

ABSTRACT
Macroautophagy/autophagy plays an important role in the immune response to invasion by intracellular pathogens such as group A Streptococcus (GAS; Streptococcus pyogenes). We previously identified RAB30, a Golgi-resident GTPase, as a novel anti-bacterial autophagic regulator in the formation of GAS-containing autophagosome-like vacuoles (GcAVs); however, the precise mechanism underlying this process remains elusive. Here, we elucidate a novel property of RAB30: the ability to recruit PI4KB (phosphatidylinositol 4-kinase beta) to the Golgi apparatus and GcAVs. We found that trans-Golgi network (TGN) vesicles were incorporated into GcAVs via RAB30 to promote GcAV formation. Moreover, depletion of phosphatidylinositol-4-phosphate (PtdIns4P), a phosphatidylinositol enriched in the TGN, by wortmannin and phenylarsine oxide, followed by subsequent repletion with exogenous PtdIns4P revealed that PtdIns4P is crucial for GcAV formation. Furthermore, we identify an interaction between RAB30 and PI4KB, in which the knockdown of RAB30 decreased the localization of PI4KB to the TGN and GcAVs. Finally, PI4KB knockout suppressed autophagy by inhibiting GcAV formation, resulting in the increased survival of GAS. Our results demonstrate a novel autophagosomal formation mechanism involving coordinative functions of RAB30 and PI4KB distinct from those utilized in canonical autophagy.
Abbreviations: GAS: group A Streptococcus; GcAVs: GAS-containing autophagosome-like vacuoles; PI4KB: phosphatidylinositol 4-kinase beta; PtdIns: phosphatidylinositol; PtdIns3P: phosphatidylinositol-3-phosphate; PtdIns4P: phosphatidylinositol-4-phosphate; PtdIns5P: phosphatidylinositol-5-phosphate; SLO: streptolysin O; TGN: trans-Golgi network; TGOLN2: trans-golgi network protein 2; PH: plekstrin homology; OSBP: oxysterol binding protein
KEYWORDS: GcAV, group A Streptococcus, PI4KB, trans-Golgi network, xenophagy
Introduction
Streptococcus pyogenes (Group A Streptococcus; GAS) is one of the most prevalent pathogens worldwide. Its ecological niche is restricted to the human throat and skin, and it is responsible for a variety of diseases, ranging from superficial infections (e.g., pharyngitis and impetigo) to systemic infections (e.g., necrotizing fasciitis and sepsis) [1]. GAS invades human epithelial cells via endocytosis and escapes into the cytoplasm by inserting streptolysin O (SLO), a cholesterol-dependent pore-forming cytolysin, into the endosomal membrane [2].
Xenophagy, a specialized form of autophagy, is the primitive defense strategy of non-phagocytic cells for halting the invasion of intracellular pathogens [3,4]. Of note, the clinically prevalent GAS strains, including the M1T1 strain, have evolved strategies to combat xenophagy [5]. The expression of SpeB, a streptococcal cysteine protease, was found to be crucial by degrading the essential xenophagic components SQSTM1/p62, CALCOCO2/NDP52, and NBR1 within the host cell cytosol [5]. Therefore the non-SpeB expressing M6 GAS JRS4 strain has been well used as a model to probe the host xenophagy response [2]. Cytosolic M6 GAS is targeted to transient double-membrane phagophores that mature into GAS-containing autophagosome-like vacuoles (GcAVs), which subsequently fuse with lysosomes, leading to the efficient degradation of GAS.
Investigating the mechanism underlying the formation of GcAVs is a compelling area of research. In contrast to the canonical autophagy that degrades self-constituents, xenophagy should be differentiated from other types of autophagy in that it is directed against foreign invaders; thus GcAVs can become 10 times larger than canonical autophagosomes to ensure the efficient eradication of bacteria [6]. This suggests that GcAVs may require additional pathways to recruit membrane sources distinct from those of canonical autophagy. In previous research, we focused on the RAB proteins, small GTPases that serve as master regulators of membrane trafficking [7], and their association with GcAV formation. RAB23 and RAB9A are crucial for GcAV formation and expansion [8], whereas RAB17 plays a role in recruiting recycling endosomes as a source of membranes for GcAVs [9]. These RAB proteins are dispensable for canonical autophagy, suggesting the presence of additional and alternative pathways for the formation of GcAVs. Thus, further research is needed to uncover the unique mechanisms of xenophagy, which have so far remained unclear.
Recently, we performed comprehensive subcellular localization analyses and revealed that RAB30 acts as a unique regulator in GcAV formation [10]. RAB30 is recruited to GcAVs in response to the induction of GAS-induced autophagy and is required for GcAV formation and the subsequent degradation of GAS [10]. However, the precise mechanism by which RAB30 contributes to the formation of autophagosomes remains unknown. RAB30 is a trans-Golgi network (TGN)-resident regulator of intracellular trafficking during Drosophila morphogenesis and is suggested to be involved in exocytosis from the Golgi and/or retrograde transport to the Golgi [11]. Therefore, in this study, we first examined changes in the localization of RAB30 and TGN during GAS infection. To our knowledge, our results establish for the first time that TGN-derived elements are recruited to GcAVs in a process dependent on GTP-active RAB30. The synthesis of phosphatidylinositol-4-phosphate (PtdIns4P), a TGN membrane-bound signaling molecule, is crucial in this pathway of GcAV formation. Furthermore, we identified a novel autophagic machinery in which RAB30 recruits PI4KB (phosphatidylinositol 4-kinase beta), the enzyme that generates PtdIns4P from phosphatidylinositol (PtdIns), resulting in the subsequent formation of GcAVs and efficient eradication of GAS.
Results
RAB30 regulates the incorporation of TGN-derived elements into GcAVs
RAB30 resides in the TGN and has been suggested to regulate post-Golgi trafficking during Drosophila morphogenesis [11]. Therefore, we first examined the localization of EmGFP-fused RAB30 protein and the TGN marker TGOLN2 during M6 GAS (strain JRS4) infection. In non-infected cells, RAB30 colocalized with TGOLN2, which was consistent with previous studies [12]. TGOLN2 was also seen to colocalize with RAB30 in GAS-infected cells (Figure 1(a,b)). Because RAB30 was shown to localize to GcAVs during GAS infection [10], we also examined the colocalization of MAP1LC3/LC3 and TGOLN2 during GAS infection. The results revealed that TGN-derived vesicles were incorporated into GcAVs (Figure 1(c)). We confirmed this result by examining the localization of another TGN-resident protein, PLEKHA3, which was also observed to surround GAS (Figure 1(d)). These results showed that TGN-derived vesicles are incorporated into GcAVs along with RAB30.
Figure 1.
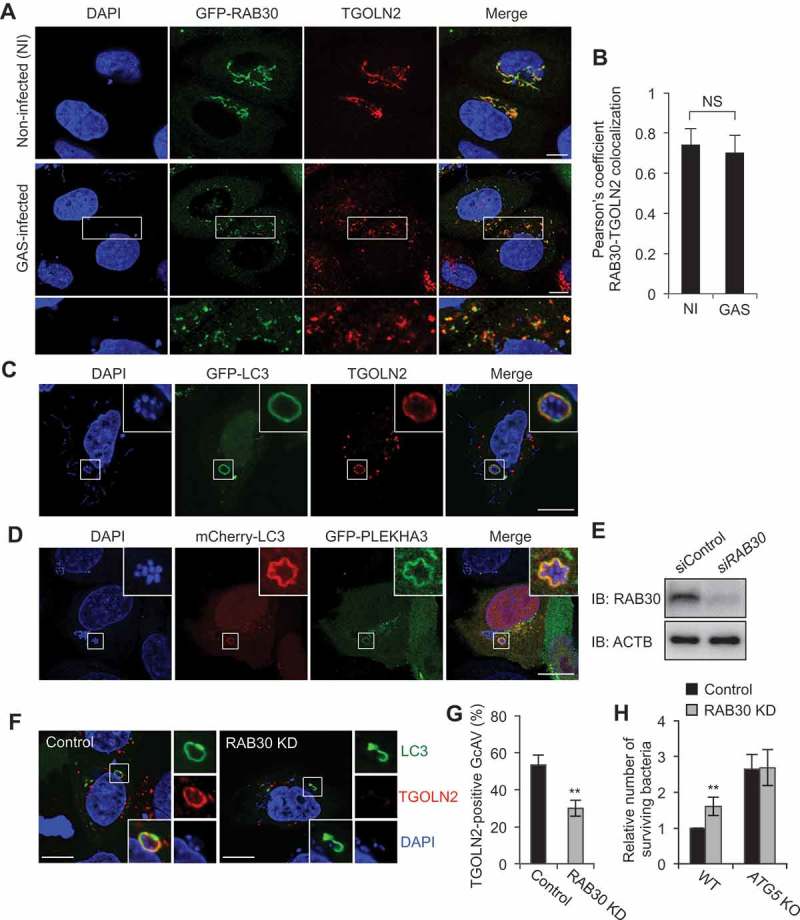
RAB30 is required for the recruitment of TGN-derived elements into GcAVs. (a) HeLa cells expressing EmGFP-RAB30 were infected with GAS for 3 h, fixed and immunostained with an anti-TGOLN2 antibody. (b) Pearson’s coefficient quantified for RAB30-TGOLN2 colocalization in noninfected (NI) or GAS-infected cells. Data represent means ± SD of 3 independent experiments (> 20 cells analyzed per condition). NS, not significant. (c) HeLa cells expressing EmGFP-LC3 were infected with GAS JRS4 for 4 h and fixed. After permeabilization, cells were stained using an anti-TGOLN2 antibody, and cellular and bacterial DNA was stained with DAPI. Images were captured using confocal microscopy. (d) HeLa cells expressing EmGFP-PLEKHA3, a resident of the TGN, were infected with GAS for 4 h. (e) HeLa cells transfected with a control siRNA or RAB30 siRNA were analyzed by immunoblotting. (f) HeLa cells were transfected with a control siRNA or RAB30 siRNA, as well as EmGFP-LC3 expression vector and then infected with GAS for 4 h. Cells were then fixed, permeabilized, and stained using an anti-TGOLN2 antibody. Bars: 10 μm. (g) Quantification of TGOLN2-positive GcAVs in control and RAB30 knockdown (KD) cells. Data represent the mean value ± SD from 3 independent experiments (> 50 GcAVs analyzed per condition). (h) HeLa WT or ATG5 knockout (KO) cells were transfected with a control siRNA or RAB30 siRNA. Two days after transfection, cells were infected with GAS for 2 and 6 h. The number of invading and surviving bacteria was measured in the GAS viability assay. *P < 0.05, **P < 0.01.
We next examined the effect of siRNA-mediated RAB30 knockdown on the recruitment of TGN-derived vesicles to GcAVs. The percentage of TGOLN2-positive GcAVs significantly decreased following RAB30 knockdown (Figure 1(e–g)), suggesting that the recruitment of TGN vesicles to the GcAV membrane is RAB30 dependent. We also compared the localization of wild-type (WT), constitutively active (QL; GTP-bound form), and dominant-negative (TN; GDP-bound form) RAB30 and quantified the colocalization of these RAB30 proteins with the TGN. The colocalization of RAB30 TN with TGOLN2-positive structures was reduced compared with that of either RAB30 WT or QL, indicating that the GTP-bound form of RAB30 preferentially colocalizes with TGOLN2 (Figure S1(a,b)). Because RAB30 also localizes to GcAVs in the GTP-bound form [10], this demonstrated that RAB30 colocalizes with both TGOLN2-positive structures and GcAVs in the GTP-bound form in GAS-infected cells.
Finally, by evaluating the number of surviving bacteria in WT and ATG5 knockout HeLa cells over time, we determined that the reduction in GcAVs observed in RAB30-knockdown cells results in decreased autophagic killing of intracellular GAS. Invading bacteria that survived were counted in colony-formation assays (bacteria viability assay). Consistent with data from our previous studies [13], GAS survival was enhanced by knockout of ATG5, indicating that ATG5-dependent autophagy degrades intracellular GAS in HeLa cells. An increase in the number of surviving GAS was observed following the knockdown of RAB30 in WT cells but not in ATG5-knockout cells (Figure 1(h)), suggesting that RAB30 is involved in the ATG5-dependent autophagic degradation of GAS. Collectively, these findings suggest that RAB30 regulates the recruitment of TGN-derived elements to GcAVs and is involved in GcAV formation required for autophagy-mediated restriction of the growth of GAS.
PtdIns4P localizes to GcAVs
As mentioned above, PLEKHA3 localized to GcAVs. PLEKHA3 contains a plekstrin homology (PH) domain that binds to PtdIns4P [14,15], a PtdIns found abundantly on the TGN membrane that plays key roles in controlling intracellular membrane trafficking [16,17]. To further determine the role of TGN vesicle recruitment to GcAVs, we next analyzed the involvement of PtdIns4P in GcAV formation.
First, we investigated the subcellular localization of PtdIns4P during GAS infection by using the PH domain of OSBP (oxysterol binding protein) as a probe for detecting PtdIns4P [14]. In non-infected cells, EmGFP-tagged OSBP-PH localized to the nucleus, plasma membrane, and TGN (Figure 2(a)). In GAS-infected cells, EmGFP-OSBP-PH additionally colocalized with GcAVs (Figure 2(b)). To further validate the localization of PtdIns4P on GcAVs, the localization of endogenous PtdIns4P was determined by immunostaining; we found that PtdIns4P showed Golgi-like localization in non-infected cells and clearly colocalized with GcAVs in GAS-infected cells (Figure 2(c)). Collectively, these results indicate that PtdIns4P localizes to GcAVs during GAS infection.
Figure 2.

GcAVs contain PtdIns4P. (a) HeLa cells expressing EmGFP-OSBP-PH were infected with GAS for 4 h and immunostained with anti-TGOLN2 antibody. (b) HeLa cells expressing EmGFP-OSBP-PH and mCherry-LC3 were infected with GAS for 4 h. (c) HeLa cells expressing mCherry-LC3 were infected with GAS for 4 h and immunostained with anti-PtdIns4P antibody. Bars: 10 μm. NI, noninfected.
Phosphorylation of phosphatidylinositol to PtdIns4P is essential for GcAV formation
PtdIns4P is synthesized at the TGN through the phosphorylation of PtdIns by PI4K (phosphatidylinositol 4-kinase). Four distinct PI4Ks have been identified in mammalian cells and categorized into types II and III according to their sensitivities to wortmannin [18]. To determine whether PtdIns4P is necessary for GcAV formation, we first treated cells with inhibitors of type II and III PI4Ks (Figure 3(a)). Wortmannin specifically inhibits phosphatidylinositol 3-kinase at low concentrations (< 200 nM) and also inhibits type III PI4Ks at high concentrations (> 5 μM) [18]. PtdIns3P is essential for starvation-induced autophagy, and we confirmed that starvation-induced autophagosome formation was inhibited following treatment with a low concentration of wortmannin (< 200 nM) (Figure S2(a,b)). Phenylarsine oxide (PAO) is an inhibitor of all types of PI4Ks (Figure 3(a)). Intriguingly, a low dose of wortmannin (100 nM or 10 μM) did not affect the efficiency of GcAV formation, but at high concentrations, wortmannin decreased GcAVs in a dose-dependent manner (Figure 3(b,c)). In contrast, PAO suppressed GcAV formation even at low concentrations (Figure 3(b,c)). To further confirm the effects of these PI4K inhibitors on GcAV formation, we examined the accumulation of LC3-II, the lipidated phagophore- and autophagosome-anchored form of LC3. As shown in Figure 3(d), addition of wortmannin or PAO suppressed LC3-II accumulation.
Figure 3.

PtdIns4P is important for autophagy during GAS infection. (a) Schematic representation of pathways for PtdIns3P and PtdIns4P synthesis and drugs targeting enzymes involved in these pathways. (b) HeLa cells expressing EmGFP-LC3 were treated with wortmannin (Wm) or PAO, and infected with GAS for 4 h. Bars: 10 μm. (c) HeLa cells expressing EmGFP-LC3 were treated with Wm or PAO at the indicated concentrations, and infected with GAS for 4 h. The percentages of GcAV-positive cells were quantified. (d) HeLa cells were treated with Wm (5 μM) or PAO (200 nM), and infected with GAS. Cells were analyzed by immunoblotting. (e) HeLa cells expressing EmGFP-LC3 were incubated with Wm (5 μM) and loaded with exogenous PtdIns4P, and infected with GAS for 4 h. The percentages of GcAV-positive cells were quantified. (f) HeLa cells treated with Wm or PAO at the indicated concentrations were infected with GAS for 2 and 6 h. The number of invading and surviving bacteria was measured in the GAS viability assay. Data in (c), (e), and (f) are the mean ± SD of 3 independent experiments. hpi, hours post infection.
Next, we assessed GcAV formation after the addition of exogenous PtdIns4P to wortmannin-treated cells [19] in order to determine whether wortmannin-induced inhibition of GcAV is a direct result of PtdIns4P depletion. The recovery of GcAV formation in wortmannin-treated cells was clearly observed following the addition of PtdIns4P in a dose-dependent manner (Figure 3(e)). These results establish conclusively that PtdIns4P directly promotes the formation of GcAVs through its conversion from PtdIns by PI4K.
Finally, we confirmed the effects of wortmannin and PAO on the killing of intracellular GAS via autophagy. These inhibitors did not directly affect the growth of GAS at the concentrations used (data not shown). The survival rate of GAS at 6 h after infection increased in a concentration-dependent manner in both wortmannin- and PAO-treated cells (Figure 3(f)), suggesting that the impairment of GcAV formation induced by these inhibitors allowed for the intracellular proliferation of GAS.
PI4KB is involved in GcAV formation
Among the 4 PI4Ks, PI4K2A, PI4K2B, and PI4KB, but not PI4KA, are thought to localize to the TGN [18], consistent with our results showing that PI4K2A, PI4K2B, and PI4KB, but not PI4KA, colocalize with GcAVs (Figure 4(a)). Therefore, we investigated which of these PI4Ks are required for PtdIns4P-dependent GcAV formation. Silencing of PI4K2A and PI4K2B slightly increased GcAVs (the number of cells containing GcAVs), whereas PI4KB knockdown resulted in a decrease of GcAV formation (Figure 4(b,c)). These results suggest that PI4KB is involved in GcAV formation.
Figure 4.
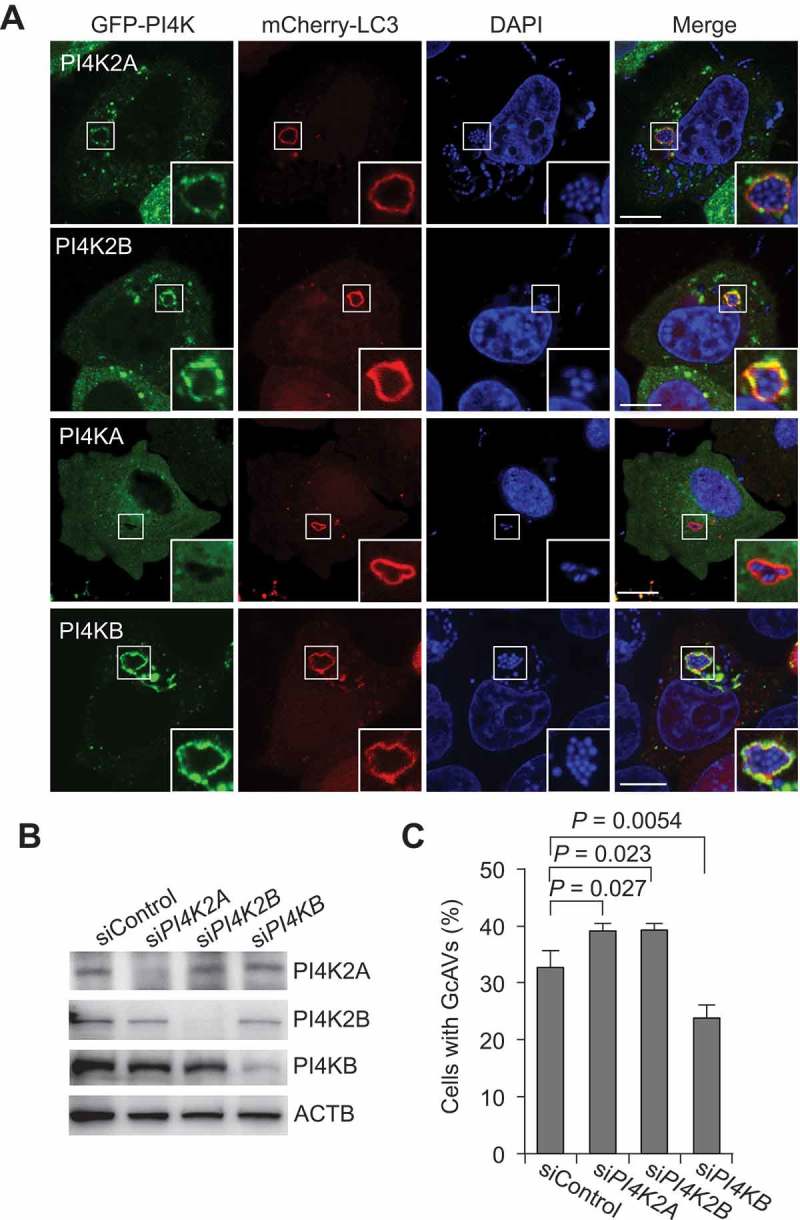
PI4KB is involved in GcAV formation. (a) HeLa cells expressing mCherry-LC3 and EmGFP-PI4Ks (PI4K2A, PI4K2B, PI4KA, or PI4KB) were infected with GAS for 4 h. Cells were fixed and stained with DAPI. Bars: 10 μm. (b) Immunoblot analysis of PI4K knockdown in HeLa cells. (c) HeLa cells transfected with mCherry-LC3 and the indicated siRNA were infected with GAS for 4 h. The percentages of cells with GcAVs were quantified. Data are the mean ± SD of 3 independent experiments (> 200 cells were analyzed per condition).
PI4KB interacts with RAB30 in a GTP-dependent manner
Next, we examined the colocalization of RAB30 with PI4K2A, PI4K2B, and PI4KB. Interestingly, while all examined PI4Ks showed a Golgi-like localization and colocalization with RAB30, PI4KB exhibited the highest colocalization with RAB30 (Figure 5(a,b)). PI4K2A and PI4K2B are heavily palmitoylated and thus localized to the membrane [20], whereas type III PI4KB is a soluble cytosolic protein that is recruited to appropriate membranes indirectly through protein-protein interactions [21]. We then investigated the interaction between PI4KB and RAB30 by conducting immunoprecipitation experiments in which FLAG-tagged RAB30 was incubated with HeLa lysate. PI4KB was shown to interact with RAB30 (Figure 5(c)). Furthermore, to examine whether the interaction between RAB30 and PI4KB was GTP-dependent, we performed affinity isolation experiments with FLAG-RAB30 WT, FLAG-RAB30 TN, and FLAG-RAB30 QL. The binding to PI4KB was strongest with RAB30 QL, followed by RAB30 WT and RAB30 TN, indicating that interactions between RAB30 and PI4KB were GTP-dependent (Figure 5(d)).
Figure 5.
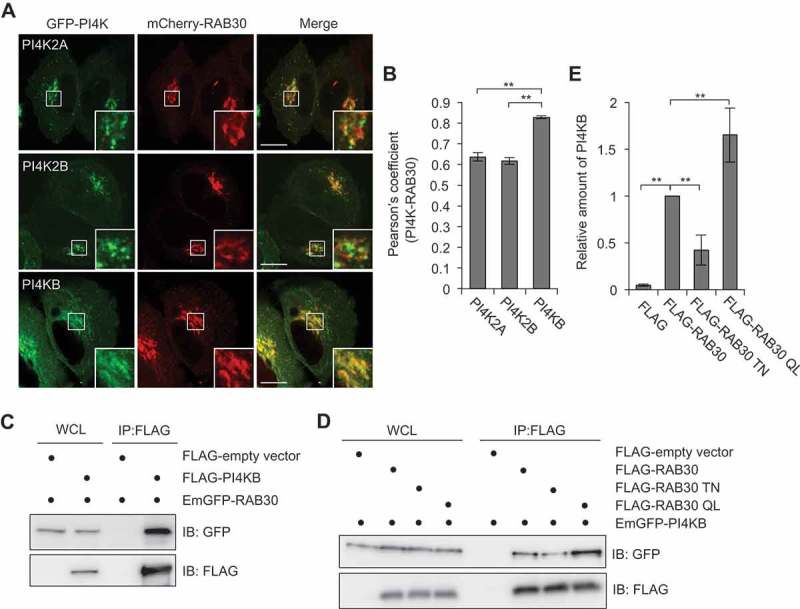
PI4KB colocalizes and interacts with RAB30. (a and b) HeLa cells expressing mCherry-RAB30 and EmGFP-PI4K (PI4K2A, PI4K2B, or PI4KB). Representative confocal images (a) and Pearson’s coefficient quantified for RAB30-PI4K colocalization (b). Bars: 10 μm. (c) HEK293T cells transfected with plasmids encoding EmGFP-RAB30 and FLAG-empty vector or FLAG-PI4KB were subjected to immunoprecipitations (IP) with an anti-FLAG antibody. The immunoprecipitated proteins and whole cell lysates (WCL) were analyzed by immunoblotting with anti-FLAG and anti-GFP antibodies. (d) HEK293T cells transfected with EmGFP-PI4KB and FLAG-empty vector, FLAG-RAB30, FLAG-RAB30 TN, or FLAG-RAB30 QL were subjected to immunoprecipitations with an anti-FLAG antibody. The immunoprecipitated proteins and whole cell lysates were analyzed by immunoblotting with anti-FLAG and anti-GFP antibodies. (e) The graph indicates a quantification of the amount of PI4KB in immunoprecipitation samples in (d). Data in (b) and (e) are the mean ± SD of 3 independent experiments.
RAB30 is necessary for the recruitment of PI4KB to TGN and GcAVs
Given the presence of a PI4KB-RAB30 complex, we hypothesized that TGN-resident RAB30 recruits PI4KB to the Golgi apparatus. As expected, we found that knockdown of RAB30 decreased the proportion of TGOLN2 signals colocalizing with EmGFP-PI4KB (Figure 6(a,b)), implying that RAB30 is necessary for PI4KB recruitment to the TGN. We also examined the colocalization of PI4K2A, PI4K2B, and PI4KB with GcAVs during GAS infection. The results revealed that RAB30-knockdown cells exhibit a reduced proportion of PI4KB-positive GcAVs without affecting the PI4K2A and PI4K2B recruitment to GcAVs (Figure 6(c,d)). These results suggest that RAB30 mediates the recruitment of PI4KB to the TGN in non-infected cells and to GcAVs in GAS-infected cells.
Figure 6.

RAB30 is involved in the recruitment of PI4KB to TGN and GcAV. (a and b) HeLa cells were transfected with EmGFP-PI4KB and a control siRNA or RAB30 siRNA. After 2 d transfection, cells were fixed and immunostained using anti-TGOLN2 antibody. Representative confocal images (a) and proportion of TGOLN2-signals colocalized with EmGFP-PI4KB from at least 30 randomly selected fields were quantified using Mander’s coefficient M1 in control and RAB30-knockdown cells (b). (c and d) HeLa cells were transfected with mCherry-LC3, EmGFP-PI4KB and a control siRNA or RAB30 siRNA. After 2 d transfection, cells were infected with GAS for 4 h. Representative confocal images (c) and the percentages of PI4K-positive GcAV. Data in (b) and (d) are the mean ± SD of 3 independent experiments. Bars: 10 μm.
Knockout of PI4KB impairs GcAV formation and GAS degradation
Finally, to test whether PI4KB is essential for antibacterial autophagy, we prepared PI4KB-knockout cells using the CRISPR-Cas9 system (Figure 7(a)) and infected them with GAS. Knockout of PI4KB did not affect GAS invasion (Figure 7(b)) but significantly decreased the number of GcAV-positive cells (Figure 7(c,d)). Moreover, supplementation of PtdIns4P to PI4KB-knockout cells recovered GcAV formation in a dose-dependent manner (Figure 7(e)), suggesting that PI4KB-mediated PtdIns4P production was involved in GcAV formation. Quantification of GAS survival also revealed a significant elevation in the GAS survival rate in PI4KB-knockout cells compared to WT cells (Figure 7(f)). Taken together, these findings suggested that PI4KB is crucial for the formation of GcAVs and the subsequent degradation of GAS (Figure 8).
Figure 7.

PI4KB is required for autophagy against GAS. (a) Immunoblot analysis of PI4KB knockout (KO) in HeLa cells. (b) HeLa WT or PI4KB KO cells were infected with GAS for 1 and 2 h. The number of invading bacteria was measured in the GAS viability assay. (c and d) HeLa wild-type (WT) and PI4KB-knockout (KO) cells were transfected with mCherry-LC3 and infected with GAS for 4 h. Representative confocal images (c) and quantification of GcAV-positive cells (d). Data are the mean ± SD of 3 independent experiments (> 200 cells were analyzed per condition). Bars: 10 μm. (e) HeLa WT or PI4KB KO cells were infected with GAS for 2 and 6 h. The number of surviving bacteria was measured in the GAS viability assay. Data in (b), (c), and (d) are the mean ± SD of 3 independent experiments.
Figure 8.

Proposed model of RAB30-mediated PI4KB regulation and GcAV formation.
Discussion
In this study, we attempted to elucidate the role of RAB30 in the formation of GcAVs. We observed that during GAS invasion, the TGN-resident RAB30 localized to GcAVs along with TGN-derived vesicles, and this process was GTP dependent. Furthermore, RAB30 depletion decreased the number of TGOLN2-positive GcAVs and increased the number of viable bacteria in cells. Together, these results demonstrate that RAB30 promotes xenophagy by incorporating TGN-derived vesicles into GcAVs. To date, many intracellular organelles, such as the endoplasmic reticulum, post-Golgi membrane, mitochondria, and plasma membrane, have been proposed as potential membrane sources for autophagosomes in mammalian cells [22–25]. Interestingly, in our study, GcAVs contained TGOLN2 and TGN-resident proteins such as PLEKHA3; these results differ from those of previous studies in which mammalian autophagosomes budded off from the TGN at the early stage of autophagy without containing any TGN-derived vesicles [26]. Moreover, in our previous study, RAB30 localized to starvation-induced autophagosomes but was not required for the formation of these autophagosomes [10]. This discrepancy suggests the presence of an alternative pathway through RAB30 to recruit the considerable membrane sources necessary for GcAV formation [6].
We also found that PtdIns4P localizes to GcAVs and that the synthesis of PtdIns4P from PtdIns is essential for GcAV formation (Figure 8). PtdIns is a key molecule involved in membrane dynamics, and PtdIns3P in particular plays a critical role in autophagosome biogenesis [27]. PtdIns5P has also been proposed to regulate autophagic initiation through PtdIns3P effectors in a PtdIns3P-independent pathway during starvation [28]. Interestingly, low concentrations of wortmannin (< 200 nM) did not affect the efficiency of GcAV formation, indicating that PtdIns3P is dispensable for GcAV formation. PtdIns4P is synthesized from PtdIns by either type II or type III PI4Ks. Although, PtdIns4P synthesis is crucial in the regulation of multiple cellular functions [29], relatively little attention has been paid to the role of PtdIns4P in the initiation of autophagy. Pik1, the yeast ortholog of PI4KB, is involved in both nonselective and selective types of autophagy [30–32]. In contrast, PtdIns4P synthesis from PI4K2A is essential for autophagosome-lysosome fusion, and thus its deficiency results in abnormally large autophagosomes [30–32]. However, no study has thus far provided insight into how PI4KB is recruited to the Golgi apparatus during the induction of autophagy.
In the present study, both wortmannin and PAO treatment inhibited GcAV formation in a dose-dependent manner. Moreover, we found that PI4KB-mediated PtdIns production is required for GcAV formation through RAB30; we therefore proposed a new model in which RAB30 recruits PI4KB to the TGN, and the subsequent phosphorylation of PtdIns to PtdIns4P is necessary for GcAV formation and efficient eradication of GAS (Figure 7). Interestingly, knockdown of PI4K2A and PI4K2B increased GcAVs. Considering the role of PI4K2A in autophagosome-lysosome fusion during starvation-induced autophagy, PI4K2A and PI4K2B might be involved in GcAV-lysosome fusion. Also, these results suggest that GcAV formation is finely controlled by local PtdIns4P levels via different PI4Ks. Of note, knockdown of RAB30 only led to a 40% reduction in the colocalization of PI4KB with TGOLN2 signals. This may be because the small GTPase ARF (ADP ribosylation factor) also recruits PI4KB to the TGN [33,34]. However, our previous study revealed that siRNA-mediated knockdown of ARF had no effect on the formation of GcAVs [10]. Hence, it is tempting to speculate that both RAB30 and ARF have the ability to recruit PI4KB to the TGN, but only the RAB30 pathway is crucial in the context of bacterial infection.
The interdependent regulation of specific RABs and PtdIns exists as a central mechanism to ensure the precision of not only membrane trafficking but also membrane tethering and fusion [35]. RAB5 recruits phosphoinositide 3-kinase to the Golgi apparatus for the maturation of early endosomes [36]. In contrast, PI4KB recruits RAB11 and its effectors to the PtdIns4P-enriched membrane, thus mediating membrane trafficking [37,38]. Also, in yeast, Ypt31/32 (yeast homologs of RAB11) and Sec4 (yeast homolog of RAB8A) have been shown to collaborate with PtdIns4P for the myosin V-dependent transport of secretory compartments [39]. To our knowledge, this is the first report that establishes a coordinative relationship between RAB proteins and PtdIns in the context of autophagy. RAB30 is required for the morphological integrity of the Golgi complex but, notably, not for anterograde or retrograde transport through the Golgi complex [12]. Also, a proteomic screen of Drosophila Rabs found that Rab30 interacts with a diverse set of Golgi proteins including Golgins and the GARP complex [40], but its precise mechanism still remains unclear. Further investigation into the coordination of RAB30 and PI4K may provide us with not only the mechanism of GcAV formation, but also insight into how the selective autophagic machinery utilizes different intracellular components for the degradation of the various intracellular components.
In summary, we demonstrated that TGN elements are recruited to GcAVs in response to GAS invasion. The findings reveal unique organelle dynamics in which RAB30 contributes to the formation of GcAVs. The results also shed light on the importance of the coordinative interaction between RAB30 and PI4KB in terms of the xenophagic machinery. Further investigation may be necessary to reveal how bacterial invasion stimulates the interaction between these 2 proteins.
Materials and methods
Cell culture and transfection
HeLa and HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (Nacalai Tesque, 18459–64) supplemented with 10% fetal bovine serum (Gibco, 26140079) and 50 μg/mL gentamicin (Nacalai Tesque, 11980–14) in a 5% CO2 incubator at 37 °C. Transfections were performed with polyethylenimine (Polyscience, 23966–2) or Lipofectamine 3000 (Invitrogen Corporation, L3000001).
Bacterial strain
The GAS strain JRS4 (M6+ F1+) was grown in Todd-Hewitt broth (BD Diagnostic Systems, 249240) supplemented with 0.2% yeast extract (Nacalai Tesque, 15838–45).
Antibodies and other reagents
The following primary antibodies were used: sheep anti-TGOLN2 (Novus Biological, NB110-40767), mouse anti-PtdIns4P (Echelon Biosciences, Z-P004), mouse anti-FLAG M2 (Sigma-Aldrich, A2220), mouse anti-GFP (GF200; Nacalai Tesque, 04363–24), rabbit anti-ACTB (13E5; Cell Signaling Technology, 4970), and rabbit anti-LC3B (D11; Cell Signaling Technology, 3868). The secondary antibodies anti-sheep IgG conjugated with Alexa Fluor 594 (A11016) and anti-mouse or anti-rabbit IgG conjugated with Alexa Fluor 488, 594, and 647 were purchased from Invitrogen Corporation (A11001, A11008, A11005, A11012, A21235, and A21244). Wortmannin was purchased from Tocris Bioscience (1232). Rapamycin was purchased from Nacalai Tesque (30037–94). PAO was purchased from Santa Cruz Biotechnology (sc-3521).
Addition of exogenous PtdIns4P to cells
Unlabeled PtdIns4P di-C6 and carrier (Echelon, P-4016 and P-9C3) were reconstituted in H2O:tert-BuOH (9:1) solution. After sonication in a bath for 1 min, carrier and lipids were combined at a 1:1 ratio for 10 min at room temperature. The mixture of lipids and carrier was diluted in media and incubated with cells for 2 h. The final concentrations used were 1–10 mM. For the negative control, DMEM was combined with carrier only and added to the cells.
Plasmids
Human PLEKHA3, OSBP, PI4K2A, PI4K2B, PI4KA, and PI4KB cDNAs were amplified by PCR from KYSE or HeLa total mRNA and cloned into pcDNA-6.2/N-EmGFP-DEST (Invitrogen Corporation, V35620) using Gateway cloning technology as described previously [8]. pcDNA-6.2/C-3xFLAG- DEST (C-terminal tagged), pcDNA-6.2/N-3xFLAG-DEST (N- terminal tagged), pcDNA-6.2/N-mCherry-DEST (N-terminal tagged) vectors were made by replacement from pcDNA-6.2- N-EmGFP to the 3xFLAG corresponding oligonucleotide or mCherry gene fragment (kindly provided from Dr. Tsien) for compatible with the Gateway system, respectively. The following Invitrogen Stealth siRNA duplexes were utilized: HSS124455 (PI4K2A), HSS124333 (PI4K2B), HSS108028 (PI4KB), and 12935200 (control).
Bacterial infection
Bacterial infection was performed as described previously [2]. Briefly, bacteria were added to cell cultures at a multiplicity of infection of 100 without antibiotics for 1 h. Infected cells were washed with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4), and antibiotic (100 μg/mL gentamicin) was added to the medium to kill extracellular bacteria.
Fluorescence microscopy
Harvested cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 20 min, and then permeabilized with 0.1% Triton X-100 (Nacalai Tesque, 35501–15) in PBS for 10 min. After washing with PBS, the cells were incubated in skim milk blocking buffer (5% skim milk, 2.5% goat serum [Sigma-Aldrich, G9023], 2.5% donkey serum [Millipore, S30], 0.1% gelatin [BD Diagnostic Systems, 214340] in PBS) or bovine serum albumin (BSA) blocking solution (2% BSA [Sigma-Aldrich, A4503], 0.02% sodium azide in PBS) for 1 h. For immunostaining, cells were incubated with primary antibodies diluted 1:100 in blocking solution at room temperature for 1 h, washed with PBS, and then probed with secondary antibody. DAPI (Nacalai Tesque, 11034–56) was diluted in blocking buffer and used to stain bacterial and cellular DNA. Fluorescence images were acquired with a confocal FV1000 laser-scanning microscope (Olympus).
Immunoprecipitation
Cells were harvested, washed with PBS, and lysed for 30 min on ice in lysis buffer containing 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1% Triton X-100, and proteinase inhibitor cocktail (Nacalai Tesque, 25955–11). Lysates were then centrifuged, and supernatants were pre-cleared for 30 min at 4°C with protein A Sepharose 4B (GE Healthcare Life Sciences, 17-1279-03). After a brief centrifugation (600 × g, 1 min), supernatants were incubated with anti-FLAG antibody for 2 h at 4°C. Protein A Sepharose 4B were then added and mixed for another 1 h. Immunoprecipitates were collected by brief centrifugation, washed 5 times with lysis buffer, and analyzed by immunoblotting.
Generation of knockout lines using CRISPR-Cas9 gene editing
In order to construct a PI4KB knockout cell line, the CRISPR-Cas9 system was used as described previously [10]. CRISPR guide RNAs (gRNAs) were designed to target an exon common to all splicing variants of the gene of interest (target sequence: TACAAGATTCTTGTGATTT). Sequencing of targeted genomic regions of knockout lines was also conducted to confirm the presence of frame-shifting indels in the genes of interest.
Statistical analysis
Quantification of colocalization and GcAV formation was performed by direct visualization with confocal microscopy. Unless otherwise indicated, at least 50 GcAVs or 200 GAS-infected cells were counted per condition for each experiment. For all graphs, at least 3 independent experiments were performed. Unless otherwise indicated, the mean ± standard deviation (SD) is shown, and P-values were calculated using two-tailed Student’s t-tests.
Funding Statement
This research was supported in part by Grants-in-Aid for Scientific Research (16H05188, 15K15130, 26462776, 17K19552), the Takeda Science Foundation (T.N.), Yakult Bio-Science Foundation, Joint Research Project of the Institute of Medical Science, the University of Tokyo (I.N.), the Research Program on Emerging and Re-emerging Infectious Diseases (18fk0108044h0202, 18fk0108073h0001) and J-PRIDE (18fm0208030h0002) from Japan Agency for Medical Research and development, AMED (I.N.).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
References
- [1].Cole JN, Barnett TC, Nizet V, et al. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–736. [DOI] [PubMed] [Google Scholar]
- [2].Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A streptococcus. Science. 2004;306:1037–1040. [DOI] [PubMed] [Google Scholar]
- [3].Levine B, Mizushima N, Virgin HW.. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kimmey JM, Stallings CL. Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol Med. 2016;22:1060–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barnett TC, Liebl D, Seymour LM, et al. The globally disseminated M1T1 clone of group A streptococcus evades autophagy for intracellular replication. Cell Host Microbe. 2013;14:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yamaguchi H, Nakagawa I, Yamamoto A, et al. An initial step of GAS-containing autophagosome-like vacuoles formation requires Rab7. PLoS Pathog. 2009;5:e1000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pfeffer SR. Rab GTPases: master regulators that establish the secretory and endocytic pathways. Mol Biol Cell. 2017;28:712–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nozawa T, Aikawa C, Goda A, et al. The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during group A streptococcus infection. Cell Microbiol. 2012;14:1149–1165. [DOI] [PubMed] [Google Scholar]
- [9].Haobam B, Nozawa T, Minowa-Nozawa A, et al. Rab17-mediated recycling endosomes contribute to autophagosome formation in response to group A streptococcus invasion. Cell Microbiol. 2014;16:1806–1821. [DOI] [PubMed] [Google Scholar]
- [10].Oda S, Nozawa T, Nozawa-Minowa A, et al. Golgi-resident GTPase Rab30 promotes the biogenesis of pathogen-containing autophagosomes. PLoS One. 2016;11:e0147061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Thomas C, Rousset R, Noselli S. JNK signalling influences intracellular trafficking during Drosophila morphogenesis through regulation of the novel target gene Rab30. Dev Biol. 2009;331:250–260. [DOI] [PubMed] [Google Scholar]
- [12].Kelly EE, Giordano F, Horgan CP, et al. Rab30 is required for the morphological integrity of the Golgi apparatus. Biol Cell. 2012;104:84–101. [DOI] [PubMed] [Google Scholar]
- [13].Minowa-Nozawa A, Nozawa T, Okamoto-Furuta K, et al. Rab35 GTPase recruits NDP52 to autophagy targets. Embo J. 2017;36:3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dowler S, Currie RA, Campbell DG, et al. Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem J. 2000;351:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kutateladze TG. Translation of the phosphoinositide code by PI effectors. Nat Chem Biol. 2010;6:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].De Matteis M, Godi A, Corda D. Phosphoinositides and the golgi complex. Curr Opin Cell Biol. 2002;14:434–447. [DOI] [PubMed] [Google Scholar]
- [17].Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. [DOI] [PubMed] [Google Scholar]
- [18].Balla A, Balla T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 2006;16:351–361. [DOI] [PubMed] [Google Scholar]
- [19].Wang J, Sun HQ, Macia E, et al. PI4P promotes the recruitment of the GGA adaptor proteins to the trans-Golgi network and regulates their recognition of the ubiquitin sorting signal. Mol Biol Cell. 2007;18:2646–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baumlova A, Chalupska D, Rozycki B, et al. The crystal structure of the phosphatidylinositol 4-kinase IIalpha. EMBO Rep. 2014;15:1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Klima M, Toth DJ, Hexnerova R, et al. Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Sci Rep. 2016;6:23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yamamoto A, Masaki R, Tashiro Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry. J Histochem Cytochem. 1990;38:573–580. [DOI] [PubMed] [Google Scholar]
- [24].Hailey DW, Rambold AS, Satpute-Krishnan P, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bejarano E, Yuste A, Patel B, et al. Connexins modulate autophagosome biogenesis. Nat Cell Biol. 2014;16:401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guo Y, Chang C, Huang R, et al. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J Cell Sci. 2012;125:1706–1715. [DOI] [PubMed] [Google Scholar]
- [27].Nascimbeni AC, Codogno P, Morel E. Phosphatidylinositol-3-phosphate in the regulation of autophagy membrane dynamics. Febs j. 2017;284:1267–1278. [DOI] [PubMed] [Google Scholar]
- [28].Vicinanza M, Korolchuk VI, Ashkenazi A, et al. PI(5)P regulates autophagosome biogenesis. Mol Cell. 2015;57:219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].D’Angelo G, Vicinanza M, Di Campli A, et al. The multiple roles of PtdIns(4)P – not just the precursor of PtdIns(4,5)P2. J Cell Sci. 2008;121:1955–1963. [DOI] [PubMed] [Google Scholar]
- [30].Albanesi J, Wang H, Sun HQ, et al. GABARAP-mediated targeting of PI4K2A/PI4KIIα to autophagosomes regulates PtdIns4P-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:2127–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yamashita S, Oku M, Wasada Y, et al. PI4P-signaling pathway for the synthesis of a nascent membrane structure in selective autophagy. J Cell Biol. 2006;173:709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang K, Yang Z, Liu X, et al. Phosphatidylinositol 4-kinases are required for autophagic membrane trafficking. J Biol Chem. 2012;287:37964–37972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Godi A, Pertile P, Meyers R, et al. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol. 1999;1:280–287. [DOI] [PubMed] [Google Scholar]
- [34].Shin HW, Nakayama K. Dual control of membrane targeting by PtdIns(4)P and ARF. Trends Biochem Sci. 2004;29:513–515. [DOI] [PubMed] [Google Scholar]
- [35].Jean S, Kiger AA. Coordination between RAB GTPase and phosphoinositide regulation and functions. Nat Rev Mol Cell Biol. 2012;13:463–470. [DOI] [PubMed] [Google Scholar]
- [36].Christoforidis S, Miaczynska M, Ashman K, et al. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol. 1999;1:249–252. [DOI] [PubMed] [Google Scholar]
- [37].Burke JE, Inglis AJ, Perisic O, et al. Structures of PI4KIIIbeta complexes show simultaneous recruitment of Rab11 and its effectors. Science. 2014;344:1035–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].de Graaf P, Zwart WT, van Dijken RA, et al. Phosphatidylinositol 4-kinasebeta is critical for functional association of rab11 with the Golgi complex. Mol Biol Cell. 2004;15:2038–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Santiago-Tirado FH, Legesse-Miller A, Schott D, et al. PI4P and Rab inputs collaborate in myosin-V-dependent transport of secretory compartments in yeast. Dev Cell. 2011;20:47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gillingham AK, Sinka R, Torres IL, et al. Toward a comprehensive map of the effectors of rab GTPases. Dev Cell. 2014;31:358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.