (e.g. coumarins,88 flavonoids89 and chelerythrine90) have good MAO inhibitory activity, and are potential lead compounds, so it is very meaningful that they are reasonably modified for finding novel potent compounds. Therefore, the optimization of parent compounds is another important approach to generate MAOIs.
4. Medicinal chemistry strategies in the target-based lead design for discovering MAO inhibitors
Target-based drug design is a challenging process that involves step-by-step knowledge acquisition (Fig. 15). Starting from the known target structures, computer studies are performed to identify potential ligands. These molecular modeling programs are followed by the synthesis of the most potent compounds. Next, the biological properties (e.g. potency, affinity and efficacy) of these synthetic compounds were evaluated using various experimental platforms.91–93 Overall, receptor-based drug design is an important component of modern medicinal chemistry.
Fig. 15. Description of the structure-based drug design process.

Coumarins' MAO inhibition activities have been identified.94 Recent studies have shown that substitution in position 3 of the coumarin nucleus can modulate its inhibitory activity against MAO-B. When an alkylamide group is introduced at this position, a strong inhibitory activity can be exerted on MAO-B.95,96 Based on the X-ray structure of MAO-B-coumarin complex, a docking molecular model of the computer-generated 2H-chromene-3-carboxamide coumarin derivative was analysed. As shown in Fig. 16, in addition to the π–π stacking interaction of coumarin with the amino acids Tyr435 and Tyr398 of MAO-B, the carbonyl oxygen atom at the 2 position of coumarin can also form H-bonding with the amino acids Tyr398, which facilitates the increase in MAO inhibitory activity. At the same time, He et al.97 found that the positions of the carbonyl groups are interchangeable, which is beneficial in forming a stable drug-MAO-B combination. So, the inhibitory activity of coumarin on MAO-B can be increased. In addition, it has been reported that the C–S–C bond has good permeability.98 In addition to the coumarin can form π–π stacking interaction with the amino acids TYR435and TYR398, the carbonyl oxygen atom at the 2 position of the coumarin can form a H-bonding with TYR398, and the carbonyl oxygen atom at the 3 position can also form another H-bonding with the amino acid Ieu-171. These two hydrogen bonds facilitate the increase in activity. Therefore, through the above analysis, He et al. have designed and synthesized a series of coumarin derivatives. Among them, compound 48 (hMAO-B IC50 = 0.2 ± 0.08 μM, hMAO-A IC50 = 39.74 ± 0.85 μM) showed the most potent activity.
Fig. 16. The flow diagram for discovering MAOIs via target structure-based approach. (PDB code: ; 2BYB).

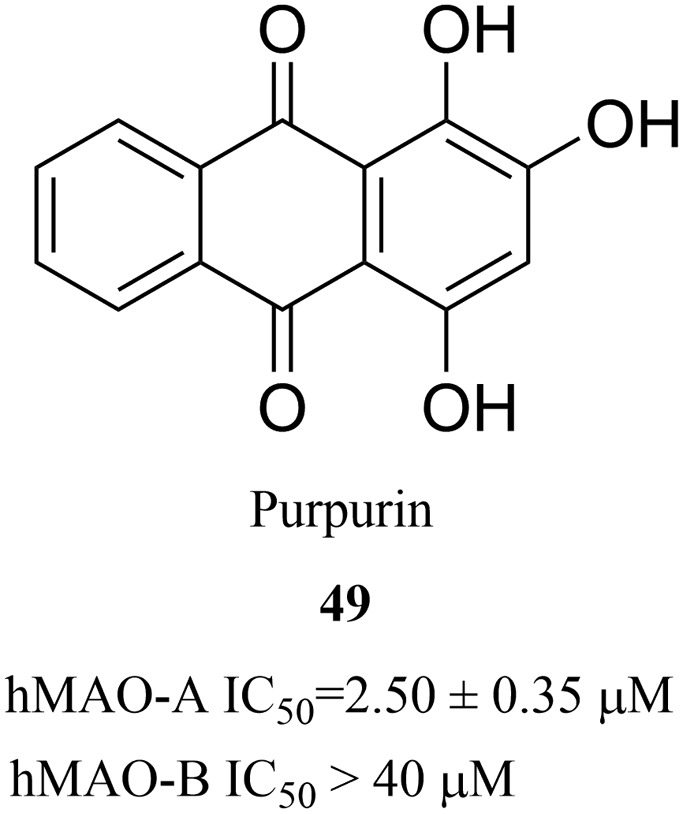
Purpurin is an anthraquinone that is found in the roots of madder, which has a wide range of biological activities, such as the inhibition of xanthine oxidase, antifungal activity and antioxidant activity.99–101 In 2017, Lee et al. found that purpurin (49) (Fig. 17) effectively and selectively inhibited MAO-A (IC50 = 2.50 μM) during the screening of target-based natural products using human MAO-A and MAO-B.102 The results of this study showed that purpurin is an effective, selective and reversible inhibitor of MAO-A, and it can be regarded as a novel potential lead compound for the development of novel reversible inhibitors of MAO-A. In addition, Yelekçi et al.103 and Kare et al.104 have also used this method to design and analyze some MAO-B inhibitors and have made considerable progress. In summary, receptor-based approach has increasingly demonstrated its advantages in the drug design process. It has been used regularly by scientists in the pharmaceutical industry as well as in academic laboratories over the past four decades. This approach is also critical for the discovery and optimization of early lead compounds.105
Fig. 17. Discovery of MAOIs via target-based screening approach.
5. Conclusions
Neuropsychiatric disorders (NDs), especially, AD, PD and Depression, have brought much economic and psychological burden on the patients. These diseases may be induced by various factors, which are closely related to monoamine oxidase. To the best of our knowledge, design strategies play a very important role in the research for drug discovery. In other words, the choice of design strategies in the development of ideal MAOIs has a pivotal role. Under these circumstances, we review here the discovery strategies, including ligand-based design strategy and receptor-based design strategy, to find ideal MAOIs.
An attempt has been made in this article to review those various strategies, which have achieved certain success. In addition, some compounds exhibit an outstanding inhibitory activity. For instance, compound 14 (hMAO-A: IC50 = 5.5 ± 1.4 nM; hMAO-B: IC50 = 150 ± 31 nM); compound 31 (hMAO-B IC50 = 0.004 ± 0.0016 μM); compound 47 (hMAO-A: IC50 = 0.304 ± 0.037 μM; hMAO-B: IC50 = 0.0014 ± 0.0001 μM). These compounds have the potential as drug leads and/or drug candidates for further development.
As mentioned earlier, neuropsychiatric disorders may be induced by various and complex factors, in which MAO is a key cause for pathogenesis. Therefore, MAO is an important target for the treatment of neuropsychiatric disorders. Hence, it is an urgent need to find the ideal MAOIs through rational strategies, and make the MAOIs key therapeutic agents or combine them with the inhibitors of other targets (that are closely related to neuropsychiatric disorders), so that the treatment of neuropsychiatric disorders will be greatly improved. Besides, from a biological point of view, a lot of research work still needs to be done in order to establish the role of MAO inhibitors in the etiology of other disorders, such as ageing, neuroprotection/neurorescue, etc. Overall, we hold the idea that a multidisciplinary coordination of methodologies from the common approaches and the emerging alternatives will lead to the discovery and development of MAOIs in the future. These approaches will help us make better use of the rational drug design strategies to find ideal MAOIs.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
Authors are thankful to the financial supports from the Natural Science Foundation of Shandong Province (No. ZR2018MH042 to X. LI), the Key Research and Development Programs of Shandong Province (No. 2016GSF201175 to X. LI; No. 2017CXGC1401 to X. Y. LIU; No. 2015ZDJS04001 to F. S. WANG), and the Fundamental Research Funds for the Central Universities–‘CACHSR for China–Australia’ (No. 2018GJ07 to X. LI).
Biographies

Renyuan Hong
obtained his B.Sc. degree in pharmaceutical science from Nanyang Normal University, China, in 2016. He is now a MS candidate at the College of Pharmacy, Shandong University. His research interests involve the design and synthesis of small molecules with anti-Alzheimer and antitumor properties.

Xun Li
obtained her M.Sc. degree at Shandong University in 2001 and obtained her Ph.D. from Tianjin University, China, in 2004. She has since worked in the College of Pharmacy, Shandong University as a senior associate professor. From 2007 to 2008, she worked as a visiting scholar in Tokyo University, Japan, funded by the Japan Medical Association. From 2012 to 2013, she further worked as a visiting scholar in the Department of Chemistry, Texas A&M University, USA. Her research interests include rational design, synthesis and evaluation of bioactive molecules with anti-Alzheimer, antitumor, antiviral and antibacterial properties.
References
- Narayan P., Ehsani S., Lindquist S. Nat. Chem. Biol. 2015;11:911–920. doi: 10.1038/nchembio.1663. [DOI] [PubMed] [Google Scholar]
- Trippier P. C., Labby K. J., Hawker D. D., Mataka J. J., Silverman R. B. J. Med. Chem. 2013;56:3121–3147. doi: 10.1021/jm3015926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thase M. E. J. Clin. Psychiatry. 2012;73:10–16. doi: 10.4088/JCP.11096su1c.02. [DOI] [PubMed] [Google Scholar]
- Kopin I. J. Ann. N. Y. Acad. Sci. 2010;648:96–104. doi: 10.1111/j.1749-6632.1992.tb24527.x. [DOI] [PubMed] [Google Scholar]
- Youdim M. B., Lavie L. Life Sci. 1994;55:2077–2082. doi: 10.1016/0024-3205(94)00388-2. [DOI] [PubMed] [Google Scholar]
- Squires R. F. Vopr. Med. Khim. 1997;43:433–439. [PubMed] [Google Scholar]
- Bortolato M., Chen K., Shih J. C. Adv. Drug Delivery Rev. 2008;60:1527–1533. doi: 10.1016/j.addr.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K., Trevor A., Castagnoli Jr N. Biochem. Biophys. Res. Commun. 1984;120:574–578. doi: 10.1016/0006-291x(84)91293-2. [DOI] [PubMed] [Google Scholar]
- Garpenstrand H., Ekblom J., Forslund K., Rylander G., Oreland L. J. Neural Transm. 2000;107:523–530. doi: 10.1007/s007020070075. [DOI] [PubMed] [Google Scholar]
- Kochersperger L. M., Parker E. L., Siciliano M., Darlington G. J., Denney R. M. J. Neurosci. Res. 1986;16:601–616. doi: 10.1002/jnr.490160403. [DOI] [PubMed] [Google Scholar]
- Fowler C. J., Tipton K. F. J. Pharm. Pharmacol. 2011;36:111–115. doi: 10.1111/j.2042-7158.1984.tb03004.x. [DOI] [PubMed] [Google Scholar]
- Ramsay R. R. Curr. Top. Med. Chem. 2012;12:2189–2209. doi: 10.2174/156802612805219978. [DOI] [PubMed] [Google Scholar]
- Carradori S., Silvestri R. J. Med. Chem. 2015;58:6717–6732. doi: 10.1021/jm501690r. [DOI] [PubMed] [Google Scholar]
- Nicotra A., Pierucci F., Parvez H., Senatori O. NeuroToxicology. 2004;25:155–165. doi: 10.1016/S0161-813X(03)00095-0. [DOI] [PubMed] [Google Scholar]
- Benedetti M. S., Dostert P. Biochem. Pharmacol. 1989;38:555–561. doi: 10.1016/0006-2952(89)90198-6. [DOI] [PubMed] [Google Scholar]
- Emilsson L., Saetre P., Balciuniene J., Castensson A., Cairns N., Jazin E. E. Neurosci. Lett. 2002;326:56–60. doi: 10.1016/s0304-3940(02)00307-5. [DOI] [PubMed] [Google Scholar]
- Hwang J. S., Lee S. A., Hong S. S., Lee K. S., Lee M. K., Bang Y. H., Ro J. S. Arch. Pharmacal Res. 2005;28:190–194. doi: 10.1007/BF02977714. [DOI] [PubMed] [Google Scholar]
- Tripathi A. C., Upadhyay S., Paliwal S., Saraf S. K. Eur. J. Med. Chem. 2018;145:445–497. doi: 10.1016/j.ejmech.2018.01.003. [DOI] [PubMed] [Google Scholar]
- Amrein R., Martin J. R., Cameron A. M. Adv. Neurol. 1999;80:509–519. [PubMed] [Google Scholar]
- Drukarch B., Muiswinkel F. L. V. Biochem. Pharmacol. 2000;59:1023–1031. doi: 10.1016/s0006-2952(99)00340-8. [DOI] [PubMed] [Google Scholar]
- Saura J., Luque J. M., Cesura A. M., Da P. M., Chanpalay V., Huber G., Löffler J., Richards J. G. Neuroscience. 1994;62:15–30. doi: 10.1016/0306-4522(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Yamada M., Yasuhara H. NeuroToxicology. 2004;25:215–221. doi: 10.1016/S0161-813X(03)00097-4. [DOI] [PubMed] [Google Scholar]
- Cooper A. J. Br. J. Psychiatry. 1989;6:38–45. [PubMed] [Google Scholar]
- Lan N. C., Johnson D. L., Abell C. W., Bembenek M. E., Kwan S. W., Seeburg P. H., Shih J. C. Proc. Natl. Acad. Sci. U. S. A. 1988;85:4934–4938. doi: 10.1073/pnas.85.13.4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T., Takamoto S., Ito A. Agric. Biol. Chem. 1990;54:253–257. [PubMed] [Google Scholar]
- Chen K., Wu H. F., Shih J. C. J. Neurochem. 2010;61:187–190. doi: 10.1111/j.1471-4159.1993.tb03554.x. [DOI] [PubMed] [Google Scholar]
- Wouters J., Ramsay R., Goormaghtigh E., Ruysschaert J. M., Brasseur R., Durant F. Biochem. Biophys. Res. Commun. 1995;208:773–778. doi: 10.1006/bbrc.1995.1404. [DOI] [PubMed] [Google Scholar]
- Wouters J. Curr. Med. Chem. 1998;5:137–162. [PubMed] [Google Scholar]
- Binda C., Newton-Vinson P., Hubalek F., Edmondson D. E., Mattevi A. Nat. Struct. Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- De Colibus L., Li M., Binda C., Lustig A., Edmondson D. E., Mattevi A. Proc. Natl. Acad. Sci. U. S. A. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar B., Gupta V., Kumar V. Curr. Drug Targets. 2016;18:87–97. doi: 10.2174/1389450117666151209123402. [DOI] [PubMed] [Google Scholar]
- Bradford M. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. J. Comput. Chem. 2010;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milczek E. M., Binda C., Rovida S., Mattevi A., Edmondson D. E. FEBS J. 2011;278:4860–4869. doi: 10.1111/j.1742-4658.2011.08386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son S. Y., Ma J., Kondou Y., Yoshimura M., Yamashita E., Tsukihara T. Proc. Natl. Acad. Sci. U. S. A. 2008;105:5739–5744. doi: 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De C. L., Li M., Binda C., Lustig A., Edmondson D. E., Mattevi A. Proc. Natl. Acad. Sci. U. S. A. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda C., Li M., Hubálek F., Restelli N., Edmondson D. E., Mattevi A. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9750–9755. doi: 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubálek F., Binda C., Khalil A., Li M., Mattevi A., Castagnoli N., Edmondson D. E. J. Biol. Chem. 2005;280:15761–15766. doi: 10.1074/jbc.M500949200. [DOI] [PubMed] [Google Scholar]
- Tsugeno Y., Ito A. J. Biol. Chem. 1997;272:14033–14036. doi: 10.1074/jbc.272.22.14033. [DOI] [PubMed] [Google Scholar]
- Son S. Y., Ma J., Kondou Y., Yoshimura M., Yamashita E., Tsukihara T. Proc. Natl. Acad. Sci. U. S. A. 2008;105:5739–5744. doi: 10.1073/pnas.0710626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colibus L. D., Li M., Binda C., Lustig A., Edmondson D. E., Mattevi A., Klinman J. P. Proc. Natl. Acad. Sci. U. S. A. 2005;102:12684–12689. doi: 10.1073/pnas.0505975102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubálek F., Binda C., Li M., Herzig Y., Sterling J., Youdim M. B. H., Mattevi A., Edmondson D. E. J. Med. Chem. 2004;47:1760–1766. doi: 10.1021/jm0310885. [DOI] [PubMed] [Google Scholar]
- Koseki Y., Aoki S. Curr. Top. Med. Chem. 2014;14:176–188. doi: 10.2174/1568026613666131113155042. [DOI] [PubMed] [Google Scholar]
- Wu X., Zhu X., Hou Y., Wu X., Zeng H. Lett. Drug Des. Discovery. 2013;10:995–1006. [Google Scholar]
- Griffen E., Leach A. G., Robb G. R., Warner D. J. J. Med. Chem. 2014;54:7739–7750. doi: 10.1021/jm200452d. [DOI] [PubMed] [Google Scholar]
- Kumar V., Chadha N., Tiwari A. K., Sehgal N., Mishra A. K. Med. Chem. Res. 2014;23:1114–1122. [Google Scholar]
- Bautista-Aguiler O. M., Esteban G., Bolea I., Nikolic K., Agbaba D., Moraleda I., Iriepa I., Samadi A., Soriano E., Unzeta M. Eur. J. Med. Chem. 2014;75:82–95. doi: 10.1016/j.ejmech.2013.12.028. [DOI] [PubMed] [Google Scholar]
- Pisani L., Farina R., Nicolotti O., Gadaleta D., Sotootero R., Catto M., Di B. M., Mendezalvarez E., Carotti A. Eur. J. Med. Chem. 2015;89:98–105. doi: 10.1016/j.ejmech.2014.10.029. [DOI] [PubMed] [Google Scholar]
- Castagnoli K., Palmer S., Anderson A., Tjerk Bueters A., Neal Castagnoli J. Chem. Res. Toxicol. 1997;10:364–368. doi: 10.1021/tx970001d. [DOI] [PubMed] [Google Scholar]
- Gnerre C., Catto M., Leonetti F., Weber P., Carrupt P. A., Altomare C., Carotti A., Testa B. J. Med. Chem. 2001;43:4747–4758. doi: 10.1021/jm001028o. [DOI] [PubMed] [Google Scholar]
- Ogunrombi M. O., Malan S. F., Terre'Blanche G., Castagnoli Jr N., Bergh J. J., Petzer J. P. Bioorg. Med. Chem. 2008;16:2463–2472. doi: 10.1016/j.bmc.2007.11.059. [DOI] [PubMed] [Google Scholar]
- Sanna M. L., Maccioni E., Vigo S., Faggi C., Cirilli R. Talanta. 2010;82:426–431. doi: 10.1016/j.talanta.2010.04.039. [DOI] [PubMed] [Google Scholar]
- Yang D. D., Wang R., Zhu J. L., Cao Q. Y., Qin J., Zhu H. L., Qian S. S. J. Mol. Struct. 2016;1128:493–498. [Google Scholar]
- Schwarzschild M. A., Agnati L., Fuxe K., Chen J. F., Morelli M. Trends Neurosci. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Bhayye S. S., Roy K., Saha A. SAR QSAR Environ. Res. 2016;27:183–202. doi: 10.1080/1062936X.2015.1136840. [DOI] [PubMed] [Google Scholar]
- Mathew B., Haridas A., Uçar G., Baysal I., Adeniyi A. A., Soliman M. E., Joy M., Mathew G. E., Lakshmanan B., Jayaprakash V. Int. J. Biol. Macromol. 2016;91:680–695. doi: 10.1016/j.ijbiomac.2016.05.110. [DOI] [PubMed] [Google Scholar]
- Viegas-Junior C., Danuello A., Da S. B. V., Barreiro E. J., Fraga C. A. Curr. Med. Chem. 2007;14:1829–1852. doi: 10.2174/092986707781058805. [DOI] [PubMed] [Google Scholar]
- Farina R., Pisani L., Catto M., Nicolotti O., Gadaleta D., Denora N., Soto-Otero R., Mendez-Alvarez E., Passos C. S., Muncipinto G. J. Med. Chem. 2015;58:5561–5578. doi: 10.1021/acs.jmedchem.5b00599. [DOI] [PubMed] [Google Scholar]
- Brühlmann C., Ooms F., Carrupt P. A., Testa B., Catto M., Leonetti F., Altomare C., Carotti A. J. Med. Chem. 2001;44:3195–3198. doi: 10.1021/jm010894d. [DOI] [PubMed] [Google Scholar]
- Mooney S., Leuendorf J. E., Hendrickson C., Hellmann H. Molecules. 2009;14:329–351. doi: 10.3390/molecules14010329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashim A., Wang L., Juneja K., Ye Y., Zhao Y., Ming L. J. Bioorg. Med. Chem. Lett. 2011;21:6430–6432. doi: 10.1016/j.bmcl.2011.08.123. [DOI] [PubMed] [Google Scholar]
- Baur J. A., Sinclair D. A. Nat. Rev. Drug Discovery. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- Lu C., Guo Y., Yan J., Luo Z., Luo H. B., Yan M., Huang L., Li X. J. Med. Chem. 2013;56:5843–5859. doi: 10.1021/jm400567s. [DOI] [PubMed] [Google Scholar]
- Park D. H., Venkatesan J., Kim S. K., Ramkumar V., Parthiban P. Bioorg. Med. Chem. Lett. 2012;22:6362–6367. doi: 10.1016/j.bmcl.2012.08.080. [DOI] [PubMed] [Google Scholar]
- Roman G. Eur. J. Med. Chem. 2015;89:743–816. doi: 10.1016/j.ejmech.2014.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Qiang X., Li Y., Luo L., Xu R., Zheng Y., Cao Z., Tan Z., Deng Y. Bioorg. Chem. 2017;71:305–314. doi: 10.1016/j.bioorg.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Hu S., Nian S., Qin K., Xiao T., Li L., Qi X., Ye F., Liang G., Hu G., He J. Chem. Pharm. Bull. 2012;60:385–390. doi: 10.1248/cpb.60.385. [DOI] [PubMed] [Google Scholar]
- Pretorius J., Malan S. F., Castagnoli Jr N., Bergh J. J., Petzer J. P. Bioorg. Med. Chem. 2008;16:8676–8684. doi: 10.1016/j.bmc.2008.07.088. [DOI] [PubMed] [Google Scholar]
- Legoabe L. J., Petzer A., Petzer J. P. Bioorg. Med. Chem. Lett. 2014;24:2758–2763. doi: 10.1016/j.bmcl.2014.04.021. [DOI] [PubMed] [Google Scholar]
- Legoabe L. J., Petzer A., Petzer J. P. Bioorg. Med. Chem. Lett. 2012;22:5480–5484. doi: 10.1016/j.bmcl.2012.07.025. [DOI] [PubMed] [Google Scholar]
- Lan J. S., Zhang T., Liu Y., Zhang Y., Hou J., Xie S., Yang J., Ding Y., Cai Z. MedChemComm. 2017;8:471–478. doi: 10.1039/c6md00586a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badavath V. N., Sinha B. N., Jayaprakash V. Int. J. Pharm. Pharm. Sci. 2015;12:272–282. [Google Scholar]
- Badavath V. N., Ucar G., Sinha B. N., Mondal S. K., Jayaprakash V. ACS Med. Chem. Lett. 2015;7:5879–5884. doi: 10.1021/acsmedchemlett.5b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani L., Farina R., Catto M., Iacobazzi R. M., Nicolotti O., Cellamare S., Mangiatordi G. F., Denora N., Sotootero R., Siragusa L. J. Med. Chem. 2016;14:6791–6806. doi: 10.1021/acs.jmedchem.6b00562. [DOI] [PubMed] [Google Scholar]
- Petzer A., Pienaar A., Petzer J. P. Life Sci. 2013;93:283–287. doi: 10.1016/j.lfs.2013.06.020. [DOI] [PubMed] [Google Scholar]
- Petzer A., Grobler P., Bergh J. J., Petzer J. P. J. Pharm. Pharmacol. 2014;66:677–687. doi: 10.1111/jphp.12193. [DOI] [PubMed] [Google Scholar]
- Vlok N., Malan S. F., Castagnoli Jr N., Bergh J. J., Petzer J. P. Bioorg. Med. Chem. 2006;14:3512–3521. doi: 10.1016/j.bmc.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Strydom B., Malan S. N. J. Bioorg. Med. Chem. 2010;18:1018–1028. doi: 10.1016/j.bmc.2009.12.064. [DOI] [PubMed] [Google Scholar]
- Batovska D. I., Todorova I. T. Curr. Clin. Pharmacol. 2010;5:1–29. doi: 10.2174/157488410790410579. [DOI] [PubMed] [Google Scholar]
- Nobre-Júnior H. V., Oliveira R. A., Maia F. D., Nogueira M. A. S., Moraes M. O. D., Bandeira M. A. M., Andrade G. M., Viana G. S. B. Neurochem. Res. 2009;34:1066–1075. doi: 10.1007/s11064-008-9876-5. [DOI] [PubMed] [Google Scholar]
- Hammuda A., Shalaby R., Rovida S., Edmondson D. E., Binda C., Khalil A. Eur. J. Med. Chem. 2016;114:162–169. doi: 10.1016/j.ejmech.2016.02.038. [DOI] [PubMed] [Google Scholar]
- Chimenti F., Fioravanti R., Bolasco A., Chimenti P., Secci D., Rossi F., Yáñez M., Orallo F., Ortuso F., Alcaro S. J. Med. Chem. 2009;52:2818–2824. doi: 10.1021/jm801590u. [DOI] [PubMed] [Google Scholar]
- Robinson S. J., Petzer J. P., Petzer A., Bergh J. J., Lourens A. C. Bioorg. Med. Chem. Lett. 2013;23:4985–4989. doi: 10.1016/j.bmcl.2013.06.050. [DOI] [PubMed] [Google Scholar]
- Glover V., Halket J. M., Watkins P. J., Clow A., Goodwin B. L., Sandler M. J. Neurochem. 1988;51:656–659. doi: 10.1111/j.1471-4159.1988.tb01089.x. [DOI] [PubMed] [Google Scholar]
- Tripathi R. K., Krishnamurthy S., Ayyannan S. R. ChemMedChem. 2015;11:119–132. doi: 10.1002/cmdc.201500443. [DOI] [PubMed] [Google Scholar]
- Manley-King C. I., Bergh J. J., Petzer J. P. Bioorg. Med. Chem. 2011;19:4829–4840. doi: 10.1016/j.bmc.2011.06.070. [DOI] [PubMed] [Google Scholar]
- Strydom B., Bergh J. J., Petzer J. P. Bioorg. Med. Chem. Lett. 2013;23:1269–1273. doi: 10.1016/j.bmcl.2013.01.003. [DOI] [PubMed] [Google Scholar]
- Jo Y. S., Huong D. T., Bae K., Lee M. K., Kim Y. H. Planta Med. 2002;68:84–85. doi: 10.1055/s-2002-20056. [DOI] [PubMed] [Google Scholar]
- Hwang J. S., Lee S. A., Hong S. S., Lee K. S., Lee M. K., Bang Y. H., Ro J. S. Arch. Pharmacal Res. 2005;28:190–194. doi: 10.1007/BF02977714. [DOI] [PubMed] [Google Scholar]
- Baek S. C., Ryu H. W., Kang M. G., Lee H., Park D., Cho M. L., Oh S. R., Kim H. Bioorg. Med. Chem. Lett. 2018;14:2403–2407. doi: 10.1016/j.bmcl.2018.06.023. [DOI] [PubMed] [Google Scholar]
- Wilson G. L., Lill M. A. Future Med. Chem. 2011;3:735–750. doi: 10.4155/fmc.11.18. [DOI] [PubMed] [Google Scholar]
- Fang Y. Expert Opin. Drug Discovery. 2012;7:969–988. doi: 10.1517/17460441.2012.715631. [DOI] [PubMed] [Google Scholar]
- Salum L. B., Polikarpov I., Andricopulo A. D. ChemInform. 2009;40:2243–2253. [Google Scholar]
- Hossain C. F., Okuyama E., Yamazaki M. ChemInform. 1996;8:1535–1539. doi: 10.1248/cpb.44.1535. [DOI] [PubMed] [Google Scholar]
- Matos M. J., Viña D., Picciau C., Orallo F., Santana L., Uriarte E. Bioorg. Med. Chem. Lett. 2009;19:5053–5055. doi: 10.1016/j.bmcl.2009.07.039. [DOI] [PubMed] [Google Scholar]
- Viña D., Matos M. J., Yáñez M., Santana L., Uriarte E. MedChemComm. 2012;3:213–218. [Google Scholar]
- He X., Chen Y. Y., Shi J. B., Tang W. J., Pan Z. X., Dong Z. Q., Song B. A., Li J., Liu X. H. Bioorg. Med. Chem. 2014;22:3732–3738. doi: 10.1016/j.bmc.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Cooper M. E. Drug Discovery Today. 2004;9:740. [Google Scholar]
- Sheu S. Y., Chiang H. C. Anticancer Res. 1900;17:3293–3297. [PubMed] [Google Scholar]
- Kang K., Fong W. P., Tsang P. W. Med. Mycol. 2010;48:904–911. doi: 10.3109/13693781003739351. [DOI] [PubMed] [Google Scholar]
- Zengin G., Degirmenci N. S., Alpsoy L., Aktumsek A. Hum. Exp. Toxicol. 2015;35:544–553. doi: 10.1177/0960327115595687. [DOI] [PubMed] [Google Scholar]
- Lee H. W., Ryu H. W., Kang M. G., Park D., Oh S. R., Kim H. Bioorg. Med. Chem. Lett. 2017;27:1136–1140. doi: 10.1016/j.bmcl.2017.01.085. [DOI] [PubMed] [Google Scholar]
- Yelekçi K., Büyüktürk B., Kayrak N. J. Neural Transm. 2013;120:853–858. doi: 10.1007/s00702-012-0954-0. [DOI] [PubMed] [Google Scholar]
- Kare P., Bhat J., Sobhia M. E. Mol. Diversity. 2013;17:111–122. doi: 10.1007/s11030-012-9420-z. [DOI] [PubMed] [Google Scholar]
- Andricopulo A. D., Salum L. B., Abraham D. J. Curr. Top. Med. Chem. 2009;9:771–790. doi: 10.2174/156802609789207127. [DOI] [PubMed] [Google Scholar]