

Abstract
Mice lacking voltage-gated Kv1.1 channels as a result of deletion of the Kcnal gene are an extensively utilized genetic model of human epilepsy and sudden unexpected death in epilepsy because of their frequent seizures and genotypic-phenotypic similarity to the human condition. Ictal behaviors, electrophysiological recordings, and gene expression studies suggest limbic circuits are critical for epilepsy in Kcna1-null mice, but the exact brain networks recruited by seizures remain unknown. In this study, Fos protein expression patterns were used to map limbic brain regions with increased neuronal activity at baseline and during spontaneous seizures in Kcna1-null mice by comparing seizing and non-seizing knockouts and wild-type controls. Basal Fos levels were unchanged in non-seizing knockout mice compared to wild types for all brain regions examined except the dentate gyrus granule cell layer which exhibited a significant decrease in Fos-positive cells. Following seizures, Kcna1-null brains exhibited significantly increased Fos labeling in the basolateral amygdala and the dentate hilus region, but not in other principal cell layers of the hippocampal formation. The selective Fos activation in the amygdala following seizures suggests that extra hippocampal limbic circuits may be critically involved with seizure generation or spread in Kcna1-null mice.
Keywords: amygdala, epilepsy, Fos, hilus, Kv1.1 channel, seizure
Kv1.1 voltage-gated potassium channel a-subunits, encoded by the Kcnal gene, act to dampen neuronal excitability by regulating action potential propagation and shape, repetitive firing properties of neurons, and neurotransmitter release (Glasscock et al. 2012; Jan and Jan 2012). Mutations in KCNA1 lead to neurological disorders associated with neuronal hyperexcitability. In patients, mutations in KCNA1 have been linked to episodic ataxia type 1, neuromyotonia, epilepsy, and sudden unexpected death in epilepsy (Browne et al. 1994; Zuberi et al. 1999; Kinali et al. 2004; Klassen et al. 2014). Mice lacking Kv1.1 channels as a result of targeted deletion of the Kcna1 gene exhibit phenotypes similar to patients, including severe epilepsy characterized by frequent spontaneous convulsive seizures that occur several times daily and often lead to sudden death (Smart et al. 1998; Glasscock et al. 2010). The high frequency of seizure activity in Kcna1 -null mice coupled with their genotypic-phenotypic similarity to human temporal lobe epilepsy has made them a preferred model for the study of anti-seizure drug and cellular gene therapies, as well as genetic modifier interactions in epilepsy (Glasscock et al. 2007; Baraban et al. 2009; Holth et al. 2013; Bomben et al. 2014).
Although the Kcna1 -null model is widely used to study epilepsy, little is known about the neuronal circuitry responsible for the generation and spread of seizures in these mice. Seizures in Kcna1 -null mice have been hypothesized to involve limbic brain circuits based on ictal behaviors, electrophysiology, and channel expression. The seizure-associated behaviors in Kcna1 -null mice resemble the well-characterized kainate and kindling-induced models of temporal lobe epilepsy in rodents, which involve amygdala and hippocampal neurocircuits (Smart et al. 1998; Rho et al. 1999; Wenzel et al. 2007). In Kcna1 -null mice, seizure-like EEG patterns sometimes occur in hippocampal depth electrodes immediately preceding ictal activity at the cortical surface, suggesting that some seizures may originate in limbic structures (Wenzel et al. 2007). In brain slice recordings from Kcnal -null mice, the CA3 hippocampal region exhibits abnormal burst firing under conditions of high extracellular K+ concentrations, GABAA antagonism, and electrical stimulation suggesting the hippocampus is particularly susceptible to epileptiform discharges in this model (Smart et al. 1998; Lopantsev et al. 2003; Glasscock et al. 2007). The increased in vivo and in vitro hippocampal excitability in Kcnal -null mice correlates with the distribution pattern of Kv1.1 subunits, which are normally expressed at high levels in the axon tracts of the hippocampus and dentate gyrus, including the perforant, mossy fiber, and Schaeffer collateral pathways, and in the cell bodies of some hilar interneurons (Wang et al. 1994; Wenzel et al. 2007). Taken together, limbic structures appear to be critically involved in the generation or spread of spontaneous seizures in Kcnal -null mice; however, the identity of the exact brain circuits activated by seizure activity in these mice remains unknown.
A widely used biochemical technique to map neuronal activity during seizures is immunohistochemical visualization of Fos protein expression. Fos protein, encoded by the immediate early gene c-fos, is rapidly and transiently translated in response to intense synaptic excitation, such as occurs during seizures, making it a useful biomarker of neuronal activation (Morgan et al. 1987). In neurons, a stimulus induces production of Fos protein, which combines with Jun, another immediate early gene protein, to form a heterodimeric transcription factor that regulates expression of late-response genes (Herrera and Robertson 1996). Fos levels peak transiently about 2 h after a neuron-activating stimulus such as intense seizure activity (Morgan et al. 1987). Fos labeling has been used extensively to identify neurons that are activated during chemically-, electrically-, or stress-induced seizures in rodent models (Herrera and Robertson 1996). However, very few studies have utilized Fos imaging to measure neuronal activation following spontaneous seizures in genetic models of epilepsy, probably owing to the inherent difficulty associated with collecting tissues within the short-lived temporal window of Fos expression when seizure occurrence is unpredictable.
Here, Fos labeling was utilized to identify brain regions activated by spontaneous seizures in the Kcna1 -null mouse for the first time. Kcna1 -null mice were video monitored in their home cages to identify behavioral seizures. Fos immunohistochemistry was then performed to map neuronal activity in brain sections, which was correlated with the presence or absence of behavioral seizures in the 4 h immediately preceding tissue harvesting. Specifically, we tested whether knockouts exhibit basal increases in Fos labeling in particular neuronal subsets that might reflect a propensity for seizure activity. We also examined whether Fos expression is increased in discrete brain regions following spontaneous behavioral seizures, suggesting neuronal activation during seizure generation or spread.
Materials and methods
Animals and genotyping
Kcna1 —/— knockout (KO) mice carry null alleles of the Kcna1 gene (chromosome 6) resulting from targeted deletion of the open reading frame, as previously described (Smart et al. 1998). The mice were bred and maintained on a Tac:N:NIHS-BC genetic background. Animals were housed at 22°C, fed ad libitum, and submitted to a 12 h light/dark cycle. All procedures were performed in accordance with the guidelines of the National Institutes of Health (NIH), as approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center-Shreveport.
For genotyping, genomic DNA was isolated by enzymatic digestion of tail clips using Direct-PCR Lysis Reagent (Viagen Biotech, Los Angeles, CA, USA). The genotypes of Kcna1 mice were determined by performing PCR amplification of genomic DNA, using allele-specific primers: a mutant specific primer (5’-CCTTCTAT CGCCTTCTTGACG-3’), a wild-type specific primer (5’-GCCTCTG ACAGTGACCTCAGC-3’), and a common primer (5’-GCTTCAGG TTCGCCACTCCCC-3’). The PCR yielded amplicons of ~337 bp for the wild-type allele and ~475 bp for the null allele.
Video monitoring of spontaneous seizures
Individually housed KO mice were video monitored in their home cages using infrared-equipped digital network cameras (DCS-942L, D-Link, Fountain Valley, CA, USA). Digital video was recorded continuously for the 24-h period immediately preceding euthanasia and tissue collection. Videos were reviewed offline to identify spontaneous convulsive seizures, which exhibit easily identifiable behavioral features similar to kindling and kainate rodent models of temporal lobe epilepsy, as described in the Results. Seizures were scored for severity using the Racine scale (1, for mouth and facial movement; 2, head nodding; 3, forelimb clonus; 4, rearing with forelimb clonus; and 5, rearing and falling with forelimb clonus) (Racine 1972). Seizure durations were approximated by measuring the time from the onset of the seizure (characterized by initial immobility coupled with head nodding) to the cessation of behavioral convulsion.
Immunohistochemistry
Age-matched (1–3 months old) KO and Kcna1++ (WT) mice of both sexes were killed within 30 min of the end of video monitoring by inhaled isoflurane overdose and intracardially perfused with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS. Animals were always killed at a similar time of day during the light phase (between 10:00 a.m. and 2:00 p.m.) to prevent potential confounding effects because of diurnal variation in neuronal activity. Following perfusion, the brains were removed and post-fixed for 2 h followed by overnight cryoprotection in 30% sucrose in PBS. Brains were then embedded in optimum cutting temperature medium and frozen at —20°C until sectioned. Coronal sections (50 im thick) were cut using a Leica cryostat maintained at —20°C and collected in PBS for free-floating immunohistochemistry. Sections were blocked for 1 h in antibody vehicle (3% bovine serum albumin, 0.3% Triton X-100 in PBS) and then incubated overnight at 22°C with rabbit polyclonal anti-c-Fos antibody (Ab-5, 1 : 20 000; Calbiochem, La Jolla, CA, USA). After washing with PBS, sections were incubated with biotinylated anti-rabbit secondary antibody (1 : 750; Vector Laboratories, Burlingame, CA, USA) for 1 h. After washing again with PBS, sections were incubated in avidin-biotin peroxidase complex (ABC kit; Vector Laboratories) for 1 h. The sections were then developed using a 3,3’-Diaminobenzidine peroxidase substrate kit (Vector Laboratories); 3,3’-Diaminobenzidine reactions were always carefully timed to ensure all sections were equally treated. Sections were mounted serially on glass slides and air-dried overnight. The slides were dehydrated using an ethanol series (75%, 85%, 95%, 100%, 100%), cleared with Histo-clear II (National Diagnostics, Atlanta, GA, USA), and then cover-slipped.
Images were captured using an Olympus BX43 light microscope outfitted with an Olympus DP72 digital camera and cellSens software. Images were analyzed for Fos-positive labeling using ImageJ software (National Institutes of Health; Bethesda, MD, USA). Immunolabeled cells were counted using the Particle Analyzer tool in ImageJ and applying the same image, threshold, size, and circularity settings for each brain region. For each animal, cell counts were obtained from two to three non-consecutive sections between Bregma −1.2 to −1.4 mm for the basolateral amygdala (BLA) and piriform cortex (layer two) and between Bregma −1.3 to −1.9 mm for the dentate granule cell layer, dentate hilus region, and the CA1/CA3 pyramidal cell layers (Paxinos and Franklin 2013). Each brain region of interest was outlined and measured using ImageJ to calculate the area. The number of Fos-positive cells for each brain region was then expressed as cells per square millimeter. All immunohistochemistry tissue processing and cell counts were performed while the observer was blinded to genotype and seizure phenotype.
Statistical analysis
Data are expressed as means ± SEM. Prism 6 software for Windows (GraphPad, La Jolla, CA, USA) was used for analyses. One-way anova (a = 0.05) was used to evaluate cell counts, followed by Tukey’s multiple comparisons test. Reported p values were generated from the multiplicity adjusted p values for each comparison as calculated by the Prism software.
Results
Behavioral seizure detection in knockout mice
To examine increases in Fos expression as a result of basal differences and seizure activity in Kcnal-null (KO) mice, three experimental groups were generated for comparisons based on genotype and seizure history: (i) seizing KO mice; (ii) non-seizing KO mice; and (iii) wild-type (WT) Kcna1+ + controls. Seizing and non-seizing KO mice were identified and scored by video monitoring knockout mice in their home cages to identify behavioral seizures during the 4-h period immediately preceding tissue harvesting. Seizures in KO mice exhibit easily identifiable behaviors facilitating accurate detection. As previously reported, the typical seizure in Kcnal-null mice usually lasts between 20 s to 2 min and includes the following pattern of behavioral progression, which is also characteristic of kainate and kindling models: initial immobility; tail extension; facial twitching, and head nodding; followed by unilateral and bilateral forelimb clonus; rearing and falling; and then synchronous forelimb and hindlimb clonus (Smart et al. 1998; Rho et al. 1999; Wenzel et al. 2007). These ictal behaviors in KO mice always correlate with seizure activity in electroencephalography (EEG) recordings (Smart et al. 1998).
Of the 13 KO mice examined in this study, 6 knockouts exhibited obvious behavioral seizures during the 4-h period immediately before tissue harvesting and were designated as the ‘seizing’ group. The observed seizures exhibited the same repertoire of behaviors as described above. Most seizures were intermediate in severity, not progressing past bilateral forelimb clonus (stage 3); however, 5 mice exhibited at least one higher grade seizure characterized by forelimb myoclonus with rearing (stage 4) or with rearing and falling (stage 5). Table 1 provides a summary of the seizures observed by animal in the seizing group, including behavioral severity, duration, and approximate onset time relative to tissue harvesting. The remaining 7 KO animals did not show obvious seizure behaviors during the 4 h prior to tissue harvesting and were designated as the ‘non-seizing’ group. Previous studies have shown that knockout mice occasionally exhibit electrographic seizures without obvious behavioral changes, raising the possibility that some seizures could have eluded detection (Smart et al. 1998). However, as described below, the Fos labeling in non-seizing Kcnal -null mice showed substantial overall similarity to wild-type controls but not to seizing Kcnal -null mice. This pattern suggests that either electrographic-only seizures were absent in the nonseizing group during the observation period or they were not sufficient to significantly activate Fos expression. Importantly, all animals were kept undisturbed in a quiet room in their home cages during video monitoring prior to euthanasia to prevent confounding effects of external stimuli, such as loud noises, novel odorants, or handling.
Table 1.
Behavioral seizures in Kcnal-null mice during the 4 h immediately before tissue harvesting
| Individual seizure characteristics |
|||||
|---|---|---|---|---|---|
| Mouse number |
Age (d) | Total number of seizures |
Approx. time between seizure onset and tissue harvesting (min) |
Seizure duration (s) |
Racine score |
| 1 | 80 | 3 | 42 | 27 | 4 |
| 120 | 43 | 4 | |||
| 240 | 34 | 3 | |||
| 2 | 79 | 4 | 143 | 60 | 4 |
| 148 | 31 | 3 | |||
| 159 | 41 | 3 | |||
| 234 | 41 | 3 | |||
| 3 | 67 | 5 | 28 | 33 | 3 |
| 67 | 27 | 3 | |||
| 108 | 25 | 3 | |||
| 145 | 34 | 3 | |||
| 193 | 27 | 4 | |||
| 4 | 36 | 2 | 89 | 60 | 3 |
| 167 | 60 | 3 | |||
| 5 | 34 | 2 | 40 | 38 | 5 |
| 213 | 37 | 5 | |||
| 6 | 30 | 2 | 73 | 33 | 5 |
| 91 | 31 | 5 | |||
Basal Fos expression appears mostly unchanged in KO mice
Since KO mice exhibit frequent interictal EEG discharges (up to 100s per hour) and intrinsic neuronal hyperexcitability in brain slice recordings, we hypothesized that Kv1.1-deficient brains would show increased basal levels of cellular Fos expression in the absence of seizures. Quantification of Fos-labeled cells was performed in limbic structures, including the BLA (Fig. 1), piriform cortex (Fig. 2), dentate gyrus (granule cell layer and hilus; Fig. 3), and hippocampus (CA1 and CA3 pyramidal cell layers; Fig. 4). In WT control mice, scattered Fos-positive cells were consistently observed in limbic and cortical regions. In non-seizing KO mice, the abundance of Fos-labeled cells was not significantly different from the levels observed in WT controls for all limbic regions examined, except for the dentate granule cell layer which exhibited a significant ~75% decrease relative to WT mice (p = 0.005; Fig. 3e). The lack of significant increases in Fos-labeling between non-seizing Kcnal--- mice and WT controls in limbic regions suggests that intrinsic hyperexcitability and interictal activity as a result of Kv1.1-deficiency are not sufficient to substantially activate basal Fos protein expression.
Fig. 1.

Fos expression in the basolateral amygdala (BLA) of a wild-type mouse (a), a non-seizing knockout (KO) mouse (b), and a seizing KO mouse (c). (d) Quantification of Fos-positive cells by group. Numbers in bars indicate number of animals analyzed. −sz, non-seizing; +sz, seizing. Scale bar: 50 μm, **p < 0.01.
Fig. 2.

Fos expression in the piriform cortex of a wild-type mouse (a), a non-seizing knockout (KO) mouse (b), and a seizing KO mouse (c). (d) Quantification of Fos-positive cells by group. Numbers in bars indicate number of animals analyzed. −sz, nonseizing; +sz, seizing. Scale bar: 50 μm
Fig. 3.
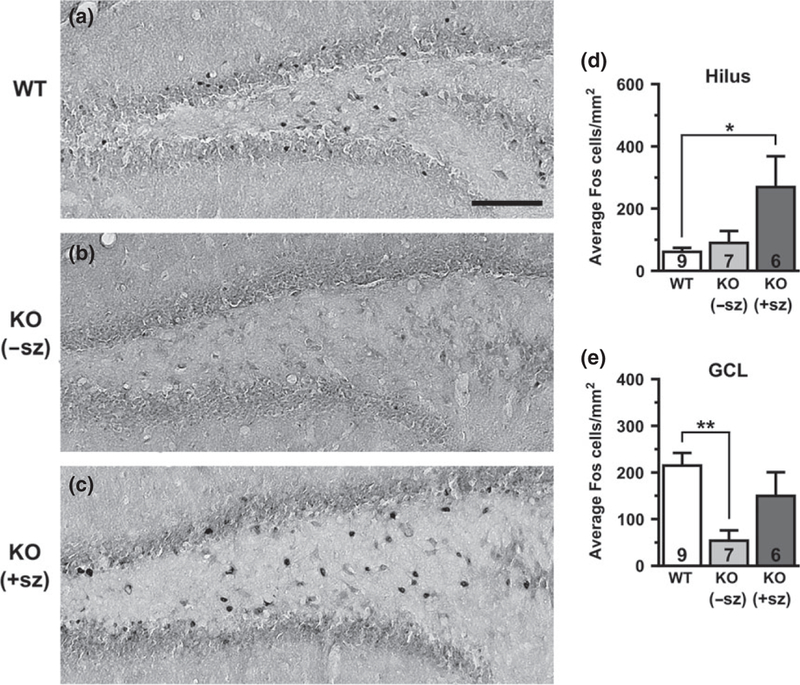
Fos expression in the dentate gyrus of a wild-type mouse (a), a non-seizing knockout (KO) mouse (b), and a seizing KO mouse (c). Quantification of Fos-positive cells by group in the hilus (d) and the granule cell layer (GCL) (e). Numbers in bars indicate number of animals analyzed. −sz, non-seizing; +sz, seizing. Scale bar: 100 μm. *p < 0.05; **p < 0.01.
Fig. 4.
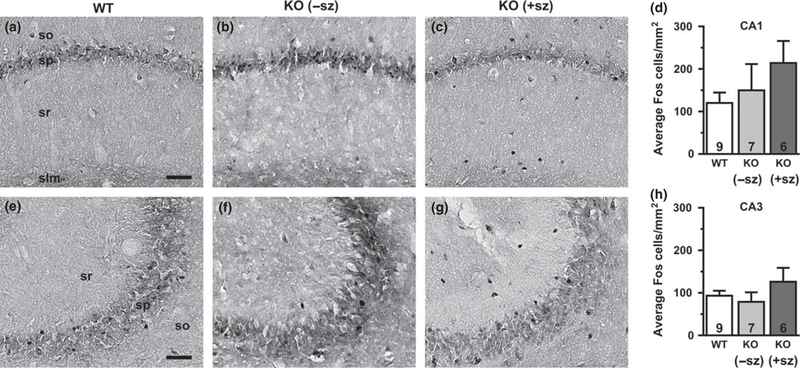
Fos expression in the hippocampal CA1 pyramidal cell layer of a wild-type mouse (a), a non-seizing knockout (KO) mouse (b), and a seizing KO mouse (c). (d) Quantification of Fos-positive cells by group in the CA1 pyramidal cell layer. Fos expression in the hippocampal CA3 pyramidal cell layer of a wild-type mouse (e), a non-seizing KO mouse (f), and a seizing KO mouse (g). (h) Quantification of Fospositive cells by group in the CA3 pyramidal cell layer. Numbers in bars indicate number of animals analyzed. −sz, non-seizing; +sz, seizing; so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum-moleculare. Scale bar: 50 μm.
Seizures activate Fos expression in select limbic regions in KO mice
Since Fos expression acts as a sensitive indicator of neuronal activity during chemically-or electrically-induced seizures, we hypothesized that Fos labeling could be used to detect brain regions recruited by spontaneous seizures in KO mice. To identify limbic structures that are abnormally activated by seizure activity, Fos labeling in seizing KO mice was compared to WT controls and to non-seizing KO mice. Overall, the brains of seizing KO mice exhibited increased numbers of Fos-labeled cells indicative of neuronal activation in several limbic regions compared to WT and nonseizing controls. In seizing KO mice, the BLA was the only limbic structure examined that exhibited a significant increase in Fos-labeling (three to fourfold) compared to both WT (p = 0.005) and non-seizing KO mice (p = 0.002; Fig. 1d). The hilus of seizing animals also showed drastically increased Fos-labeling of at least fourfold compared to WT (p = 0.027) and about threefold compared to non-seizing KO animals, but the latter comparison did not reach statistical significance (p = 0.076; Fig. 3d). The Fos-positive cells in the hilus are presumed to be predominantly inhibitory interneurons, but excitatory mossy cells also localize to this region (Freund and Buzsaki 1996). The piriform cortex of seizing KO mice exhibited a twofold increase in Fos-positive cells compared to WT and non-seizing KO mice, but these changes were not statistically significant (p = 0.110 and p = 0.090, respectively; Fig. 2d). The number of Fos-labeled cells in the dentate granule cell layer and the CA1 and CA3 pyramidal cell layers of seizing KO mice did not show significant differences compared to WT and non-seizing KO mice (Figs 3 and 4). Notably, 5/6 seizing KO mice and 1/7 non-seizing KO mice exhibited numerous intensely labeled Fos-positive cells in the stratum radiatum subfield of CA1 and CA3, which is home to several types of GABAergic interneurons (Fig. 4c and g) (Freund and Buzsaki 1996). In WT mice, sporadic Fos-labeled cells were rarely observed in the CA1/CA3 stratum radiatum layer (Fig. 4a and e). The robust Fos activation in the amygdala and hilus of seizing KO mice suggests these structures are critical components of the seizure circuitry in these animals. Furthermore, the increased Fos labeling in seizing KO mice compared to nonseizing KO mice suggests that seizures are responsible for the Fos activation and not baseline increases in neuronal activity.
Discussion
Previous work in Kcna1 -null mice has focused on the hippocampus as the primary onset zone of seizures, but the seizure-related Fos pattern in KO mice suggests the possibility of an extrahippocampal contribution by the BLA. Although the role of the amygdala in epilepsy is not as well-recognized as the hippocampal formation, it has been implicated in seizure initiation and spread in both patients and animal models (Kullmann 2011). Normally, the amygdala mediates emotional processing of sensory information and fear-related responses, learning, and memory (Sah et al. 2003). Anatomically, the amygdala possesses reciprocal connections with other limbic nuclei (including the hippocampus and piriform cortex), which can facilitate coactivation and seizure spread, respectively (Loscher and Ebert 1996; Davis and Whalen 2001; Sah et al. 2003). In patients, surgical resection of the amygdala has been used as an effective treatment for temporal lobe epilepsy (Feindel and Rasmussen 1991; Schramm 2008). Resected amygdala tissue from temporal lobe epilepsy patients shows the capacity for spontaneous interictal discharges when recorded in vitro, as well as alterations in neurotransmitter receptor densities, which together suggest evidence of disinhibition of amygdala circuitry that could contribute to seizure initiation (Graebenitz et al. 2011).
In rodent models, the amygdala exhibits extreme susceptibility to electrical kindling-induced seizures (Goddard et al. 1969). Similar to temporal lobe epilepsy patients, the amygdala in epileptic pilocarpine-treated rats exhibits spontaneous neuronal activity and altered inhibitory synaptic properties suggesting hyperexcitability (Benini and Avoli 2006). In addition, chronic spontaneous seizures in pilocarpine-treated rats induce Fos expression in a pattern similar to seizing KO mice, including Fos-labeling in the amygdala, piriform cortex, and interneurons of the hippocampus and dentate gyrus (Harvey and Sloviter 2005). However, the principal cells of the hippocampus and dentate gyrus remain largely Fos-negative, as in Kcna1-null mice, suggesting that spontaneous seizures in pilocarpine-treated rats originate outside of the hippocampus (Harvey and Sloviter 2005). At baseline, the significantly reduced Fos protein expression in the granule cell layer of non-seizing KO mice is consistent with reports of persistent down-regulation of basal c-fos mRNA levels in the dentate gyrus of young KO mice between the ages of P10-P34 (Rho et al. 1999). Similarly, chronically epileptic pilocarpine-treated mice also exhibit decreased Fos-labeling in granule cells between 4–8 h after a single spontaneous seizure, which is comparable to the postseizure interval of > 4 h in our non-seizing KO group (Peng and Houser 2005). Reductions in basal Fos levels may be because of a temporary refractory period for expression reinduction or a long-term adaptive response in a particular brain region (Morgan et al. 1987; Mello et al. 1996; Rho et al. 1999).
In addition to the Fos labeling patterns described in this work, previous neuropathological studies provide additional evidence for excessive activation of the amygdala and hilus during seizures in Kcna1 -null mice. In KO mice experiencing status epilepticus, histological experiments reveal extensive neuronal cell death, terminal degeneration, and astrogliosis in the hilus, amygdala, piriform cortex, and hippocampus (Wenzel et al. 2007). Interestingly, however, neuronal architecture in the BLA nucleus is relatively preserved (Wenzel et al. 2007). Magnetic resonance imaging of Kcna1 -null brains reveals significantly enlarged hippocampus and ventral cortex, including the amygdala and piriform region (Persson et al. 2007). Megencephaly (mceph) mice, which carry a mutation in Kcna1 causing truncated and non-functional Kv1.1 channels, also exhibit spontaneous seizures coupled with brain enlargement in these areas (Diez et al. 2003; Persson et al. 2005). Brain overgrowth because of Kv1.1-deficiency correlates with seizure activity since seizure suppression by either anticonvulsant therapy or genetic modifiers normalizes brain volume (Lavebratt et al. 2006; Holth et al. 2013).
Although Fos activation is a widely used biomarker of neuronal activity, the technique has some inherent limitations. Since Fos immunoreactivity provides only a snapshot of neuronal activity, it does not allow determination of the sequence of neuronal recruitment during seizures. Furthermore, it is possible that the threshold and timing of Fos activation varies between cell types, such that not all neurons recruited during seizures exhibit increased Fos labeling. In our study, mice in the seizing group exhibited different seizure frequencies and post-seizure intervals which could potentially lead to variability in the degree or extent of Fos activation.
In summary, our work uses Fos expression patterns to provide the first map of brain regions recruited by seizures in Kcna1 -null mice. Our finding that spontaneous seizures in KO mice significantly activate Fos expression in extrahippocampal networks, namely the amygdala, suggests critical involvement of these limbic circuits in epilepsy in Kcna1- null mice.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R00HL107641).
Abbreviations used:
- KO
knockout
- PBS
phosphate-buffered saline
Footnotes
conflict of interest disclosure
The authors declare no conflicts of interest.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Baraban SC, Southwell DG, Estrada RC, Jones DL, Sebe JY, Alfaro-Cervello C, García-Verdugo JM, Rubenstein JLR and Alvarez-Buylla A (2009) Reduction of seizures by transplantation of cortical GABAergic interneuron precursors into Kv1.1 mutant mice. Proc. NatlAcad. Sci. USA 106, 15472–15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benini R and Avoli M (2006) Altered inhibition in lateral amygdala networks in a rat model of temporal lobe epilepsy. J. Neurophysiol. 95, 2143–2154. [DOI] [PubMed] [Google Scholar]
- Bomben V, Holth J, Reed J, Cramer P, Landreth G and Noebels J (2014) Bexarotene reduces network excitability in models of Alzheimer’s disease and epilepsy. Neurobiol. Aging 35, 2091–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P and Litt M (1994) Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat. Genet. 8, 136–140. [DOI] [PubMed] [Google Scholar]
- Davis M and Whalen PJ (2001) The amygdala: vigilance and emotion.Mol. Psychiatry 6, 13–34. [DOI] [PubMed] [Google Scholar]
- Diez M, Schweinhardt P, Petersson S, Wang F-H, Lavebratt C, Schalling M, Hokfelt T and Spenger C (2003) MRI and in situ hybridization reveal early disturbances in brain size and gene expression in the megencephalic (mceph/mceph) mouse. Eur. J. Neurosci. 18, 3218–3230. [DOI] [PubMed] [Google Scholar]
- Feindel W and Rasmussen T (1991) Temporal lobectomy with amygdalectomy and minimal hippocampal resection: review of 100 cases. Can. J. Neurol. Sci. 18, 603–605. [DOI] [PubMed] [Google Scholar]
- Freund TF and Buzsaki G (1996) Interneurons of the hippocampus. Hippocampus 6, 347–470. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Yoo JW and Noebels JL (2007) Masking epilepsy by combining two epilepsy genes. Nat. Neurosci. 10, 1554–1558. [DOI] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL and Noebels JL (2010) Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J. Neurosci. 30, 5167–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Qian J, Kole MJ and Noebels JL (2012) Transcompartmental reversal of single fibre hyperexcitability in juxtaparanodal Kv1.1-deficient vagus nerve axons by activation of nodal KCNQ channels. J. Physiol. 590, 3913–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard GV, McIntyre DC and Leech CK (1969) A permanent change in brain function resulting from daily electrical stimulation. Exp. Neurol. 25, 295–330. [DOI] [PubMed] [Google Scholar]
- Graebenitz S, Kedo O, Speckmann E-J, Gorji A, Panneck H, Hans V, Palomero-Gallagher N, Schleicher A, Zilles K and Pape H-C (2011) Interictal-like network activity and receptor expression in the epileptic human lateral amygdala. Brain 134, 2929–2947. [DOI] [PubMed] [Google Scholar]
- Harvey BD and Sloviter RS (2005) Hippocampal granule cell activity and c-Fos expression during spontaneous seizures in awake, chronically epileptic, pilocarpine-treated rats: implications for hippocampal epileptogenesis. J. Comp. Neurol. 488, 442–463. [DOI] [PubMed] [Google Scholar]
- Herrera DG and Robertson HA (1996) Activation of c-fos in the brain. Prog. Neurobiol. 50, 83–107. [DOI] [PubMed] [Google Scholar]
- Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J and Noebels JL (2013) Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 33, 1651–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan LY and Jan YN (2012) Voltage-gated potassium channels and the diversity of electrical signalling. J. Physiol. 590, 2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinali M, Jungbluth H, Eunson LH, Sewry CA, Manzur AY, Mercuri E, Hanna MG and Muntoni F (2004) Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia. Neuromuscul. Disord. 14, 689–693. [DOI] [PubMed] [Google Scholar]
- Klassen TL, Bomben VC, Patel A et al. (2014) High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia 55, e6–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM (2011) What’s wrong with the amygdala in temporal lobe epilepsy? Brain 134, 2800–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavebratt C, Trifunovski A, Persson A-S, Wang F-H, Klason T, Ohman I, Josephsson A, Olson L, Spenger C and Schalling M (2006) Carbamazepine protects against megencephaly and abnormal expression of BDNF and Nogo signaling components in the mceph/mceph mouse. Neurobiol. Dis. 24, 374–383. [DOI] [PubMed] [Google Scholar]
- Lopantsev V, Tempel BL and Schwartzkroin PA (2003) Hyperexcitability of CA3 pyramidal cells in mice lacking the potassium channel subunit Kv1.1. Epilepsia 44, 1506–1512. [DOI] [PubMed] [Google Scholar]
- Loscher W and Ebert U (1996) The role of the piriform cortex in kindling. Prog. Neurobiol. 50, 427–481. [DOI] [PubMed] [Google Scholar]
- Mello LE, Kohman CM, Tan AM, Cavalheiro EA and Finch DM (1996) Lack of Fos-like immunoreactivity after spontaneous seizures or reinduction of status epilepticus by pilocarpine in rats. Neurosci. Lett. 208, 133–137. [DOI] [PubMed] [Google Scholar]
- Morgan JI, Cohen DR, Hempstead JL and Curran T (1987) Mapping patterns of c-fos expression in the central nervous system after seizure. Science 237, 192–197. [DOI] [PubMed] [Google Scholar]
- Paxinos G and Franklin KBJ (2013) The Mouse Brain in Stereotaxic Coordinates. Elsevier Academic Press, New York. [Google Scholar]
- Peng Z and Houser CR (2005) Temporal patterns of fos expression in the dentate gyrus after spontaneous seizures in a mouse model of temporal lobe epilepsy. J. Neurosci. 25, 7210–7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A-S, Klement G, Almgren M, Sahlholm K, Nilsson J, Petersson S, Arhem P, Schalling M and Lavebratt C (2005) A truncated Kv1.1 protein in the brain of the megencephaly mouse: expression and interaction. BMC Neurosci. 6, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A-S, Westman E, Wang F-H, Khan FH, Spenger C and Lavebratt C (2007) Kv1.1 null mice have enlarged hippocampus and ventral cortex. BMC Neurosci. 8, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Rho JM, Szot P, Tempel BL and Schwartzkroin PA (1999) Developmental seizure susceptibility of kv1.1 potassium channel knockout mice. Dev. Neurosci. 21, 320–327. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ESL, Lopez De Armentia M and Power J (2003) The amygdaloid complex: anatomy and physiology. Physiol. Rev. 83, 803–834. [DOI] [PubMed] [Google Scholar]
- Schramm J (2008) Temporal lobe epilepsy surgery and the quest for optimal extent of resection: a review. Epilepsia 49, 1296–1307. [DOI] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A and Tempel BL (1998) Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron 20, 809–819. [DOI] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Schwartzkroin PA and Tempel BL (1994) Localization of Kv1.1 and Kv1.2, two K channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J. Neurosci. 14, 4588−4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Vacher H, Clark E, Trimmer JS, Lee AL, Sapolsky RM, Tempel BL and Schwartzkroin PA (2007) Structural consequences of Kcnal gene deletion and transfer in the mouse hippocampus. Epilepsia 48, 2023–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi SM, Eunson LH, Spauschus A, Silva R. De., Tolmie J, Wood NW, McWilliam RC et al. (1999) A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 122 (Pt 5), 817–825. [DOI] [PubMed] [Google Scholar]