

ABSTRACT
Macroautophagy/autophagy is a conserved catabolic process that maintains cellular homeostasis under basal growth and stress conditions. In cancer, autophagy can either prevent or promote tumor growth, at early or advanced stages, respectively. We screened public databases to identify autophagy-related somatic mutations in cancer, using a computational approach to identify cancer mutational target sites, employing exact statistics. The top significant hit was a missense mutation (Y113C) in the MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) protein, which occurred at a significant frequency in cancer, and was detected in early stages in primary tumors of patients with known tumor lineage. The mutation reduced the formation of GFP-LC3B puncta and attenuated LC3B lipidation during Torin1-induced autophagy. Its effect on the direct physical interaction of LC3B with each of the 4 proteins that control its maturation or lipidation was tested by applying a protein-fragment complementation assay and co-immunoprecipitation experiments. Interactions with ATG4A and ATG4B proteases were reduced, yet without perturbing the cleavage of mutant LC3B. Most importantly, the mutation significantly reduced the interaction with the E1-like enzyme ATG7, but not the direct interaction with the E2-like enzyme ATG3, suggesting a selective perturbation in the binding of LC3B to some of its partner proteins. Structure analysis and molecular dynamics simulations of LC3B protein and its mutant suggest that the mutation changes the conformation of a loop that has several contact sites with ATG4B and the ATG7 homodimer. We suggest that this loss-of-function mutation, which attenuates autophagy, may promote early stages of cancer development.
KEYWORDS: ATG4B, ATG7, autophagy, LC3B, Protein-Fragment Complementation Assay, cancer associated somatic mutation
Introduction
Autophagy is a catabolic pathway, highly conserved in all eukaryotic cells. By this process, cytoplasmic components are recycled by double-membrane vesicles that form in the cytoplasm, engulf cytoplasmic components and organelles, and deliver them to the lysosome for degradation [1]. Autophagy serves as a survival mechanism in response to stress conditions, such as nutrient starvation or hypoxia, restoring homeostasis by recycling amino acids for energy production and synthesis of new proteins [2]. Due to its important activity in cells, autophagy is involved in the etiology of many human diseases, including cancer, where it plays a dual role [3]. At early stages of tumor development, autophagy acts as tumor suppressor by removing damaged proteins and organelles. Attenuation of autophagy at this stage leads to accumulation of defective mitochondria, promoting the production of reactive oxygen species (ROS) and DNA damage, which leads to tumorigenesis [4,5]. Thus, loss-of-function mutations in autophagy genes may promote early stages of cancer development. Conversely, in advanced tumor stages, in the absence of extensive angiogenesis, the inner tumor cells are under hypoxic stress and nutrient starvation conditions [6]. Therefore, activation of autophagy at this stage is essential for the survival of tumor cells. Moreover, even under fed conditions, established tumors such as those driven by the Ras oncogene, become addicted to autophagy to maintain their mitochondrial metabolic function [7]. Thus, unlike the early stages, inhibition of autophagy at late stages of cancer may be therapeutically exploited [8].
This interesting dualistic relationship between autophagy and cancer prompted us to look for hotspot somatic cancer mutations in autophagy genes that could cause either gain- or loss-of-function of the corresponding proteins. Due to the typical large number of tumor mutations, it is very hard to distinguish those that drive cancer from those that occur due to the breakdown of cellular gene-repair and maintenance mechanisms. However, mutations that have an adaptive value for tumors are expected to be repeatedly fixed in the clonal population of different tumors, showing a significant recurrence. Thus, given a large enough dataset of somatic variations and proper statistics, we can identify gene positions and segments that show higher than expected mutation densities relative to the overall mutations distribution across the gene. To address this issue, we developed a bioinformatics approach to detect gene sites with significant recurrent somatic mutations in cancer samples. Using this approach we identified 20 point-mutations in 12 out of 33 examined autophagy genes in the COSMIC database of somatic mutations in human cancer (cancer.sanger.ac.uk) [9]. The top significant hit, found in liver and melanoma tumors was the tyrosine to cysteine at position 113 (Y113C) missense mutation within the MAP1LC3B gene. The mutation reduced the formation of GFP-LC3B puncta and attenuated LC3B lipidation. Functional interaction analysis with 4 proteins that regulate LC3B, combined with 3D-structural modeling and molecular dynamics simulations, suggested that this single amino acid substitution reduces autophagy by decreasing LC3B’s binding to the E1-like ATG7 protein. The identification of this loss-of-function mutation in the primary tumors of patients with known tumor lineage is consistent with a tumor suppressive role of autophagy at these stages.
Results
Computational analysis identifies the Y113C mutation in LC3B as a candidate target site in cancer samples
To identify cancer mutational target sites in autophagy genes, we undertook a computational approach, looking for significant recurring somatic mutational events during tumorigenesis. All possible currently reported mutations in 33 autophagy genes (Table S1) from the whole genome screen (WGS) section of the ‘Catalogue Of Somatic Mutations In Cancer (COSMIC)’ database were analyzed. The COSMIC database includes entries from primary tumor sites, together with their metastases. Notably, in some cases we have found that some entries taken from the same donor appear under different individual identifiers, leading to redundant event counting. To overcome this problem, several steps were taken, as detailed in Materials and Methods, to ensure that the scored mutations were derived from independent events. To compute all possible nucleotide substitution outcomes in a gene, we first calculated the expected probabilities for each mutation type (e.g. non-synonymous) per gene position or gene interval [10]. Adapting the Poisson distribution, we then calculated the probabilities for the observed number of independent mutations per interval, in the context of the overall number of observed mutations in each gene. Finally, we manually inspected the sources of significant recurring mutation events in a gene position, verifying their independence.
In this manner, we identified 20 positions in 12 autophagy genes with significant (P ≤ 0.01) recurring mutation events (Table 1). The most significant recurrence was in coding position 338 of the MAP1LC3B gene (P = 1.18x10−7) (Figure 1(a)). An A to G mutation, leading to a Y113C substitution in the LC3B protein, was identified in 5 different individuals, 3 with liver carcinoma and 2 with melanoma. From the 2 melanoma patients, several samples had been collected over time and further sequenced [11], including the primary tumor and different late metastatic lesions. Importantly, the LC3BY113C mutation was present in the primary tumors of both patients and in all, or most, of the tested metastatic samples (4/4 and 2/3 in each patient), suggesting that the mutation occurred at early stages of tumor development and was further maintained in the metastases. The Y113 residue is highly conserved in the LC3 protein family, including the closely related members of the GABARAP family of mammalian orthologs of yeast Atg8 (formerly including GABARAPL2/GATE-16 separately, as a family) [12] (Figure 1(b)). It is highly conserved in evolution from yeast to mammals (Figure 1(c)). Notably, we did not identify any report of germline mutations in this position in the available public databases [13].
Table 1.
Autophagy gene positions with significant accumulation of non-synonymous mutations.
| Gene Name | Number-mutations | CDS length | Mutation CDS | Mutation AA | Mutation genome position | FATHMM prediction | P-value | P(BY) |
|---|---|---|---|---|---|---|---|---|
| MAP1LC3B | 5 | 378 | c.338A > G | p.Y113C | 16:87436663–87436663 | PATHOGENIC | 4.78E-11 | 1.18E-07 |
| RB1CC1 | 5 | 4785 | c.2312C > T | p.A771V | 8:53570077–53570077 | PATHOGENIC | 3.99E-11 | 1.73E-06 |
| 4 | c.4349A > G | p.H1450R | 8:53548618–53548618 | PATHOGENIC | 4.30E-09 | 9.31E-05 | ||
| 3 | c.2096C > T | p.T699M | 8:53570293–53570293 | PATHOGENIC | 3.86E-07 | 5.57E-03 | ||
| ATG16L1 | 4 | 1824 | c.1745C > T | p.A582V | 2:234202917–234202917 | PATHOGENIC | 5.87E-09 | 4.33E-05 |
| UVRAG | 4 | 2100 | c.443G > A | p.R148Q | 11:75599883–75599883 | PATHOGENIC | 9.11E-09 | 1.57E-04 |
| ATG5 | 3 | 828 | c.697G > A | p.E233K | 6:106634546–106634546 | NA | 2.19E-07 | 1.33E-03 |
| ULK2 | 3 | 3111 | c.2105C > T | p.P702L | 17:19689396–19689396 | PATHOGENIC | 1.66E-07 | 2.23E-03 |
| 3 | c.2461C > T | p.R821W | 17:19685380–19685380 | PATHOGENIC | 1.66E-07 | 2.23E-03 | ||
| ULK1 | 3 | 3153 | c.373G > A | p.A125T | 12:132393245–132393245 | PATHOGENIC | 6.24E-07 | 4.25E-03 |
| 3 | c.950C > T | p.S317F | 12:132396488–132396488 | PATHOGENIC | 6.24E-07 | 4.25E-03 | ||
| RUBCN | 3 | 2919 | c.1624C > T | p.R542W | 3:197421306–197421306 | PATHOGENIC | 5.25E-07 | 4.37E-03 |
| ATG7 | 3 | 2112 | c.1630C > T | p.P544S | 3:11402205–11402205 | PATHOGENIC | 5.42E-07 | 4.72E-03 |
| 3 | c.811C > T | p.R271C | 3:11374489–11374489 | PATHOGENIC | 5.42E-07 | 4.72E-03 | ||
| DAPK1 | 3 | 4293 | c.1573G > A | p.D525N | 9:90264980–90264980 | PATHOGENIC | 1.06E-06 | 5.08E-03 |
| 3 | c.173G > A | p.R58H | 9:90219979–90219979 | PATHOGENIC | 1.06E-06 | 5.08E-03 | ||
| AMBRA1 | 3 | 3627 | c.1985G > A | p.R662H | 11:46529825–46529825 | PATHOGENIC | 9.45E-07 | 5.57E-03 |
| 3 | c.2504C > T | p.S835F | 11:46456446–46456446 | PATHOGENIC | 9.45E-07 | 1.00E-02 | ||
| 3 | c.3381G > T | p.E1127D | 11:46419246–46419246 | NEUTRAL | 9.45E-07 | 1.00E-02 | ||
| WDFY3 | 3 | 10,581 | c.7778G > C | p.R2593T | 4:85638146–85638146 | PATHOGENIC | 5.08E-07 | 1.00E-02 |
Figure 1.
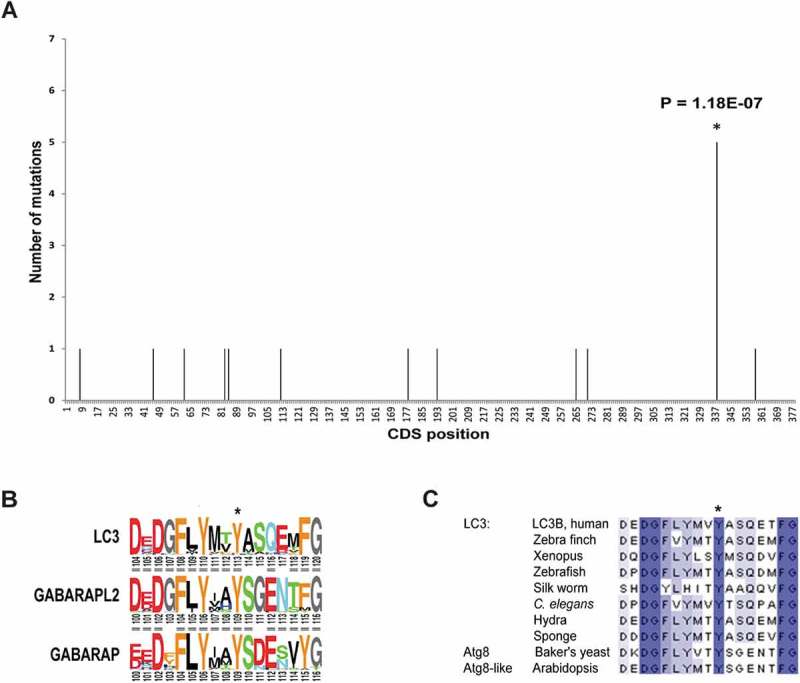
LC3B Tyr113 is highly conserved among eukaryotes and Y113C mutation is significantly present in cancer. (a) Histogram of the mutations on MAP1LC3B gene found in cancer tumors, taken from the COSMIC database. (b) Conservation of the C-terminal end of LC3B family with its homologs GABARAP and GABARAPL2. Tyr113 is marked with *. (c) Multiple sequence alignment of the C’-terminal end of LC3B with its orthologs. Color gradient represents level of conservation.
LC3BY113C mutation reduces its lipidation and GFP-LC3B puncta formation
To assess whether the mutation has any functional effects on autophagy, a phenotypic GFP-LC3B puncta assay was conducted (Figure 2(a,b)). To this end, HeLa cells were transiently transfected with GFP-LC3 plasmids expressing LC3B WT, LC3BY113C or LC3BG120A, and autophagy levels were measured under basal growth conditions or upon 2-h treatment with the MTOR (mammalian target of rapamycin kinase) inhibitor Torin1. GFP-LC3BG120A was used as a reference for a known loss-of-function mutation, as it cannot undergo cleavage by the ATG4 proteases and the subsequent steps leading to PE conjugation [14]. Cells transfected with GFP-LC3BY113C plasmid following Torin1 treatment showed a significantly lower number of puncta, compared to those transfected with GFP-LC3B WT (52% reduction). The effect of the Y113C mutation on the ability to produce puncta was nearly as strong as the G120A mutation, and both mutants exhibited attenuated responses to Torin1 treatment relative to GFP-LC3B WT. Western blotting indicated equal expression of the GFP-LC3 variants, excluding the trivial possibility that the differences in autophagy levels were due to differences in protein levels (Figure 2(c)). In a second assay, we examined the effects of the mutation on LC3B–PE conjugation on western blots (Figure 2(d)). To this end, HEK293A cells were transfected with MYC-LC3B variants and then treated with Torin1 to induce autophagy. The lipidated LC3-II form induced in Torin1-treated cells expressing LC3BY113C was lower than cells expressing the LC3B WT, suggesting that Y113C mutation impairs the lipidation process.
Figure 2.
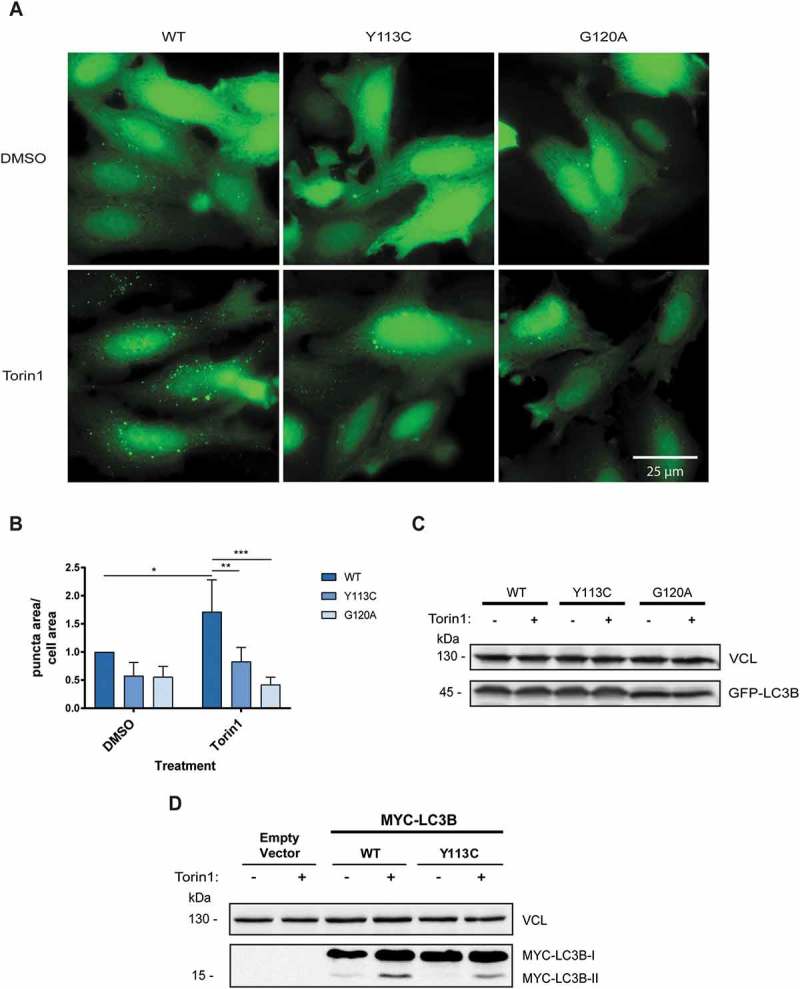
LC3BY113C mutation reduces autophagy levels in cells. (a) Fluorescence micrographs of HeLa cells transfected with plasmids encoding GFP-LC3B (LC3B WT, LC3BY113C, or LC3BG120A), and treated with DMSO or Torin1 for 2 h. (b) Autophagy levels measured in GFP-LC3B variants treated with DMSO or Torin1. The results were normalized to GFP-LC3B WT treated with DMSO. Autophagy levels were measured as puncta area/total cell area. The calculated results are an average of 3 independent biological experiments. Statistical significance was determined by two-way analysis of variance (ANOVA), *P < 0.05; **P < 0.01; ***P < 0.001. (c) Samples from B were western blotted for GFP and VCL, as loading control. (d) Western blots of HEK293A cells transfected with MYC-LC3B variants (LC3B WT and LC3BY113C) and treated with Torin1 or DMSO for 4 h, blotted with anti-MYC antibodies.
LC3BY113C mutation reduces the physical interaction with ATG4B without perturbing its cleavage by the protease
The first processing step of LC3B is cleavage at Gly120 by ATG4A or ATG4B, which exposes it for the next steps of covalent binding to E1-like ATG7, E2-like ATG3, and lipidation [14]. We first assessed whether the Y113C mutation affects the physical interaction of LC3B with ATG4A or ATG4B using the protein-fragment complementation assay (PCA) reporters, which were previously generated in our lab [15]. The PCA strategy is based on the fusion of unfolded and inactive fragments of a reporter protein (in our case, Gaussia luciferase, GLuc) to 2 proteins of interest. When these proteins interact, the fragments (e.g. GLuc [Fr1] and GLuc [Fr2]) are brought into proximity, allowing the reporter to fold into its active conformation and regain its activity[16]. ATG4A-GLuc (Fr2) or ATG4B-GLuc (Fr2) was coexpressed in HEK293T cells with GLuc (Fr1)-LC3B WT or mutant GLuc (Fr1)-LC3BY113C, and the relative luminescence, which represents the interaction strength, was measured in cell lysates (Figures 3(a) and S1A). The GLuc (Fr1)-LC3BY113C interactions with ATG4A-GLuc (Fr2) and ATG4B-GLuc (Fr2) were reduced by 22% and 27%, respectively, in comparison to GLuc (Fr1)-LC3B WT. The expression levels of GLuc (Fr1)-LC3B WT and GLuc (Fr1)-LC3BY113C were similar when coexpressed with each of the partners, confirming that the difference in luminescence is a direct effect of the protein-protein interaction and not due to changes in expression levels (Figure 3(b)). The reduced direct interaction of LC3B with ATG4B by the Y113C mutation was then further confirmed in co-immunoprcipitaion experiments where binding to endogenous ATG4B protein was assessed. To this end, HEK293T cells were transfected with HA-LC3B WT or HA-LC3BY113C mutant and 24 h later, the LC3B variants were immunoprecipitated with anti-HA antibodies and immunoblotted for ATG4B. The mutation reduced the binding of LC3B to ATG4B, consistent with the PCA results (Figure 3(c)). Figure 3(d) shows the quantification of 3 independent experiments, further documenting the effect that the mutation exerts on binding to the ATG4B protease.
Figure 3.
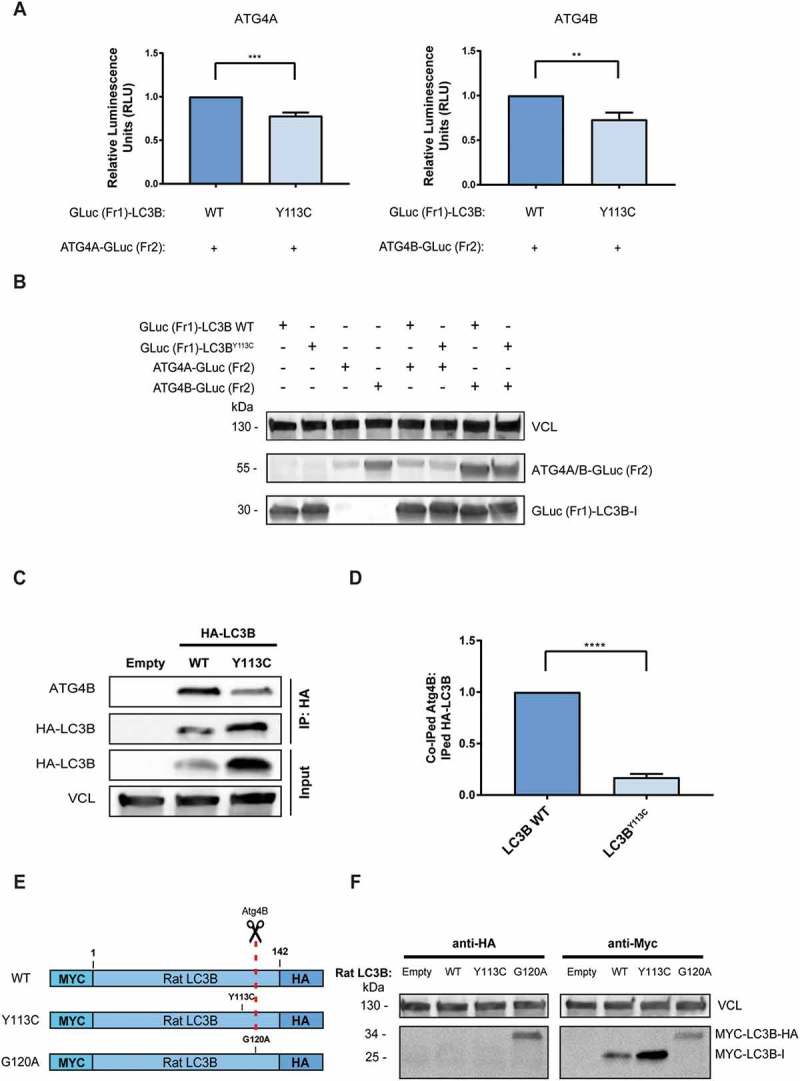
LC3BY113C mutation reduces binding to ATG4B yet does not impair its maturation by cleavage. (a) The relative interaction of GLuc (Fr1)-LC3B with ATG4A-GLuc (Fr2) and ATG4B-GLuc (Fr2) measured by PCA. The calculated results are the mean of 3 independent experiments and normalized to GLuc (Fr1)-LC3B WT. Statistical significance determined by the two-tailed unpaired Student t test, **P < 0.01; ***P < 0.001 (b) Western blot of the PCA samples, reacted with anti-Gaussia antibodies. VCL was used as a loading control. (C) Co-immunoprecipitation of HA-tagged LC3B variants with endogenous ATG4B. VCL was used as loading control for total cell extract. (d) The relative coIPed levels of endogenous ATG4B with LC3B variants (LC3B WT and LC3BY113C). IP levels were quantified using densitometry, and coIPed endogenous ATG4B levels were normalized to IPed HA-LC3B variants. The results are the mean of 3 independent experiments and normalized to the HA-LC3B WT variant. Statistical significance determined by the two-tailed unpaired Student t test, using the Holm-Sidak correction, ****P < 0.0001. (e) Schema of Rat MYC-LC3B-HA constructs used for the LC3B cleavage assay. Red dashed line indicates Gly120 cleavage site by ATG4B protease. (f) HEK293T cells transfected with MYC-LC3B-HA variants (LC3B WT, LC3BY113C, or LC3BG120A were subjected to western blotting with anti-MYC and anti-HA antibodies. Vinculin was used as a loading control.
Next, we examined whether the mutation impaired LC3B cleavage by its proteases, by expressing rat LC3B tagged with the MYC epitope at the N terminus and the HA epitope at the C terminus [17] in HEK293T cells (see the schema in Figure 3(e)). Proper cleavage at Gly120 should remove the C-terminal tail, including the HA epitope, and reduce the migration size of the lipidated LC3B. Probing the western blot with anti-MYC antibodies indicated that the Y113C mutant migrated on gels at the same size position as LC3B WT (Figure 3(f), right panel), suggesting that the mutant was properly cleaved. As expected, and in contrast to the Y113C mutant, the G120A mutant, which lacks the cleavage site, retained its full-length size (Figure 3(f), right panel). The anti-HA antibodies only detected the uncleavable LC3BG120A mutant, and not LC3B WT or the LC3BY113C mutant (Figure 3(f), left panel). This supports the conclusion that the Y113C mutant underwent complete cleavage by the endogenous proteases. Taken together, these results imply that while the LC3B mutant displays reduced physical interactions with the ATG4A/B proteases, the extent of reduction is not functionally rate-limiting, as it continues to be processed towards its lipidated form.
The LC3BY113C mutation reduces the direct physical interaction with ATG7, and not with ATG3
Next, we tested the effect of the mutation on the direct physical interactions with the 2 other proteins that interact with LC3B, the E1-like ATG7 and the E2-like ATG3, using the PCA system (Figures 4(a) and S1B). To this end, the GLuc (Fr1)-LC3B WT or mutant GLuc (Fr1)-LC3BY113C reporters were coexpressed in HEK293T cells either with ATG3-GLuc (Fr2) or with ATG7-GLuc (Fr2), and the luminescence, which represents the interaction strength, was measured in cell lysates. The interaction of the LC3BY113C mutant with ATG7 was reduced by 54% in comparison to LC3B WT, whereas the interaction with ATG3 did not change significantly. Western blot analysis indicated that the expression levels of WT and mutant GLuc (Fr1) -LC3B were similar when coexpressed with each of the partners, confirming that the difference in luminescence is a direct effect of the protein-protein interaction and not due to changes in expression (Figure 4(b)). The differential effects of the mutation on the direct interaction of LC3B with its partners were then also confirmed in co-immunoprecipitation experiments as described in the previous section, where binding to endogenous ATG3 and ATG7 proteins was assessed in mild buffer conditions to maintain non-covalent protein-protein interactions with LC3B. Notably, the mutation reduced the co-immunoprcipitaion of LC3B to ATG7 and did not interfere with the capability of the mutant LC3B to physically interact with ATG3, consistent with the PCA results (Figure 4(c)). Figure 4(d) shows the quantification of 3 independent experiments, further documenting the differential effect that the mutation exerts on the binding to the different LC3B interacting proteins. These results prompted us to perform 3D-structural modeling and MD simulations as described below.
Figure 4.
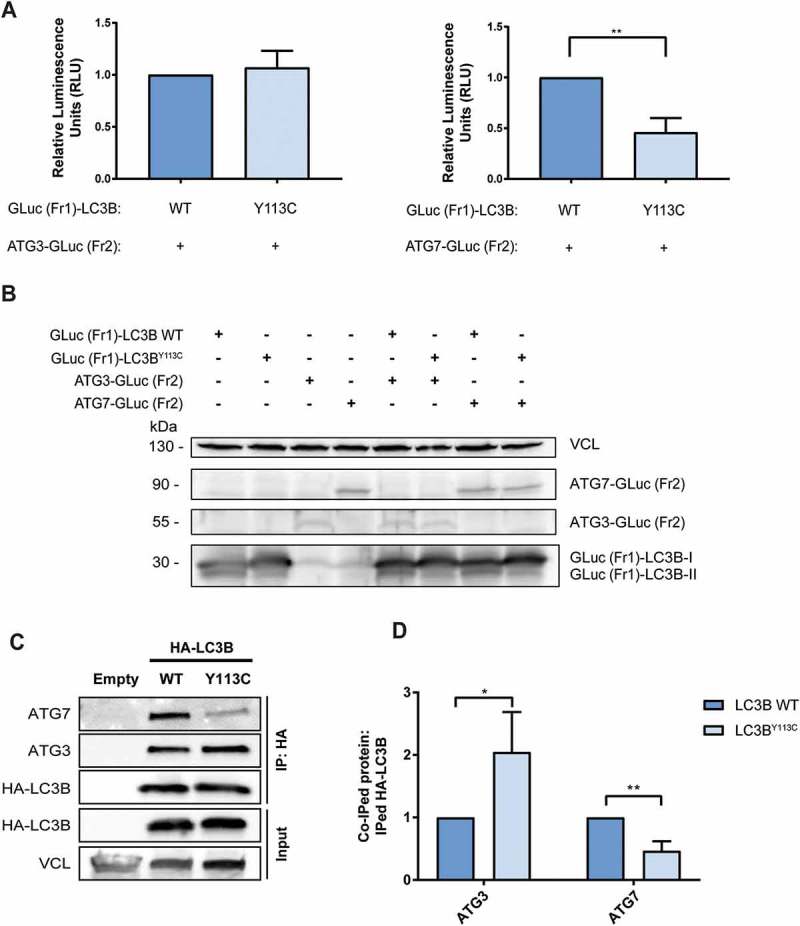
LC3BY113C mutation reduces physical interaction with ATG7, but not ATG3. (a) The relative interaction of GLuc (Fr1)-LC3B with ATG3-GLuc (Fr2) and ATG7-GLuc (Fr2) measured by PCA. The results calculated are the mean of 3 independent experiments and normalized to the GLuc (Fr1)-LC3B WT variant. Statistical significance determined by the two-tailed unpaired Student t test, **P < 0.01. (b) Western blot of the PCA samples, blotted with anti-Gaussia antibodies. VCL was used as a loading control. (c) Co-immunoprecipitation of HA tagged LC3B variants with endogenous ATG3 and ATG7. VCL was used as loading control for total cell extract. (d) The relative coIPed levels of endogenous ATG3 and ATG7 with LC3B variants (LC3B WT and LC3BY113C). IP levels were quantified using densitometry and coIPed endogenous ATG7 and ATG3 levels were normalized to IPed HA-LC3B variants. The results calculated are the mean of 3 independent experiments and normalized to the HA-LC3B WT variant. Statistical significance determined by the two-tailed unpaired Student’s t-test, using Holm-Sidak correction, *P < 0.05; **P < 0.01.
Structural analysis suggests conformation change in LC3B loop 38 to 50 that affects LC3B-ATG7 and LC3B-ATG4B interaction
In order to estimate the effect of the Y113C mutation on the structure and dynamics of free LC3B, we first purified recombinant LC3B WT and LC3BY113C proteins and analyzed them by performing size exclusion chromatography (Figure S2). Both LC3B WT and LC3BY113C showed a single narrow peak at the same size suggesting similar overall folding of the 2 variants. Next, we calculated molecular dynamics (MD) trajectories for WT and mutant LC3B. We first compared and analyzed experimental 3D-structures of free and bound LC3 and of the closely related yeast- and potato-Atg8. The superposition of selected structures (listed in Materials and Methods), shown in Figure 5(a), highlights the stability of the fold, with significant divergence of only the N- and C-terminal segments and loops 38 to 50 and 71 to 78. Loop 38 to 50 connects 2 β-strands in the central β-sheet of the molecule. The fold is one residue shorter in Atg8 (residues 36 to 47) compared to LC3B, yet its conformation is similar. The location of Y113 (Y109 in Atg8) is very similar in all the superposed structures. It resides at the end of a β-strand and is completely buried inside the protein core. The OH-moiety of Y113 is hydrogen-bonded to the backbone atoms of P45 and L47 (human LC3B numbering), and the aromatic ring contacts the side chains of I35, and the hydrophobic part of R37, L44, L47, and occasionally, L71 (Figure 5(b)). These contacts are observed in the majority of the structures, including solution NMR structures, which reflect the mobility of the proteins. The hydrogen bonds and the contacts of Y113 with L44 and L47 stabilize the structure of loop 38 to 50, and the replacement of Y with the much smaller C residue is likely to lead to structural changes to compensate for the change in residue size and the loss of stabilizing hydrogen bonds.
Figure 5.
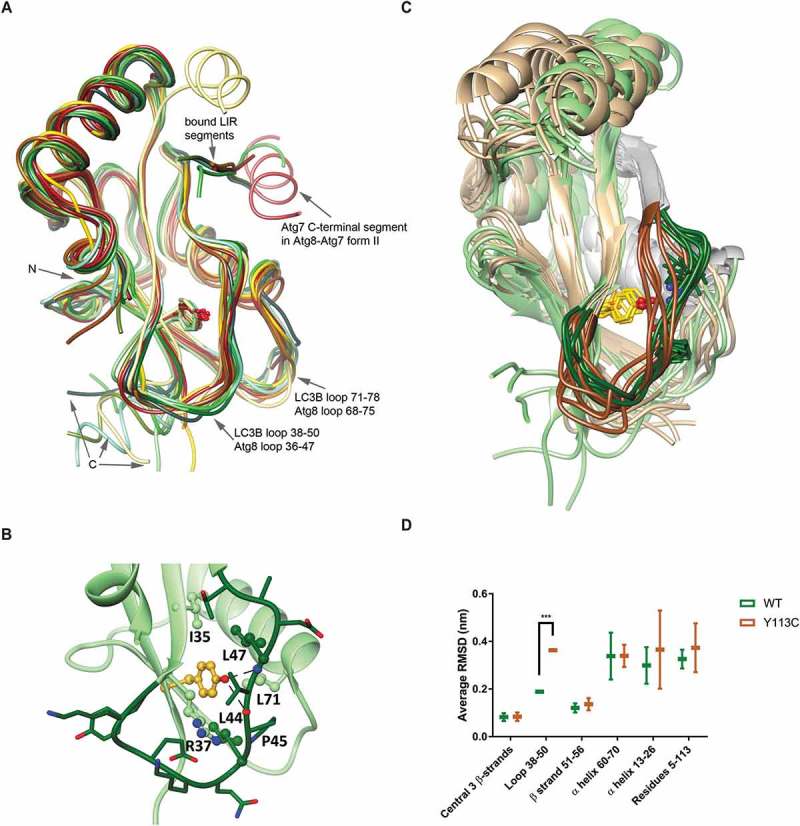
Loop 38 to 50 conformation is changed in LC3BY113C. (a) Superposition of experimental structures of LC3B and yeast Atg8. Free LC3B structure (1UGM) is shown in dark green and bound structures from PDB entries 2Z0D, 2ZJD, 5D94, 5MS2 and 5GMV are in other shades of green; bound Atg8 structures from PDB entries 3RUI, 2ZPN, 3VXW, 2LI5, 2KQ7, 5I83 and 5YEC are shown in different yellow-to-brown shades. The binding positions of LIR motifs and of ATG7 in the form-II LC3B-ATG7 complex are indicated. (b) The intramolecular interactions of LC3B residue Y113 (LC3B was extracted from the LC3-ATG4B complex, PDB code 2Z0D). Y113 is emphasized in gold and loop 38 to 50 is shown as all-atom stick diagram in dark green. The hydrogen bonds between Y113-OH and the back-bone atoms of P45 and L47 are indicated by the dashed lines. Y113 also makes hydrophobic contacts with the side chains of I35, R37, L44, L47 and L71, shown as ball and stick and labeled. (c) Super-positioned screenshots of LC3B WT (green) and LC3BY113C (beige) from the MD trajectories. Loop 38 to 50 is labeled in dark shades of green and brown. Tyr113 and Cys113 are marked in yellow. The LIR motif binding segments are in gray. (d) Average RMSD of different LC3B segments in 2 independent trajectories. Statistical significance determined by the two-tailed unpaired Student t test, ***P < 0.001.
Figure 5(c) presents selected snapshots from the 160 ns production stage of the MD simulations for each, LC3B WT and LC3BY113C, superposed using the Cα atoms of the central β-sheet (root mean square deviations, RMSDs, along the 4 trajectories and snapshots at 5 ns interval are shown in Figures S3 and S4). It appears that while the central β-sheet is stable, the N-terminal helix, C-terminal segment 114 to 120, loops 38 to 50 and 71 to 78, and to a lesser extent, helix 60 to 70, are mobile, in line with the variance in the experimental structures (Figure 5(a)). The position of Y113 and the H-bond interactions between Y113-OH and the backbone O and N atoms of P45 and L47, respectively, are maintained throughout the trajectories, with only occasional disruptions (Figure S5), emphasizing the importance of the conserved Y113 for the stabilization of loop 38 to 50. In the mutant, loop 38 to 50 shifts to fill the void created by the replacement of Y with C (Figure 5(c)), and it is more mobile, showing larger RMSD from the energy-minimized starting structure, compared with LC3B WT (Figure 5(d) and S6 and Table S2). The conformation change in loop 38 to 50 may also affect the downstream LIR motif binding site, which consists of β-strand 51 to 56 and helix 60 to 70 [18], but these segments appear to be as stable in the Y113C mutant as in LC3B WT (Table S2 and Figure S6). These results suggest that the Y113C mutation might affect intermolecular interactions that involve loop 38 to 50, but not interactions through the LIR-motif binding site, including the proposed first step of LC3/Atg8 and ATG7/Atg7 recognition [19] (Figure 5(a)).
The contribution of the LC3B loop 38 to 50 to the interaction with ATG4B (PDB codes 2Z0D, 2Z0E and 2ZZP for complexes of rat LC3B with human ATG4B) is relatively small, burying 117Å2 of the surface of residues 38 to 50 (average for the 3 structures) (Figure 6(a)). The major point of contact, between LC3B residue Y38 and ATG4B residues L232 and T233, is observed in all 3 structures, and it was shown to be important for the specificity of LC3B-ATG4B binding [20]. The backbone of Y38 is hydrogen-bonded to the backbone of Y113, and a shift of loop 38 to 50 may have some effect on the interaction. This analysis fits well with the experimental data showing that Y113C affects the physical interaction with ATG4B. Yet, the perturbation is not sufficient to interfere with the subsequent proteolytic cleavage.
Figure 6.
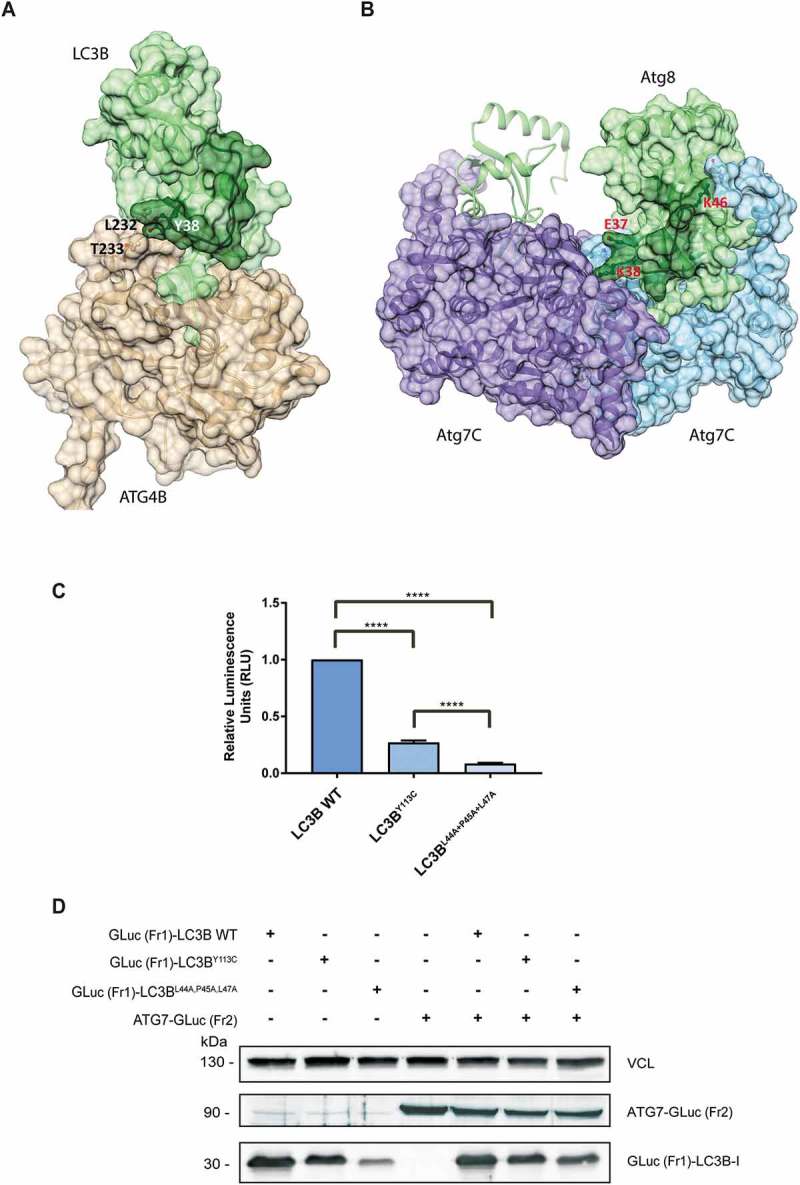
Loop 38 to 50 is directly involved in the interaction of LC3B with ATG4B and ATG7. (a) The structure of the LC3B-ATG4B complex (PDB code 2Z0D): Shown are the solvent accessible surfaces of ATG4B in beige and of LC3 in green, with loop 38 to 50 in dark green. The contact between LC3B residue Y38 and ATG4B residues L232-and T233 is labeled. (b) The structure of the Atg8-Atg7C hetero-tetramer (PDB code 3RUI): Shown is the solvent accessible surface of Atg7C homodimer in light blue and purple. Atg8 is colored green with the loop 36 to 47 (corresponding to LC3 loop 38 to 50) in dark green; the surface of only one Atg8 monomer is shown for clarity. Atg8 residues that interact with Atg7C are labeled. Note that Atg8 residue K38 interacts with both monomers of the Atg7C dimer (see text). (c) The relative interaction of GLuc (Fr1)-LC3B WT, GLuc (Fr1)-LC3BY113C and GLuc (Fr1)-LC3BL44A,P45A,L47A with ATG7-GLuc (Fr2) measured by PCA. The results calculated are the mean of 3 independent experiments and normalized to GLuc (Fr1)-LC3B WT variant. Statistical significance was determined by the two-tailed unpaired Student t test, ****P < 0.0001. (d) Western blot of the PCA samples, blotted with anti-Gaussia antibodies. VCL was used as a loading control.
The effect of the Y113C mutation on the interaction with ATG7 could be estimated by analyzing the structure of the hetero-tetramer [Atg8-Atg7C]2 (PDB codes 3RUI and 3VH3). In this complex, loop 36 to 47 of yeast Atg8 (corresponding to loop 38 to 50 of LC3B) interacts with both monomers of Atg7C, burying 188Å2 and 73Å2 of its surface, respectively (Figure 6(b)) (calculated for structure 3RUI, which includes all the atoms). Contacts include electrostatic interactions between Atg8 residues E37, K38 and K46, and Atg7C residues H554, D465 and E620, respectively, and between Atg8 K38 and the backbones of G543 and L542 and side chain of L314 of the other Atg7C monomer. Thus, structural changes brought about by the Y113C mutation may have a larger effect on ATG7 binding and subsequently on ATG7’s E1 activity.
Superposition of Atg7C from the Atg7C-Atg8 complex (3RUI) on the Atg7-Atg3 crosslinked complex (4GSL[21]) places Atg8 loop 36 to 47 away from Atg3 (not shown), consistent with the PCA and affinity-isolation experiments showing uninterrupted interactions between LC3B and ATG3.
In order to validate the importance of loop 38 to 50 to the interaction between LC3B and ATG7, we performed additional PCA experiments, in which we mutated loop 38 to 50 in LC3B and assessed the effect on the interaction with ATG7. We chose to mutate residues L44, P45 and L47, which make direct intramolecular contacts with Y113 (Figure 5(b)), but are not involved in direct intermolecular contacts with either ATG4B or ATG7. These triple mutations are likely to destabilize loop 38 to 50 and affect its conformation, but not the direct contacts with ATG4B or ATG7, thus imitating the conformational effect of mutation Y113C. HEK 293T cells were cotransfected with GLuc (Fr1)-LC3B variants (LC3B WT, LC3BY113C or LC3BL44A,P45A,L47A triple mutants) along with ATG7-GLuc (Fr2) (Figure 6(c)). The interaction of the GLuc (Fr1)-LC3BL44A,P45A,L47A variant was dramatically reduced in comparison to GLuc (Fr1)-LC3B WT, and also GLuc (Fr1)-LC3BY113C. This experiment validates the importance of the stabilization of loop 38 to 50, to the binding of LC3B with ATG7. Western blot analysis of the cell lysates showed equal levels of expression among the different LC3B variants (Figure 6(d)).
Discussion
The proposed tumor-suppressing features of autophagy in early stages of cancer [4,5], and its tumor-promoting features at later stages [6–8], raise challenging concepts concerning the relationships between autophagy and cancer. Loss-of-function mutations in autophagy genes could confer tumor-promoting features at early stages of cancer only if they reduce, but not completely eliminate, autophagy in cells, since some level of autophagy should remain exploitable later in the growing tumor. Indeed, in previous studies revealing recurrent mutations in autophagy genes in human cancer patient samples, the most frequent gene mutations are in autophagy regulators and pathway interactors, rather than mutations in the core machinery of autophagy [22,23]. Among the few reports on the latter group is a cancer-associated mutation in MAP1LC3A [24], which results in the protein’s reduced cleavage by ATG4B and reduced autophagy levels. Our computational analysis shown here confirmed that mutations in core-machinery autophagy genes are indeed rare in different types of cancer. Among the few detectable hits, the LC3BY113C mutation received the highest score. Notably, in the 2 melanoma patients carrying the LC3BY113C mutation, it was found in the primary tumors and persisted in the metastases, suggesting that the mutation potentially plays a role in early cancer development [11]. Information as to the primary tumors was unavailable for the other 3 liver cancer patients. This mutation has also been recently identified by an in silico screen of 4 autophagy genes based on the NCBI dbSNP database on Hepatocellular Carcinoma [25].
The exact statistic we provide herein indicates that recurring somatic fixations of the LC3BY113C mutation provide some adaptive advantage in the cancerous environment. Based on our various experimental analyses, we suggest that the potential tumor-promoting effect of the LC3BY113C mutation emerges from its reduced autophagic capacity, as shown by the GFP-LC3B puncta assay and by the LC3B lipidation results, under stress conditions (represented by Torin1 treatment). The reduction in autophagy levels could lead to increases in reactive oxygen species and DNA damage, thus promoting cancer at initial stages. As the effect of the mutation on LC3B lipidation is partial, rather than a complete block, it could still support the continuous growth of tumor cells at later stages, in particular under stressful conditions. Notably, it has been reported that downregulation of ATG5 expression by promoter methylation contributes to tumorigenesis in early-stage cutaneous melanoma, supporting the hypothesis that reduced autophagy accelerates cancer formation at these stages [26]. In addition, it has been found by data mining that selective allelic loss of ATG5 increases the risk for metastases in human melanoma, while complete loss of Atg5 in mouse model systems reduces tumor onset, suggesting that melanoma cannot sustain a complete blockage of autophagy [27]. It would be interesting to check the functional outcome of the other significant mutations we discovered in our bioinformatics screen (Table 1).
The maturation of LC3B by proteolytic cleavage, and its processing by ubiquitin-like conjugation reactions, prompted us to examine whether the reduction in autophagy levels results from a malfunction in one of the stages of the process. Despite the detected reduction in the physical interaction between LC3B and ATG4A/B, LC3B cleavage by its cellular proteases still occurs, signifying that the partial decline in the interaction caused by the Y113C mutation does not affect the LC3B cleavage stage of the process. It is therefore assumed that the reduction in the physical interaction of mutant LC3B with the E1-like ATG7 protein, detected by the PCA reporters and by the co-immunoprecipitation experiments, may be the direct cause for the reduction in LC3B–PE conjugation and lipidation – the final stage of LC3B processing. Notably, no reduction in the physical interaction with ATG3 was detected, thus supporting specificity towards ATG7 binding, and excluding a general effect of the mutation on the overall 3D-protein structure of LC3B. Obviously, the data emerging from the PCA and affinity-isolation experiments correspond to the non-covalent protein-protein interactions within each pair of proteins, independent of the ubiquitin-like conjugating cascades.
The high conservation of Tyr113 (Y113) in all LC3 orthologs and in other Atg8-family proteins, together with it being buried in the core of the protein, indicates the importance of the residue to LC3B 3D-structure and function [11,28]. The MD trajectories consist of the coordinates of every atom in the system and their change over time; hence, the combined 160 ns trajectories provide a useful, though possibly incomplete, picture of the structural changes and mobility within the molecule. The predicted effect of the mutation on the structure of the protein is local, predominantly modifying loop 38 to 50, which contributes to the interaction with ATG4B, and even more effectively to the binding to ATG7. This loop is flexible also in LC3B WT, but the flexibility is controlled by the interactions with Y113. In contrast, the LIR-motif binding segments maintain a similar structure in the mutant, suggesting that interactions through this site are not likely to be affected by mutation Y113C. Notably, the conclusions emerging from the analyses of the experimental structures and the molecular dynamics trajectories were further validated experimentally, showing that a triple mutation in the 38 to 50 loop affecting the intramolecular interactions with Y113, but not the direct intermolecular interactions with ATG7, strongly reduced the binding to ATG7. Altogether, we hypothesize that this well-studied structural outcome of the Y113C mutation, which limits the direct physical interaction with ATG7, is responsible for the observed reduced LC3B lipidation and GFP-LC3B puncta formation. The significant recurrence of this mutation in cancer, especially its detection in primary tumors, suggests that the mutation has the potential to promote early cancer development.
Materials and methods
Data collection
Human cancer somatic mutations were retrieved from the whole genome screening data of the Catalogue of Somatic Mutations in Cancer, COSMIC, version 78 (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic). The human coding exome coordinates and sequences were retrieved from the Ensembl GRCh37 release [29]. The AnnoVar software package [30] was used to annotate all possible nucleotide substitutions (relative to the GRCh37; h19 reference genome) in every human protein-coding (CDS) position documented in GRCh37 as previously described [10]. For each variation we determined its specific protein-transcript consequences (e.g., non-synonymous), its population frequency, and its predicted functional likelihood, using both SIFT and PolyPhen algorithms [31,32]. This analysis calculates the distribution of all possible nucleotide substitution consequences (i.e., non-synonymous, synonymous, and stop-gain) for each protein-coding (CDS) position.
Calculation of mutation density clusters
To identify gene target sites in cancer, we first quantified the amount of independent mutational events per gene per site in the COSMIC database (cancer.sanger.ac.uk). To avoid multiple counting of mutations that are identical by descent (as in cases of primary tumor and metastases) we used the COSMIC ‘individual identifiers’ to join all mutations per gene site per individual as one call. Nevertheless, we still found that in some cases, data from a single patient were present in 2 or more different individuals’ identifiers, leading to mutation redundancies. To identify such cases and further reduce the redundancy, we also removed all individuals with 3 or more mutations that had all of their mutations fully included in another individual. (i.e., remove A if A#m ≥ 3, and Am/(Am ∩ Bm) = 1, where A and B are samples with different individual identifiers, A#m is the number of mutations in A, and Am and Bm are the mutations in A and B.
To identify gene positions/segments with high mutational density, we used the Poisson distribution. For each gene we calculated the Poisson probability distribution of the number of observed mutations in each of the gene position/interval as follows:
P(xgi; (ngi/sgi)l) = (e–(ngi/sgi)l) ([(ngi/sgi)l]xgi)/xgi!, where xgi, the number of events in an interval, is the number of the observed non-redundant somatic mutation from type i (i being non-synonymous or stop-gain) in specific gene g interval length l, as obtained from the COSMIC version 78 database; n is the number of all observed mutations from type i in gene g interval 1; and s is the number of all possible computed substitutions sites of type i (e.g., non-synonymous sites) in gene g. The Poisson P values were then corrected for multiple testing by the Benjamini Yekutieli correction [33], corresponding to the number of tested intervals in gene g (for example, when testing intervals of single nucleotide positions the number of tests is the CDS length). The intervals we examined were 1, 3, 9, 15, 30, 45, and 60 bases, using a step of one base for the interval of 1 (i.e., when examining every base) and a step of 3 for all other intervals.
DNA constructs
The PCA plasmids used in this study (GLuc [Fr1]-LC3B, ATG7-GLuc [Fr2], ATG3-GLuc [Fr2], ATG4A-GLuc [Fr2] and ATG4B-GLuc [Fr2]) were previously generated by our lab [15]. All mutant plasmids were generated by restriction-free cloning using the KAPA HiFi HotStart ReadyMixPCR Kit (Kapa Biosystems, KK2601) according to the manufacturer’s protocol. The triple mutated GLuc (Fr1)-LC3BL44A,P45A,L47A plasmid was generated in two sequential steps. L44A and P45A mutations were generated at first, followed by L47A mutation in the second step. pCI-neo-Myc-LC3-HA (Addgene, 45244) and pCI-neo-myc-LC3G120A-HA (Addgene, 45245) were a gift from Tamotsu Yoshimori. The pEGFPC1-LC3 plasmid was a kind gift from N. Mizushima (The University of Tokyo, Tokyo, Japan) and T. Yoshimori (Osaka University, Osaka, Japan).
GFP-LC3 punctate staining
HeLa cells were grown in Dulbecco modified Eagle medium (DMEM; Biological Industries, 01-055-1A) on 15-cm plates (6.5 × 105 cells per plate) containing 13-mm glass cover-slips. The cells were transfected with 18 μg of GFP-LC3 (LC3B WT, LC3BY113C, LC3BG120A) and 12 μg of empty pcDNA3 vector (Thermo Fisher Scientific, V79020), using jetPEI transfection reagent (Polyplus, 101-10N) according to the manufacturer’s protocol. After 24 h, the cells were washed with Dulbecco phosphate-buffered saline (PBS; Biological Industries, 02-023-1A) and autophagy was induced with 200 nM Torin1 (BioVision incorporated, 2273) in DMEM for 2 h. The slides were then removed from the plate, washed with cold PBS, fixed with 3.7% paraformaldehyde (Electron Microscopy Sciences, 15710) and viewed by fluorescence microscopy (Olympus BX41, Tokyo, Japan) with x60 (N.A. 1.25) UPlan-Fl oil immersion objective, and digital images obtained with a DP72 CCD camera (Olympus, Tokyo, Japan) using CellSens Standard software (Olympus). The percentage of puncta area per total cell area was determined using MetaMorph imaging software (Molecular Devices). The remaining cells on the plates were harvested for protein extraction and western blot analysis.
Protein-fragments complementation assay (PCA)
HEK 293T cells grown in DMEM on 10-cm plates (1.5x106) were cotransfected with 10-μg GLuc -fragments plasmids using calcium-phosphate transfection reagents [34]. After 24 h, the cells were harvested and lysed in luciferase lysis buffer (25 mM Tris, pH 8.5, 150 mM NaBr, 5 mM EDTA, 0.1% NP40 [Sigma-Aldrich, CA-630], 5% glycerol [Bio-Lab, 000712022300], 65 µM sodium oxalate [Thermo Fisher Scientific, AC207725000], 0.5 mM reduced glutathione [Thermo Fisher Scientific, AC120000050], 0.5 mM oxidized glutathione [Thermo Fisher Scientific, AC320220050]). Native coelenterazine (Nanolight, 303) was diluted in luciferase assay buffer (25 mM Tris, pH 7.75, 1 mM EDTA, 0.5 mM reduced glutathione, 0.5 mM oxidized glutathione, 75 mM urea [MP Biomedicals, 821527]) to a final concentration of 20 µM. The lysates were diluted to a concentration of 2 μg/μl in a 96-well microplate (35 μl per well) and each well was injected with 100μl of coelenterazine before reading. Luminescence signal (integrated over 10 sec) was read using a Veritas microplate luminometer (Turner BioSystems, USA).
Protein extraction and analysis
Cells were lysed in B buffer (20 mM HEPES, pH 7.6, 100 mM KCl, 0.5 mM EDTA, 0.4% NP40, 20% glycerol) or in luciferase lysis buffer (25 mM Tris, pH 8.5, 150 mM NaBr, 5 mM EDTA, 0.1% NP40, 5% glycerol, 65 µM sodium oxalate, 0.5 mM reduced glutathione, 0.5 mM oxidized glutathione). The buffers were supplemented with 10 μl/ml 0.1M PMSF (Sigma-Aldrich, 93482) and 1% protease (Sigma-Aldrich, P8340) and phosphatase (Sigma-Aldrich, P0044) inhibitor cocktails. Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes, which were incubated with antibodies to MYC (a kind gift from Moshe Oren, Weizmann Institute of Science), LC3B (Cell Signaling Technology, 3868), GFP (Roche, 1814460), VCL (vinculin; Sigma-Aldrich, V9131) and HA (BioLegend, 901502) or Gaussia luciferase (NanoLight Technology, 401). Detection was done with either horseradish peroxidase (HRP)-conjugated goat anti-mouse (Jackson ImmunoResearch, 115-035-003) or anti-rabbit (Jackson ImmunoResearch, 111-165-144) secondary antibodies, followed by enhanced chemiluminescence using EZ-ECL (Biological Industries, 20-500).
Co-immunoprecipitation
HEK 293T cells were grown on 15-cm plates (4.5 × 106 cells) and transfected with 13.5 μg HA-LC3B WT or HA-LC3BY113C plasmids using calcium-phosphate transfection reagents. Empty pcDNA3 vector was used as a control. After 24 h, the cells were harvested and lysed in luciferase lysis buffer. The protein levels were quantified using Bradford reagent (Bio-Rad, 500-0006) and equal quantities of the lysates were taken for immunoprecipitation. HA beads (Sigma-Aldrich, A2095) were added to the lysates and incubated at 4°C overnight. The beads were washed 3 times with lysis buffer and the protein was eluted using excess HA peptides (Sigma-Aldrich, I2149). Co-immunoprecipitation of the LC3B-interacting proteins was evaluated using the following antibodies: ATG7 (Sigma-Aldrich, A2856), ATG3 (MBL International Corporation, M133-3) and ATG4B (Sigma-Aldrich, A2981).
Cloning, expression and purification of recombinant LC3B WT and LC3BY113C
Human LC3B WT and LC3BY113C (both carrying G120A substitution followed by the deletion of 5 residues in the C terminus) were cloned into pET28-bdSUMO based on the pK151 vector containing an N-terminal poly His-tag (His14) (obtained from Dirk Görlich, Max-Planck-Institute, Göttingen, Germany) upstream of the bdSUMO fusion [35]. The His14-bdSUMO fusion contains a bdSENP1 recognition site, allowing the complete cleavage of the fusion with this protease. This plasmid was expressed in E. coli BL21 (DE3) cells upon induction with 200 μM IPTG (Thermo Fisher Scientific, R0392) for ~18 h at 15°C. The clarified lysate was incubated for 1 h with Ni-NTA beads (Adar Biotech, 1018-25) at 4°C. The beads were then washed extensively with buffer A (20 mM Tris, pH 8, 1 M NaCl) followed by washes with the same buffer containing 0.2 M NaCl and 1 mM EDTA (buffer B). bdSENP1 protease expressed and purified at the Structural Proteomics Unit at the Weizmann Institute using a plasmid obtained from Dirk Görlich, Max-Planck-Institute, Göttingen, Germany, following the protocol previously described [35]. Cleavage of the fusion using bdSENP1 protease was performed on the beads by adding the enzyme and shaking for 1 h at 25°C. The purified cleaved proteins were analyzed on an analytical Superdex75 HR 10/30 (GE Healthcare Bio-Sciences Ltd, 17-5174-01) column equilibrated with 20 mM Tris, pH 8, 0.2 M NaCl, exhibiting in each case a single peak.
Structural analysis and MD simulations
The relevant structures were downloaded from the Protein Data Bank [36], specifically, free and bound LC3B, PDB codes 1UGM [37], 2Z0D, 2Z0E, 2ZZP [20], 2ZJD [38], 5D94 [18], 5WRD [39], 5MS2 [40], 5GMV [41] and 2K6Q [42]; yeast Atg8, 2KQ7 [43], 2KWC [44], 3RUI [45], 2ZPN [42], 3VXW [46], 2LI5 and 3VH3 [47]; potato ATG8, 5L83 [48]; human ATG4B complexed to rat LC3B, 2Z0D, 2Z0E and 2ZZP; the hetero-tetramer [Atg8-Atg7C]2, 3RUI, 3VH3 and 5YEC [19]; and NMR structures of Atg8 and LC3B proteins, 2KQ7, 2KWC, 2LI5 and 2K6Q.
Molecular dynamics simulations were done with the Gromacs software package [49]. The starting structure of LC3B was extracted from the experimental structure of the ATG4B-LC3B complex (PDB code 2Z0D, reference 18). The starting structure of the Y113C mutant was obtained by replacing Y113 with C. Each of the molecules was immersed in a box of water, the system was neutralized and energy minimized. Each MD simulation started with 200 ps of water organization, in which all the non-hydrogen atoms of the protein were restrained to their initial positions whereas the solvent molecules (water and neutralizing ions) and the hydrogen atoms of the protein were free to move. Next, all the restraints were removed and the simulation continued for 100 ns. Two trajectories, with different starting velocities, were calculated for each, WT and mutant LC3B. Figure S2 presents the RMSD from the energy minimized structure, calculated for all the Cα atoms of the protein (Cα RMSD), showing that the systems have equilibrated after 20 ns or less. Thus, the last 80ns of each simulation (production stage) were used in the analyses of the trajectories, providing an overall of 160 ns trajectory for each, WT and mutant LC3B.
RMSD and distance distributions were calculated with the Gromacs package. UCSF-Chimera [50] was used for structure analyses and preparation of the figures.
Statistical methods
All the statistical analyses, excluding the calculation of mutation density clusters (Described separately) were performed using Prism 7.02 software (GraphPad Software).
Funding Statement
This work was supported by a grant from the European Research Council (ERC) under the European Union’s Seventh Framework Program (FP7/2007-2013/ERC, grant agreement 322709).
Abbreviations
- ATG
autophagy-related
- COSMIC
catalogue of somatic mutations in cancer
- Fr1/2
fragment ½
- GABARAP
GABA type A receptor-associated protein
- GABARAPL2
GABA type A receptor-associated protein like 2
- GFP
green fluorescent protein; GLuc: Gaussia Luciferase
- HA
human influenza hemagglutinin
- HEK
human embryonic kidney
- IP
immunoprecipitation
- LIR
LC3-interacting region
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 beta
- MD
molecular dynamics
- NMR
nuclear magnetic resonance
- PCA
protein-fragment complementation assay
- PDB
protein data bank
- PE
phosphatidylethanolamine
- RMSD
root mean square deviations
- SUMO
small ubiquitin-like modifier
- VCL
vinculin.
Acknowledgments
We would like to thank Shira Albeck and Yoav Peleg for the cloning, expression and purification of the recombinant LC3B variants, as well as for the size exclusion chromatography analysis of the proteins. We would also like to thank Santosh K Dasari, Aya Shkedy, Naama Yaeli Slonim, Ruth Shiloh, Naama Dekel, Lital Povodovski, Rivi Halimi, Maya David, Liat Hammer and Vered Solomon for providing significant assistance and crucial advisory along all the experimental procedures and writing of this paper. This work was supported by a grant from the European Research Council under the European Union’s Seventh Framework Program (FP7/2007-2013/ERC grant agreement 322709). AK is the incumbent of the Helena Rubinstein Chair of Cancer Research.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. [Internet]. 2014;24:24–41. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3879710&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yorimitsu T, Klionsky DJ.. Autophagy: molecular machinery for self-eating. Cell Death Differ. [Internet]. 2005;12 Suppl 2:1542–1552. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16247502%5Cnhttp://www.nature.com/cdd/journal/v12/n2s/pdf/4401765a.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levine B, Kroemer G.. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chen H-Y, White E. Role of autophagy in cancer prevention. Cancer Prev Res (Phila). [Internet]. 2011;4:973–983. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3136921&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. [Internet]. 2011;25:795–800. Available from: http://genesdev.cshlp.org/content/25/8/795.short [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Guo JY, Karsli-Uzunbas G, Mathew R, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27:1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. [Internet]. 2012;12:401–410. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3664381&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Forbes SA, Beare D, Gunasekaran P, et al. COSMIC : exploring the world ’ s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gershoni M, Pietrokovski S. The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol. [Internet]. 2017;15:7 Available from:: http://bmcbiol.biomedcentral.com/articles/10.1186/s12915-017-0352-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sanborn JZ, Chung J, Purdom E, et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S. [Internet]. 2015;112:10995–11000. Available from: http://www.pnas.org/content/112/35/10995.full [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shpilka T, Weidberg H, Pietrokovski S, et al. Atg8 : an autophagy-related ubiquitin-like protein family. Genome Biol. 2011;12:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. [Internet]. 2016;536:285–291. Available from: 10.1038/nature19057%0Ahttp://10.0.4.14/nature19057%0Ahttp://www.nature.com/nature/journal/v536/n7616/abs/nature19057.html#supplementary-information [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kabeya Y. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gilad Y, Shiloh R, Ber Y, et al. Discovering protein-protein interactions within the programmed cell death network using a protein-fragment complementation screen. Cell Rep. [Internet]. 2014;8:909–921. [DOI] [PubMed] [Google Scholar]
- [16].Remy I, Michnick SW. A highly sensitive protein- protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3:977–979. [DOI] [PubMed] [Google Scholar]
- [17].Fujita N, Hayashi-Nishino M, Fukumoto H, et al. An Atg4B mutant hampers the lipidation of LC3 Paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Olsvik HL, Lamark T, Takagi K, et al. FYCO1 contains a C-terminally extended, LC3A/B-preferring LC3-interacting region (LIR) motif required for efficient maturation of autophagosomes during basal autophagy. J Biol Chem. 2015;290:29361–29374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yamaguchi M, Satoo K, Suzuki H, et al. Atg7 activates an autophagy-essential ubiquitin-like protein Atg8 through multi-step recognition. J Mol Biol. [Internet]. 2017;430:249–257. [DOI] [PubMed] [Google Scholar]
- [20].Satoo K, Noda NN, Kumeta H, et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. Embo J. [Internet]. 2009;28:1341–1350. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2683054&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kaiser SE, Mao K, Taherbhoy AM, et al. Noncanonical E2 recruitment by the autophagy E1 revealed by Atg7 – atg3 and Atg7 – atg10 structures. Nat Struct Mol Biol. [Internet]. 2012;19:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lebovitz CB, Robertson AG, Goya R, et al. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy. 2015;11:1668–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Costa JR, Prak K, Aldous S, et al. Autophagy gene expression profiling identifies a defective microtubule-associated protein light chain 3A mutant in cancer. Oncotarget. [Internet]. 2016;7:41203–41216. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27256984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Awan F, Obaid A, Ikram A, et al. Mutation-structure-function relationship based integrated strategy reveals the potential impact of deleterious missense mutations in autophagy related proteins on hepatocellular carcinoma (HCC): a comprehensive informatics approach. Int J Mol Sci. [Internet]. 2017;18:139 Available from:: http://www.mdpi.com/1422-0067/18/1/139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu H, He Z, von Rutte T, et al. Down-Regulation of Autophagy-Related Protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med. [Internet]. 2013;5:202ra123–202ra123. Available from: http://stm.sciencemag.org/cgi/doi/10.1126/scitranslmed.3005864 [DOI] [PubMed] [Google Scholar]
- [27].García-Fernández M, Karras P, Checinska A, et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy. [Internet]. 2016;12:1776–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kouno T, Mizuguchi M, Tanidal I, et al. Solution structure of microtubule-associated protein light chain 3 and identification of its functional subdomains. J Biol Chem. 2005;280:24610–24617. [DOI] [PubMed] [Google Scholar]
- [29].Herrero J, Muffato M, Beal K, et al. Ensembl comparative genomics resources. Database. 2016;2016:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- [32].Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. [Internet]. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under depencency. Ann Stat. 2001;29:1165–1188. [Google Scholar]
- [34].Kingston RE, Chen CA, Rose JK. Calcium phosphate transfection. Curr Protoc Mol Biol [Internet]. 2003;63:9.1.1–9.1.11. [DOI] [PubMed] [Google Scholar]
- [35].Frey S, Görlich D. A new set of highly efficient, tag-cleaving proteases for purifying recombinant proteins. J Chromatogr [Internet]. 2014;1337:95–105. [DOI] [PubMed] [Google Scholar]
- [36].Rose PW, Prli A, Altunkaya A, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017;45:D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sugawara K, Suzuki NN, Fujioka Y, et al. The crystal structure of microtubule-associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes to Cells. 2004;9:611–618. [DOI] [PubMed] [Google Scholar]
- [38].Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283:22847–22857. [DOI] [PubMed] [Google Scholar]
- [39].Sakurai S, Tomita T, Shimizu T, et al. The crystal structure of mouse LC3B in complex with the FYCO1 LIR reveals the importance of the flanking region of the LIR motif. Acta Crystallogr. Sect F Struct Biol Commun. [Internet]. 2017;73:130–137. Available from: http://scripts.iucr.org/cgi-bin/paper?S2053230X17001911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yang A, Pantoom S, Wu YW. Elucidation of the anti-autophagy mechanism of the legionella effector ravz using semisynthetic LC3 proteins. Elife. 2017;6:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lv M, Wang C, Li F, et al. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell. 2017;8:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Noda NN, Kumeta H, Nakatogawa H, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes to Cells. 2008;13:1211–1218. [DOI] [PubMed] [Google Scholar]
- [43].Schwarten M, Stoldt M, Mohrlüder J, et al. Solution structure of Atg8 reveals conformational polymorphism of the N-terminal domain. Biochem. Biophys Res Commun. [Internet]. 2010;395:426–431. [DOI] [PubMed] [Google Scholar]
- [44].Kumeta H, Watanabe M, Nakatogawa H, et al. The NMR structure of the autophagy-related protein Atg8. J Biomol NMR. 2010;47:237–241. [DOI] [PubMed] [Google Scholar]
- [45].Hong SB, Kim B-W, Lee K-E, et al. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol. [Internet]. 2011;18:1323–1330. [DOI] [PubMed] [Google Scholar]
- [46].Kondo-Okamoto N, Noda NN, Suzuki SW, et al. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J Biol Chem. 2012;287:10631–10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Noda NN, Satoo K, Fujioka Y, et al. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol Cell. [Internet]. 2011;44:462–475. [DOI] [PubMed] [Google Scholar]
- [48].Maqbool A, Hughes RK, Dagdas YF, et al. Structural basis of host autophagy-related protein 8 (ATG8) Binding by the irish potato famine pathogen effector protein PexRD54. J Biol Chem. 2016;291:20270–20282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Van Der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. [DOI] [PubMed] [Google Scholar]
- [50].Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.