

ABSTRACT
Hepatocellular carcinoma (HCC) is the second cause of death for cancer worldwide, justifying the urgent need for novel therapeutic approaches. Immunotherapeutic strategies based on triggering and/or rescuing tumor antigen-specific T cells may be promising particularly if combined together. As preliminary step toward this goal, we have investigated the expression of antigen presenting molecules (HLA class I and class II) and immune checkpoints (PD-1 and PD-L1) in 43 HCC samples from distinct patients and in HCC cell lines. While normal hepatocytes did not express HLA class I and II, HCC cells strongly upregulated HLA class I while remaining negative for HLA class II. The absence of HLA class II expression in HCC cell lines correlated with lack of expression of the HLA class II transactivator, CIITA, which could not be rescued even after interferon-gamma treatment. This was due to high methylation levels of interferon-gamma-sensitive CIITA promoter IV strongly suggesting a biologically relevant developmental silencing of HLA-II expression in liver cell lineage. HCC tumor tissues showed a variable degree of leukocyte infiltration. Infiltrating lymphocytes expressed PD-1, while PD-L1 was expressed in cells with monocyte-macrophage morphology mostly localized at the tumor margin, but not in tumor cells. De novo expression of HLA class I, instrumental for presenting tumor antigens to cytotoxic T lymphocytes, and the correct characterization of the cells expressing checkpoint inhibitors in the tumor tissue should be the ground for setting novel strategies of combined approaches of immunotherapy in HCC based on tumor peptide vaccines and anti-checkpoint inhibitor antibodies.
Keywords: HCC, CIITA, HLA, PD-1, PD-L1
Introduction
Hepatocellular carcinoma (HCC) accounts for 90% of primary liver cancers and is one of the deadliest cancers ranking sixth in global incidence and second in terms of cancer deaths in the world, after lung cancer, with a tribute of 0,8 million people in 2012 (WHO, IARC http://globocan.iarc.fr)1. Current curative approaches for HCC involve partial liver resection, liver transplantation, chemotherapy, and transarterial chemoembolization.2 Despite important advances in the diagnosis and treatment of liver cancer in the recent decades, the 5-year survival rate remains dismal, (less than 10%), because of the high frequency of intrahepatic recurrence after hepatectomy and the low effect of systemic therapy by the single drug Sorafenib, the current standard of care for advanced disease.3 Thus, novel complementary strategies of treatment are urgently required for this deadly disease.
Recently, immunotherapeutic approaches, such as treatment with antibodies specific for immune checkpoints expressed on effector T cells (CTLA-4, PD-1) and antigen presenting cells (PD-L1), have revitalized the enthusiasm in the field of cancer therapy, particularly for the preliminary success obtained in the treatment of cancers such as melanomas and NSCLC.4 Based on the fact that blocking immune checkpoints is a way to unleash preexisting immunity to tumors, somehow inhibited in tumor patients, in principle this approach could be applied to a wide variety of cancers; within this line similar treatments are presently in clinical trials also for HCC.5,6 It is of interest that the ligand of PD-1, the PD-L1 molecule, can be expressed also in tumor cells. This is relevant because not only antigen presenting cells (APC) but also tumor cells can potentially block the activity of effector T cells during their functional lifespan, particularly in the tumor microenvironment, and result in worst prognosis for cancer patients.7–9 Indeed, recent results have raised the possibility of a correlation, in terms of bad prognosis, between expression of PD-L1 in HCC cells and patient’s outcome,10–12 supporting the notion that blocking these checkpoints molecules with inhibitor antibodies may be a promising way to increase the tumor patient’s capacity to fight cancer.13
As outlined above, however, targeting the immune checkpoints requires the presence of previously triggered anti-tumor T cells, both CD4 and CD8, whose recognition of tumor antigens by their clonotypically distributed receptors depends on antigen presentation on HLA-encoded class II and class I cell surface molecules, respectively.
It is apparent that immunotherapeutic strategies against cancer, and thus against HCC as well, could not rely on a single approach but on a variety of combination therapies which should take into account primarily the mechanisms at the basis of the triggering of the anti-tumor immune response. As adaptive immune response to tumors is essentially a T cell response, a crucial step of this response is the initial recognition of tumor-associated antigens (TAA), here defined as antigens derived by both over-expressed proteins or mutated proteins (neoantigens) in tumor cells. This process is accomplished in two steps: a)- antigen processing of TAA by specialized cells, such as dendritic cells and macrophages (APC) and presentation of appropriate TAA peptides within the context of HLA class II molecules; b)- recognition of MHC class II-bound peptides displayed on the cell surface by HLA class II-restricted CD4 + T cells, designated T helper (TH) cells.14 TH cells are fundamental for optimal induction of both humoral and cellular effector mechanisms,15 particularly for the maturation of HLA class I-restricted CD8+ naïve T cells, their clonal expansions and acquisition of cytotoxic function.16 The latter function is of relevance in the context of anti-tumor immunity, since CD8+ cytotoxic T cells (CTL) are believed to be the major lymphocyte effectors against cancer cells.17 In physiological conditions, expression of HLA class II molecules is confined to B cells, APCs and other few cell types. While B cells constitutively express HLA class II molecules, other cell types may express these molecules under the induction of certain inflammatory cytokines, particularly IFNγ. Both constitutive and inducible HLA class II expression are under the control of the class II transactivator, also designed CIITA, discovered in our laboratory.18–20 In tumors, particularly in tumors of epithelial origin, HLA class II expression is not a common event but when present is always under the control of CIITA,21 and is generally associated with better prognosis both on primary tumors22,23 and in tumors with poor metastatic potential.24 Moreover, since the final effectors of the adaptive anti-tumor immune response are primarily the CD8+ CTLs, it is expected that expression of MHC class I molecules in tumor cells is one of the most important parameters associated with the efficacy of immune response against the tumors. Indeed, lack or reduced expression of HLA class I molecules in tumor cells is often associated with tumor escape from the immune system.25 In HCC cells, early investigation on the expression of HLA class I and class II molecules, was performed by two groups. Paterson et al. also showed low expression of HLA class I in normal hepatocytes and increased expression in HCC cells. Very faint and scattered expression of HLA class II cell surface antigens was found in about 40% of HCC cells.26 In partial agreement, Sung et al. showed no expression of HLA class I on normal hepatocyte and expression of HLA class I in HCC.27 Normal liver cells were negative for HLA class II and only 3 out of 11 HCC samples in those original studies showed faint HLA class II expression.27
Due to their mutual importance in the immune regulation of anti-cancer response, it is crucial to study the comparative expression of HLA cell surface molecules (both class I and class II) and immune checkpoints such as PD-1 and PD-L1 in tumor tissues if one wants to integrate possible synergistic immunotherapeutic strategies such as tumor-specific peptide vaccination and checkpoint blockade. Within this frame we decided to investigate by immunohistochemistry the expression of the above markers in a significant number of HCC tumor tissues. Moreover, we analyzed by immunofluorescence and cytofluorometry, HCC cell lines for their expression of HLA class I, class II, PD-1 and PD-L1 in steady state conditions, and upon stimulation by IFNγ, a crucial inflammatory cytokine often produced by lymphocytes infiltrating the tumor and capable of stimulating the expression of both HLA (class I and class II) and PD-L1 molecules. One peculiar reproducible finding was the lack of HLA class II expression in HCC cells even after IFNγ treatment which correlated with the lack of expression of CIITA mRNA, in contrast to an up-regulation of HLA class I expression. Further experiments showed that absence of CIITA transcription correlated with high methylation levels of IFNγ-sensitive CIITA promoter IV, and not to defects of the IFNγ-dependent signal transduction pathway.
The results obtained clearly indicate a dichotomy in the expression of HLA class I and class II molecules due a developmental silencing of CIITA in hepatocytes that seems to be maintained in neoplastic transformation. Moreover, HCC tumor tissues presented distinct degree of PD-1-expressing infiltrating lymphocytes. PD-L1 was expressed in cells with monocyte-macrophage morphology localized at the tumor margin, but not in tumor cells. These results are discussed within the context of possible strategies for more suitable protocols of combined immunotherapeutic approaches in HCC.
Results
Histopathological grading and etiopathogenesis of HCC under study
We have analyzed 43 distinct HCC samples, each from distinct patients who underwent clinical treatment in our hospital. As far as pathogenesis, laboratory analysis showed that 18 hepatocarcinomas arosed in patients positive for Hepatitis C virus (HCV) infection, one HCC developed in a patient positive for Hepatitis B virus (HBV) infection and two HCC arose in a patient positive for both HBV and HCV. Remaining patients were either negative (10 patients) or not assessed for previous HBV/HCV infection (Table 1). Two patients developed HCC on a previous cirrhosis with a history of alcohol abuse. In terms of histopathological differentiation stage of the disease, samples were classified as Grade 1, 2 or 3 following the progressive malignant alterations of the tumor cells, respectively, Grade 3 being the most anaplastic tumor. In relation to the presence of cellular infiltrate, tumors were classified as 0 (no infiltrate), 1+, 2+ and 3+, on the basis of progressively higher number of infiltrating cells, mostly lymphocytic cells, as described in Materials and Methods. Of notice, degree of infiltration did not strictly correlate with either HCV/HBV infection or the grading of malignancy in terms of histopathological feature. Indeed, tumors with the highest infiltrate could be either negative or positive for HCV infection, and similarly tumors with no infiltration could be again negative or positive for HCV infection.
Table 1.
Expression of HLA class I and class II in HCC tumors from 43 distinct patients and correlation with clinico-pathological findings.
| Cases |
HLA-I Expression |
HLA-II Expression |
||||||
|---|---|---|---|---|---|---|---|---|
| Variables | Total 43 | High (≥ 70%)a |
Medium (20% ≤ x < 70%) |
Low (< 20%) |
p value | Positive 1 1% ≤ x < 3%a |
Negative | p value |
| Gender | ||||||||
| M/F | 35/8 | 18/2 | 13/6 | 4/0 | 6/1 | 29/7 | ||
| Age Mean (years) |
67 ± 11 | 68/67 | 62/70 | 69/0 | 0.370 | 64/73 | 67/69 | 0.573 |
| Grading | 0.582 | 0.311 | ||||||
| G3 | 1 | 2 | 0 | 1 | 2 | |||
| G2 | 17 | 12 | 3 | 6 | 26 | |||
| G1 | 2 | 5 | 1 | 0 | 8 | |||
| Infiltration | 0.586 | 0.137 | ||||||
| 3+ | 2 | 0 | 0 | 1 | 1 | |||
| 2+ | 6 | 6 | 1 | 1 | 12 | |||
| 1+ | 9 | 9 | 1 | 5 | 14 | |||
| 0 | 3 | 4 | 2 | 0 | 9 | |||
| CD8b | 43.5 | 18.8 | 2.5 | 0.040* | 50 | 34.6 | 0.344 | |
| CD4 | 28 | 19.2 | 2.5 | 0.138 | 19.5 | 21.8 | 0.445 | |
| Infection | 0.556 | 0.815 | ||||||
| HCV and/or HBV | 9 | 11 | 1 | 4 | 17 | |||
| No infection | 7 | 3 | 2 | 2 | 10 | |||
| Not documented | 4 | 5 | 1 | 1 | 9 | |||
a percent values (in parenthesis) indicate the number of cells expressing HLA-I or HLA-II molecules
b mean of CD8 or CD4 positive infiltrating cells as obtained by counting the stained cells in at least three representative fields of the corresponding High, Medium and Low HLA class I-expressing cells
*statistically significant with p < 0.05
De novo expression of HLA class I cell surface molecules in HCC tumor cells and correlation with lymphocyte infiltration
The expression of HLA class I and class II molecules was then assessed in HCC tumors and compared with the surrounding, unaffected normal liver of the same patient. Moreover, additional normal liver tissues, from individuals undergoing liver surgery from cancer-unrelated pathology, were analyzed. As common feature, HLA class I cell surface molecules were not detectable in normal liver parenchymal cells (see as an example Figure 1, panel b). Expression of HLA class I in normal liver tissue was essentially confined to liver sinusoidal epithelial cells (LSEC) and Kupffer cells (KC). Similarly, HLA class II (DR and DQ) molecules were not expressed in normal liver parenchymal cells, whereas they were expressed in LSEC and KC cells (Figure 1, panels c and d, respectively). In HCC, irrespective of the absent, low or high inflammatory infiltrate, the majority of tumor cells were clearly positive for HLA class I expression (Table 1, and Figure 1, panels f, j and n). In most cases, the percentage of HLA class I positive tumor cells was higher than 50%. Only in two cases, we found 5% or less HLA class I-positive tumor cells, respectively.
Figure 1.
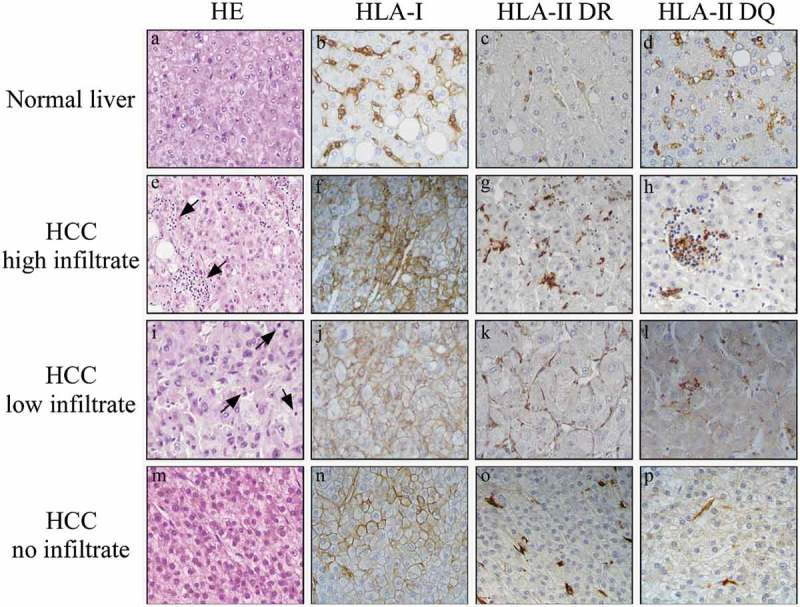
HLA class I, but not HLA class II, is highly expressed on HCC tumor cells.
Immunohistochemical staining for both HLA class I and HLA class II in paraffin-embedded blocks of HCC tissue samples. The upper panels (a-d) show normal liver tissue with HLA class I and HLA class II expression (here assessed for both HLA-DR and HLA-DQ) confined to LSEC and KC cells. In contrast, the HCC tumor tissues, classified as having high infiltrate (panel e, arrowheads), low infiltrate (panel i, arrowheads), or no infiltrate (panel m), show strong membrane expression of HLA class I (panels f, j, n), but no expression HLA class II (panels g, h, k, l, o, p) in tumor cells. Original magnification X 400.
Nevertheless, differences were observed in the amount of expression of HLA class I at single tumor cell level, usually with higher expression in those tumor cells accompanied by higher mono-lymphocytic infiltration. (Figure 1, compare panel f with panels j and n). Interestingly, lymphocyte infiltration was mostly represented by CD8 + T cells and to lesser extent by CD4 + T cells (Table 1, and Figure 2). The degree of CD8 + T cell infiltration significantly correlated with the intensity of HLA class I expression (Table 1). As far as the expression of HLA class II molecules, it was not detected in most of the tumor cells, irrespective of the level of infiltration of tumor tissues (Figure 1, panels g,h,k,l,o,p), while it was detected again in LSEC and KC, and in tumor infiltrating lymphocytes (Figure 1, panels g,h,k,l,o,p). When we compared clinico-pathological parameters (gender, age, tumor grading, rate of infiltration and infection), with low (≤ 20%), medium (20% to 70%) and high (≥ 70%) number of tumor cells expressing HLA-I or HLA-II molecules, we observed no significant correlation (Table 1). An analysis of correlation between outcome and marker expression could be done in relation to the expression of HLA class I for a number of patients (31 patients) included in the high (n.17) and medium (n.14) HLA expression groups. Supplementary Figure 1 shows that in the analyzed groups, although there is no statistical difference in the final outcome, a clear difference was observed up to 60 months where the two curves of high HLA class I expression group (A, black squares) and medium HLA class I expression group (B, grey squares) show 50% and 23% survival, respectively.
Figure 2.
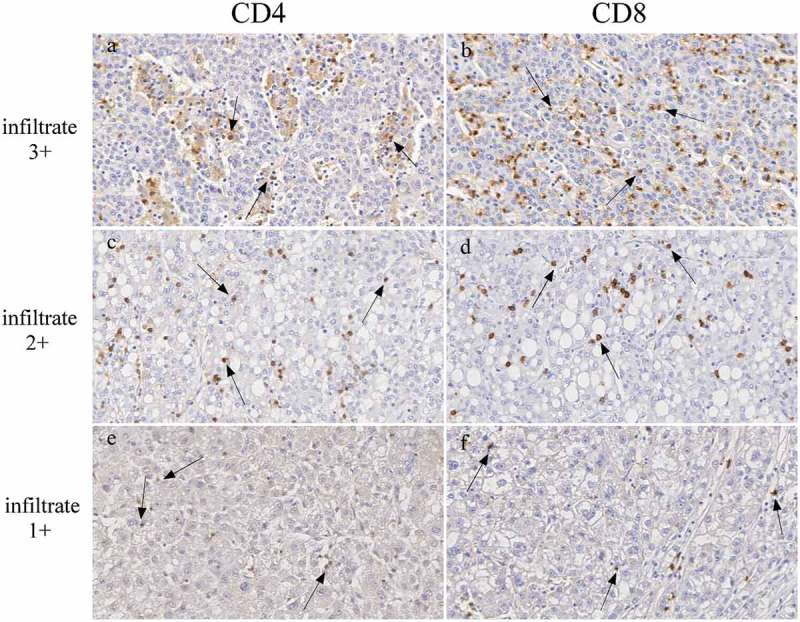
CD4 and CD8 infiltration in HCC tumors.
Immunohistochemical staining for both CD4 and CD8 in paraffin-embedded blocks of HCC tissue samples. Panels show representative HCC tumor tissues with very high (top, infiltrate 3+), high (middle, infiltrate 2+) and low (bottom, infiltrate 1+) CD4 (left panels, a,c,e) and CD8 (right panels, b,d,f) infiltration (original magnification, x200). Examples of CD4 or CD8 positive lymphocytes are indicated by arrows.
Expression of HLA cell surface molecules in HCC tumor cell lines
In order to compare the expression of human MHC cell surface molecules in tumor cells from patients’ tumor tissues and HCC tumor cell lines, we analyzed by immunofluorescence and flow cytometry the HLA class I and HLA class II cell surface phenotype of 4 HCC cell lines, namely Alex, Hep3B, HepG2 and HUH7. Interestingly, and at variance with the results in HCC in tumor tissues, the 4 HCC cell lines expressed very low HLA class I molecules with two of them (HepG2 and HUH7) expressing levels approaching negative values (Figure 3(a), HLA-I, solid lines). Thus, HCC cell lines did not mimic the behavior of hepatocarcinoma cells from cancer tissues. HLA class II molecules were not expressed in any HCC cell line (Figure 3(a), HLA-DR, solid lines), thus mirroring the phenotype of HCC tumor cells from tumor tissues. Thus, the adaptation of HCC tumor cells in vitro and the generation of cell lines was accompanied by a substantial modification of the HLA class I expression, while the HLA class II phenotype was unchanged. The expression of HLA genes and particularly of HLA class II genes can be rescued by treatment with the inflammatory cytokine IFNγ which can also up-regulate pre-existing expression of HLA class I molecules. As far as HLA class II genes, IFNγ does not directly activate them but it does so via the transcriptional activation of CIITA which, in turn, activates HLA class II gene transcription. Interestingly, after IFNγ treatment, the expression of HLA class I was upregulated in all 4 cell lines whereas the expression of HLA class II remained negative (Figure 3(a), bold lines). THP-1 cells, which are known to have a functional IFNγ signaling pathway, were used as a control for HLA class I and class II expression.
Figure 3.
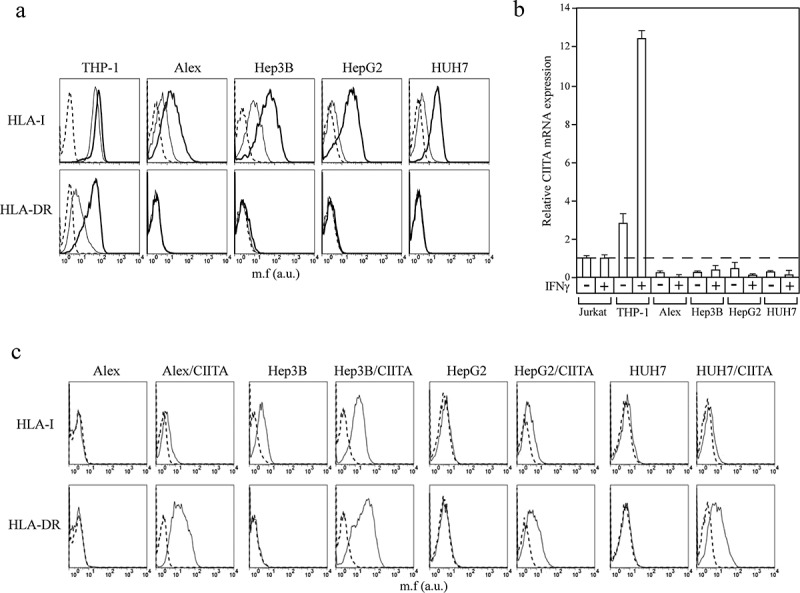
Lack of CIITA expression is responsible for the absence of HLA-II expression in HCC tumor cell lines.
(a) HLA class I (HLA-I) and HLA class II (HLA-DR) cell surface phenotype of Alex, Hep3B, HepG2 and HUH7 HCC cell lines were carried out by immunofluorescence and FACS analysis. The monocytic THP-1 cell line was used as a positive control for both baseline and IFNγ-induced HLA-I and HLA-DR expression. Histograms represent fluorescence profiles of the cells indicated on the top incubated with specific anti-HLA-I or HLA-DR mAbs followed by incubation with FITC-conjugated F(ab)2 anti mouse antibody as second reagent. Cells were either untreated (solid line) or treated with IFNγ (bold line). Controls (dashed line) are cells incubated with the second reagent only. Mean fluorescence (m.f.) values are expressed in the abscissa as arbitrary units (a.u.). A representative experiment is shown out of three independent experiments with very similar results. (b) CIITA mRNAs expression was assessed by qRT-PCR in cells treated or not with IFNγ. The results of a representative experiment, out of at least three experiments with similar results, performed in triplicates are shown. CIITA mRNA levels are expressed as values relative to those of untreated Jurkat T cells set to 1. THP-1 cells were used as a positive control for both basal and IFNγ-induced- CIITA mRNA expression. Error bars represent the standard deviation. (c) The stable expression of CIITA in Alex, Hep3B, HepG2 and HUH7 cells restores the HLA-DR expression. HLA-I and HLA-DR cell surface expression was carried out by immunofluorescence and FACS analysis. Histograms represent fluorescence profiles of the cells indicated on the top incubated with specific anti HLA-I or HLA-DR mAbs (solid line) followed by incubation with FITC-conjugated F(ab)2 anti mouse antibody as second reagent. Controls (dashed line) are cells incubated with the second reagent only. Mean fluorescence (m.f.) values are expressed in the abscissa as arbitrary unit (a.u). A representative experiment is shown out of three independent experiments with very similar results.
We next investigated whether lack of HLA class II expression was dependent from a primary defect in HLA class II transcription and/or protein synthesis or from a defect in CIITA expression. Real time-quantitative PCR showed that HCC cell lines treated with IFNγ did not express CIITA mRNA at all (Figure 3(b)). Moreover, genetic transfer of a CIITA expressible cDNA into HCC cell lines resulted in a consistent and stable expression of HLA class II genes and corresponding cell surface molecules (Figure 3(c)). Taken together, these results show that the defect of HLA class II expression in HCC cells was dependent on lack of transcription of endogenous CIITA even after treatment with IFNγ and not to an intrinsic defect of HLA class II genes.
Lack of expression of CIITA in HCC is not due to defects of IFNγ signaling pathway
To investigate, in more detail, whether the lack of IFNγ-dependent expression of CIITA in HCC could be associated to a defect in the IFNγ signaling pathway we analyzed the integrity of the engaged intracellular factors after interaction of IFNγ with its cell surface receptor. This interaction recruits Jak1 and Jak2 kinases which in turn phosphorylate and activate the transcription factor STAT1 (p-STAT1) that dimerizes, migrates to the nucleus where it binds to the promoter of IRF1, and activates its transcription. IRF1 and dimerized p-STAT1 bind to CIITA pIV together with the ubiquitous USF1 transcription factor and activate the transcription of CIITA.28
We therefore evaluated the level of p-STAT1 in CIITA-negative HepG2 and, as control, in IFNγ-mediated CIITA-inducible RA cell lines (Supplementary Fig. 2). Results in Figure 4(a) show that STAT1 was phosphorylated in HepG2 following IFNγ exposure. As expected, high levels of STAT1 phosphorylation were also observed in RA cells after treatment with IFNγ but not in untreated cells (Figure 4(a)). We next assessed by real time PCR the expression of IRF1 in HepG2 cells exposed to IFNγ. As shown in Figure 4(b) (right panel), IFNγ treatment determined a significant induction of IRF1 mRNA expression in HepG2 compared to the untreated cells. Similar results were obtained in RA glioma cells (Figure 4(b), left panel).
Figure 4.
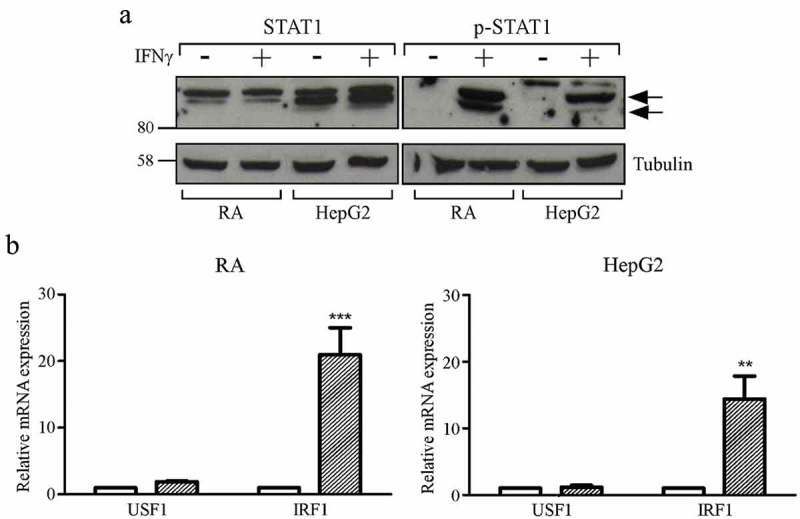
STAT1 phosphorylation and significant increase of IRF1 mRNA levels after treatment with IFNγ in HepG2 hepatocarcinoma and RA glioma cell lines.
(a) Total cell extracts of RA and HepG2 cell were collected after 72 hours treatment with IFNγ (+) or its vehicle (-) and analyzed by western blotting using anti-STAT1 and anti-phosphorylated-STAT1 (p-STAT1) antibodies. Tubulin was used as a loading control. The two black arrows on the right indicate the bands corresponding to the phosphorylated STAT1. A representative experiment is shown out of three independent experiments with very similar results. (b) USF1 and IRF1 mRNAs expression in RA (left panel) and HepG2 (right panel) cells treated with IFNγ (dashed colums) or with its vehicle (white colums) for 72 were assessed by qRT-PCR. The results of three representative experiments performed in triplicates are shown. USF1 and IRF1 mRNA levels in IFNγ treated cells are expressed as values relative to those of untreated cells set to 1. Two-WAY ANOVA test has been performed (** p ≤ 0.01 for Hep-G2 and *** p ≤ 0.001 for RA as compared to corresponding untreated cells). Error bars represent the standard deviation.
Finally, we evaluated the expression of USF1 in both HepG2 and RA cells. As shown in Figure 4(b), USF1 was expressed in both cell lines and IFNγ did not affect its expression level. Taken together these findings confirmed that all the transcription factors required for a correct stimulation of CIITA gene expression were enrolled after IFNγ treatment in HCC cells.
Hypermethylation of the CIITA promoter IV region, including the DNA binding motifs for STAT1, USF1 and IRF1, correlates with lack of CIITA expression in HepG2 cells
The above results suggest that chromatin accessibility of STAT1, USF1 and IRF1 may be hampered at the level of CIITApIV in HCC cells. To verify this hypothesis, we investigated the presence of methyl-CpG sites in the region of the CIITA promoter IV that extends 350 bp upstream of the transcription start site, and subdivided it for PCR amplification in two shorter regions (a and b) schematically represented in Figure 5(a). DNA sequencing analysis showed 11 potential CpG island sites for methylation, of which 8 are located in region b which includes GAS, E-box and IRF consensus sequences for STAT1, USF1 and IRF1, respectively (Figure 5(a), upper diagram). Bisulfite sequencing of the CIITApIV in HepG2 revealed a high concentration of methylated CpG dinucleotides as compared to CIITApIV of RA, all along the sequence analyzed, particularly enriched at the region spanning nucleotides −42 to + 26 (Figure 5(b)). Consistent with the data discussed above, the level of CpG methylation was not affected by IFNγ treatment. Differently from HepG2, in RA cells the DNA segments analyzed were unmethylated or hypomethylated, independently of stimulation with IFNγ (Figure 5(b)), supporting the idea that the methylation status of CIITA pIV could influence the accessibility of specific transcription factors to the promoter, thus affecting CIITA gene transcription.
Figure 5.
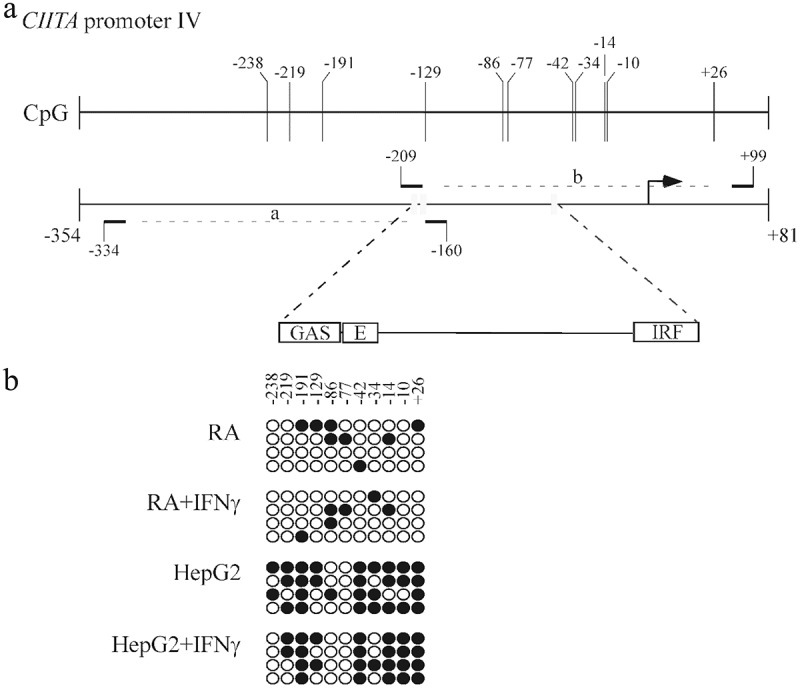
CIITA promoter IV is highly methylated in HepG2 hepatocarcinoma cells.
(a) Schematic representation of the human CIITA promoter IV region used for PCR amplification of bisulfite converted DNA. The sequence, including GAS, E-box and IRF1 consensus elements, was amplified by PCR in two sub regions (a: from position −334 to −160; b: from position −209 to + 99), using specific primers as indicated in Materials and Methods. The numbers indicate the relative distance upstream of the transcriptional start site (black arrow). Vertical bars in the upper line indicate CpG dinucleotide. (b) CpG methylation of the human CIITA promoter IV selected region in RA and HepG2 cells treated with 500U/ml of IFNγ or its vehicle for 3 days. The methylation status of CpGs in the promoter region was monitored by bisulfite analysis. The filled and open circles represent the methylated and unmethylated CpG dinucleotides, respectively. A mean of 4 separate clones was analyzed for each region. High methylation was observed in HepG2 cells, independently of IFNγ treatment.
Taken together these data strongly suggest that lack of HLA-II expression in hepatocarcinoma cells is due to lack of CIITA transcription most likely generated by CpG island hypermethylation of CIITApIV.
Expression of PD-1 and PD-L1 checkpoint inhibitors in HCC tumor tissues
To further investigate the expression of additional relevant markers involved in the control of immune response against HCC and relevant for immunotherapeutic strategies, we next assessed the expression of checkpoint inhibitor PD-1 molecule and its major receptor PD-L1 in HCC tumor tissues. Figure 6 shows the analysis by immunohistochemistry of four representative HCC tumor tissues from distinct patients characterized by very high (infiltrate 3+), high (infiltrate 2+), low (infiltrate 1 +), or no leukocyte infiltration. Clear expression of PD-1 was detected in infiltrating cells. PD-1+ cells were interspersed within the HCC tumor tissue and were mainly represented by lymphocytes (Figure 6, panels a, c, e, arrows). On the other hand, in HCC tumor tissues with relatively very high and high degree of leukocyte infiltration, PD-L1 cell surface marker was expressed in cells with monocyte-macrophage morphology (Figure 6, panels b, d, f, arrows). In HCC tumor tissues with low or absent leukocyte infiltration, PD-L1 was usually expressed at very low level or not expressed (Figure 6, panel f and h, respectively). In most instances, positive cells were seen at the margin of the tumor tissue. These cells displayed the morphology of KC (Figure 6, panel b, arrowheads) or hepatic stellate cells (Figure 6, panel b, arrows). Interestingly, we found that the number of infiltrating cells in HCC was significantly correlated with both PD-1 and PD-L1 expression (Table 2, p = 9,73x10 – 5 and p = 0.019, respectively). Although the vast majority of HCC tumor cells were not stained for the PD-L1 marker, some tumor cells (less than 3% and only in 3 out of 43 HCC cases analyzed) displayed a low but distinct positivity for PD-L1 expression (Figure 6, panel b, arrows). These findings were confirmed by the use of two distinct commercially available reagents specific for PD-L1, a rabbit and a mouse monoclonal antibody. Due to the partially conflicting results with previous reports 10–12 we further investigated in more detail several cases with the highest number of PD-L1 positive cells to clearly delineate the cell population(s) expressing PD-L1. Figure 7 shows the results of such analysis. Left series and right series of panels are taken at x100 and x200 magnification, respectively. As outlined above, the majority of PD-L1 positive cells were located at the margin of the tumor mass, with few but evident PD-L1 positive cells also within the tumor mass (Figure 7, panels a and b). The tumor mass was clearly positive for cytokeratin 8 and cytokeratin 18 (Figure 7, panels c and d), two intermediate filament proteins expressed in normal simple and glandular epithelia as well as in adenocarcinomas, including HCC,29 but not expressed in hematopoietic cells.30 Again, most of the PD-L1 positive cells at the margin of the tumor mass and among the HCC cells displayed a KC and/or a dendritic morphology (Figure 7, panel b, arrows) and were not stained for cytokeratin 8/18 (Figure 6, panel d, arrows). To further confirm that the vast majority of PD-L1 positive cells were not the HCC tumor cells, tissue sections were stained with an antibody specific for the CD68 (KP-1) molecule, a lysosomal/endosomal transmembrane glycoprotein expressed mainly in the myeloid cell lineage, including monocytes and tissue macrophages, in certain non-myeloid cells such as fibroblasts31 and in some tumor cells but not in HCC cells (http://www.proteinatlas.org/ENSG00000129226-CD68/cancer).32 Results clearly showed that the PD-L1 positive cells found at the margin of the tumor tissue mostly overlapped with CD68-positive cells (Figure 7, compare panels a and e, and panels b and f). Interestingly, a diffuse staining with the anti-CD68 reagent was observed within the tumor mass, indicating that a substantial number of monocyte-macrophage and/or fibroblast cells were infiltrating the tumor (Figure 7, panel f)
Figure 6.
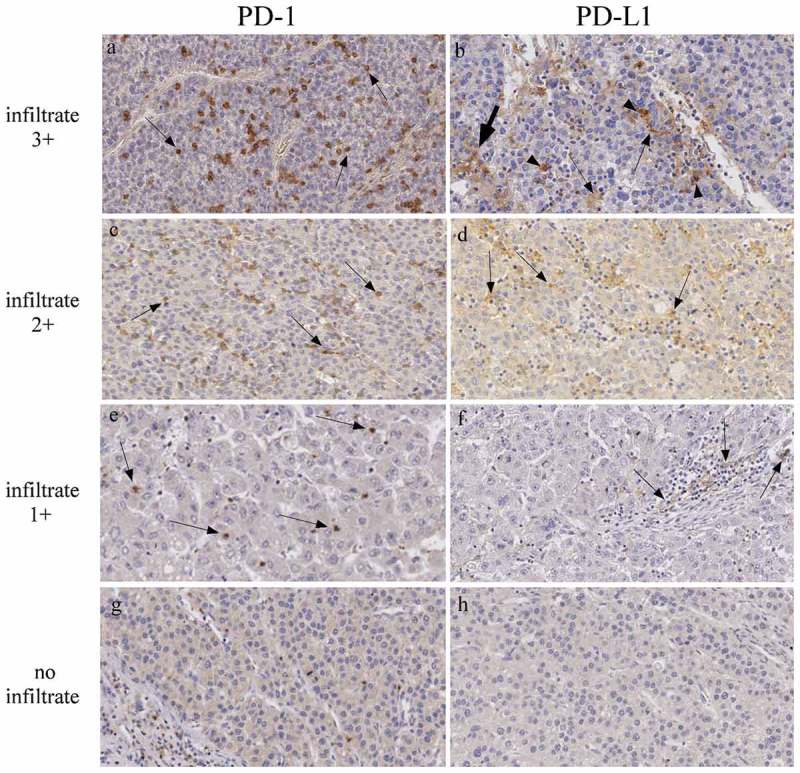
PD-1 and PD-L1 expression in HCC tumor tissues.
Immunohistochemical staining for PD-1 and PD-L1 in formalin-fixed paraffin-embedded sections of HCC. The left and right panels show representative HCC tumor tissues with very large (infiltrate 3+), large (infiltrate 2+), low (infiltrate 1+), or absent (no infiltrate), lympho-monocyte infiltration (original magnification, x200). Representative of infiltrating PD-1-positive lymphocytes, largely detectable in panel a,c and e, are indicated with arrows. Similarly, representative PD-L1 positive infiltrating cells are indicated by arrows (panels b, d, f). Arrowheads and bold arrow (panel b) indicate liver resident cells, most likely Kupffer cells and hepatic stellate cells, respectively.
Table 2.
Correlation between both PD-1 and PD-L1 expression on the tumor infiltrating cells and the grade of tumor infiltration.
| Level of Infiltration |
|||||
|---|---|---|---|---|---|
| Variables | 3+ | 2+ | 1+ | 0 | p value |
| PD-1 Expression | 9.73*10–5 | ||||
| High | 1 | 2 | 0 | 0 | |
| Medium | 0 | 7 | 2 | 0 | |
| Low | 0 | 2 | 9 | 0 | |
| None | 1 | 2 | 8 | 9 | |
| PD-L1 Expression | 0.019 | ||||
| High | 1 | 0 | 0 | 0 | |
| Medium | 0 | 4 | 0 | 0 | |
| Low | 1 | 2 | 1 | 0 | |
| None | 1 | 6 | 18 | 9 | |
Figure 7.
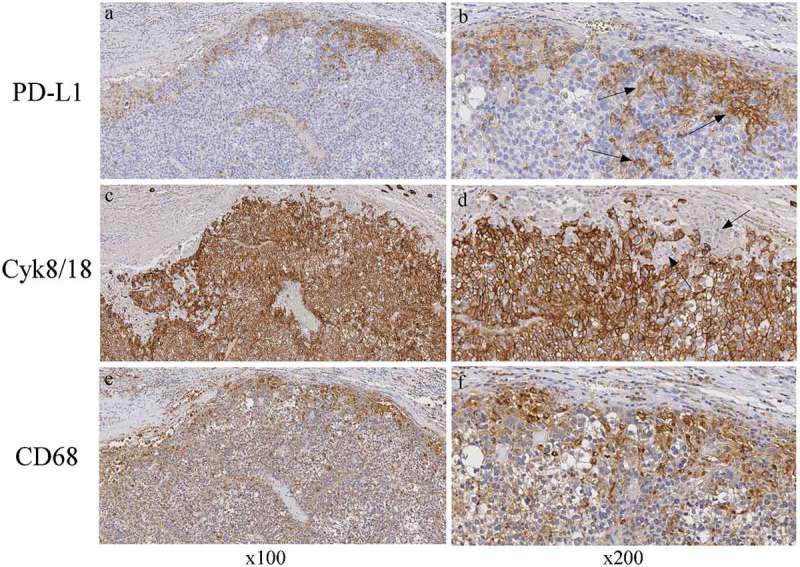
CD68-positive cells but not Cyk8/18-positive HCC tumor cells co-express PD-L1.
Immunohistochemical staining for PD-L1, CyK8/18 and CD68-positive cells. Photographs showing representative paraffin-embedded blocks of HCC tumor tissue samples with large numbers of CD68-positive myeloid cells concentrated at the margin of the tumor and high PD-L1 expression in the same area (right bottom and top panels, respectively). CyK8/18-positive tumor cells (large brown area in panels c, d) do not overlap with PD-L1-positive cells (panel d, arrows). Left panels original magnification, x100; right panels, original magnification x200.
PD-1 and PD-L1 expression was also assessed in the HCC cell lines. As expected, PD-1 was not expressed in any of the cell lines analyzed. Similarly, PD-L1 was not expressed in the 4 HCC cell lines (Figure 8). Noteworthy, treatment with IFNγ, while not modifying the expression of PD-1, resulted instead in a clear induction of expression of PD-L1 in three out of 4 HCC cell lines analyzed in this study (Figure 7).
Figure 8.
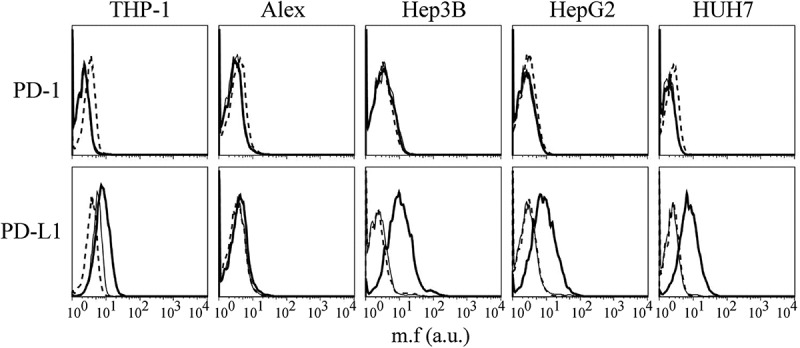
PD-L1, but not PD-1, expression is induced by IFNγ in HCC cell lines.
PD-1 and PD-L1 cell surface expression in Alex, Hep3B, HepG2 and HUH7 HCC cell lines were assessed by immunofluorescence and FACS analysis before and after treatment with IFNγ for 72 hours. Histograms represent fluorescence profiles of the cells indicated on the top incubated with mouse specific anti PD-1 or PD-L1 mAbs followed by FITC-conjugated F(ab)2 anti-mouse antibody. Untreated (thin solid line) or treated with IFNγ (bold line). Cells incubated with FITC-conjugated F(ab)2 anti mouse antibody only (dashed line) were used as negative controls. Mean fluorescence (m.f.) values are expressed in the abscissa as arbitrary units (a.u.). A representative experiment is shown out of three independent experiments with very similar results.
Discussion
Hepatocellular carcinoma constitutes the most frequent malignant neoplasia of the liver and unfortunately still one of the relatively untreatable forms of cancer. Recently immunotherapeutic approaches have fueled new hope in several forms of cancers, by providing the framework for new strategies of anti-tumor treatment. Two complementary approaches, based on the stimulation of the tumor-specific lymphocytes, both CD4+ and CD8 + T cells, with relevant tumor associated peptides (therapeutic vaccines), and rescuing tumor-specific T cells exhausted and/or blocked in their function, via antibodies against inhibitory checkpoints and checkpoint ligands (i.e. CTLA-4/B7 and PD-1/PD-L1), are actively pursued in clinical trials. The success of these approaches is strictly dependent on three major elements: 1) the characterization of the tumor-specific antigens presented within the context of HLA class I and class II molecules and thus the expression of HLA molecules in tumor cells; 2) the presence of infiltrating lymphocytes and APC within and/or around the tumor; 3) the expression of inhibitory molecules on lymphocytes and their ligands on APC.
In this study, we have analyzed some of these aspects on a series of 43 distinct HCC tumors by assessing the expression of HLA class I and class II, PD-1 and PD-L1 cell surface molecules on tumor cells, the cell distribution of the markers within the tumor tissue and their comparative expression in HCC tumor cell lines.
One important finding of our analysis was the differential expression of HLA class I molecules in HCC tumor cells with respect to normal hepatocytes. While normal hepatocytes did not express HLA class I, HCC cells were mostly positive. Positivity did not correlate, however, with previous HCV infection or with the presence of the leukocyte infiltration although stronger expression was observed in highly infiltrated HCC.
Infiltrating cells showed a prevalence of CD8+ and, to lesser extent, of CD4+ lymphocyte subpopulation, and the degree of CD8 + T cell infiltration correlated indeed with high expression of HLA class I in tumor cells. This may suggest that, although most tumor cells express HLA class I molecules, the degree of expression may be regulated by the presence of inflammatory cytokines, such as IFNγ, in the tumor microenvironment produced by CD8+ and CD4 + T cells.
The biological reason underlying the de novo expression of HLA class I molecules in HCC tumor cells is not known at the present. It should be stressed that a similar behavior was also reported for pancreatic adenocarcinomas.33 It may rely on the specific tumor microenvironment present in HCC with an inflammatory milieu providing specific cytokine stimuli inducing HLA class I expression. This idea is supported by the finding presented in this study that in vitro adapted HCC cell lines do not express HLA class I cell surface molecules but they can be induced to do so when treated with IFNγ. This finding is of interest because in many cases tumor cells tend to lose expression of HLA class I molecules and this has been correlated with mechanisms of escape from immune recognition by tumor-specific CTL.25,34 Moreover, loss of HLA class I expression in tumor cells can have also a negative impact on immunotherapies aimed at blocking immune checkpoints molecules.35 Thus, the fact that HCC express HLA class I molecules could indeed be beneficial not only for approaches of tumor vaccination with tumor-specific peptides, but also in combined therapies with peptide vaccines and antibodies to checkpoint inhibitors.
HLA class II expression, although positive in resident liver KC and LSEC, was negative both on normal hepatocytes and tumor cells. This finding was expected as HLA class II expression is a rare event on tumor cells. It should be underlined, however, that when HLA class II molecules are expressed in tumor cells, this often results in a better prognosis. 22,23,36,37 On the other hand, it was relatively unexpected that HLA class II expression in HCC cells could not be rescued by treatment with IFNγ as many cell types, including both normal and tumor cell types, can do so after exposure to the cytokine.38–40 This was not due to an overall defect in the pathway of IFNγ signaling since, as shown here, HLA class I expression was upregulated in these cells after treatment with IFNγ. Further analysis showed that lack of expression of HLA class II molecules was not due to a structural defect of HLA class II genes but rather to the lack of expression of CIITA, the crucial transcriptional activator of these genes. Indeed, the introduction of a CIITA expression plasmid into different HCC cell lines restored class II gene expression, indicating that the inhibition of CIITA expression prevents class II transcription in these cells. To gain further insights into the mechanism of lack of CIITA expression in HCC we focused our attention on the specific steps of IFNγ signaling leading to the activation of transcription of the HLA class II transactivator. These steps include initial recruitment of Jak-1 and Jak-2 kinases on the cytoplasmic tail of the IFNγ receptor with consequent phosphorylation and homodimerization of STAT1, migration of STAT1 dimers into the nucleus, binding to the promoter and consequent expression of IRF-1 transcrition factor, subsequent binding of IRF1, STAT1 and USF1 to promoter IV (pIV) of CIITA and activation of CIITA transcription. We found the integrity of the IFNγ signaling since STAT1 was phosphorylated, IRF1 was induced and USF1 was present. We then reasoned that CIITA pIV could be unresponsive to otherwise normal specific transcription factors because of possible epigenetic modification at level of pIV DNA sequence. Previous studies showed that alteration in chromatin structure and methylation of CIITA promoter may in fact affect the accessibility of transcription factors contributing to transcriptional silencing of CIITA particularly in tumor cells.41,42 Indeed, we found that the pIV DNA region including the consensus elements for the key transcription factors essential for the optimal activation of CIITA gene transcription, was highly methylated, thus preventing promoter occupancy and IFNγ inducible transcription.
Within the context of HCC pathology this finding may be of relevance as a putative mechanism of tumor escape because tumor cells, having not the capacity to express HLA class II molecules even in an inflammatory environment, are prevented to serve even as surrogate antigen-presenting cells for their own putative tumor antigens, as instead it has been shown in various in vivo experimental systems 21,43–47 and for HCC cell lines in vitro.48 Conversely, if CIITA under the control of an ubiquitous expressable promoter could be genetically transferred into HCC cell lines (as shown here) or primary HCC tumor cells, and induce HLA class II expression, this may offer the possibility to expose relevant HLA class II-bound TAA peptides, otherwise impossible to detect, for a better stimulation of tumor-specific TH cells in tumor vaccination approaches Indeed this strategy is presently pursued in a combined effort of the European Hepavac Consortium (Hepavac-Cancer Vaccine development for Hepatocellular Carcinoma: www.hepavac.eu/).48–51
The intrinsic impossibility to express HLA class II molecules in hepatocytes may bear even more importance in the normal physiology of the liver because the danger of normal hepatocytes potentially becoming antigen presenting cells of pinocytosed foreign antigens derived from the massive uptake of nutrients, waste material and pathogens from the blood, or even self antigens, is prevented. This observation may thus help explaining at least in part the peculiar “tolerogenic environment” found in the liver.52–54
Another relevant finding of our study was the demonstration that there is an important variability in the number of tumor-infiltrating lymphocytes. Interestingly, the intensity of infiltration correlated with PD-1 expression in infiltrating cells. HCC tumors with either absent, low, medium or high number of PD-1-positive cell infiltration were observed with apparently no significant correlation with a previous condition of HCV/HBV infection. While the presence of PD-1 expression in tumor-infiltrating lymphocytes argues in favour of stimulated but exhausted and/or blocked lymphocytes, the variable number of these cells certainly should be seriously taken into consideration to evaluate the possible outcome of anti-PD-1 immunotherapy. First, anti-PD1 therapy may work better on those cases in which a clear infiltration of PD-1-positive lymphocytes with high level of PD-1 expression is present. In the best hypothesis, according to our data this would apply to 10/43 case (3+, very high expressing: 1 case; 2+, high expressing: 9 cases). Among the remaining cases, anti-PD-1 may not work in 9/43 (no infiltrating cells at all) and in those cases with low levels of infiltration and low (9/43) or absent (8/43) expression of PD-1.
The degree of PD-1-positive cell infiltration correlated with the presence of PD-L1 positive cells in the tumor tissue. In this case, however, most of the PD-L1- positive cells were not infiltrating the tumor mass but they were localized mostly at the margin of the tumor mass. Analysis of the morphology of these cells indicated that the majority of them were both resident monocyte-macrophage KC and blood-derived monocytic cells. Furthermore PD-L1 was expressed in other liver resident cells such as stellate cells and LSEC. This interesting correlation between the number of PD-1-positive and PD-L1-positive cells suggest that, in HCC, tumor-infiltrating lymphocytes not only represent late-stimulated effectors but they are probably proportionally engaged in a state of efficient functional block by their interaction with PD-L1-positive cells. While PD-1 is essentially expressed in functionally mature immune cells, PD-L1 can be expressed in APC but also in other cells, including tumor cells. Given the function of PD-L1 molecule in blocking the activity of immune effector cells via its interaction with PD-1, it has been hypothesized that PD-L1 expression in tumors can contribute to the anergic state of the anti-tumor immune response in cancer patients.7-9 It was therefore important to assess whether PD-L1 was expressed in HCC tumor cells. In this study we report the unprecedented finding that HCC tumor cells, at variance with other tumors, do not express this relevant marker. These results were obtained by using two different monoclonal antibodies against distinct epitopes of PD-L1 molecule. This finding was paralleled by the similar finding obtained in HCC cell lines in which PD-L1 was not expressed in 4 out of 4 cell lines analyzed, although its expression could be rescued by IFNγ treatment. Our findings are thus at variance with those reported by other groups10-12 showing a higher percentage of PD-L1 expression in HCC tumor cells. The reasons for this discrepancy are unknown and may be partially related to the patients’ sample analyzed, the ethnic derivation of patients and different reagents used for detection.
Therefore, within this frame the potential use of adjuvant anti-PD-L1 antibody immunotherapy to block negative signals mediated by tumor PD-L1 on anti-tumor effector lymphocytes has no practical support. Furthermore, considering both the level of infiltrating cells and their expression of PD-L1, we found that only in 5/43 cases there was a substantial infiltrate with cells expressing high/medium levels of the marker. Thus, our concern is that a careful choice of HCC-bearing patient should be operated based on their immunohistological pattern of expression of PD1 and/or PD-L1 if anti-checkpoint blockade immunotherapy has to be applied.
In conclusion, the results presented in this study expand our knowledge on the expression of immune relevant molecules in HCC and may help to envisage better combination strategies of immunotherapeutic intervention based on the analysis of the HCC HLA class I- and class II-specific immunopeptidome, consequent identification of relevant TAA for inclusion in multi-peptide vaccines, together with the appropriate identification of those patients more prone to respond to immune checkpoint blockade.
Materials and methods
Patient series and immunohistochemistry
Formalin fixed-paraffin embedded samples of 43 HCC that underwent surgical resection from 1999 to 2013 in our University hospital were retrieved from the files of the Anatomopathology Unit of our Department. All the cases, classified according to WHO criteria55 were classical HCCs with trabecular, pseudo-glandular or solid pattern. The histological grade, based on tumor differentiation, was: a)- well differentiated in 8 cases, showing tumor cells with mild atypia organized in trabeculae, b)- moderately differentiated in 32 cases characterized by trabecular structures of three or more cells in thickness, and c)- poorly differentiated in 3 cases composed by tumor cells with moderate to marked pleomorphism, growing mostly in a solid pattern.
The immune infiltration, evaluated in hematoxylin-eosin, was scored from 0 to 3, with score 0 = no inflammatory cells; score 1 = rare lymphoid cells dispersed among tumor cells; score 2 = a moderate amount of lymphocytes along the growing margins and in the sinusoid-like blood spaces; score 3 = marked lymphocyte infiltration along the scarce connective stroma and intermingled with tumor cells in the center of the tumor and along the tumor edge.
Immunohistochemical study was performed on 3 μm formalin-fixed, paraffin embedded sections deparaffinised and rehydrated through alcohol series to water, as previously described.56 Briefly, endogenous activity was blocked with 3% aqueous hydrogen peroxide for 10 min, antigen retrieval was performed for each antigen in a domestic 750 KW microwave oven with different solutions (EDTA or citrate buffer pH 6,0) as detailed in Table 3. Primary antibodies were applied overnight at 4°C followed by a polymeric detection system (Ultravision DAB Detection System, LabVision, Värmdö Sweden) according to the manufacturer’s protocol. The immunoreaction was developed with 3.3ʹ-diaminobenzidine tetrahydrochloride (DAB) (Sigma Aldrich, St. Louis, MO, USA) as chromogen.
Table 3.
Conditions and reagents used for immunohistochemistry studies.
| Antibody specificity | Source | M/R (Clone)a | Antigen retrievalb | Working Dilution |
|---|---|---|---|---|
| PD-1 | Origene | M (UMAB199) | E (20min) | 1/500 |
| PD-L1 | Cell Signalling | M(405.9A11) | E (20min) | 1/100 |
| PD-L1 | Spring Bioscience | R (SP142) | E (20min) | 1/100 |
| HLA-I | AbCam | R (EP1395Y) | TC (10min) | 1/250 |
| HLA-II (DR) | ThermoFisher | M (LN-3) | TC (10min) | 1/300 |
| HLA-II (DQ) | AbCam | M (ab55158) | TC (10min) | 1/50 |
| CD68 | DAKO | M (1G12) | TC (10min) | 1/100 |
| Cytocheratin 8/18 | Ventana, | B22.1 B23.1 | TC (10min) | 1/1 |
| CD4 | Ventana | R (SP35) | E (10min) | 1/1 |
| CD8 | Ventana | R (SP57) | TC (10min) | 1/1 |
a M, mouse; R, rabbit.
b E, EDTA pH 8,0; TC, citrate buffer pH 6,0; in parenthesis, incubation time.
HCC cell lines and generation of transfectants stably expressing CIITA
Four HCC cell lines, Alex, Hep3B, HepG2 and HuH7 were used in this study. The cells lines were kindly donated by Prof. Massimo Levrero, University of Rome “La Sapienza”. HCC cell lines were grown in RPMI 1640 medium (Lonza BioWhittakerTM, Catalog number: BE12-702F) supplemented with 10% heat-inactivated fetal calf serum (FCS) without antibiotics. THP-1 and Jurkat are a mielomonocytic and a T cell line, respectively, used in experiments of phenotypic and molecular characterization of HLA and CIITA gene expression. RA is a glioblastoma cell line kindly donated by Prof. Pierre Robe, University of Utrecht, The Netherlands. THP1, Jurkat and RA cell lines were maintained in culture with the same modalities of the HCC cell lines .
HCC tumor cells were transfected with 5μg of flag-CIITA (pcfCIITA) expression vector57 or pCDNA3.1 empty plasmid by using FugeneHD (Promega, catalog number E2311), as previously described58.
CIITA-transfected HCC cells underwent G418 selection 0.5 mg/ml (Sigma Aldrich, catalog number A-1720). MHC-II- positive cells were enriched by fluorescence-activated cell sorting with a BD FACS ARIA II cell sorter (Becton-Dickinson, catalog number 95131) and subjected to limiting-dilution cloning. The glioblastoma RA cell line was kindly donated by Prof. Pierre Robe, University Medical Center of Utrecht, The Netherlands.
FACS analysis and ifnγ treatment
The cell surface expression of HLA class I, HLA class II, PD-1 and PD-L1 molecules was assessed by immunofluorescence and flow cytometry (BD FACSAriaTM II Cell Sorter, BD Biosciences). Briefly, cells were washed twice with PBS, dissociated with trypsin-EDTA, and resuspended in complete medium (RPMI, 10% FCS). Cells (3 × 105/tube) were pelletted at 800 × g for 5 min, washed, the supernatant was discarded and the pellet resuspended in PBS for FACS analysis. The following monoclonal antibodies were used as primary antibodies: B9.12.1 (HLA class I), D1-12 (HLA class II)33, CD279 (PD-1) (clone EH12.2H7) PE (Biologend, catalog number 329906) and CD274 (PD-L1) (clone 29E.2A3) PE (Biologend, catalog number 329706) as previously described.59
3x105 HCC cells (Alex, Hep3B, HepG2 and HUH7) were plated in 6 multi-well plates and treated with 500 U/ml of IFNγ (Origene, catalog number TP723162) or with its vehicle. Seventy-two hours after treatment the cells were collected and analyzed by immunofluorescence and flow cytometry, as indicated above. Incubation with IFNγ for 72 hrs was carried out because 48–72 hours are required to achieve the highest cell surface expression and accumulation of HLA class I and class II molecules in response to the cytokine, as assessed by preliminary experiments. The data were analyzed by using FlowJo 9.5.2 software
Quantification of mrna by real-time PCR (qRT-PCR)
Total RNA, was extracted from cells using TRIzol reagent (Thermo Fisher Scientific, catalog number 15596026), as previously described.60 cDNA was synthesized from 0.5 μg total RNA using iScript cDNA Synthesis Kit (Bio-Rad, catalog number 170–8890). 0.5 μg of cDNA were amplified by PCR by using an ABI Prism 7000 sequence detection system (Thermo Fisher Scientific) with IQSYBR Green PCR master mix (Bio-Rad, catalog number 172–5122) according to the manufacturer protocol. All of the reactions were performed in an ABI PRISM 7700 apparatus (PE Applied Biosystems). The PCR conditions were 50 °C for 2 min, 95 °C for 15 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. A 20 min dissociation protocol was also applied. The copy numbers of the CIITA, USF1 and IRF1 transcripts were calculated using the comparative Ct method (also known as the 2−[delta] [delta]Ct method), where [delta]Ct, sample = Ct, CIITA/USF1/IRF1 – Ct, RPS7, and [delta]Ct, sample is the Ct value for any sample normalised to the RPS7 endogenous housekeeping transcripts and [delta] [delta]Ct = [delta]Ct, sample – [delta]Ct, wt. In all of the samples, 2−[delta] [delta]Ct refers to an N- fold increase in the CIITA/USF1/IRF1 copy numbers relative to the untreated cells (vehicle). Each reaction was performed in triplicate.. The following primer pair sets were used: CIITA forward 5′-ggatcctcacggcctttt-3′; reverse 5′-ccccgatcttgttctcactc-3′; IRF1, forward 5ʹ-atgcccatcactcggatgc-3ʹ; reverse 5ʹ-ccctgcttttatcggcctg-3ʹ; USF1, forward 5ʹ-tgttactacccagggctcaga-3ʹ; reverse 5ʹ-acatcatcacaaagaattgacca-3ʹ; RPS7, forward 5′-tggagatgaactcggacctc-3′, reverse 5′-cgaccaccaccaacttcaa-3′.
Western blotting
Cell lysate of RA and HepG2 either treated with IFNγ or its vehicle were analyzed for the expression of STAT1 and phospho-STAT1 (p-STAT1) by SDS-PAGE and Western blotting with anti-STAT1 polyclonal antibody (Invitrogen, catalog number 710078) and anti-Phospho-STAT1 (Tyr701) monoclonal antibody (ThermoFisher Scientific, catalog number MA5-15071), respectively. Endogenous tubulin was detected by using anti-tubulin monoclonal antibody (Sigma, catalog number T5168). Horseradish peroxidase (HRP)-conjugated anti-mouse immunoglobulin or anti-rabbit immunoglobulin secondary antibodies (ThermoFisher Scientific, catalog number 31430 and 31460, respectively) were used. Blots were developed by chemiluminescence assay (SuperSignalTM West Pico Chemiluminescent Substrate; ThermoFisher Scientific, catalog number 34080)
DNA methylation status and sequencing analysis
DNA methylation status of CIITA promoter IV, the IFNγ-inducible promoter of the class II transactivator, was assessed by bisulfite treatment of 0.5 μg genomic DNA isolated from both RA and HepG2 cells using the Bisulfite Conversion kit (Active Motif, catalog number 55016) following the manufactures’ protocol. Five microliters of bisulfite-treated DNA were amplified by PCR. Primers used for this analysis were designed using the MethPrimer software61 and the forward and reverse sequences were: (a) 5ʹ-aaatagagatttatttaggggtggg-3′ and 5ʹ-caaacacctactataaccacca-3′; (b) 5ʹ- ttgggatgttatttttgataaagta-3′ and 5′-acaaaaaaaactttaatcacctacc-3′. PCR products were cloned into the pGEM-T Easy vector (Promega, catalog number A1360) and 4 clones from each condition were sequenced for analysis of the percentage of methylated CpGs. The DNA was sequenced using T7 universal primer 5´- taatacgactcactataggg-3´ by Barcode sequencing (TIB Mol biol s.r.l.)
Statistical analysis
The hypothesis of independence between variables was tested by analysing contingency tables (see Tables 1 and 2) by a Chi-squared test with (rows – 1) (columns – 1) degrees of freedom or by Mann-Whitney test. Variables were considered dependent when p < 0.05. The other statistical analysis was performed using the GraphPad Prism software v. 6.0 (GraphPad Software, http://www. graphpad.com). Comparison between the groups was performed by using the Two-WAY ANOVA test. p values ≤ 0.01 were considered significant.
Funding Statement
This work was supported by the European Community [FP7 Grant no. 602893]; University of Insubria [FAR 2016 and 2017]; University of Insubria and Regione Lombardia [Consortium Project DOTE-UNIRE TumVac]; University of Insubria [The Institutional Grant 2017];University of Insubria [FAR 2016 and 2017].
Abbreviations
- HCC
hepatocellular carcinoma
- CIITA
class II transactivator
- TAA
tumor-associated antigens
- APC
antigen presenting cells
- IFNγ
interferon gamma
- PD-1
programmed cell death protein1
- PD-L1
programmed death-ligand 1
- CTLA4
cytotoxic T-lymphocyte-associated protein 4.
Acknowledgments
This work was supported by the following grants: European Community FP7 Grant no. 602893 “Cancer Vaccine Development for Hepatocellular Carcinoma-HepaVAC” http://www.hepavac.eu (to RSA and GF); University of Insubria “FAR 2016 and 2017” (to RSA and GF); by The Institutional Grant 2017, University of Insubria, to RA; GF was supported by University of Insubria and Regione Lombardia Consortium Project DOTE-UNIRE: TumVac, New Strategies of Anti-Tumor Vaccination.
Disclosure of Potential Conflicts of Interest
The Authors declare that they have no conflict of interest.
Supplementary material
Supplementary data for this article can be accessed here.
References
- 1.WHO IARC 2012http://globocan.iarc.fr.
- 2.Ulahannan SV, Duffy AG, McNeel TS, Kish JK, Dickie LA, Rahma OE, McGlynn KA, Greten TF, Altekruse SF.. Earlier presentation and application of curative treatments in hepatocellular carcinoma. Hepatology. 2014;60:1637–1644. doi: 10.1002/hep.27288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng AL, Guan Z, Chen Z, Tsao CJ, Qin S, Kim JS, Yang TS, Tak WY, Pan H, Yu S, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: subset analyses of the phase III Sorafenib Asia-Pacific trial. Eur J Cancer. 2012;48:1452–1465. doi: 10.1016/j.ejca.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Sharma P, Allison JP.. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 5.Greten TF, Wang XW, Korangy F. Current concepts of immune based treatments for patients with HCC: from basic science to novel treatment approaches. Gut. 2015;64:842–848. doi: 10.1136/gutjnl-2014-307990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kudo M. Immune checkpoint inhibition in hepatocellular carcinoma: basics and ongoing clinical trials. Oncology. 2017;92:50–62. doi: 10.1159/000451016. [DOI] [PubMed] [Google Scholar]
- 7.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Annals Oncol. 2016;27:409–416. doi: 10.1093/annonc/mdv615. [DOI] [PubMed] [Google Scholar]
- 9.Wang Q, Liu F, Liu L. Prognostic significance of PD-L1 in solid tumor: an updated meta-analysis. Medicine (Baltimore). 2017;96: e6369. doi:10.1097/MD.0000000000006369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, Yang YP, Tien P, Wang FS. PD-1 and PD-L1 upregulation promotes CD81 T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer. 2011;128:887–896. doi: 10.1002/ijc.25397. [DOI] [PubMed] [Google Scholar]
- 11.Gao Q, Wang X-Y, Qiu S-J, Yamato I, Sho M, Nakajima Y, Zhou J, Li B-Z, Shi Y-H, Xiao Y-S, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15. doi: 10.1158/1078-0432.CCR-09-0547. [DOI] [PubMed] [Google Scholar]
- 12.Calderaro J, Rousseau B, Amaddeo G, Mercey M, Charpy C, Costentin C, Luciani A, Zafrani ES, Laurent A, Azoulay D, et al. Programmed Death Ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology. 2016;64:2038–2046. doi: 10.1002/hep.28710. [DOI] [PubMed] [Google Scholar]
- 13.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Germain RN, Margulies DH. The biochemistry and cellular biology of antigen processing and presentation. Annu Rev Immunol. 1993;11:403–450. doi: 10.1146/annurev.iy.11.040193.002155. [DOI] [PubMed] [Google Scholar]
- 15.Pardoll DM, Topalian SL. The role of CD4+ T cell responses in anti-tumor immunity. Curr Opin Immunol. 1998;10:588–594. doi: 10.1016/S0952-7915(1098)80228-80228. [DOI] [PubMed] [Google Scholar]
- 16.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll DM, Levitsky H. The central role of CD4+ T cells in the antitumor immune response. J Exp Med. 1998;188:2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boon T, Cerottini JC, Van Den Eynde B, Van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. 1994;12:337–365. doi: 10.1146/annurev.iy.1112.040194.002005. [DOI] [PubMed] [Google Scholar]
- 18.Accolla RS, Scarpellino L, Carra G, Guardiola J. Trans-acting element(s) operating across species barriers positively regulate the expression of major histocompatibility complex class II genes. J Exp Med. 1985;162:1117–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Accolla RS, Jotterand-Bellomo M, Scarpellino L, Maffei A, Carra G, Guardiola J. Air-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. J Exp Med. 1986;164:369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or Bare Lymphocyte Syndrome). Cell. 1993;75:135–146. [PubMed] [Google Scholar]
- 21.Accolla RS, Lombardo L, Abdallah R, Raval G, Forlani G, Tosi G. Boosting the MHC class II-restricted tumor antigen presentation to CD4+ T helper cells: a critical issue for triggering protective immunity and re-orienting the tumor microenvironment toward an anti-tumor state. Frontiers Oncol. 2014;4:32. doi: 10.3389/fonc.2014.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabrera T, Ruiz-Cabello F, Garrido F. Biological implication of HLA-DR expression in tumors. Scand J Immunol. 1995;41(398):406. doi: 10.1111/j.1365-3083.1995.tb03584.x. [DOI] [PubMed] [Google Scholar]
- 23.Samuels S, Spaans VM, Osse M, Peters LA, Kenter GG, Fleuren GJ, Jordanova ES. Human Leukocyte Antigen-DR Expression is significantly related to an increased disease-free and disease-specific survival in patients with cervical adenocarcinoma. Int J Gynecol Cancer. 2016;26:1503–1509. doi: 10.1097/IGC.0000000000000783. [DOI] [PubMed] [Google Scholar]
- 24.Shi B, Vinyals A, Alia P, Broceño C, Chen F, Adrover M, Gelpi C, Price JE, Fabra A. Differential expression of MHC class II molecules in highly metastatic breast cancer cells is mediated by the regulation of the CIITA transcription Implication of CIITA in tumor and metastasis development. Int J Biochem Cell Biol. 2006;38:544–562. doi: 10.1016/j.biocel.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Del Campo AB, Carretero J, Aptsiauri N, Garrido F. Targeting HLA class I expression to increase tumor immunogenicity. Tissue Antigens. 2112;79:147–154. doi: 10.1111/j.1399-0039.2011.01831.x. [DOI] [PubMed] [Google Scholar]
- 26.Paterson AC, Sciot R, Kew MC, Callea F, Dusheiko GM, Desmet VJ. HLA expression in human hepatocellular carcinoma. Br J Cancer. 1988;57:369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sung CH, Hu CP, Hsu HC, Ng AK, Chou CK, Ting LP, Su TS, Han SH, Chang CM. Expression of class I and class II major histocompatibility antigens on human hepatocellular carcinoma. J Clin Invest. 1989;83:421–429. doi: 10.1172/JCI113900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muhlethaler-Mottet A, Di Berardino W, Otten LA, Mach B. Activation of the MHC Class II Transactivator CIITA by Interferon-γ Requires Cooperative Interaction between Stat1 and USF-1. Immunity. 1998;8:157–166. [DOI] [PubMed] [Google Scholar]
- 29.Strnad P, Paschke S, Jang KH, Ku NO. Keratins: markers and modulators of liver disease. Curr Opin Gastroenterol. 2012;28:209–216. doi: 10.1097/MOG.0b013e3283525cb8. [DOI] [PubMed] [Google Scholar]
- 30.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10:6897–6904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 31.Gottfried E, Kunz‐Schughart LA, Weber A, Rehli M, Peuker A, Müller A, Kastenberger M, Brockhoff G, Andreesen R, Kreutz M. Expression of CD68 in non‐myeloid cell types. Scand J Immunol. 2008;67:453–463. doi: 10.1111/j.1365-3083.2008.02091.x. [DOI] [PubMed] [Google Scholar]
- 32.The human protein atlas. http://www.proteinatlas.org/ENSG00000129226-CD68/cancer.
- 33.Scupoli MT, Sartoris S, Tosi G, Ennas MG, Nicolis M, Cestari T, Zamboni G, Martignoni G, Lemoine NR, Scarpa A, et al. Expression of MHC class I and class II antigens in pancreatic adenocarcinomas. Tissue Antigens. 1996;48:301–311. [DOI] [PubMed] [Google Scholar]
- 34.Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. doi: 10.1016/j.coi.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park J, Thomas S, Munster PN. Epigenetic modulation with histone deacetylase inhibitors in combination with immunotherapy. Epigenomics. 2015;7:641–652. doi: 10.2217/epi.15.16. [DOI] [PubMed] [Google Scholar]
- 36.Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7. doi: 10.1038/ncomms10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sconocchia G, Eppenberger-Castori S, Zlobec I, Karamitopoulou E, Arriga R, Coppola A, Caratelli S, Spagnoli GC, Lauro D, Lugli A, et al. HLA class II antigen expression in colorectal carcinoma tumors as a favorable prognostic marker. Neoplasia (New York, NY). 2014;16:31–42. doi: 10.1593/neo.131568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steimle V, Siegrist C, Mottet A, Lisowska-Grospierre B, Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265:106–109. [DOI] [PubMed] [Google Scholar]
- 39.Rigaud G, De Lerma Barbaro A, Nicolis M, Cestari T, Ramarli D, Riviera AP, Accolla RS. Induction of CIITA and modification of in vivo HLA-DR promoter occupancy in normal thymic epithelial cells treated with IFN-gamma: similarities and distinctions with respect to HLA-DR-constitutive B cells. J Immunol. 1996;156:4254–4258. [PubMed] [Google Scholar]
- 40.Accolla RS, De Lerma Barbaro A, Mazza S, Casoli C, De Maria A, Tosi G. The MHC class II transactivator: prey and hunter in infectious diseases. Trends Immunol. 2001;22:560–563. doi: 10.1016/S1471-4906(01)02003-8. [DOI] [PubMed] [Google Scholar]
- 41.Morris AC, Spangler WE, Boss JM. Methylation of class II trans-activator promoter IV: a novel mechanism of MHC class II gene control. J Immunol. 2000;164:4143–4149. [DOI] [PubMed] [Google Scholar]
- 42.Radosevich M, Song Z, Gorga JC, Ksander B, Ono SJ. Epigenetic silencing of the CIITA gene and posttranscriptional regulation of class II MHC genes in ocular melanoma cells. Invest Ophthalmol Vis Sci. 2004;45:3185–3195. doi: 10.1167/iovs.04-0111. [DOI] [PubMed] [Google Scholar]
- 43.Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol. 2003;33:1183–1192. doi: 10.1158/1078-0432.CCR-1106-0165. [DOI] [PubMed] [Google Scholar]
- 44.Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbaro A, Procopio FA, Comes A, Zardi L, Ferrini S, Accolla RS. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin Cancer Res. 2006;12:3435–3443. doi: 10.1158/1078-0432.CCR-06-0165. [DOI] [PubMed] [Google Scholar]
- 45.Frangione V, Mortara L, Castellani P, De Lerma Barbaro A, Accolla RS. CIITA-driven MHC-II positive tumor cells: preventive vaccines and superior generators of anti-tumor CD4+ T lymphocytes for immunotherapy. Int J Cancer. 2010;127:1614–1624. doi: 10.1002/ijc.25183. [DOI] [PubMed] [Google Scholar]
- 46.Ekkirala CR, Cappello P, Accolla RS, Giovarelli M, Romero I, Garrido C, Garcia-Lora AM, Novelli F. Class II transactivator-induced MHC class II expression in pancreatic cancer cells leads to tumor rejection and a specific antitumor memory response. Pancreas. 2014;43:1066–1072. doi: 10.1097/MPA.0000000000000160. [DOI] [PubMed] [Google Scholar]
- 47.Bou Nasser Eddine F, Forlani G, Lombardo L, Tedeschi A, Tosi G, Accolla RS. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4+ TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cell. Oncoimmunology. 2017. doi: 10.1080/2162402X.2016.1261777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sartoris S, Valle MT, De Lerma Barbaro A, Tosi G, Cestari T, D’Agostino A, Megiovanni AM, Manca F, Accolla RS. HLA class II expression in uninducible hepatocarcinoma cells after trasfection of the AIR-1 gene product CIITA. Acquisition of antigen processing and presentation capacity. J Immunol. 1998;161:814–820. [PubMed] [Google Scholar]
- 49.Bou Nasser Eddine F, Ramia E, Tosi G, Forlani G, Accolla RS. Tumor Immunology meets…Immunology: modified cancer cells as professional APC for priming naïve tumor-specific CD4+ T cells. Oncoimmunology. 2017;6:e1356149. doi: 10.1080/2162402X.2017.1356149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herkel J, Jagemann B, Wiegard C, Lazaro JF, Lueth S, Kanzler S, Blessing M, Schmitt E, Lohse AW. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocytes. Hepatology. 2003;37:1079–1085. doi: 10.1053/jhep.2003.50191. [DOI] [PubMed] [Google Scholar]
- 51.Hepavac cancer vaccine development for hepatocellular carcinoma. http://www.hepavac.eu.
- 52.Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 53.Makarova-Rusher OV, Medina-Echeverz J, Duffy AG, Greten TF. The yin and yang of evasion and immune activation in HCC. J Hepatol. 2015;62:1420–1429. doi: 10.1016/j.jhep.2015.02.038. [DOI] [PubMed] [Google Scholar]
- 54.Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol. 2012;12:201–213. doi: 10.1038/nri3169. [DOI] [PubMed] [Google Scholar]
- 55.Theise ND, Curado MP, Franceschi S, Hytiroglou P, Kudo M, Park YN, Sakamoto M, Torbenson M, Wee A. Hepatocellular carcinoma In: Bosman FT, Carneiro F, Hruban RH, Theise ND, editors. WHO classification of tumours of the digestive system. Geneva (Switzerland): WHO Press; 2010. 4th ed. p. 205–216. [Google Scholar]
- 56.Chiaravalli AM, Longhi E, Vigetti D, De Stefano FI, Deleonibus S, Capella C, Solcia E, Parravicini C. Gastrointestinal cancers reactive for the PAb416 antibody against JCV/SV40 T-Ag lack JCV DNA sequences while showing a distinctive pathologic profile. J Clin Pathol. 2012;66:44. doi: 10.1136/jclinpath-2012-200963. [DOI] [PubMed] [Google Scholar]
- 57.Forlani G, Abdallah R, Accolla RS, Tosi G. The major histocompatibility complex class II transactivator CIITA inhibits the persistent activation of NF-κB by the Human T Cell Lymphotropic Virus Type 1 Tax-1 Oncoprotein. J Virol. 2016;90:3708–3721. doi: 10.1128/JVI.03000-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tosi G, Forlani G, Andresen V, Turci M, Bertazzoni U, Franchini G. Major histocompatibility complex class II transactivator CIITA is a viral restriction factor that targets human T-cell lymphotropic virus type 1 Tax-1 function and inhibits viral replication. J Virol. 2011;85. doi: 10.1128/JVI.00813-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raval GU, Bidoia C, Forlani G, Tosi G, Gessain A, Accolla RS. Localization, quantification and interaction with host factors of endogenous HTLV-1 HBZ protein in infected cells and ATL. Retrovirology. 2015;12:59. doi: 10.1186/s12977-015-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Forlani G, Turrini F, Ghezzi S, Tedeschi A, Poli G, Accolla RS, Tosi G. The MHC-II transactivator CIITA inhibits Tat function and HIV-1 replication in human myeloid cells. J Transl Med. 2016;14:94. doi: 10.1186/s12967-016-0867-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;11:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.