

Abstract
Approximately 30% of metastatic breast cancers harbor estrogen receptor α (ERα) mutations associated with resistance to endocrine therapy and reduced survival. Consistent with their constitutive proliferation, T47D and MCF7 cells in which wild-type ERα is replaced by the most common mutations, ERαY537S and ERαD538G, exhibit partially estrogen-independent gene expression. A novel invasion/dissociation/rebinding assay demonstrated that the mutant cells have a higher tendency to dissociate from invasion sites and rebind to a second site. Compared to ERαD538G breast tumors, ERαY537S tumors exhibited a dramatic increase in lung metastasis. Transcriptome analysis showed that the ERαY537S and ERαD538G mutations each elicit a unique gene expression profile. Gene set enrichment analysis showed Myc target pathways are highly induced in mutant cells. Moreover, chromatin immunoprecipitation showed constitutive, fulvestrant-resistant, recruitment of ERα mutants to the Myc enhancer region, resulting in estrogen-independent Myc overexpression in mutant cells and tumors. Knockdown and virus transduction showed Myc is necessary and sufficient for ligand-independent proliferation of the mutant cells but had no effect on metastasis-related phenotypes. Thus, Myc plays a key role in aggressive proliferation-related phenotypes exhibited by breast cancer cells expressing ERα mutations.
Keywords: breast cancer, ERαY537S, ERαD538G, Myc, RNAseq, metastasis
1. INTRODUCTION
At diagnosis, approximately 75% of breast cancers are estrogen receptor alpha (ERα) positive1. Endocrine therapies for ERα positive tumors include aromatase inhibitors, tamoxifen, fulvestrant (ICI-182,780/Faslodex) and other selective estrogen receptor modulators, and degraders, 2–4. Although endocrine therapies are effective initially, resistance often develops5. While resistance mechanisms are diverse, approximately 30% of metastatic tumors harbor ERα ligand binding domain (LBD) mutations, most commonly ERαD538G and ERαY537S6–8. These mutations are rare in primary tumors and increase after endocrine therapy9, 10.
To characterize these aggressive tumors, we, and others used CRISPR/Cas9, long-term-estrogen-deprived selection and other methods to generate cell lines bearing ERα mutations11–16. Consistent with their estrogen-independent proliferation and tamoxifen resistance11, structural and modeling studies suggest ERαY537S and ERαD538G mutants are locked in active conformations and exhibit reduced affinity for 4-hydroxytamoxifen (OHT)17. Moreover, ESR1 mutations increase breast cancer stem cell activity16. Since invasiveness of these cell lines, was largely unstudied, we used a novel, invasion-dissociation-rebinding (IDR) assay to analyze metastasis-related properties. Compared to parental cells, T47D-ERαY537S (TYS) and T47D-ERαD538G (TDG) cells11 exhibit increased invasiveness and TYS and TDG cells and MCF7-derived MCF7-Y537S and MCF7-D538G cells all exhibit increased dissociation and rebinding at a second site. Patients whose breast cancers express the ERαY537S and ERαD538G mutations have 1-year and 6-month shorter lifespans, respectively, than patients whose tumors express wild type ERα18. Notably, compared to the ERαD538G (TDG) tumors, the more lethal ERαY537S (TYS) tumors exhibited greatly increased lung metastases; wild type ERα (T47D) tumors did not metastasize. Thus, the aggressive phenotypes of cells expressing ERαY537S and ERαD538G include resistance to drugs targeting estrogen production and binding to ERα, estrogen-independent proliferation and an increase in stemness and metastasis-related properties.
How these aggressive phenotypes link to the mutant cells transcriptome was largely unknown. A few studies began transcriptome characterization12, 14, 19, and recent studies identified specific coactivators and unique binding sites of mutant ERα15, 20. Remaining to be done were detailed transcriptome comparisons of ERαD538G and ERαY537S mutant cells, analysis of invasiveness, and identification and analysis of specific genes contributing to the aggressive phenotypes of mutation-bearing tumors.
To identify pathways that might play a role in these aggressive phenotypes, we performed unbiased RNAseq analysis of global gene expression profiles. Compared to parental T47D cells, TYS and TDG cells exhibit distinct patterns of gene expression. Gene set enrichment analysis (GSEA) of T47D and MCF7 RNAseq data showed Myc targets are highly induced in mutant cells. Myc is important in diverse cellular processes including proliferation, biosynthesis and global metabolism21, 22. Dysregulated Myc expression contributes to malignant transformation, tumor progression and reduced responsiveness to anticancer drugs23–28. In breast cancer, Myc overexpression is associated with tamoxifen resistance in vitro and in patients5.
Chromatin immunoprecipitation (ChIP) demonstrated estrogen-independent, fulvestrant-resistant, recruitment of ERαY537S and ERαD538G to the Myc enhancer. Moreover, cell and tumor studies demonstrated estrogen-independent Myc expression in the mutants is higher than in estrogen-treated controls. Myc knockdown blocked estrogen-independent growth of TYS and TDG cells. Notably, expression of Myc in estrogen-deprived T47D cells partially reproduces the estrogen-independent proliferation and antiestrogen resistance, but not the increased invasiveness, dissociation and rebinding, displayed by mutant cells. Our identification of a role for Myc in a sub-set of the aggressive phenotypes displayed by ERα mutant cells illustrates the utility of these cell models and transcriptome data as tools for identifying pathways that contribute to the aggressiveness of ESR1 mutant cells.
2. Materials and methods
2.1. Cell culture and proliferation assays
Media and conditions were previously described29. T47D, MCF7 and the mutant clones were generated and cultured as described11, 14. Cells were authenticated at University of Arizona Genetics Core. E2, fulvestrant and z-OHT were from Sigma. JQ1 was from Selleck. Cells proliferation assays were as described29.
2.2. Generation of luciferase-expressing cell lines
The pcDNA3-Luc vector was transfected into T47D, TYS clone 4 and TDG clone 1 cells, respectively. Colonies were selected for 2 weeks in G418.
2.3. qRT-PCR and RNAseq data analysis
Cells were cultured and plated as described29. For RNAseq, T47D, TYS and TDG cells were treated with vehicle (EtOH) or 10 nM E2. Total RNA of three biological replicates was collected and cDNA library were prepared using TruSeq Stranded mRNAseq Sample Prep kit (Illumina). Single-end RNA sequencing was performed by the University of Illinois High-Throughput Sequencing Unit (HiSeq 4000 (Illumina)). Software used for data analysis is in Supplementary Table S1. Raw and processed data of RNAseq were deposited in NCBI GEO [GSE108304].
2.4. Western blot
Whole cell extracts were prepared and western blots were performed as described29. Antibodies are in Supplementary Table S2.
2.5. siRNA knockdowns
siRNA knockdowns were performed using DharmaFECT1 and 100 nM ON-TARGET plus non-targeting pool or SMARTpools for ERα and c-Myc (Dharmacon). Transfection conditions were as described29.
2.6. Chromatin immunoprecipitation (ChIP)
Chromatin was prepared from three biological replicates incubated 30 min in 10 nM E2 or pretreated with 500 nM fulvestrant for 10 min before E2 addition. Samples were sheared using an M220 Focused-ultra sonicator (Covaris). ChIP assays were as described30.
2.7. Lentivirus infection
Lentivirus was produced by cotransfecting pCDH-puro-cMyc (Addgene #46970) or pHIV-Luciferase vector (Addgene #21375) with packaging vectors pCI-VSVG (Addgene #1733) and psPAX2 (Addgene #12260) into HEK293 cells using Lipofectamine 3000 (Thermo Fisher).
2.8. Cell invasion assay
Millipore polycarbonate cell culture inserts (12 µm) were coated with 25 µg/ml collagen or Matrigel (Corning). 100,000 luciferase-expressing cells in 0.5 ml medium containing 0.1% BSA were placed in the upper chamber and 0.55 ml medium containing 20% CD-FBS were in bottom chamber31, 32. After 24h, upper chamber cells were removed. 150 µl Bright-Glo™ (Promega) was added into the wells and luciferase activity was measured using a PHERAStar plate-reader (BMG Labtech).
2.9. Mouse xenograft
All animal studies were approved by the University of Illinois Institutional Animal Care (IACUC) committee. Five female mice were used for each cell line. Estrogen pellets (90 day; Innovative Research of America) were implanted subcutaneously 30 days prior to T47D-Luc cell injection; a second estrogen-release pellet was implanted 3 months after the first pellet. No estrogen supplementation was used in the TYS-Luc and TDG-Luc mice. 5 × 10 6 T47D, TYS and TDG cells in Matrigel stably expressing the luciferase gene (T47D-Luc, TYS-Luc and TDG-Luc) were grafted orthotopically into ovariectomized NSG mice. Mice were anesthetized, injected with luciferin substrate and tumor bioluminescence was monitored using an IVIS Spectrum CT live-animal imaging system (PerkinElmer). Based on the growth of the primary breast tumors, mice were sacrificed about 2 months (TYS-Luc), about 3 months (T47D-Luc) and about 4 months (TDG-Luc), after initiating tumor growth. Consistent with in vitro results for the cell lines, the TDG-Luc tumors have about 12-fold higher luciferase emission per mg excised breast tumor weight than the TYS-Luc tumors. Therefore, the TYS-Luc and TDG-Luc lung metastases data in Fig. 2E was normalized for the difference in luciferase emission. Since no lung metastases were observed for T47D-Luc, normalization was not relevant.
Fig. 2.
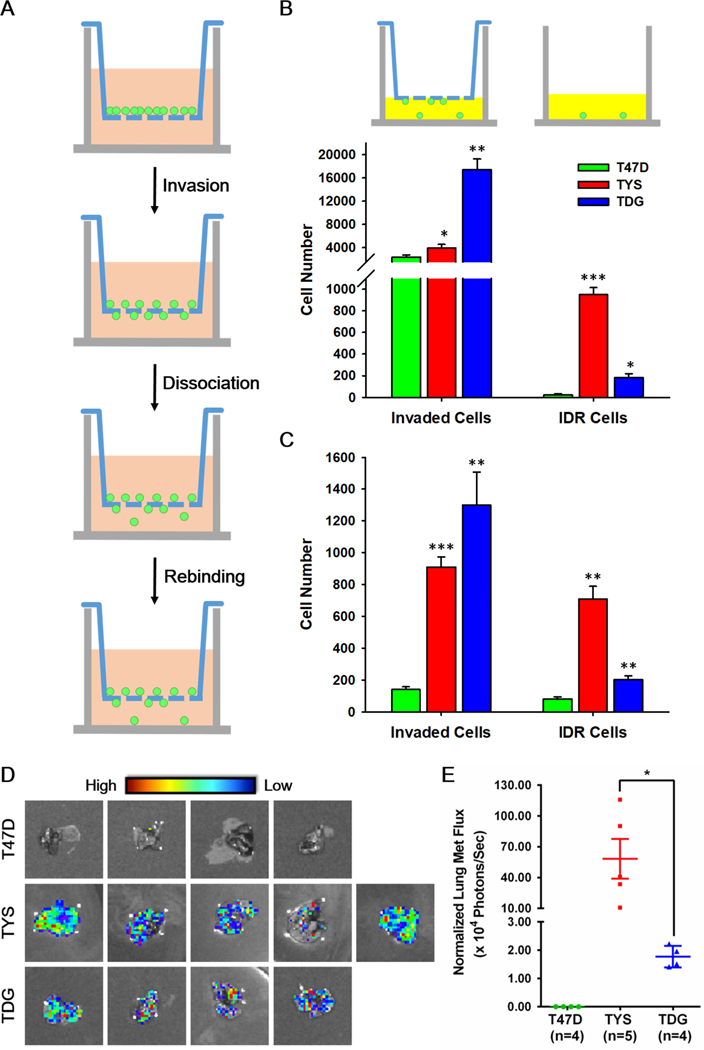
ERα Y537S and D538G mutations increase breast cell invasiveness and promote a metastatic phenotype. A Schematic of the invasion-dissociation-rebinding (IDR) assay. B The upper panel illustrates measurement of total invaded cells and IDR (invaded then dissociated and rebound) cells. IDR assay of T47D, TYS and TDG cells with collagen- B or matrigel-coated C membranes and chambers (n=5). Cell number calculated from a standard curve of light units (luciferase activity) versus cell number for each cell line. D Ex vivo imaging of excised lungs from xenograft bearing mice. As shown in the High to Low spectrum band, white dots are not bioluminescent signals. E Quantification of the bioluminescent signal in the lung areas shown in Fig. 2D. Shown as mean flux ± s.e.m.. The fluxes of lung metastases were normalized for the emission efficiency determined by tumor flux/ tumor weight. Similar results were obtained when the data was calculated as lung flux/ primary breast tumor flux. * Indicates a significant difference among groups using student t test. *P<0.05, **P<0.005, ***P<0.001. ns, not significant.
2.10. Statistics
Each in vitro experiment was performed at least three times. Statistical significance was determined by an unpaired two-tailed Student’s t-test or ANOVA using SPSS statistics (IBM). Data were presented as mean ± s.e.m and a p value of <0.05 was defined as statistically significant.
3. RESULTS
3.1. T47D-ERαY537S (TYS) and T47D-ERαD538G (TDG) cells display a constitutive gene expression pattern
To evaluate the effect of ERα mutations on gene expression, we performed RNAseq in T47D, TYS and TDG breast cancer cells. Without estrogen, T47D, TYS and TDG cells exhibit very different gene expression patterns. Expression of 3669 and 2592 genes were altered in TYS and TDG cells, respectively (Table S3); 2020 of these genes were shared by TYS and TDG cells (Supplementary Fig. 1A). To evaluate expression of direct and indirect ERα target genes, we chose 4- and 24-hour E2 treatment. After 4h E2 treatment, 317 genes were differentially regulated in T47D cells; 272 of these genes are shared by one or both mutant cells (Fig. 1A). A heatmap of over 13,000 genes and a multi-dimensional scaling (MDS) plot show a modest effect of 4h E2 treatment on the T47D transcriptome, with minimal effects on the mutants (Supplementary Fig. 1B, Fig. 1D).
Fig. 1.

TYS and TDG cells exhibit unique gene expression patterns. A,B Venn diagrams comparing genes with absolute fold change >2 and false discovery rate (FDR) q-value <0.05 in T47D, TYS and TDG cells after 4h A and 24h B E2 treatment. C Heatmap showing log counts per million (CPM) of genes that have CPM >1 in at least 3 samples after 24h E2 treatment. D,E Multi-dimensional scaling (MDS) plot of RNAseq samples 4h D and 24h E after E2 addition. Similarities of gene expression patterns were calculated and mapped for T47D (T), TYS (Y) and TDG (D) cells treated with vehicle (V) or estrogen (E). Data from each of the three biological replicates is shown. F Real-time PCR analysis of GREB1, PGR, CXCL12, IL1R1, PI9 and EGR3 in T47D, TYS and TDG cells after addition of vehicle or E2 for 4h or 24h. (mean ± s.e.m., n=3). Different letters indicate a significant difference among groups (P<0.05) using one-way ANOVA followed by Duncan’s post hoc test.
24h E2 incubation dramatically increased differentially expressed T47D cell genes; most of these genes (1509/1748) were differentially expressed in one or both mutant cells (Fig. 1B). Compared to vehicle, the heatmap and MDS plot display a large shift of the E2-treated T47D transcriptome toward the TDG cells (Fig. 1C,E). To further compare T47D, TYS and TDG cells, we used qPCR to analyze expression of well-characterized ERα target genes. For all genes tested, estrogen responses were robust in T47D, reduced in TDG and minimal in TYS cells (Fig. 1F).
To test for off-target effects of CRISPR/Cas9, we evaluated expression of these genes both in additional clones of T47D-ERαY537S (clone 39) and T47D-ERαD538G (clone 28) and in MCF7 cell lines containing ERαY537S and ERαD538G mutations generated through virus infection, not CRISPR. Consistently, ERαY537S cells responded less to estrogen than ERαD538G and parental cells (Supplementary Fig. 2, 3A). Overall, the data demonstrate estrogen-independent gene expression in the mutant cells.
3.2. ERαY537S and ERαD538G cells have distinct gene expression profiles and exhibit aggressive phenotypes
Although TYS and TDG cells both display ligand-independent gene expression, their gene expression profiles differ. MDS shows that after 24h E2 treatment T47D cells shifted towards TDG cells, indicating that E2-stimulated T47D and TDG cells have more closely related gene expression patterns. Notably, E2 treatment had almost no effect on TYS gene expression (Fig. 1E). To further address potential off-target effects of CRISPR/Cas933, we generated an MDS plot from raw RNAseq datasets of MCF7, and virus-generated MCF7-Y537S and MCF7-D538G [SRA: SRP093386]14 (Supplementary Fig. 3C). Consistent with the T47D data, the estrogen-treated MCF7 gene expression profile is closer to the MCF7-D538G cell profile and MCF7-Y537S cells respond least to estrogen.
To begin to explore the interplay between gene expression patterns and the aggressive phenotypes induced by ERα mutations, we examined proliferation and resistance to endocrine therapies. Parental T47D cells did not grow in estrogen-depleted medium, while TYS and TDG cells (Supplementary Fig. 4A), other mutant T47D clones and MCF7-Y537S and MCF7-D538G cells all exhibited robust estrogen-independent proliferation (Supplementary Fig. 4B,C). We used dose-response studies to evaluate resistance to endocrine therapies. In T47D cells, proliferation was nearly abolished by a 25- to 100-fold molar excess over estrogen of z-4-hydroxytamoxifen (OHT), or fulvestrant/ICI. In contrast, TYS and TDG cells exhibited partial resistance to OHT and fulvestrant (Supplementary Fig. 4A). Notably, in MCF7 cells, a 50-fold molar excess of OHT or fulvestrant abolished estrogen induced cell proliferation, but had no effect on proliferation of MCF7-Y537S and MCF7-D538G cells (Supplementary Fig. 4C). Thus, both T47D- and MCF7-derived cell lines containing ERαY537S and ERαD538G exhibit estrogen-independent proliferation and resistance to OHT and fulvestrant.
Although ESR1 mutations are observed in metastatic breast cancers, metastasis is difficult to model in cell culture. To probe steps in metastasis beyond invasion, we developed a quantitative invasion-dissociation-rebinding (IDR) assay (Fig. 2A). Using T47D, TYS and TDG cell lines stably expressing luciferase, we quantified both total cells that invaded through collagen- or Matrigel-coated membranes (Fig. 2B; Invaded Cells) and cells that invaded and then dissociated from their membrane invasion site and rebound to a second site (Fig. 2B; IDR cells). ERα mutations significantly increased invasiveness. Even though more TDG cells invaded through membranes, more TYS cells dissociated and rebound (Fig. 2B,C, Supplementary Fig. 4D). Using lentiviral transduction of luciferase, we performed IDR assays on MCF7, MCF7-Y537S and MCF7-D538G cells. Compared to MCF7 cells, ERα mutations did not increase invasion, but strongly elevated dissociation and rebinding (Supplementary Fig. 4E). Increased ability to dissociate and rebind at a second site is a previously unexplored metastasis-related property of ERα mutant cells.
We evaluated the ability of orthotopic breast tumors derived from TYS-Luc (ERαY537S) and TDG-Luc (ERαD538G) cells to metastasize to lung and investigated whether invasion or dissociation-rebinding correlated with in vivo metastatic potential. As a control, we used estrogen-supplemented T47D-Luc, which expresses wild type ERα. All the mice grew primary tumors (Supplementary Fig. 4F). Because the light output from the large primary breast tumors masks signals from lung metastases, we evaluated the extent of lung metastasis using ex vivo bioluminescent imaging (BLI) of lungs excised from tumor bearing mice. Consistent with earlier reports34, 35, lung metastases were not detected in mice harboring T47D-Luc xenografts (Fig. 2D). All mice harboring primary breast tumors expressing ERαY537S and ERαD538G developed lung metastases (Fig. 2D). Since light emission per mg of TDG-Luc breast tumor is 12-fold higher than from TYS-Luc breast tumors, the moderately lower visualized level of metastasis seen in the lungs of TDG-Luc tumors compared to TYS-Luc tumors (Fig. 2D) reflects a dramatically lower level of lung metastasis. Normalized for this difference in light emission, lung metastases from mice with primary TYS-Luc breast tumors averaged 32 times higher signals than lung metastases from mice harboring primary TDG breast tumors (Fig. 2E). A >20-fold increase in metastases in mice harboring TYS-Luc breast tumors compared to TDG-Luc tumors is also observed if the metastases data is calculated as the ratio of light emission by each lung metastases relative to light emission from the primary breast tumor in that mouse.
3.3. ERαY537S and ERαD538G mutations elicit constitutive hormone-independent pathway alterations
To probe pathways related to therapy resistance and metastatic potential in ERαY537S and ERαD538G cells, we used the RNAseq datasets to perform GSEA. Highly upregulated pathways were largely consistent across T47D and MCF7 mutant cells, with little estrogen dependence (Fig. 3, Supplementary Fig. 5). Consistent with estrogen-independence, without estrogen, estrogen response pathways were upregulated in mutant cells (Fig. 3A). Cell cycle related pathways were constitutively elevated in mutant cells. Confirming our earlier observation11, the tumor protective unfolded protein response (UPR) was upregulated in T47D and MCF7 mutant cells.
Fig. 3.

Estrogen upregulated pathways are constitutively activated in TYS and TDG cells. Gene set enrichment analysis (GSEA) using the ‘hallmark gene set’ as the reference dataset. The bar chart shows the normalized enrichment scores (NES) of pathways that are significantly up- or down-regulated in pair-wise comparisons between vehicle A 4h E2 B and 24h E2 C treated T47D, TYS and TDG cells and vehicle-treated T47D cells. All pathways had a false discovery rate q<0.0005 except those marked *, which were q<0.005.
Consistent with direct regulation by ERα, in T47D cells after 4h estrogen treatment, “Estrogen_Response” pathways were the top upregulated pathways, (Fig. 3B). After 24h estrogen treatment, most pathways highly upregulated in mutant cells were also upregulated in parental T47D cells.
3.4. The Myc pathway exhibits constitutive and elevated activation in mutant cells
At 24 hours, Myc targets were the top upregulated pathway in mutant cells (Fig. 3, Supplementary Fig. 5). There is a strong correlation between Myc targets and cancer stemness, which can lead to therapy resistance and metastasis36. Compared to T47D cells, Myc target genes important in DNA replication, protein synthesis and cell cycle progression are constitutively overexpressed in TYS and TDG cells (Fig. 4A,B).
Fig. 4.
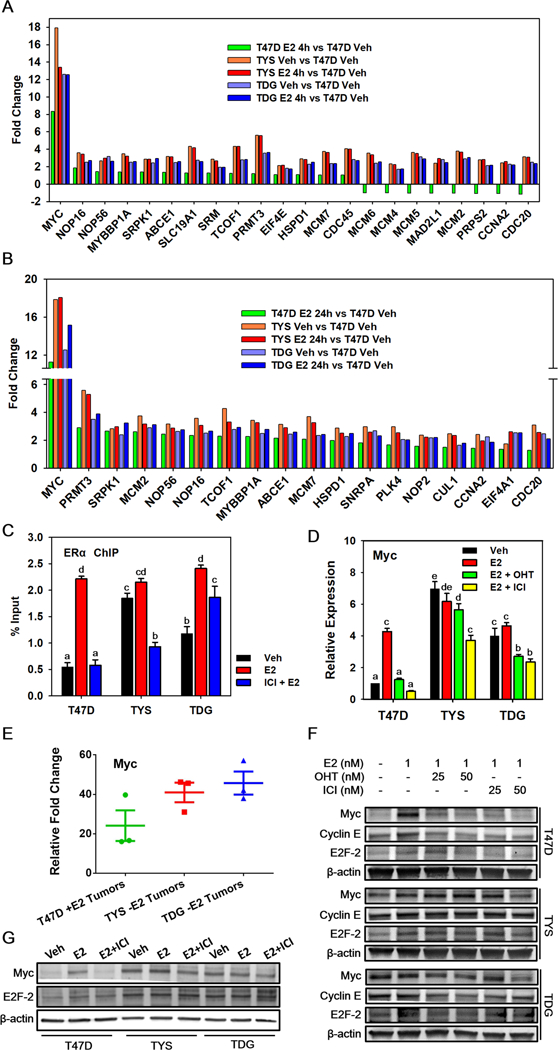
Myc is overexpressed and contributes to antiestrogen resistance in TYS and TDG cells. A,B Bar charts showing the RNAseq mean fold change of Myc target genes in T47D, TYS and TDG cells after addition of E2 for 4h A or 24h B. C ChIP was performed in T47D, TYS and TDG cells treated with or without 500 nM fulvestrant/ICI for 10 min before adding 10 nM E2 for 30 min. Real-time PCR was used to analyze the enrichment of ERα binding sites at the Myc enhancer region.51 D qRT- PCR analysis of Myc mRNA levels in T47D and mutant cells after treatment for 24h with 1 nM E2, 1 nM E2 + 25 nM OHT, or 1 nM E2 + 25 nM fulvestrant/ICI. E qRT- PCR analysis of Myc mRNA levels in tumors induced with estrogen (T47D) or without added estrogen (TYS and TDG). C,D,E Mean ± s.e.m., n=3. Different letters indicate a significant difference among groups (p <0.05) using one-way ANOVA followed by Tukey’s post hoc test. F Western blot analysis of Myc, Cyclin E and E2F-2 levels in T47D, TYS and TDG cells after 24h in the indicated concentrations of vehicle, E2, OHT and fulvestrant. G Western blot analysis of Myc and E2F-2 protein levels following 24h treatment with vehicle, or 1 nM E2, with or without 50 nM fulvestrant.
As expected, ERα knockdown blocked estrogen-stimulated proliferation of T47D cells and proliferation of TYS and TDG cells with and without E2 (Supplementary Fig. 6A). Consistent with a role for Myc, ERα knockdown also blocked estrogen stimulated Myc expression in T47D cells and reduced Myc expression in mutant cells (Supplementary Fig. 6B).
We next assessed whether Myc expression in the TYS and TDG cells was constitutive and resistant to antagonists. Chromatin immunoprecipitation (ChIP) in T47D cells showed that E2 stimulates, and a 50X excess of ICI/fulvestrant blocks, recruitment of ERα to the Myc enhancer region. Recruitment of ERαY537S was constitutive with little effect of E2; binding of ERαD538G was partially constitutive and was further increased by E2. ICI reduced, but did not eliminate binding of ERα mutants to Myc enhancer (Fig. 4C).
Moreover, compared to T47D cells, Myc mRNA expression in TYS and TDG cells was constitutive and elevated (Fig. 4D). Notably, compared to estrogen-treated T47D tumors, vehicle-treated TYS and TDG tumors exhibited higher Myc mRNA expression (Fig. 4E). Consistent with the ChIP, OHT or ICI abolished E2-induction of Myc mRNA in T47D cells, but reduced Myc levels less than 50% in TYS and TDG cells (Fig. 4D). Western blot analysis confirmed that, in T47D cells, Myc protein exhibits fulvestrant-sensitive is E2-induction. In contrast, in TYS and TDG cells, Myc levels are high in vehicle-treated cells, show little increase in response to E2 and are only modestly sensitive to OHT and fulvestrant (Fig. 4F,G). Constitutive Myc expression and antiestrogen resistance were also observed in other T47D mutant clones and in MCF7 mutant cells (Supplementary Fig. 6C,D)
In T47D cells, but not in the mutant cells, antiestrogens downregulated induction of mRNA and protein encoding Myc cell cycle effector targets, Cyclin E and E2F-2 (Supplementary Fig. 6E,F, Fig. 4F,G). Although OHT and fulvestrant blocked E2 induction of E2F-2 mRNA in mutant cells, it was still expressed at levels >5 fold higher than in T47D cells (Supplementary Fig. 6E). These data help explain why TYS and TDG cells continue to proliferate during endocrine therapy.
3.5. Myc expression is required for estrogen-independent proliferation of TYS and TDG cells and is sufficient to confer partial resistance to endocrine therapy
To further evaluate Myc’s role, we altered its level. After RNAi knockdown, levels of Myc protein in mutant cells and vehicle treated T47D cells were similar (Fig. 5A). Myc knockdown greatly reduced E2 stimulated proliferation of T47D cells and abolished estrogen-independent proliferation of mutant cells (Fig. 5B). Targeting Myc expression through the bromodomain inhibitor JQ1 is an emerging therapeutic strategy37, 38. JQ1 reduced Myc expression by ~50% (Fig. 5C) and abolished proliferation of T47D, TYS and TDG cells (Fig. 5D). As a bromodomain inhibitor, JQ1 acts on diverse targets. These data suggest other pathways promote proliferation in TYS and TDG cells by working synergistically with Myc.
Fig. 5.
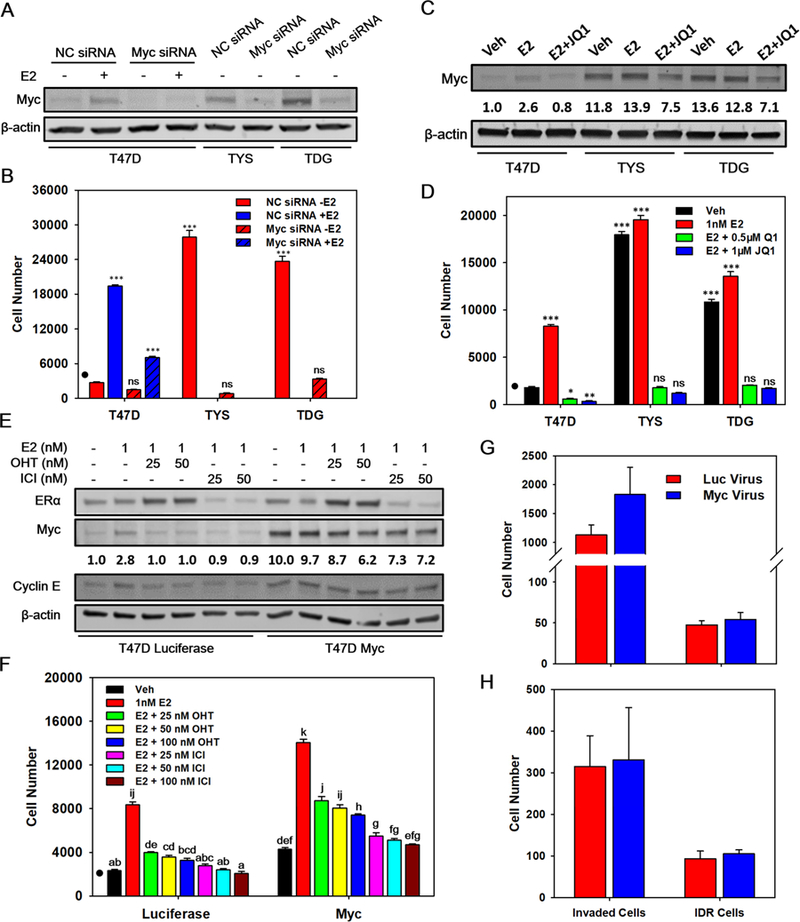
Myc is necessary and sufficient for estrogen-independent cell proliferation and antiestrogen resistance, but does not affect invasiveness. A Western blot showing Myc protein levels after treatment of T47D, TYS and TDG cells with 100 nM non-coding (NC) or Myc SMARTpool siRNA for 24h, followed by treatment with vehicle or 1 nM E2 for 24h. B Proliferation of T47D, TYS and TDG cells treated with 100 nM NC or Myc SMARTpool siRNA, followed by treatment with vehicle or 1 nM E2 for 96 h (mean ± s.e.m., n=6). C Western blot showing Myc levels following treatment of cells with vehicle or 1 nM E2 with, or without, 1 µM JQ1 for 8h. D Proliferation of T47D, TYS and TDG cells treated with vehicle, E2, or E2 plus 0.5 or 1 µM JQ1 for 96 h (mean ± s.e.m., n=6). E Western blot showing ERα, Myc and Cyclin E levels in T47D, TYS and TDG cells treated with Myc-lentivirus or control luciferase-lentivirus, followed by the indicated concentrations of E2, OHT and ICI for 24h. F Proliferation of T47D, TYS and TDG cells, after transduction with Myc or control lentivirus containing medium for 24h, followed by treatment with the indicated concentrations of E2, OHT and ICI for 4 days. (mean ± s.e.m., n=6). B,D,F ‘•’ denotes cell number at day 0. C,E Myc protein levels were quantitated using a PhosphorImager and ImageQuant. B,D * indicates a significant difference among groups using one-way ANOVA followed by Tukey’s post hoc test. *P<0.05, **P<0.005, ***P<0.001. ns, not significant. F Different letters indicate a significant difference among groups (P<0.05) using one-way ANOVA followed by Tukey’s post hoc test. G,H IDR assay of T47D cells transduced with Myc-lentivirus or luciferase-lentivirus with collagen- G or matrigel-coated H membrane and chamber (mean ± s.e.m., n=5). Cells were transduced with virus and after 1 day total invaded cells and cells that invaded, then dissociated and rebound (IDR) were measured using their luciferase activity and a standard curve for each cell line of luciferase activity versus cell number.
Since reducing constitutive Myc expression abolished estrogen-independent proliferation of TYS and TDG cells, we explored whether constitutive Myc expression in estrogen-deprived T47D cells could recapitulate their aggressive phenotypes. T47D cells were infected with lentivirus expressing Myc, or a luciferase control. After infection, Myc protein levels in T47D cells were increased to levels similar to those in TYS and TDG cells (Fig. 5C,E). Myc overexpression in OHT- and fulvestrant-treated T47D cells did not alter ERα levels, but increased levels of the downstream effector, Cyclin E (Fig. 5E). Constitutive Myc expression facilitated both E2-independent and E2-dependent proliferation of T47D cells (Fig. 5F). Notably, compared to control cells, Myc overexpression increased resistance to OHT and fulvestrant (Fig. 5F). However, Myc overexpression had no effect on the number of invaded cells, or on the number of dissociated and rebound cells (Fig. 5G,H). Thus, while estrogen-independent expression of Myc in T47D cells does not reproduce the metastasis-related phenotypes exhibited by ERα mutant cells, Myc expression partially recapitulates their proliferation-related phenotypes of increased growth, estrogen-independent proliferation and resistance to OHT and fulvestrant.
4. DISCUSSION
Clustered mutations in the ERα LBD occur in 20–40% of ERα positive metastatic breast cancers7, 10, 39. While all metastatic ERα positive breast cancers display resistance to endocrine therapy and ultimately to standard chemotherapy, patients with tumors harboring the common ERαY537S and ERαD538G mutations exhibit 1-year and 6-month shorter median survival than patients whose metastatic tumors contain wild type ERα18. To identify roles of these mutations, transient transfection, CRISPR/Cas9 and other techniques were used to express ERα LBD mutations in breast cancer cells7, 9, 13–15.
Transcriptome level studies of cells bearing ESR1 mutations are limited and were either restricted to a single mutation13, or lack extensive functional analysis14. We compared T47D cells harboring ERαY537S and ERαD538G mutations and parental cells at two time points and confirmed several key observations with the MCF7 dataset. Surprisingly, the MDS results show that the mutations are not simply constitutively active ERα, and instead exhibit unique gene expression patterns.
These ERα LBD mutations occur primarily after patients have received endocrine therapy7, 9, 10, 40. Very recently, naturally occurring ESR1 Y537C and Y537S mutations were identified by selection in long-term-estrogen-deprived MCF7 cells12. Taken together, these studies, and the data we present, show that the changes in the Y537 and D538 ESR1 mutations are sufficient to confer on breast cancer cells partial resistance to antiestrogens and estrogen-independent gene expression and cell proliferation. Across different clones, different parental cell lines and zygosity status, expression of most mRNAs tested exhibited a higher basal level and lower estrogen response in ERαY537S cells than in ERαD538G cells. Compared to cells expressing ERαD538G, we (Supplementary Fig. 4A), we and others, observe increased antiestrogen resistance in cell lines expressing ERαY537S11, 14. These data suggest enhanced estrogen-independent gene expression in breast cancer cells expressing ERαY537S may confer unique properties that influence tumor behavior and response to therapy.
Previous studies focused almost entirely on proliferation-related properties of ESR1 mutations. However, ESR1 mutations were detected at higher allele frequency in metastases than in the primary breast cancers41, 42. To probe the role of ESR1 mutations in metastasis-related properties we improved conventional transwell assays32 which fail to detect invaded cells that partially recapitulate the metastasis-related property of cell dissociation from the membrane and reattachment at a second site. Using T47D, TYS and TDG cells that stably express luciferase and luciferase-expressing lentivirus in MCF7 cell lines, we used our invasion-dissociation-rebinding assay to quantitate both the number of invaded cells and the number of invaded cells that then dissociate from the membrane and rebind on the bottom of the well. In the in vitro invasion-dissociation-rebinding assay, ERα mutations significantly increase both T47D cell invasion and dissociation-rebinding. Consistent with the cell-based data, our in vivo data shows that the ERα mutations drive metastasis in otherwise non-metastatic T47D tumors. These data illustrate the value of the IDR assay (Fig. 2A). The dramatic increase in lung metastasis of TYS tumors compared to TDG tumors (Fig. 2E) was not predicted by the widely used Matrigel and collagen invasion assays, which show increased invasion by TDG cells (Fig. 2C). In contrast, the dissociation rebinding assay, which shows strongly increased dissociation and rebinding by the TYS cells compared to the TDG cells (Fig. 2C), is much more consistent with the in vivo lung metastasis data (Fig. 2E). While metastasis is an exceptionally complex multi-step process that cannot be fully modeled with cell-based assays, our data shows that, compared to the traditional Matrigel invasion assay, our simple in vitro IDR assay explores cell properties that correlate with in vivo metastatic frequency. Thus, the IDR assay provides a useful in vitro model for investigation of metastasis-related properties.
To identify pathways driving these aggressive phenotypes of ERα mutant cells, we performed GSEA using T47D and MCF7 RNAseq datasets. Compared to parental cells, Myc target genes were highly enriched in T47D and MCF7 mutant cells; other upregulated pathways like “E2F targets” and “G2M checkpoint” are tightly correlated with Myc expression. Myc directly induces expression of cell cycle regulators, including Cyclin D, Cyclin E and E2Fs.43, 44 Myc also promotes cell cycle progression by regulating CDK phosphorylation and antagonizing cell cycle inhibitor expression.45, 46 In estrogen-deprived ERαY537S and ERαD538G cells, Myc was highly induced, suggesting Myc might play an important role in their E2-independent proliferation. Myc knockdown demonstrated that Myc is necessary for E2-independent proliferation of the TYS and TDG cells. Constitutive Myc expression in E2-deprived T47D cells was sufficient to induce moderate E2-independent proliferation. Moreover, TYS and TDG cells developed estrogen-independent tumors in ovariectomized mice; Myc expression was highly elevated in these tumors. Myc overexpression in tumors has been correlated with cancer stemness, which leads to reduced responsiveness to anticancer drugs and increased metastatic potential24, 25, 36. In breast cancer cells, overexpression of Myc and its downstream targets Cyclin E1 and Cyclin D1 results in decreased sensitivity to antiestrogens47, 48. An analysis of Myc in 399 patients with ERα positive breast cancer showed that higher levels of Myc expression were associated with shorter relapse free survival48. Notably, while these studies and our data demonstrate an important role for Myc in proliferation-related phenotypes and therapy resistance, Myc expression in T47D cells had no effect on metastasis-related invasion, dissociation and rebinding.
In addition to Myc upregulation, these cell lines exhibit alterations in protective pathways associated with resistance to cell death. The UPR was upregulated in E2-treated T47D and MCF7 cells and in ERαY537S and ERαD538G mutants. UPR upregulation is consistent with our recent work demonstrating E2-activation of the anticipatory UPR in ERα containing T47D and MCF7 breast cancer cells49, in PEO4 ovarian cancer cells50, and estrogen-independent UPR activation in TYS and TDG cells11. Since increased expression of a UPR gene index was tightly correlated with reduced time to tumor recurrence, tamoxifen resistance and reduced survival49, these pro-survival changes may contribute to the pathology of tumors expressing ERα mutations.
Supplementary Material
Highlights.
Cells bearing ERα Y537S and D538G mutations exhibit unique gene expression profiles.
ERα mutations promote increased invasiveness and drive breast cancer metastasis.
The Myc pathway is constitutively upregulated in cells expressing ERα mutations.
Myc overexpression has no effect on metastasis related tumor cell invasiveness.
Myc overexpression recapitulates ERα mutation endocrine therapy resistant cell proliferation.
ACKNOWLEDGEMENTS
We thank laboratory members for reading the manuscript.
Funding: This work was supported by NIH [RO1DK071909], DOD [BCRPW81XWH-13] and the E. Howe Scholar Award to DS, ODS, NCCIH and NCI [P50AT006268] to WH, BCRF/Pfizer [IIDRP-16–006] to BHP and a C.F. Kade fellowship to LY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST The authors declare no conflicts of interest.
REFERENCES
- 1.Clark GM, Osborne CK, McGuire WL (1984). Correlations between estrogen receptor, progesterone receptor, and patient characteristics in human breast cancer. J Clin Oncol 2: 1102–1109. [DOI] [PubMed] [Google Scholar]
- 2.Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM et al. (1998). Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst 90: 1371–1388. [DOI] [PubMed] [Google Scholar]
- 3.Howell A, Robertson JF, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR et al. (2002). Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol 20: 3396–3403. [DOI] [PubMed] [Google Scholar]
- 4.Dowsett M, Cuzick J, Ingle J, Coates A, Forbes J, Bliss J et al. (2009). Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J Clin Oncol 28: 509–518. [DOI] [PubMed] [Google Scholar]
- 5.Musgrove EA, Sutherland RL (2009). Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 9: 631–643. [DOI] [PubMed] [Google Scholar]
- 6.Alluri PG, Speers C, Chinnaiyan AM (2014). Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res 16: 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson DR, Wu Y-M, Vats P, Su F, Lonigro RJ, Cao X et al. (2013). Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 45: 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu G, Fuqua SA (2016). ESR1 mutations in breast cancer: proof-of-concept challenges clinical action. Clin Cancer Res 22: 1034–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM et al. (2014). Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor–positive breast cancer. Clin Cancer Res 20: 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sefrioui D, Perdrix A, Sarafan‐Vasseur N, Dolfus C, Dujon A, Picquenot JM et al. (2015). monitoring ESR1 mutations by circulating tumor DNA in aromatase inhibitor resistant metastatic breast cancer. Int J Cancer 137: 2513–2519. [DOI] [PubMed] [Google Scholar]
- 11.Mao C, Livezey M, Kim JE, Shapiro DJ (2016). Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor α Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Scientific Reports 6: 34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin L-A, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T et al. (2017). Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun 8: 1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrod A, Fulton J, Nguyen VT, Periyasamy M, Ramos-Garcia L, Lai C-F et al. (2017). Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 36: 2286–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L et al. (2017). Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res 19: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A et al. (2018). Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 33: 173–186. e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gelsomino L, Panza S, Giordano C, Barone I, Gu G, Spina E et al. (2018). Mutations in the estrogen receptor alpha hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett 428: 12–20. [DOI] [PubMed] [Google Scholar]
- 17.Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ et al. (2016). Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 5: e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D et al. (2016). Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA oncology 2: 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF et al. (2017). Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 36: 2286–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gates LA, Gu G, Chen Y, Rohira AD, Lei JT, Hamilton RA et al. (2018). Proteomic proiling identiies key coactivators utilized by mutant ERα proteins as potential new therapeutic targets [DOI] [PMC free article] [PubMed]
- 21.Gabay M, Li Y, Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 4: a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kress TR, Sabò A, Amati B (2015). MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15: 593–607. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Osdal T, Ho Y, Chun S, McDonald T, Agarwal P et al. (2014). SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute Myeloid Leukemia stem cells. Cell stem cell 15: 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X, Cai H, Liang Y, Chen L, Wang X, Si R et al. (2015). Inhibition of c-Myc by let-7b mimic reverses mutidrug resistance in gastric cancer cells. Oncol Rep 33: 1723–1730. [DOI] [PubMed] [Google Scholar]
- 25.Shajahan-Haq AN, Cook KL, Schwartz-Roberts JL, Eltayeb AE, Demas DM, Warri AM et al. (2014). MYC regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol Cancer 13: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ladanyi M, Park CK, Lewis R, Jhanwar SC, Healey JH, Huvos AG (1993). Sporadic amplification of the MYC gene in human osteosarcomas. Diagn Mol Pathol 2: 163–167. [PubMed] [Google Scholar]
- 27.Schneider‐Stock R, Boltze C, Jäger V, Epplen J, Landt O, Peters B et al. (2003). Elevated telomerase activity, c ‐MYC ‐, and hTERT mRNA expression: association with tumour progression in malignant lipomatous tumours. J Pathol 199: 517–525. [DOI] [PubMed] [Google Scholar]
- 28.Escot C, Theillet C, Lidereau R, Spyratos F, Champeme M-H, Gest J et al. (1986). Genetic alteration of the c-myc protooncogene (MYC) in human primary breast carcinomas. Proc Natl Acad Sci 83: 4834–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu L, Andruska N, Zheng X, Shapiro DJ (2016). Anticipatory Activation of the Unfolded Protein Response by Epidermal Growth Factor is Required for Immediate Early Gene Expression and Cell Proliferation. Mol Cell Endocrinol 422: 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krieg AJ, Krieg SA, Ahn BS, Shapiro DJ (2004). Interplay between estrogen response element sequence and ligands controls in vivo binding of estrogen receptor to regulated genes. J Biol Chem 279: 5025–5034. [DOI] [PubMed] [Google Scholar]
- 31.Pedraz-Cuesta E, Fredsted J, Jensen HH, Bornebusch A, Nejsum LN, Kragelund BB et al. (2016). Prolactin Signaling Stimulates Invasion via Na+/H+ Exchanger NHE1 in T47D Human Breast Cancer Cells. Mol Endocrinol 30: 693–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Platet N, Garcia M (1998). A new bioassay using transient transfection for invasion-related gene analysis. Invasion and Metastasis 18: 198–208. [DOI] [PubMed] [Google Scholar]
- 33.Kosicki M, Tomberg K, Bradley A (2018). Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol [DOI] [PMC free article] [PubMed]
- 34.Murthy MS, Scanlon EF, Jelachich ML, Klipstein S, Goldschmidt RA (1995). Growth and metastasis of human breast cancers in athymic nude mice. Clin Exp Metastasis 13: 3–15. [DOI] [PubMed] [Google Scholar]
- 35.Walsh MD, Luckie SM, Cummings MC, Antalis TM, McGuckin MA (1999). Heterogeneity of MUC1 expression by human breast carcinoma cell lines in vivo and in vitro. Breast cancer research and treatment 58: 253–264. [DOI] [PubMed] [Google Scholar]
- 36.Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN et al. (2018). Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 173: 338–354. e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146: 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA et al. (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci 108: 16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R et al. (2013). D538G mutation in estrogen receptor-α: a novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 73: 6856–6864. [DOI] [PubMed] [Google Scholar]
- 40.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M et al. (2013). ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 45: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S et al. (2015). Droplet digital polymerase chain reaction assay for screening of ESR1 mutations in 325 breast cancer specimens. Transl Res 166: 540–553. e542. [DOI] [PubMed] [Google Scholar]
- 42.Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ et al. (2016). Sensitive detection of mono-and polyclonal ESR1 mutations in primary tumors, metastatic lesions, and cell-free DNA of breast cancer patients. Clin Cancer Res 22: 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sears R, Ohtani K, Nevins JR (1997). Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol Cell Biol 17: 5227–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pérez-Roger I, Solomon DL, Sewing A, Land H (1997). Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27 Kip1 binding to newly formed complexes. Oncogene 14. [DOI] [PubMed] [Google Scholar]
- 45.Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F et al. (2001). Myc represses the p21 (WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci 98: 4510–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bretones G, Delgado MD, León J (2015). Myc and cell cycle control. Biochim Biophys Acta 1849: 506–516. [DOI] [PubMed] [Google Scholar]
- 47.Venditti M, Iwasiow B, Orr FW, Shiu RP (2002). C‐myc gene expression alone is sufficient to confer resistance to antiestrogen in human breast cancer cells. Int J Cancer 99: 35–42. [DOI] [PubMed] [Google Scholar]
- 48.Miller TW, Balko JM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M et al. (2011). A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin Cancer Res 17: 2024–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andruska N, Zheng X, Yang X, Helferich WG, Shapiro DJ (2015). Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor alpha-positive breast cancer. Oncogene 34: 3760–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng X, Andruska N, Lambrecht MJ, He S, Parissenti A, Hergenrother PJ et al. (2018). Targeting multidrug-resistant ovarian cancer through estrogen receptor α dependent ATP depletion caused by hyperactivation of the unfolded protein response. Oncotarget 9: 14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roderick JE, Tesell J, Shultz LD, Brehm MA, Greiner DL, Harris MH et al. (2014). c-Myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric T-ALL cells. Blood 123: 1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.