

ABSTRACT
SAGA and TFIID are related transcription complexes, which were proposed to alternatively deliver TBP at different promoter classes. Recent genome-wide studies in yeast revealed that both complexes are required for the transcription of a vast majority of genes by RNA polymerase II raising new questions about the role of coactivators.
KEYWORDS: RNA polymerase II, coactivator, SAGA complex, TFIID complex, nascent RNA
Introduction
RNA-polymerase II (Pol II)-mediated transcription is a highly regulated process that determines cellular function and cell identity through the accurate synthesis of mRNAs. Although its regulation occurs at all stages of transcription, regulation at the stage of initiation is a key mechanism to control gene expression. For initiation, the basal transcription machinery composed of Pol II and the general transcription factors (GTFs), nucleates pre-initiation complex (PIC) formation on gene promoters [1,2]. TFIID, composed of the TATA-box binding protein (TBP) and TBP-associated factors (TAFs) is the first GTF that binds promoter sequences. Once bound to the different promoter elements, GTFs enable correct positioning of Pol II relative to the transcription start site (TSS) and facilitate the transition to productive elongation. However, the compact structure of chromatin has been shown to act as a barrier for PIC formation. Thus, other transcription factors are required to specifically modulate the chromatin landscape at proximity of promoters for productive PIC assembly.
Coactivators are recruited to the vicinity of gene promoters through their interaction with gene-specific activators bound to mammalian enhancers or yeast Upstream Activating Sequences (UAS). Different activities facilitating transcription were found associated with coactivators, namely chromatin remodelers, histone modifiers or adaptors that link activators to the transcription machinery. While most coactivator complexes were initially thought to regulate specific subsets of genes, some were reported to have a more global role in transcription. One important example is the Mediator complex which was described as an integral part of the basal transcription machinery, required for nearly all Pol II mediated transcription [3–6]. In this point-of-view, we will summarize recent insights into the role of two coactivator complexes, TFIID and SAGA (Spt-Ada-Gcn5-acetyltransferase), in global Pol II transcription in S. cerevisiae. Furthermore, we will discuss potential global functions for other coactivators and whether similar mechanisms exist in metazoans.
Role of SAGA and TFIID in pol II transcription
An extensively characterized coactivator is the evolutionary conserved SAGA complex organized in distinct functional and structural modules (reviewed in [7]). SAGA activates transcription through histone modifying activities (acetylation and deubiquitination) and by recruiting TBP to promoters. Early genome-wide analyses of SAGA function in Pol II transcription in budding yeast by Pugh and colleagues, showed that upon deletion of the TBP-interacting subunit Spt3, the steady-state RNA levels of ~10% of genes were decreased by more than 2-fold [8]. Meanwhile, 90% of the genes were affected upon conditional depletion of the TFIID subunit Taf1, also involved in TBP-recruitment to promoters. Although this seminal study cautiously concluded that “TFIID and SAGA make overlapping contribution to the expression of all genes”, the proposed classification of genes as either SAGA-dominated or TFIID-dominated was oversimplified over time, categorizing each gene as dependent exclusively on one or the other coactivator. It was further shown that the SAGA-dominated genes were highly enriched in stress-regulated genes containing consensus TATA elements in their core promoters, while the TFIID-dominated genes tended to be more constitutively expressed and lack a strong consensus TATA [9]. In good agreement with these findings, early studies emphasized that SAGA is recruited to its target genes through the interaction of its Tra1 subunit with a set of activators predominantly stimulating stress-responsive genes, Gcn4 and Gal4 among others [10,11]. Together, these observations pointed towards a specific role for SAGA in the transcription of highly regulated genes. Importantly, these findings suggested that genes can be differentially regulated depending on their promoter sequence by utilizing specific sets of transcription factors and coactivators.
TFIID is a general transcription factor, composed of TBP and several TAFs. Only its TBP subunit but not TAFs is necessary and sufficient for PIC assembly and transcription in vitro. TAFs are targeted by several activators and potentiate their activities suggesting that TFIID has coactivator functions [12]. Unlike SAGA, TFIID directly contacts DNA and interacts with other components of the basal transcription machinery. TBP, as part of the TFIID complex, tends to bind promoters lacking a consensus TATA-box sequence, whereas TATA-containing promoters are bound by TBP but are relatively depleted of TAFs [13]. These observations further supported a predominant role for TFIID in the regulation of TATA-less genes, enriched with housekeeping functions.
Integration of binding profiles and transcriptional effects
The analysis of the genome-wide localization of SAGA by chromatin immunoprecipitation (ChIP) indicated that SAGA is recruited to a limited subset of genes, in agreement with its requirement for the expression of only ~10% of the yeast genome [14]. However, subsequent comparison of localization and expression studies showed a weak correlation between chromatin binding sites and transcriptional effects [15]. Similar findings were made for several other coactivators in S. cerevisiae. Indeed, transcriptome analyses of mutant strains for different chromatin modifiers including Set1, Set2 and Dot1, catalyzing H3K4, H3K36 and H3K79 methylation respectively, revealed limited effects of these enzymes on transcription. These results were surprising as the histone marks deposited by these factors are localized at nearly all active genes, suggesting a more global role for Pol II transcription [16]. Similar observations for SAGA showed that its histone acetylation and deubiquitination activities act on the entire transcribed genome [17]. Thus, the contrast between broad enzymatic activities and restricted transcriptional effects appears as a general feature of chromatin altering complexes.
As a very large number of different proteins are known to bind each promoter, a functional redundancy between these factors is likely to explain at least partially, the discrepancy between factor location and expression effect. However, it seems possible that for some factors, the observed differences could instead result from limitations of the methodologies used. Indeed, ChIP approaches are antibody dependent and may be insensitive to transient chromatin interactions which might be problematic for coactivators with low ChIP efficiency [18,19]. In addition, steady-state mRNA analyses might be inaccurate to measure Pol II activity. Indeed, several studies revealed that a global decrease in Pol II transcription is compensated by a simultaneous and global decrease in mRNA decay, thereby buffering steady-state mRNA levels [20–23]. The use of improved methodologies was highly warranted to re-examine the role of SAGA and TFIID in Pol II transcription, in light of recent observations showing that TFIID is equally recruited at promoters of both SAGA- and TFIID-dominated genes and that SAGA inactivation decreases Pol II recruitment at both classes of genes [17,18].
SAGA and TFIID are generally required for pol II transcription
Two recent studies aimed at providing a more detailed analysis of the genome-wide occupancy and the role in Pol II transcription of the SAGA and TFIID complexes [24,25]. They used chromatin endogenous cleavage coupled with high-throughput sequencing (ChEC-seq), a formaldehyde- and antibody-independent approach to determine the binding profiles of dynamic factors such as coactivators [19]. ChEC-seq was previously used to clarify the genome-wide binding profile of Mediator, revealing an association with the UASs at a majority of genes, whereas TFIID was recruited at core promoters, to which it binds cooperatively with Mediator [18]. Using ChEC-seq SAGA was exclusively detected at the UASs of both SAGA- and TFIID-dominated genes, in agreement with the idea that SAGA is recruited to UASs by sequence-specific DNA-binding transcription factors [24].
To quantify nascent transcription upon inactivation of SAGA or TFIID, these studies used native Pol II ChIP or metabolic labeling with 4-thiouracil (4tU) followed by quantification of the purified newly transcribed mRNAs. These analyses were done on SAGA deletion strains or using inducible depletion systems (auxin-inducible degradation or anchor away technology) and revealed that nearly all Pol II transcribed genes are dependent on TFIID, SAGA and Mediator. Importantly, TATA-containing and TATA-less genes were similarly affected upon inactivation of either of these three complexes.
These analyses of nascent transcription indicate that SAGA, TFIID and Mediator make important contributions to Pol II transcription. Each of these coactivators appears to be absolutely required for gene expression as an inducible depletion of Mediator subunits or TAFs caused a dramatic decrease in genome-wide Pol II recruitment by about 8-fold for Med14 or by about 3 to 4-fold for different TAFs [25]. Similarly, nascent mRNA transcription was reduced by about 10-fold in a double SAGA mutant strain (SPT3 and GCN5 deletions) [24]. Such large transcriptional effects indicate that the activities of these three coactivators on Pol II transcription are not functionally redundant but rather suggest that SAGA, TFIID and Mediator function at different rate-limiting steps. Earlier work indicated a reciprocal dependency in genome-wide recruitment of TFIID and Mediator and cooperativity between SAGA and Mediator has also been suggested [1,18]. The broad genome-wide recruitment of SAGA, Mediator and TFIID at most active genes support the idea that different coactivators work cooperatively to assemble the PIC. However, a more detailed description of coactivator interactions and the mechanisms of cooperativity remain to be elucidated.
Interestingly, the average Pol II occupancy was decreased to a comparable extent, by about 3- to 4-fold, upon depletion of four different TFIID subunits [25]. However, the depletion of these TAFs did not significantly alter the complex architecture, indicating that most TAFs are individually important for TFIID function. These observations strikingly contrast with the results for SAGA, in which suppression of different activities results in highly variable effects in Pol II transcription. The mRNA synthesis rates were unaffected by the loss of the deubiquitinase Ubp8, or the TBP-interacting protein Spt8, but were significantly decreased upon loss of Spt3 (2-fold change) or the histone acetyltransferase Gcn5 (1.5-fold change). Strikingly, global mRNA synthesis was decreased by about 10-fold in a SPT3 and GCN5 double deletion strain, suggesting that the functional modules of SAGA make different contributions but act in a synergistic manner on Pol II transcription.
The intriguing observation that some coactivators are recruited at most expressed genes and have a global contribution to Pol II transcription, raises multiple interesting considerations. Beyond TFIID and SAGA, are other coactivators globally required for Pol II transcription? If these coactivators each occupy the regulatory regions of most genes, which factors define the specific expression levels for each gene? How do coactivators contribute to gene expression changes in response to variations in transcription factor recruitment? How do SAGA and TFIID mediate transcription from TATA-containing and TATA-less promoters? Do these complexes have similar broad distributions and functions in metazoans?
Do other coactivators have a global role in pol II transcription?
As a global role in Pol II transcription is now proposed for three different coactivator complexes (Mediator, SAGA and TFIID), it is tempting to speculate that other coactivators might also have a broader function than anticipated. Particularly, histone-modifying complexes such as NuA4 or COMPASS/Set1C deposit marks (H4 acetylation and H3K4 trimethylation) that are enriched at most active promoters. These factors may have broader effects on transcription that were overlooked when analyzing steady-state mRNA. Chromatin remodeling complexes are also expected to have broad genome-wide activities. Indeed, the RSC complex was previously shown to act at a majority of yeast promoters to slide or evict nucleosomes thus positioning nucleosomes flanking the nucleosome depleted region (NDR) [26,27]. Similarly to our observations on SAGA, RSC was shown to be required for global transcription, although localization studies revealed a limited number of RSC binding sites [28,29]. The authors suggested that the interaction of RSC with many binding sites might be too transient to be readily detected by ChIP. A re-analysis using ChEC-seq may reveal more a widespread localization of RSC across the genome. Similarly, at the majority of yeast promoters, the first nucleosome found downstream of the NDR (+1 nucleosome) often contains the histone variant H2A.Z where it is deposited by the SWR complex [27]. Here again, the broad distribution of H2A.Z and of the SWR complex contrasted with limited gene expression changes detected by transcriptome studies in the corresponding mutant strains [14,30]. Although these differences might be explained by factor redundancy or by gene-specific features accounting for dependencies on certain factors only, it would be important to analyze the role of the above-mentioned factors through a direct characterization of Pol II activity.
Differential gene sensitivity to specific coactivators
Although SAGA, TFIID and Mediator can be considered as general cofactors for Pol II in yeast, it does not imply that each complex makes equal contributions to the expression of every individual gene. Although genome-wide Pol II occupancy or mRNA synthesis rates were consistently decreased upon depletion of these coactivators, a range of gene expression changes was observed in each mutant strain suggesting variable requirements of genes on certain coactivator complexes [24,25,31]. These variable dependencies on coactivators are indicative of gene-specific properties of varying importance that would determine the relative contribution of each coactivator complex to PIC formation. Gene-specific features are diverse in nature, corresponding to DNA sequence elements (e.g. presence and position of a consensus TATA-box, number and diversity of transcription factor binding sites) or specific chromatin architecture (e.g. nucleosome positioning and occupancy around the promoter, patterns of histone modifications or transcription factor occupancy) [32,33].
Genes are often divided in two categories corresponding to different strategies for transcriptional regulation (Figure 1). Housekeeping genes are constitutively expressed with little influence from external or internal signals and were proposed to be more dependent on TFIID. In contrast, highly regulated genes have a higher transcriptional plasticity and were suggested to be more dependent on SAGA. However, the above-described observations clearly demonstrate that these two classes are equally sensitive to the loss of TFIID or SAGA and thus cannot be distinguished by their dependency on these coactivators. Nevertheless, these two gene classes can be differentiated by gene specific features including promoter organization or chromatin architecture [27].
Figure 1.
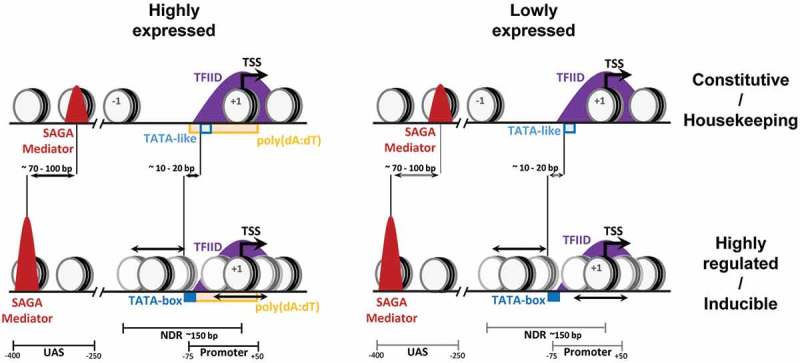
Different combinations of gene-specific features in constitutively active and stress responsive genes lead to the characterization of distinct mechanisms for transcription regulation of these two gene categories.
Promoters with a consensus TATA-box (TATAWAWR sequence), are more often found in highly regulated genes, whereas housekeeping genes are predominantly lacking a TATA-box in their promoters [9]. Further analyses identified TATA-like elements, having 1 or 2 mismatches from the consensus, at the sites of PIC assembly, in the majority of TATA-less promoters in yeast [13]. Interestingly, the localization of SAGA and Mediator analyzed by ChEC-seq revealed a higher occupancy of these complexes at the UASs of TATA-containing genes than at TATA-less genes [18,24]. In addition, the average location of both SAGA and Mediator was found more upstream (by 70–100 bp) relative to the TSS at TATA-containing than at TATA-less genes [24]. As these two gene categories are equally sensitive to the loss of SAGA or Mediator subunits, the PIC formation is likely differently regulated at promoters containing either a consensus TATA-box or a TATA-like element. These gene classes also differ by their respective distances between the TATA-element and the TSS, being 10–20 bp longer at TATA-containing than at TATA-less promoters [13]. In contrast, ChEC-seq signals for Taf1 were highly similar at both promoter categories suggesting that TFIID is similarly recruited at the NDR of both TATA-containing and TATA-less promoters [18].
It is not clear whether the presence of a consensus TATA-box or a TATA-like element is the only sequence element which determines the PIC architecture at these two gene classes. Along these lines, it was recently shown that TAFs interact with downstream promoter elements to facilitate transcription re-initiation [34]. Downstream binding events occurred specifically at TFIID-dominated genes although these genes did not have higher TAF occupancy. These observations suggest that the ability of a promoter to drive TAF-dependent re-initiation events might better define which genes are more sensitive to TAF mutation. Another recent study used an in vitro system to assess transcription from TATA-containing and TATA-less promoters and revealed that both promoter classes are TFIID-dependent, in agreement with in vivo observations described earlier [35]. On TATA-containing promoters, TBP could complement the loss of TFIID only in vitro, but not in vivo. These data together suggest that most PICs assembled in vivo contain the TFIID complex. This work also revealed other promoter sequence features in addition to TATA-elements that distinguish TATA-less from TATA-containing promoters. Several studies indicated that T-richness upstream and A-richness downstream of the TSS distinguish highly from lowly expressed genes [27,33,36,37]. Indeed, highly expressed genes were found to be more sensitive to SAGA mutations and displayed higher Mediator occupancy than lowly expressed genes [18,24]. Although it is unclear how the T- and A-richness would be mechanistically linked with the requirement for certain coactivators, A/T rich sequences are known to negatively influence nucleosome occupancy, thereby potentially reducing the requirement of chromatin regulators [32].
Housekeeping/TATA-less and highly regulated/TATA-containing genes were also associated with differential chromatin organization and sensitivity to chromatin regulators [27,32]. Constitutively expressed genes often display a broad NDR with well positioned flanking nucleosomes. At these promoters, transcription factor binding sites lie within the NDR which may explain their lower sensitivity to disruption of chromatin regulators. In contrast, promoters of highly regulated genes have higher nucleosome occupancy upstream of the TSS with less defined positioning. At these gene promoters, transcription factor binding sites are more distal relative to the TSS and are often occupied by nucleosomes. A putative competition between nucleosomes and transcription factors at these promoters may account for their higher sensitivity to chromatin regulation, in agreement with their higher histone turnover. Although SAGA, TFIID and Mediator similarly affect the expression of these two gene classes, different functions of these coactivators could be used depending on promoter elements and chromatin architecture characteristic of housekeeping genes or genes with high transcriptional plasticity.
Openings and perspectives
Improvements in genome-wide approaches to analyze gene expression and chromatin binding have conciliated conflicting data concerning the role of transcriptional coactivators. The arising findings challenge the established paradigm stating that coactivators act on specific gene subsets and start to shift it towards a more global role of many coactivators in Pol II transcription in S. cerevisiae [18,24,25,31]. These results suggest that the combinatorial activities of these factors are necessary for accurate transcription. However, to match the various transcriptional requirements of all genes, coactivators might act to different extents to facilitate the expression of each individual gene. Thus, the combination of activators binding to UASs, nucleosome occupancy, promoter architecture and sequence elements in core promoters like TATA elements or others, seem to participate in the fine-tuning of transcription by properly coordinating the activities of each coactivator on every gene according to its expression needs.
These discoveries raise new questions regarding the mechanisms of coactivator recruitment to all active genes. Either each coactivator can interact with a wide variety of activators or recruitment can be also mediated through activator-independent interactions. Interestingly, the loss of the Tra1 subunit which is expected to mediate SAGA interaction with activators, has limited phenotypic and transcriptional effects in S. pombe [38]. Thus, SAGA recruitment likely relies on other subunits that can interact with either DNA-bound transcription factors or directly to chromatin. Indeed, many coactivators have been shown to contain a variety of protein domains that recognize histone marks found at most active promoters [39]. For example, SAGA contains a Tudor domain and bromodomains reported to interact with methylated and acetylated histones which could stabilize the binding of SAGA to active gene promoters [40].
The increased complexity of gene expression programs and regulation in mammalian cells is linked with an expansion of activators and coactivators repertoire and further diversification of gene promoter and enhancer features. Most coactivators were highly conserved through evolution, with similar complexes found in yeast and metazoans. Nevertheless, several duplication events led to the expansion of the coactivator repertoire, potentially due to the increased complexity of multicellular organisms. For example, the HAT module of the SAGA complex in yeast is incorporated in both the SAGA and ATAC coactivator complexes in metazoans [7]. Similarly, the yeast COMPASS complex diverged into at least seven different complexes in mammalian cells [41]. Thus, the presence of related activities in different complexes with redundant functions, complicates the analysis of coactivator requirements for Pol II transcription in mammalian cells. However, a recent study using quantification of newly synthesized mRNAs, proposed that the bromodomain-containing protein BRD4 acts as a general coactivator for Pol II transcription, demonstrating that this approach is also feasible in mammalian cells [42]. However, deciphering the genome-wide action of each coactivator will likely require sophisticated experimental set-ups and approaches.
Funding Statement
This work was supported by the Agence Nationale de la Recherche [15-CE11-0022]; European Research Council [2013-340551].
Acknowledgments
We thank Farrah El Saafin for critically reading the manuscript. V.F. was recipient of the IDEX-University of Strasbourg international PhD programme. Work in the authors laboratory was supported by Agence Nationale de la Recherche (ANR-15-CE11-0022 SAGA2) and the European Research Council (ERC) Advanced grant (ERC-2013-340551, Birtoaction). This study was also supported by ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Hahn S, Young ET.. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics. 2011;189:705–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sainsbury S, Bernecky C, Cramer P. Structural basis of transcription initiation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16:129–143. [DOI] [PubMed] [Google Scholar]
- [3].Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Takagi Y, Kornberg RD. Mediator as a general transcription factor. J Biol Chem. 2006;281:80–89. [DOI] [PubMed] [Google Scholar]
- [5].Holstege FC, Jennings EG, Wyrick JJ, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. [DOI] [PubMed] [Google Scholar]
- [6].Thompson CM, Young RA. General requirement for RNA polymerase II holoenzymes in vivo. Proc Natl Acad Sci USA. 1995;92:4587–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Helmlinger D, Tora L. Sharing the SAGA. Trends Biochem Sci. 2017;42:850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Huisinga KL, Pugh BF. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell. 2004;13:573–585. [DOI] [PubMed] [Google Scholar]
- [9].Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. [DOI] [PubMed] [Google Scholar]
- [10].Brown CE, Howe L, Sousa K, et al. Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science. 2001;292:2333–2337. [DOI] [PubMed] [Google Scholar]
- [11].Bryant GO, Ptashne M. Independent recruitment in vivo by Gal4 of two complexes required for transcription. Mol Cell. 2003;11:1301–1309. [DOI] [PubMed] [Google Scholar]
- [12].Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. [DOI] [PubMed] [Google Scholar]
- [13].Rhee H, Pugh FB. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Venters BJ, Wachi S, Mavrich TN, et al. A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces. Mol Cell. 2011;41:480–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lenstra TL, Holstege FC. The discrepancy between chromatin factor location and effect. Nucleus. 2012;3:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pokholok DK, Harbison CT, Levine S, et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. [DOI] [PubMed] [Google Scholar]
- [17].Bonnet J, Wang CY, Baptista T, et al. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 2014;28:1999–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grunberg S, Henikoff S, Hahn S, et al. Mediator binding to UASs is broadly uncoupled from transcription and cooperative with TFIID recruitment to promoters. EMBO J. 2016;35:2435–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zentner GE, Kasinathan S, Xin B, et al. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun. 2015;6:8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Munchel SE, Shultzaberger RK, Takizawa N, et al. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell. 2011;22:2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Rodriguez-Molina JB, Tseng SC, Simonett SP, et al. Engineered covalent inactivation of TFIIH-kinase reveals an elongation checkpoint and results in widespread mRNA stabilization. Mol Cell. 2016;63:433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sun M, Schwalb B, Pirkl N, et al. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol Cell. 2013;52:52–62. [DOI] [PubMed] [Google Scholar]
- [23].Sun M, Schwalb B, Schulz D, et al. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 2012;22:1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Baptista T, Grünberg S, Minoungou N, et al. SAGA is a general cofactor for RNA polymerase II transcription. Molecular Cell. 2017;68:130–14300000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Warfield L, Ramachandran S, Baptista T, et al. Transcription of nearly all Yeast RNA polymerase II-transcribed genes is dependent on transcription factor TFIID. Mol Cell. 2017;68:118–129.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hartley PD, Madhani HD. Mechanisms that specify promoter nucleosome location and identity. Cell. 2009;137:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rando OJ, Winston F. Chromatin and transcription in Yeast. Genetics. 2012;190:351–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Parnell TJ, Huff JT, Cairns BR. RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. EMBO J. 2008;27:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ng HH, Robert F, Young RA, et al. Genome-wide location and regulated recruitment of the RSC nucleosome-remodeling complex. Genes Dev. 2002;16:806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lenstra TL, Benschop JJ, Kim T, et al. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell. 2011;42:536–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Plaschka C, Larivière L, Wenzeck L, et al. Architecture of the RNA polymerase II-mediator core initiation complex. Nature. 2015;518:376–380. [DOI] [PubMed] [Google Scholar]
- [32].Levo M, Segal E. In pursuit of design principles of regulatory sequences. Nat Rev Genet. 2014;15:453–468. [DOI] [PubMed] [Google Scholar]
- [33].Lubliner S, Keren L, Segal E. Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res. 2013;41:5569–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Joo Y, Ficarro SB, Soares LM, et al. Downstream promoter interactions of TFIID TAFs facilitate transcription reinitiation. Genes Dev. 2017;31:2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Donczew R, Hahn S. Mechanistic differences in transcription initiation at TATA-less and TATA-containing promoters. Mol Cell Biol. 2017;38:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lubliner S, Regev I, Lotan-Pompan M, et al. Core promoter sequence in yeast is a major determinant of expression level. Genome Res. 2015;25:1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Maicas E, Friesen JD. A sequence pattern that occurs at the transcription initiation region of yeast RNA polymerase II promoters. Nucleic Acids Res. 1990;18:3387–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Helmlinger D, Marguerat S, Villén J, et al. Tra1 has specific regulatory roles, rather than global functions, within the SAGA co-activator complex. EMBO J. 2011;30:2843–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hassan AH, Prochasson P, Neely KE, et al. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. [DOI] [PubMed] [Google Scholar]
- [40].Musselman CA, Lalonde ME, Cote J, et al. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Muhar M, Ebert A, Neumann T, et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science. 2018;360:800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]