

ABSTRACT
RNA processing encompasses the capping, cleavage, polyadenylation and alternative splicing of pre-mRNA. Proper muscle development relies on precise RNA processing, driven by the coordination between RNA-binding proteins. Recently, skeletal muscle biology has been intensely investigated in terms of RNA processing. High throughput studies paired with deletion of RNA-binding proteins have provided a high-level understanding of the molecular mechanisms controlling the regulation of RNA-processing in skeletal muscle. Furthermore, misregulation of RNA processing is implicated in muscle diseases. In this review, we comprehensively summarize recent studies in skeletal muscle that demonstrated: (i) the importance of RNA processing, (ii) the RNA-binding proteins that are involved, and (iii) diseases associated with defects in RNA processing.
KEYWORDS: RNA processing, skeletal muscle, alternative splicing, transcription, epigenetics
Introduction
Skeletal muscle
In healthy humans, skeletal muscle accounts for approximately 35–45% of body composition [1,2]. Skeletal muscle is a highly organized contractile organ that converts chemical energy into mechanical energy to allow for functional movement. Skeletal muscle serves a variety of other biological functions besides maintaining posture and body position. It protects the openings of the urinary and digestive tracts, regulates body temperature, and regulates nutrient homeostasis by acting as a major storage site for lipids and carbohydrates [3,4]. Throughout the body, skeletal muscles come in various shapes and sizes; however, the basic anatomy is the same. Bundles of multinucleated muscle cells (myofibers) make up a whole muscle and within the myofibers are thousands of myofibrils that contain billions of myofilaments [1,3] (Figure 1(a)). Skeletal muscle consists of 80% protein, most of which are the myofilament proteins, actin and myosin. Myofilament proteins form the sarcomere and are the major contractile unit of a skeletal muscle [1,3]. Sarcomeres are organized into parallel and overlapping thin (actin) and thick (myosin) filaments that undergo cycles of actin-myosin cross-bridge formation [3,5] (Figure 1(a), bottom right).
Figure 1.
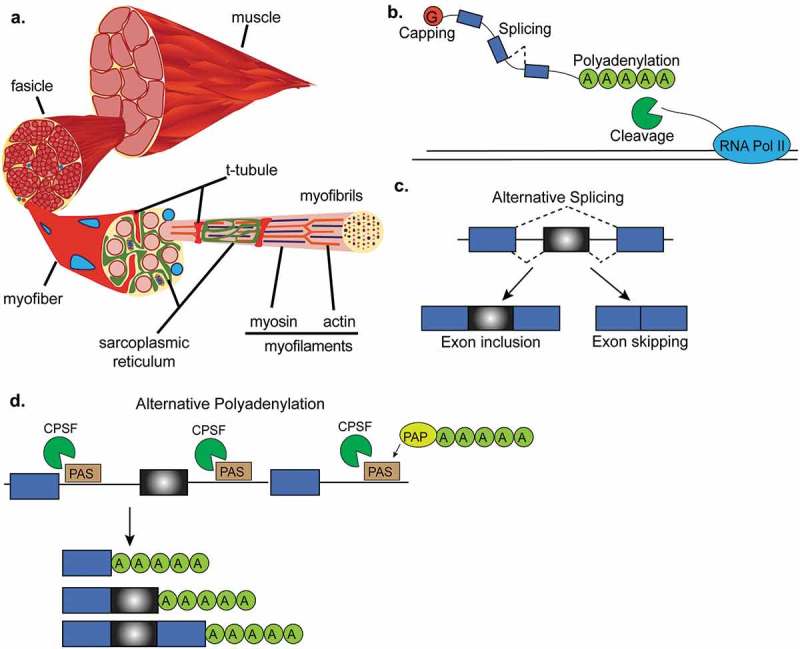
RNA processing in skeletal muscle. (a). Cross-section of a representative skeletal muscle. Individual multinucleated muscle fibers (blue dots) are bundled into fascicles. Each muscle fiber is made of myofibrils that contain the myofilaments, actin and myosin. T-tubules penetrate the muscle fiber and come into close proximity to the sarcoplasmic reticulum which surround the myofilaments. (b). Interactive view of RNA processing. After RNA polymerase-II (RNA Pol-II) transcribes DNA, newly synthesized mRNA is capped, cut and polyadenylated by the cleavage and polyadenylation factor, and then spliced. (c). Alternative splicing of a cassette exon that can either be included or skipped in the final mRNA transcript. (d). Alternative polyadenylation site selection. The 3ʹUTRs of some genes contain multiple polyadenylation signals that can be recognized by the polyadenylation and cleavage factor (CPSF). After the cleavage and polyadenylation factor binds to a polyadenylation signal (PAS), the polyA polymerase (PAP) adds the string of adenosines to the end of the mRNA transcript. This panel shows an example where three mRNA transcripts are generated by different polyadenylation site selection.
The unique architecture of skeletal muscle ensures for rapid and synchronous conduction of an action potential and subsequent release of calcium from the sarcoplasmic reticulum [6] which is essential for the excitation-contraction coupling needed to generate force. The transverse tubular system within muscle cells is formed by the transverse tubules (T-tubules) which are deep invaginations of the plasma membrane allowing for the close connection of the extracellular environment with the sarcoplasmic reticulum. Both T-tubules and the sarcoplasmic reticulum mature within the first three postnatal weeks in mouse skeletal muscle and are essential for adult muscle function [6,7]. Muscle contraction occurs when membrane depolarization and subsequent calcium release from the sarcoplasmic reticulum facilitates cross-bridge formation between actin and myosin. Myosin acts as the motor of the muscle because of an ATPase domain in the myosin head. The calcium-dependent troponin-complex and tropomyosin associate with the actin filament to prevent it from binding to myosin until excitation-contraction occurs [5,8]. Thus, muscle contraction is a result of sliding of the filaments and shortening of the sarcomere.
Myofiber types and metabolism
The ability of skeletal muscle to perform a very diverse array of functions throughout the body is afforded by a heterogeneity in the types of myofibers that differ in their contractile ability and metabolic requirements. Classically, myofibers are categorized by their expression of the myosin heavy chain ATPase isoforms [9]. In general, myofibers expressing the slow type I myosin ATPase (slow-twitch myofibers) rely on oxidative metabolism, are resistant to fatigue, and have a slow rate of contraction. Type II myofibers express a fast myosin heavy chain, contract and fatigue rapidly, and rely on glycolytic metabolism [9]. Muscle fiber types are further sub-classified based on developmental stage, contractile speed, color, oxidative activity, pH lability of actomyosin ATPase, motor unit innervation, and fatigability among others [3,8].
Extensive RNA processing in muscle biology
Decades of research have demonstrated that skeletal muscle is one of the most dynamic tissues in the body. Muscle has an inherent ability to fine tune its response to environmental and physiological changes, including but not limited to exercise, diet, disuse, and disease [10–13]. Many adaptations of skeletal muscle are through transcriptional programs that modulate patterns of gene expression. However, mechanisms that directly target RNA also have the potential to modulate muscle physiology, suggesting that posttranscriptional mechanisms have important regulatory functions in skeletal muscle biology.
RNA processing
RNA processing encompasses four main processes: (a) capping, (b) alternative splicing, (c) cleavage, and (d) polyadenylation of the pre-mRNA to produce a mature mRNA (Figure 1(b)). Capping of the 5ʹ end of the RNA is necessary for mRNA translation, the recruiting of proteins involved in alternative splicing [14] and polyadenylation, and to prevent transcript degradation by 5ʹ to 3ʹ exonucleases [15] (Figure 1(b)). Alternative splicing of pre-mRNA is a co- and posttranscriptional mechanism of gene regulation by which one gene can encode multiple protein isoforms due to the inclusion or exclusion of alternative regions (Figure 1(c)). Alternative splicing increases proteome diversity from a limited number of protein-coding genes. mRNA cleavage and polyadenylation are regulated by the cleavage and polyadenylation factor (CPSF). The 3ʹ end of newly transcribed pre-mRNAs are cleaved from RNA polymerase-II (Pol-II) followed by the addition of a polyadenine tail (polyA) to the 3ʹ end of a pre-mRNA transcript to prevent mRNA degradation (Figure 1(d)). Importantly, alternative polyadenylation creates different 3ʹ untranslated regions (3ʹUTR) which can impact transcript stability and thus further contribute to gene expression regulation.
Capping in muscle biology
Skeletal muscle contains 50–75% of total body protein [1] and the balance between protein synthesis and degradation governs muscle mass [12]. Thus, research focused on the molecular mechanisms regulating mRNA translation has been instrumental in understanding how skeletal muscle responds to environmental and physiological challenges. The terminal 5ʹ nucleotide of all Pol-II transcribed eukaryotic mRNAs contains an N7-methylated guanosine cap structure (5ʹm7G) [15]. The capping process is coupled to transcription and occurs within the first 25–30 nucleotides of newly transcribing RNAs [15–17]. The m7G cap serves a central role in the life cycle and biological function of an mRNA and is largely regulated by the binding of protein factors within the nucleus and cytoplasm. In the nucleus, the heterodimeric cap binding complex comprised of CBP80 and CBP20 proteins [18], interacts with a variety of other protein complexes to influence pre-mRNA splicing [19], polyadenylation processing [20], exosomal mediated RNA degradation [21], and nuclear export [22–24]. Once an mRNA is exported to the cytoplasm, the cap binding complex, still bound to the m7G cap, recruits eukaryotic initiation factors (eIF) to initiate translation of the mRNA [25–27].
Possibly the most well studied mechanism controlling mRNA translation in skeletal muscle is the mechanistic target of rapamycin complex-1 (mTORC1) pathway. mTORC1 activity is positively regulated by anabolic signals like growth factors, mechanical loading, and nutrients following feeding [28–31] and negatively regulated by catabolic conditions like starvation, aging, and disuse, among others [32–34]. Upon its activation, mTORC1 phosphorylates two known substrates that regulate cap-dependent translation – eIF4E binding proteins 1 and 2 (4EBP1 and 4EBP2) and the 70 kDa ribosomal S6 protein kinase (p70S6K1, also known as S6K1). Hyperphosphorylation of 4EBP1 or 4EBP2 releases eIF4E and permits its interaction with eIF4G to promote the formation of eIF4F complex and subsequent 5ʹ cap recognition, ribosome loading, and translation initiation [25,35,36] (Figure 2, left). S6K1 phosphorylates several substrates involved with the 5ʹ mRNA cap and translation initiation, including programmed cell death-4 (PDCD4) and eIF4B [37,38] (Figure 2, right).
Figure 2.
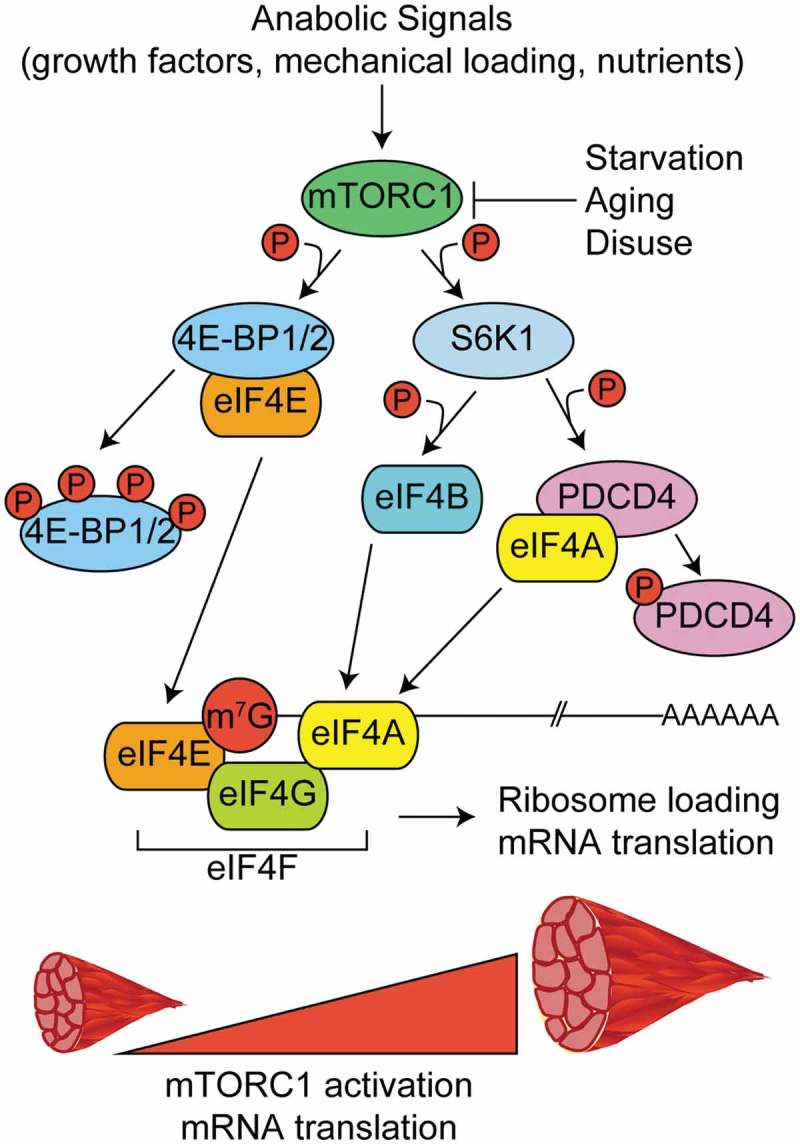
Regulation of mRNA capping and translation initiation by mTORC1. In response to anabolic stimuli, mTORC1 phosphorylates multiple downstream targets to govern the formation of the multi-protein eukaryotic initiation factor 4F complex (eIF4F) and subsequent translation initiation. The eIF4F complex is comprised of eIF4E (m7G mRNA cap binding protein), eIF4G (initiation factor scaffold protein), and eIF4A (RNA helicase) and its formation is the rate limiting step in translation initiation. Phosphorylation of eIF4E binding proteins 1 and 2 (4EBP1 and 4EBP2) frees eIF4E and allows it to bind to the m7G cap. The kinase activity of the 70 kDa ribosomal S6 protein kinase (S6K1) is increased in response to phosphorylation by mTORC1. S6K1 phosphorylates programmed cell death-4 (PDCD4) and relieves its inhibitory effect on eIF4A. S6K1 also phosphorylates eIF4B which further promotes helicase activity of eIF4A.
S6K1 regulation of skeletal muscle
Myotubes that express a constitutively active form of S6K1 have increased fiber diameter indicative of skeletal muscle hypertrophy [39]. Several studies have demonstrated reductions in total body and muscle mass in S6K1 knockout mice compared to their wildtype counterparts [40,41]. The reduction in skeletal muscle mass has been attributed to a reduction in myofiber cross-sectional area and not to a loss in myofiber number [41]. Furthermore, muscle fiber cross-sectional area was increased in S6K1 knockout mice when electroporated with a constitutively active AKT, an upstream activator of mTORC1, suggesting that S6K1 is not necessary for muscle hypertrophy in vivo [42]. Cotransfection of a constitutively active AKT and an non-phosphorylatable form of 4EBP1 also induced muscle hypertrophy in wildtype animals; however, muscle hypertrophy was reduced in S6K1 knockout mice, suggesting that 4EBP1 compensates for the loss of S6K1 in muscle growth [42]. Functional studies also demonstrated that loss of S6K1 reduced muscle force normalized to the muscle weight [42]. Collectively, these data suggest that there are multiple pathways regulating skeletal muscle growth and furthermore that S6K1 plays a role in regulating muscle hypertrophy and muscle force.
Protein synthesis and 4EBP1 and 4EBP2
Relatively little is known about the role of 4EBP1 and 4EBP2 in the regulation of skeletal muscle mass. The 4EBP1 and 4EBP2 double knockout mouse revealed no effect on body mass, lean mass, or the absolute mass of the gastrocnemius or soleus muscle under basal or septic conditions [43]. Sepsis, but not loss of 4EBP1 and 4EBP2 reduced in vivo rates of protein synthesis, the magnitude of reduction was similar between wild type and knockout mice, and these changes were concurrent with reductions in the phosphorylation of 4EBP1 and S6K1 [43]. Sepsis also caused a 55% reduction in eIF4E bound to eIF4G in the wild type mice; however, the double knockout mice showed no difference even though protein synthesis was decreased [43]. These findings suggested that other mechanisms control translation initiation after 5ʹ cap recognition. Combined, these data demonstrate that mTORC1 governs translation initiation through its kinase activity toward factors that regulate 5ʹm7G cap binding.
Alternative splicing in skeletal muscle
Alternative splicing allows single genes to give rise to multiple mature mRNAs (Figure 1(c)). Skeletal muscle, together with brain and heart, are the tissues that exhibit the highest levels of tissue-specific and conserved alternative splicing [44]. Skeletal muscle undergoes extensive physiological changes during development to produce fully functional adult muscles [45]. At the molecular level, these changes are accompanied by numerous transcriptional and posttranscriptional changes, including those controlled by alternative splicing mechanisms [46,47]. Alternative splicing changes are controlled mostly by RNA-binding proteins (RBPs) that change their expression levels during development. Below we describe some examples of the functional importance of alternative splicing in muscle patho-physiology.
BIN1 mis-splicing, t-tubule defects, and muscle weakness
Bridging integrator-1 (BIN1), also known as amphiphysin-2, is a BAR-domain protein involved in membrane curvature dynamics and endocytosis [48]. In skeletal muscles, BIN1 is involved in the formation of T-tubules and membrane tubular invaginations [48–51]. Several exons of the Bin1 gene are regulated by alternative splicing in different tissues, developmental stages, and pathological conditions [49,51,52]. Exon 11 is only expressed in skeletal muscles and encodes a phosphoinositide binding domain [49,51]. Phosphoinositides bind membrane proteins like BIN1 to aid in membrane trafficking, reorganization of the cytoskeleton, and cell signaling [53,54]. The inclusion of exon 11 is developmentally regulated in skeletal muscles mostly between birth and adulthood [49], a postnatal time window that overlaps the formation of the T-tubules. When exon 11 is skipped, BIN1 lacks the phosphatidylinositol 5-phosphate binding site and thus this isoform is not able to regulate membrane tubulation and formation of the T-tubules [49] (Figure 3(a)). In humans with myotonic dystrophy (DM), exon 11 is lacking in adulthood leading to severe alterations in T-tubule organization and muscle weakness. When Bin1 splicing is manipulated in mouse models to force the skipping of exon 11, animals develop defects in T-tubule organization and muscle weakness, similar to what is observed in humans suffering with DM [49]. A mutation that affects exon 11 splicing has been identified in a family with rapidly progressive centronuclear myopathy [55]. Interestingly, a mutation in the same acceptor splice site is the genetic cause of inherited centronuclear myopathy in dogs. In both humans and dogs, the mutations led to the skipping of exon 11, ultrastructural defects in muscle, muscle weakness, and atrophy [55]. Collectively, these data demonstrate that splicing regulation of exon 11 of the Bin1 gene is evolutionary conserved and is implicated in T-tubule biogenesis and maintenance and ultimately in muscle strength in health and disease.
Figure 3.
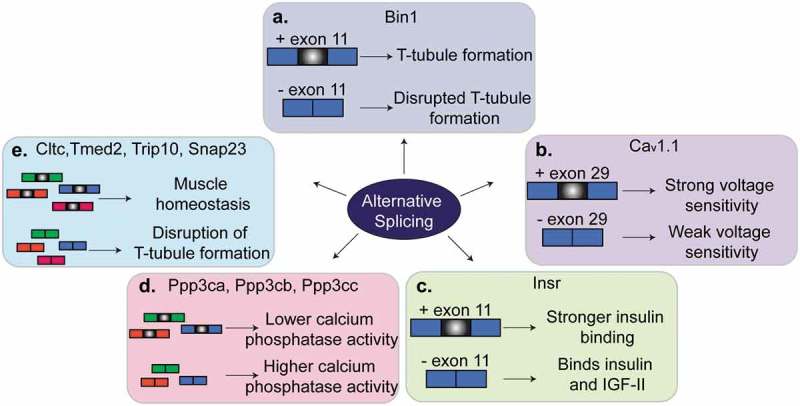
Regulation of alternative splicing in muscles. Several genes that are alternatively spliced produce different phenotypes in muscle depending on whether the alternative exon is included or skipped. (a). Alternative splicing of exon 11 of Bin1 pre-mRNA contributes to T-tubule formation and maintenance. (b). Skipping of exon 29 in Cav1.1 results in weak voltage sensitivity in adult skeletal muscle. (c). Inclusion of exon 11 of Insr pre-mRNA allows for stronger binding of insulin by the insulin receptor and thus insulin sensitivity. Skipping of exon 11 in adult muscles results in insulin resistance. (d). A switch from the adult to the fetal splicing isoforms of Ppp3ca, Ppp3cb and Ppp3cc leads to higher calcium phosphatase activity. (d). A switch in the splicing isoform of Cltc, Tmed2, Trip10 or Snap23 contributes to muscle homeostasis and T-tubule maintenance.
Cav1.1 mis-splicing and muscle wasting
The CACNA1S gene encodes the Cav1.1 protein which is a calcium channel and voltage sensor that controls excitation-contraction coupling in muscle cells. Inclusion of exon 29 of the CACNA1S gene is developmentally regulated and encodes for amino acids that connect two transmembrane domains of the protein [56]. Skipping of exon 29 shortens the loop connecting the two domains and causes weak voltage sensitivity [56,57] (Figure 3(b)). In healthy adult muscles, exon 29 is mostly included, while in individuals with DM exon 29 is absent [58]. The characteristic muscle weakness and centralized nuclei in humans with DM is also observed in mice administered an antisense oligonucleotide (ASO) to block inclusion of exon 29 [58]. Alternative splicing of CACNA1S is antagonistically regulated by two RBPs, muscle blind like-1 (MBNL1) and CUGBP Elav-Like Family Member-1 (CELF1), which are involved in DM and will be discussed later in this review [58].
Insulin receptor splicing and metabolism
The INSR gene encodes the insulin receptor and exon 11 is alternatively spliced giving rise to two protein isoforms, INSR-A and INSR-B. INSR-A and INSR-B exist in a ratio that depends on tissue contexts, developmental stage, or disease conditions [59–61]. The INSR-A form lacks exon 11, is prevalent in fetal stages, and is highly expressed in the nervous system, whereas the INSR-B form includes exon 11, is prevalent in adulthood, and is highly expressed in adipose tissue, liver, and muscle [60,61]. The INSR-A splice variant prefers binding of insulin growth factor-2 (IGF2) over insulin growth factor-1 (IGF1) and insulin, while the INSR-B is more sensitive to insulin [61,62] (Figure 3(c)). Alternative splicing of the INSR gene is regulated by multiple RBPs, including CELF1, MBNL1, and the heterogenous ribonucleoprotein family (HNRNP) family [63–65]. In skeletal muscle, insulin and IGF1 activate the PI3K/AKT/mTOR pathway which determines myofiber size [66]. Mis-splicing of INSR pre-mRNA is observed in diabetes and muscular dystrophies [67] where there is a switch from the expression of the INSR-B variant to the INSR-A isoform in muscle tissues. This transition alters PI3K/AKT signaling and leads to insulin resistance [59,67].
Alternative splicing of calcineurin-A and activation of NFATC signaling
Calcium plays a key role in muscle contraction and force generation [68]. Several genes encoding proteins involved in calcium homeostasis are alternatively spliced in skeletal muscle development [69]. Calcineurin is a calcium-dependent phosphatase that controls calcium handling and skeletal muscle hypertrophy by transducing external signals to the nucleus [70]. Indeed, skeletal muscle growth is governed by calcineurin-dependent signaling events [68]. During skeletal muscle development, there are three genes encoding the catalytic subunit of calcineurin-A (Ppp3ca, Ppp3cb, and Ppp3cc) that are regulated by alternative splicing transitioning from the expression of fetal to adult isoforms [47]. The fetal isoform proteins encoded by these genes have a higher calcineurin phosphatase activity which induces the nuclear localization of NFATC, a family of transcription factors involved in muscle fiber-specificity [47,70–72] (Figure 3(d)). Experiments using ASOs in adult mice and differentiated myotubes in culture, revealed that when the fetal isoforms were forced to be expressed in adult muscles, there was a prolonged muscle relaxation time, the calcineurin target NFATC3 showed a more nuclear localization, and transcription of several NFATC targets was affected.
Alternative splicing of trafficking genes and muscle cell architecture
Skeletal muscle is a complex tissue composed of membrane compartments and tubular invaginations that are important for excitation-contraction coupling. Adult skeletal muscle cells are larger than fetal and neonatal ones and thus need specialized membrane components for proper muscle contraction. Membrane trafficking proteins regulate internalization, recycling and degradation of ion channels, receptors and membrane components. Numerous genes encoding membrane trafficking and endocytosis proteins are alternatively spliced during mouse skeletal muscle development to aid in the transition of fetal to adult muscles [47,73]. Reversion of four trafficking genes (Cltc, Tmed2, Trip10, and Snap23) to the fetal alternative splicing patterns led to enlarged muscle fibers and disruption of T-tubule organization which demonstrated the importance of adult alternative splice isoforms in the maintenance of muscle homeostasis [73] (Figure 3(e)).
In summary, alternative splicing is important for the development of fully functional skeletal muscles. Alterations in alternative splicing contribute to the development of muscle weakness, irregular metabolism, and defects in intracellular architecture. However, a vast number of splice isoforms have not yet been studied and the functional consequences of alternative splicing are still under-investigated.
Alternative polyadenylation in muscle biology
Polyadenylation is the terminal step of mRNA processing, during which immature mRNA is cleaved from Pol-II and a string of 100–250 adenosine monophosphates are added to the transcript (polyA tail) (Figure 1(d)). The addition of the polyA tail prevents enzymatic cleavage of the transcript, allows for nuclear export, and promotes efficient translation initiation. A highly-conserved set of proteins comprises the polyadenylation complex and includes the cleavage and polyadenylation specificity factor (CPSF) and the cleavage stimulation factor (CstF). CPSF recognizes the polyadenylation signal (a cis AAUAAA motif) and cleaves 10–30 nucleotides downstream of a CA-site, releasing Pol-II from the nascent mRNA. The polyadenylation complex is regulated by surrounding elements, typically found within the 3ʹUTRs (for recent reviews [74,75]).
Numerous genes contain multiple polyadenylation sites. Commonly, alternative polyadenylation (APA) sites are found within the 3ʹUTRs and, therefore, their selection does not alter the translated polypeptide sequence but can critically affect stability. Such sites are termed UTR-APAs [76]. Alternative polyadenylation sites can also be located upstream of a coding element and therefore, site selection results in alternative transcripts and polypeptides. These sites are termed coding region-alternative polyadenylation (CR-APA) [76]. In CR-APA, a single gene can give rise to multiple transcripts and the resulting proteins may have different functions. APA at these locations may impact mRNA stability, translation efficiency, nuclear export, and protein function [75,76].
The regulation of polyadenylation site selection is highly dynamic and is influenced by numerous RBPs. The advent of next generation sequencing has allowed for increased understanding of alternative polyadenylation site selection in a tissue-specific manner. RNA-sequencing analysis in C. elegans has revealed widespread use of alternative polyadenylation sites in muscle tissue [77]. Furthermore, muscle tissue has distinct polyadenylation signatures with approximately 30% of transcripts using tissue-specific alternative polyadenylation [77]. Tissue and developmental-specific expression of RBPs, microRNAs (miRNA), and other regulatory factors contribute to alternative polyadenylation site selection during muscle development and are discussed in detail later in the review.
Posttranscriptional regulation of muscle-specific genes by alternative polyadenylation and miRNAs
Several of the alternatively polyadenylated 3ʹUTRs identified in C. elegans contain miRNA binding sites, suggesting that alternative polyadenylation is a tissue-specific posttranscriptional regulatory mechanism [77]. One specific example of such regulation has been demonstrated for the Paired box gene-3 (PAX3) which is a major regulator of muscle stem cell development [78]. Upon myogenic differentiation, PAX3 mRNA expression is significantly downregulated via the miRNA miR-206 and miR-1 [79,80]. Quiescent muscle stem cells express PAX3 at varying degrees with particular subpopulations paradoxically expressing high PAX3 and high miR-206 [78]. Myogenic progenitors, preferentially utilize an upstream polyadenylation site in PAX3 which shortens the 3ʹUTR and allows transcripts to escape miRNA-induced silencing [78] (Figure 4(a)).
Figure 4.
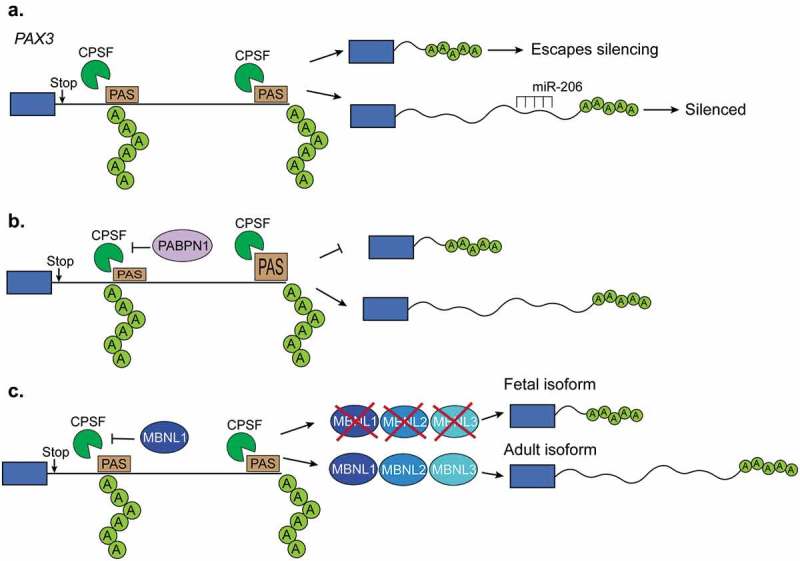
Regulation of polyadenylation in muscles. (a). The length of Pax3 3ʹUTR determines whether the transcript can be silenced by the microRNA miR-206. When a proximal polyadenylation site is used, the 3ʹUTR is shorter and lacks the miR-206 binding site and, in consequence, the transcript can escape the miR-206-induced silencing. In contrast, when a distal polyA site is used, the 3ʹUTR contains those binding sites leading to the silencing. (b). The polyA binding protein-1 (PABPN1) suppresses weak polyadenylation and cleavage sites by blocking the cleavage and polyadenylation factor (CPSF) from binding to these sites. Therefore, PABPN1 promotes the usage of a distal polyA site and thus longer transcripts by this mechanism. (c). Triple knockout of MBNL proteins in mice leads to the reactivation of fetal polyadenylation programs that preferentially use distal sites and thus generates short 3ʹUTRs. MBNL proteins bind similar sites as the CPSF does. In this way, binding of MBNL proteins close to the proximal polyA sites prevents CPSF from binding and promotes the selection of more distal polyA sites.
RNA-binding proteins in skeletal muscle physiology
During skeletal muscle development, temporal regulation of transcriptional and posttranscriptional networks are essential for proper function [45] (Figure 5). RBPs control alternative splicing, mRNA stability, and polyadenylation in skeletal muscle. In terms of alternative splicing regulation, RBPs exert this regulatory role by helping the spliceosome during splicing reactions, recruiting other splicing factors or adaptor molecules, or directly promoting skipping or inclusion of the alternative region [81]. The regulation of mRNA stability determines whether the final mRNA transcript is degraded before translation occurs. Synthesis and decay rates of mRNA transcripts are determined by the transcript itself and its interaction with RBPs [82,83]. Polyadenylation site selection is also controlled in muscle by several RBPs and this regulation contributes to an overall muscle-specific polyadenylation profile [84]. The resulting transcripts can then be regulated by miRNAs, ultimately allowing for muscle-specific gene expression programs. In the next paragraphs, we describe several families of RBPs that are involved in RNA processing in skeletal muscles.
Figure 5.
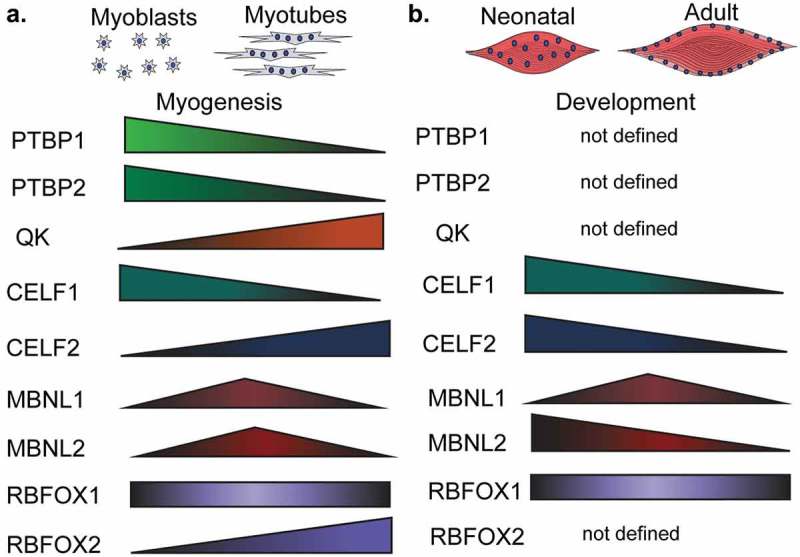
Expression of RNA-binding proteins during muscle development and myogenesis. The expression levels of several RNA-binding proteins (RBPs) change during muscle cell differentiation (myogenesis) (a) and skeletal muscle development (b). CELF: CUGBP Elav-like family member, MBNL: muscleblind like protein, QK: quaking, PTBP: polypyrimidine binding proteins, RBFOX: RNA binding fox-1 homolog.
HNRNPs
Members of the HNRNP family lack a serine-arginine rich domain and tend to promote exon skipping; however, some members of the family can act as both splicing repressors and activators [85,86]. HNRNPs often compete with serine-arginine rich domain proteins for binding sites on pre-mRNAs. HNRNPF and HNRNPA1 regulate, for example, alternative splicing of the INSR gene by binding to intronic and exonic splicing regulatory elements [63]. Additionally, HNRNPH1 controls alternative splicing of the RNA binding fox-1 homolog-2 (RBFOX2) [87], a critical splicing regulator in skeletal muscle. Indeed, HNRNPH1 depletion caused splicing changes in RBFOX2 downstream targets [87]. HNRNPH1 is upregulated in myotonic dystrophy type-1 (DM1) and by interacting with two other RBPs that are misregulated in this disease (CELF1 and MBNL) contributes to mis-splicing of the INSR [88]. The preferential expression of INSR-A over INSR-B ultimately leads to insulin resistance in individuals with DM1 [88].
Two HNRNPs that have been implicated extensively in alternative splicing regulation are the polypyrimidine binding proteins, PTBP1 and PTBP2. These two proteins are very similar in their sequences and RNA-binding abilities [89]. PTBP1 and PTBP2 bind preferentially to CUCUCU-motifs [90] and promote exon inclusion or exon skipping depending on their binding location relative to the alternative exon [91–93]. Even though PTBP1 and PTBP2 are similar, some alternative splicing events respond more to one protein than the other [94]. PTBP1 protein levels decrease during myotube formation in the C2C12 mouse muscle cell line [46], a well-established model of myogenesis in vitro (Figure 5(a)). To the best of our knowledge, PTBP2 has not been yet investigated in the context of muscle cell differentiation or skeletal muscle development.
Quaking protein
QK protein levels increase during myogenesis [95] (Figure 5(a)) and QK regulates alternative splicing decisions by binding to ACUAA-motifs, which are known to be enriched in regions downstream of muscle-specific exons. QK also binds to UAAY-motifs located 1–20 nucleotides after the ACUAA-motif [96]. When QK binds upstream of the alternative region, it promotes its inclusion. When QK binds downstream, it promotes skipping [95]. Together with PTBP1, QK co-regulates alternative splicing of an important set of muscle-specific genes during myogenesis [95]. Almost 40% of the genes regulated by alternative splicing during myogenesis are controlled by one or both of these RBPs [95].
CELF proteins
CELF proteins bind to GU-rich sequences and are involved in different aspects of RNA processing [97]. In vitro, CELF1 is upregulated during myogenesis (Figure 5(a)) [46] and its depletion leads to stabilization of GU-rich transcripts [98]. CELF1 and CELF2 are elevated in fetal muscle but are downregulated during postnatal development (Figure 5(b)) [46,47]. Constitutive CELF1 knockout mice exhibited growth, viability, and spermatogenesis defects [99] and several splicing and mRNA stability CELF1 targets were identified in the hearts of these animals [100]. CELF1 and MBNL1 compete for similar 3ʹUTR binding sites and act antagonistically in skeletal muscles and hearts [101]. Indeed, CELF proteins can destabilize mRNAs while MBNL proteins aid in their translation [98,102].
PABPN1
The poly-adenine-binding protein nuclear-1 (PABPN1) is a major component of the alternative polyadenylation complex [103]. PABPN1 blocks weak polyadenylation sites and PABPN1 depletion has been shown to cause genome-wide 3ʹUTR shortening [104], (Figure 4(b)). Muscle expression of PABPN1 decreases during aging and human mutations in the PABPN1 gene are associated with oculopharyngeal muscular dystrophy (OPMD) [105] An OPMD-associated PABPN1 mutant displayed a general preference for proximal polyadenylation cleavage sites [104]. More recently, studies have investigated the role of PABPN1 in polyadenylation site selection within internal gene regions [105]. When PABPN1 function is lost, alternative polyadenylation site selection displays an overall preference for distal polyadenylation sites, indicating a mechanism for transcriptome changes in aged muscle and in OPMD [105]. Interestingly, transcripts that use the distal polyadenylation site are sequestered in the nucleus and, thus, not efficiently translated [105]. Therefore, PABPN1 appears to have multiple roles in polyadenylation site selection and may regulate translational silencing in muscle during aging and OPMD.
MBNL proteins
The muscleblind-like family of proteins, MBNL1 and MBNL2 extensively regulate alternative splicing during muscle development [84]. MBNL proteins also bind the 3ʹUTR of nascent mRNA transcripts and are key regulators of polyadenylation site selection [84]. MBNL proteins tend to bind to YGCY-motifs [106] and are downregulated during mouse skeletal muscle development [47] (Figure 5(b)). MBNL1 depletion in mice causes several of the symptoms, splicing changes, and polyadenylation switches observed in humans suffering with DM [84,107,108] Similarly, MBNL1 depletion in mouse embryonic fibroblasts altered polyA site selection for thousands of genes and induced a global reversion in polyadenylation site selection back to a fetal profile which is present in DM both in humans and mice (Figure 4(c)) [84]
RBFOX proteins
Fox homolog proteins (RBFOX) are highly expressed in adult skeletal muscles (Figure 5(b)) and are required for tissue development and muscle cell differentiation [46,109–111]. RBFOX proteins tend to bind to GCATG-sequences at a location greater than 500 nucleotides from the regulated alternative region [112]. RBFOX1 can also bind to expanded CCUG-repeats that are a hallmark of myotonic dystrophy type-2 (DM2) [113]. MBNL proteins compete with RBFOX1 for binding to CCUG-repeats. In fact, overexpression of RBFOX1 results in less MBNL1 sequestration and less of a DM2 phenotype possibly due to the correction of alternative splicing changes caused by MBNL1 sequestration [113]. Deletion of RBFOX1 in mice causes extensive alternative splicing changes, loss of muscle function, and reduced force production [110]. RBFOX2 depletion prevents myoblast fusion partially through splicing regulation of the transcription factor known as myocyte-specific enhancer factor-2D (Mef2d) [111,114]. Furthermore, depletion of both RBFOX1 and RBFOX2 in mice resulted in a severe loss of muscle mass and alteration of splicing of numerous transcripts [109]. Among these changes, these mice exhibit mis-splicing of the calpanin-3 expressing an active form of this protease that leads to an increase in protein degradation by the proteasome [109]. Combined, these data demonstrate the importance of RBFOX proteins in RNA processing in skeletal muscle physiology.
In summary, RBPs are key regulators of RNA processing in skeletal muscles and misregulation of their expression, localization, or functions leads to defects in mRNA stability, alternative splicing, and polyadenylation. In the next section, we focus on the epigenetic regulation of myogenesis and further in the review we detail muscle disorders caused by defects in RNA processing and RNAbased therapies.
Epigenetic regulation of myogenesis
RBPs have long been known to influence muscle architecture. However, only within the last 20 years has the role of epigenetics and myogenesis and lineage determination been uncovered. Perhaps the most well-known regulator of myogenesis is the myoblast determination protein (MYOD1) which associates with histone-modifiers to promote muscle cell differentiation. Specifically, MYOD1 initiates a feed-forward myogenic program that facilitates the differentiation of myoblasts into myotubes [115]. Early studies revealed that histone deacetylase I (HDAC I) associates with MYOD1 in myoblasts, thereby inhibiting transcription of MYOD1 target genes [116,117]. In contrast, a myogenic program of gene expression is induced in differentiating muscle cells when MYOD1 is associated with histone acetyl transferases (HATs) [116–118], (Figure 6). Indeed, genome-wide studies have demonstrated that myogenesis leads to changes in global histone modifications [119] While the overall distribution of histone modifications appear to be conserved during differentiation, there is a marked reduction of histone 3 (H3) acetylation in myotubes compared with myoblasts [119]. In addition, H3 trimethylation of lysine 27 (H3K27me3) is associated with gene repression in myotubes [119].
Figure 6.

Regulation of myogenesis by MyoD1 activation of muscle genes. In myoblasts, MyoD1 associates with histone deacetylases (HDACs), leading to repression of muscle genes. During differentiation, MyoD1 interacts with histone acetyl transferases (HATs) to deposit acetylation (Ac) on histones of MyoD1 target genes. Acetylation of these muscle target genes promotes muscle fate determination.
The importance of histone acetylation in striated muscle identity has recently been expanded. Particularly, the nucleosome remodeling and deacetylase (NuRD) complex regulates tissue identity. Loss of Chd4, the catalytic subunit of the NuRD complex in cardiomyocytes activates skeletal muscle lineage genes. Interestingly, deletion of Chd4 in skeletal muscle cells promotes the expression of cardiac genes [120]. Therefore, epigenetic mechanisms, particularly histone acetylation, are important for myogenesis and muscle specification. Still, the contribution of histone modifications and epigenetics in general to RNA processing in muscle cells is unknown.
Although the relationship between RNA processing and epigenetics has yet to be investigated in the context of skeletal muscle, evidence from neuronal and cancer cell lines suggests that histone modifications can mediate alternative splicing [121–126]. Membrane depolarization of neuroblastoma cells hyperacetylated the region surrounding exon 18 in the neural cell adhesion molecule (NCAM) gene resulting in exon skipping [121,122]. Creating a more permissive chromatin environment via treatment with an HDAC inhibitor increased exon skipping whereas enrichment of repressive marks around exon 18 promoted its inclusion [121,122]. Histone marks have also been shown to interact with RNA binding proteins directly or through adaptor proteins [125,126]. For example, the MORF-related gene 15 (MRG15) mediates the interaction between H3K36me3 and the splicing factor PTBP1 [125]. This suggests a model by which histone marks recruit splicing machinery and in turn regulate exon inclusion or skipping. Future studies investigating the connection between splicing and epigenetics in a muscle context may elucidate important regulatory mechanisms that control muscle development and regeneration.
RNA processing in muscle diseases and therapies
DM
Myotonic dystrophy is a disease characterized by muscle wasting, relaxation and damage to the nervous system [84,113,127–130]. There are two main forms of adult onset myotonic dystrophy: DM1 and DM2. DM1 is caused by 50–3,000 CTG repeats in the 3ʹUTR of the DM1 protein kinase (DMPK) gene that accumulate over time; however, full penetrance of the disease occurs at 50 repeats [131,132] (Figure 7(a)). DM2 is caused by 75–11,000 CCUG repeats within intron 1 of the CCHC-type zinc finger nucleic acid binding protein gene (CNBP, also known as ZNF9) (Figure 7(b)) [133]. Both of these repeat expansions cause the formation of toxic hairpin RNAs that sequester RBPs, like MBNL proteins [134]. This results in misregulation of RNA processing. For example, sequestration of MBNL leads to its loss-of-function and thus a reversion of adult splicing and polyadenylation programs back to fetal ones [135,136]. DM1 is more severe than DM2 because CUG-repeats both sequester MBNL proteins (loss-of-function) and induce CELF hyperactivation (gain-of-function) [113,137]. The more CUG-repeats present appear to positively correlate with MBNL protein sequestration and severity of the disease [130]. The lower severity of DM2 compared to DM1 can be attributed to the fact that the CCUG-repeats do not sequester MBNL proteins as well as CUG-repeats do [113]. Furthermore, RBFOX proteins bind to CCUG-repeats but not to CUG-repeats which allows RBFOX proteins to compete with MBNL proteins in DM2, preventing complete MBNL1 sequestration. In this manner, the phenotype is less severe in DM2 than in DM1 [110].
Figure 7.
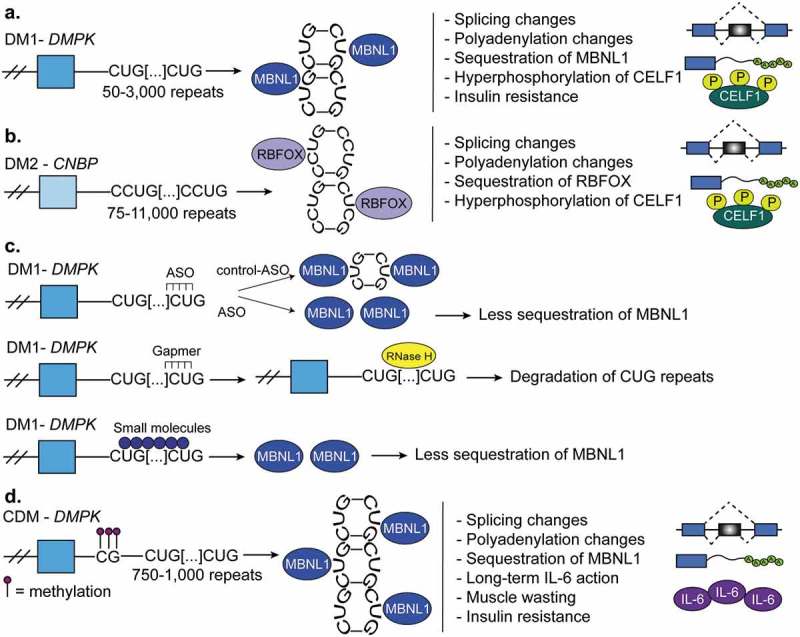
Myotonic dystrophy. (a). Myotonic dystrophy type-1 (DM1) is characterized by 50–3,000 CUG-repeats in the 3ʹUTR of DM1 protein kinase (DMPK) gene. The CUG-repeats cause RNA hairpins that sequester muscleblind like protein-1 (MBNL1) which leads to global changes in alternative splicing, polyadenylation, and mRNA stability. In DM1, the CUGBP Elav-like family member-1 (CELF1) is hyperphosphorylated and this leads to its gain-of-function. (b). Myotonic dystrophy type-2 (DM2) is characterized by 75–11,000 CCUG-repeats in the intron 1 of the CCHC-type zinc finger nucleic acid binding protein (CNBP, also known as ZNF9) gene. CCUG-hairpins sequester RNA binding fox-1 homolog proteins (RBFOX) which leads to extensive changes in alternative splicing and polyadenylation. CELF1 also becomes highly expressed and hyperphosphorylated in DM2. (c). Therapies for DM1. First, antisense oligonucleotides (ASOs) are used to block the transcription of portions CUG-repeats and in this manner MBNL sequestration is corrected. Second, gapmers that bind upstream of the CUG-repeats recruit RNAse-H which degrades the CUG-repeats and corrects the molecular defects of the disease. A third therapy is not RNA-based and instead uses various small molecules that block MBNL1 sequestration. This prevents the global misregulation of alternative splicing programs that occurs in DM1. (d). Congenital myotonic dystrophy (CDM) is characterized by 750–1,000 CUG-repeats at the 3ʹUTR of DMPK and hypermethylation of CpG islands upstream of the repeats. This results in sequestration of MBNL proteins as in DM1 and therefore, misregulation of alternative splicing, polyadenylation, and mRNA stability. Severe CDM results in muscle immaturity due to the improper activation of the cytokine, IL-6, an inhibitor of myoblast differentiation.
Several therapeutics are used in the treatment of DM (Figure 7(c)). ASOs are used to block the transcription of portions of CUG-repeats in the 3ʹUTR of the DMPK gene and in this manner correct MBNL sequestration. DMSXL mice have a mutant form of the human DMPK gene and a similar amount of CUG-repeats compared to humans with DM and treating these mice with the ASOs led to weight gain, improvement in muscle strength, and a 70% decrease in the abundance of CUG-repeat RNA in skeletal muscle [128]. A second therapy uses ASO gapmers that bind upstream of the CUG repeats. Gapmers recruit RNAse-H which degrades the CUG-repeats and corrects the molecular defects of the disease [138]. This is an effective therapy because mutant DMPK and RNAse-H normally accumulate in the nucleus. A third therapy is not RNA-based and instead uses various small molecules that block MBNL1 sequestration. This prevents the global misregulation of alternative splicing programs that occurs in DM1 [139].
Congenital myotonic dystrophy (CDM)
Congenital myotonic dystrophy (CDM) is a form of DM1 that is present prenatally and inherited in an autosomal dominant manner [129,130]. CDM is caused by the presence of 750–1,000 CUG-expansions in the 3ʹUTR of the DMPK gene [130] (Figure 7(d)). Interestingly, CDM exhibits unique hypermethylation of CpG sites near the CUG-repeats but the full implications of this CpG hypermethylation are still unknown [140] The degree of methylation upstream of the repeat prevents the transcriptional regulator CTCF from binding to the site [130] and the loss of CTCF regulation results in transcriptional dysregulation and transcription of more CTG-repeats [140]. Changes in alternative splicing and alternative polyadenylation are altered in tissues affected by CDM [130]. A Mbnl1/Mbnl2/Mbnl3 triple knockout mouse recapitulated numerous features of CDM and the reactivation of the fetal splicing and polyadenylation patterns [130] (Figure 4(c)), demonstrating the role of MBNL proteins in the disease. Finally, severe CDM results in muscle immaturity due to the improper activation of the cytokine, IL-6, an inhibitor of myoblast differentiation [140,141].
Duchenne muscular dystrophy (DMD)
DMD is an X-linked disease resulting in progressive muscle loss. The majority of DMD cases are caused by a reading frame mutation in the DMD gene which is 79 exons long and encodes the dystrophin protein which in muscles links actin with the extracellular matrix [142–144] (Figure 8(a)). Mis-splicing of some exons within the DMD gene has also been implicated in DMD; however, splicing variants contribute to less than 7% of all disease-causing variants. In 2016, the FDA approved the ASO known as Eteplirsen to promote the skipping of exon 51 in DMD pre-mRNA [143]. Skipping of exon 51 restores the reading frame and thus produces a partially functional DMD protein [143,144] (Figure 8(b)). Since Eteplirsen is applicable to approximately 14% of all DMD cases [143], other ASO technologies are currently in clinical trials [145]. Small molecule therapy has been used to generate compounds that increase exon 51 skipping [146]. Dantrolene is a small molecule that targets the ryanodine receptor and has been used to treat hyperthermia but also works to treat DMD [146]. Dantrolene promotes exon 51 skipping in the DMD mouse model (mdx) and also reprogrammed myotubes from DMD patients. The mode of action is suspected to be linked to the ryanodine receptor but the complete mechanism is not known [146].
Figure 8.
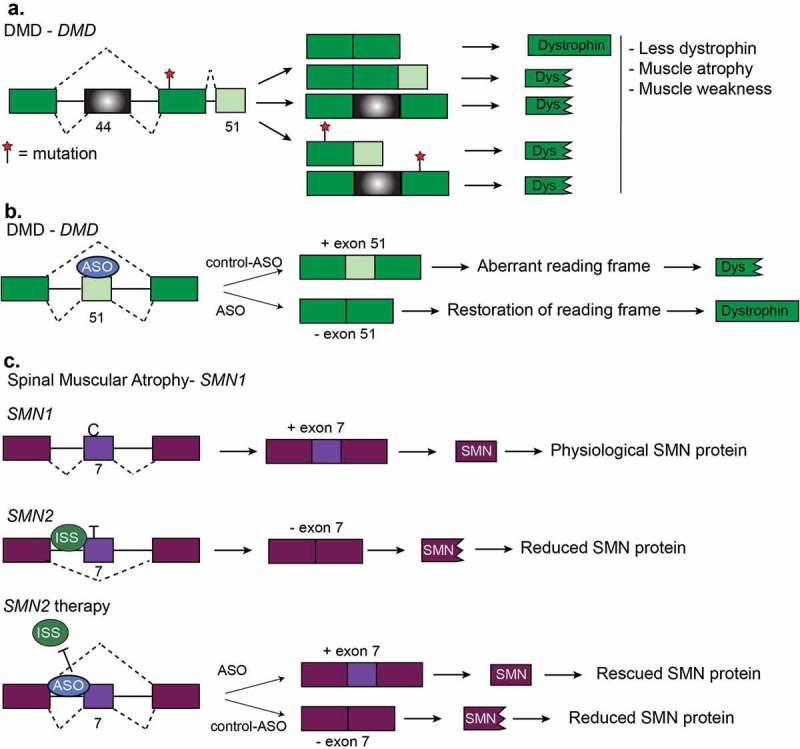
Other diseases and RNA-based therapies. (a). Duchenne muscular dystrophy (DMD) is characterized by mutations which disrupt the reading frame resulting in truncated dystrophin proteins. Truncated dystrophin leads to muscle atrophy and muscle weakness. Two frequent frameshift mutations are in exon 44 and exon 51. (b). Restoration of the DMD reading frame is possible using antisense oligonucleotides (ASOs). Here, ASOs are used to induce the skipping of exon 51 of DMD pre-mRNA. Skipping of this exon restores the reading frame and results the expression of functional dystrophin protein. (c). Generation of functional SMN protein in Spinal Muscular Atrophy (SMA). SMA is caused by a lack of survival motor neuron (SMN) protein. This happens when there is a deletion or mutation in the survival motor neuron-1 (SMN1) gene or increased copy number of SMN2. SMN2 produces much less functional SMN protein because of a C/T transition that prevents inclusion of exon 7. Multiple therapies have been devised for SMA that corrects for the loss of SMN1 by promoting exon inclusion in SMN2 via ASOs to increase SMN protein levels.
Amyotrophic lateral sclerosis (ALS)
ALS is a devastating and progressive neuromuscular disease characterized by motor neuron death. In recent years, the importance of RNA processing, and especially RBPs in the ALS pathology has been elucidated [147–149]. Numerous mutations associated with ALS give rise to protein or RNA aggregates that sequester RBPs outside of the nucleus and prevents them from regulating their targets. C9orf72 is the most commonly mutated gene in familial and sporadic ALS, accounting for approximately 40% and 10% of cases, respectively [150]. ALS-associated C9orf72 mutations (GGGGCC-repeat expansions) lead not only to transcriptional defects in the C9orf72 gene itself, but also to downstream RNA processing abnormalities [151,152]. Mutated C9orf72 RNA forms distinct aggregates in the cytosol which are thought to contribute to RNA toxicity via the sequestration of the HNRNP proteins outside of the nucleus [153]. A nearly universal clinical feature of ALS is the presence of cytoplasmic inclusion bodies composed of the DNA binding protein of 43 kDa (TDP43) [154,155]. In individuals with ALS, misfolded TDP43 aggregates in the cytosol of spinal motor neurons and glial cells leading to frontotemporal lobe degeneration [154]. Nuclear TDP-43 binds to thousands of RNAs and primarily is enriched at introns where it regulates alternative splicing [156,157]. The cytoplasmic aggregation of TDP43 depletes nuclear TDP43 and, thus, dysregulation of its targets is commonly observed in ALS [157]. TDP43 also plays a role in RNA stability and homeostasis [158], and whole genome RNA instability has been recently demonstrated in fibroblasts from individuals with ALS [159]. TDP43 interacts with other RNA processing factors, such as Matrin-3 (MATR3) and Fused-in-sarcoma (FUS) [160,161], that are frequently mutated in ALS [161–164]. While MATR3 is involved in polyadenylation site selection and intron retention via interactions with PABPN1 [162–164], FUS is a RBP that colocalizes with wild-type TDP43 in cytoplasmic inclusion bodies in models of ALS [161]. However, studies in autopsy human tissues do not support the idea that FUS and TDP43 overlap [165], indicating the need for additional studies to explore the potential interaction between FUS and TDP43 [166]. In summary, ALS-associated mutations in RBPs lead to dysregulation of RNA processing and stability. Furthermore, enrichment of mutations in multiple RBPs in the pathology of ALS indicates that RNA processing may play a vital role in the development of the disease.
Spinal muscular atrophy (SMA)
SMA is a progressive motor neuron disease that affects young children. SMA is caused by a lack of survival motor neuron (SMN) protein. This happens when there is a deletion or mutation in the survival motor neuron-1 (SMN1) gene or increased copy number of SMN2. SMN1 and SMN2 produce the same protein because SMN2 arose from a gene duplication event of SMN1. However, SMN2 produces less functional SMN protein because of a C/T transition that prevents an exonic enhancer from including exon 7 [167]. Multiple therapies have been devised for SMA that corrects for the loss of SMN1 by promoting exon inclusion in SMN2 via ASOs to increase SMN protein levels [168–170] (Figure 8(c)). This approach rescues the phenotype of SMA observed in a SMN mouse model and, in 2016, the FDA approved the ASO Nusinersen to treat SMA in humans [168,169]. A second therapy for SMA is to provide a copy of SMN via adenoassociated virus and this treatment resulted in a longer survival time of humans with SMA and increased the expression of SMN protein in mice [169,171]. Finally, small molecules are also efficacious in promoting SMN2 exon 7 inclusion [172], thus, several therapies targeted to RNA processing seem promising to treat SMA.
Perspectives
RNA processing contributes to skeletal muscle development and function and its misregulation can lead to devastating muscular diseases. Several RBPs regulate RNA processing in muscles and also contribute to the pathogenesis of diseases. However, there are numerous questions that still need to be answered about how RNA processing is regulated in skeletal muscle. First, there are likely thousands of splice variants and alternative polyadenylated transcripts with unexplored functions. Second, more research is needed to fully understand the functional consequences of global networks of alternative splicing and polyadenylation in muscles. Third, thorough mechanisms elucidating how RBPs are regulated during skeletal muscle development and diseases have not been completely defined. Fourth, cellular processes such as epigenetics and lifestyle could also play a role in RNA biology which can be explored in skeletal muscle. A greater understanding of the mechanisms controlling RNA processing in skeletal muscle will facilitate the development of new therapeutics to treat muscular diseases.
Funding Statement
The authors are funded by the following funding sources: a Junior Faculty Development Award from UNC-Chapel Hill (to J.G), a Pilot & Feasibility Research Grant (P30DK056350) from the Nutrition and Obesity Research Center (NORC, UNC-Chapel Hill) (to J.G), startup funds from UNC-Chapel Hill (to J.G), a NCTraCs Pilot Grant (550KR181805) from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through Grant Award Number UL1TR002489 (to J.G) and in part by the March of Dimes Foundation (5-FY18-36) (Basil O’Connor Starter Scholar Award) (to J.G). E.R.H is supported in part by the NIH-NIGMS training award T32 GM007092. H.J.W. is part of the MiBio (Mechanistic, interdisciplinary studies of Biological systems) institutional training program and is supported in part by the NIH-NIGMS training award T32 GM119999. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Frontera WR, Ochala J.. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int. 2015;96:183–195. [DOI] [PubMed] [Google Scholar]
- [2].Janssen I, Heymsfield SB, Wang Z, et al. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J Appl Physiol. 2000;89:81–88. [DOI] [PubMed] [Google Scholar]
- [3].Schiaffino S, Reggiani C.. Fiber types in mammalian skeletal muscles. Physiol Rev. 2011;91:1447–1531. [DOI] [PubMed] [Google Scholar]
- [4].Zierath JR, Hawley JA. Skeletal muscle fiber type: influence on contractile and metabolic properties. PLoS Biol. 2004;2:e348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Trovato F, Imbesi R, Conway N, et al. Morphological and functional aspects of human skeletal muscle. J Funct Morphol Kinesiol. 2016;1:289–302. [Google Scholar]
- [6].Takekura H, Flucher BE, Franzini-Armstrong C. Sequential docking, molecular differentiation, and positioning of T-tubule/SR junctions in developing mouse skeletal muscle. Dev Biol. 2001;239:204–214. [DOI] [PubMed] [Google Scholar]
- [7].Franzini-Armstrong C. Simultaneous maturation of transverse tubules and sarcoplasmic reticulum during muscle differentiation in the mouse. Dev Biol. 1991;146:353–363. [DOI] [PubMed] [Google Scholar]
- [8].Greising SM, Gransee HM, Mantilla CB, et al. Systems biology of skeletal muscle: fiber type as an organizing principle. Wiley Interdiscip Rev Syst Biol Med. 2012;4:457–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bassel-Duby R, Olson EN. Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem. 2006;75:19–37. [DOI] [PubMed] [Google Scholar]
- [10].Black AJ, Ravi S, Jefferson LS, et al. Dietary fat quantity and type induce transcriptome-wide effects on alternative splicing of pre-mRNA in rat skeletal muscle. J Nutr. 2017;147:jn254482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Clarke K, Ricciardi S, Pearson T, et al. The role of Eif6 in skeletal muscle homeostasis revealed by endurance training co-expression networks. Cell Rep. 2017;21:1507–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Phillips SM, Glover EI, Rennie MJ. Alterations of protein turnover underlying disuse atrophy in human skeletal muscle. J Appl Physiol. 2009;107:645–654. [DOI] [PubMed] [Google Scholar]
- [13].Sylow L, Kleinert M, Richter EA, et al. Exercise-stimulated glucose uptake — regulation and implications for glycaemic control. Nat Rev Endocrinol. 2017;13:133–148. [DOI] [PubMed] [Google Scholar]
- [14].Lenasi T, Peterlin BM, Barboric M. Cap-binding protein complex links pre-mRNA capping to transcription elongation and alternative splicing through positive transcription elongation factor b (P-TEFb). J Biol Chem. 2011;286:22758–22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ramanathan A, Robb GB, Chan SH. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44:7511–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moteki S, Price D. Functional coupling of capping and transcription of mRNA. Mol Cell. 2002;10:599–609. [DOI] [PubMed] [Google Scholar]
- [17].Shatkin AJ, Manley JL. The ends of the affair: capping and polyadenylation. Nat Struct Biol. 2000;7:838–842. [DOI] [PubMed] [Google Scholar]
- [18].Gonatopoulos-Pournatzis T, Cowling VH. Cap-binding complex (CBC). Biochem J. 2014;457:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pabis M, Neufeld N, Steiner MC, et al. The nuclear cap-binding complex interacts with the U4/U6·U5 tri-snRNP and promotes spliceosome assembly in mammalian cells. Rna. 2013;19:1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Flaherty SM, Fortes P, Izaurralde E, et al. Participation of the nuclear cap binding complex in pre-mRNA 3ʹ processing. Proc Natl Acad Sci USA. 1997;94:11893–11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Andersen PR, Domanski M, Kristiansen MS, et al. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol. 2013;20:1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Izaurralde E, Adam S. Transport of macromolecules between the nucleus and the cytoplasm. Rna. 1998;4:351–364. [PMC free article] [PubMed] [Google Scholar]
- [23].Nojima T, Hirose T, Kimura H, et al. The interaction between cap-binding complex and RNA export factor is required for intronless mRNA export. J Biol Chem. 2007;282:15645–15651. [DOI] [PubMed] [Google Scholar]
- [24].Daneholt B. Assembly and transport of a premessenger RNP particle. Proc Natl Acad Sci USA. 2001;98:7012–7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Grüner S, Peter D, Weber R, et al. The structures of eIF4E-eIF4G complexes reveal an extended interface to regulate translation initiation. Mol Cell. 2016;64:467–479. [DOI] [PubMed] [Google Scholar]
- [26].Jackson RJ, Hellen CUT, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pestova TV, Kolupaeva VG, Lomakin IB, et al. Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci USA. 2001;98:7029–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Frey JW, Jacobs BL, Goodman CA, et al. A role for Raptor phosphorylation in the mechanical activation of mTOR signaling. Cell Signal. 2014;26:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gordon BS, Williamson DL, Lang CH, et al. Nutrient-induced stimulation of protein synthesis in mouse skeletal muscle is limited by the mTORC1 repressor REDD1. J Nutr. 2015;145:708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gordon BS, Steiner JL, Lang CH, et al. Reduced REDD1 expression contributes to activation of mTORC1 following electrically induced muscle contraction. Am J Physiol Endocrinol Metab. 2014;307:E703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Rossetti ML, Fukuda DH, Gordon BS. Androgens induce growth of the limb skeletal muscles in a rapamycin-insensitive manner. Am J Physiol Integr Comp Physiol. 2018; 315:R721-R729. [DOI] [PubMed] [Google Scholar]
- [32].Kelleher AR, Kimball SR, Dennis MD, et al. The mTORC1 signaling repressors REDD1/2 are rapidly induced and activation of p70S6K1 by leucine is defective in skeletal muscle of an immobilized rat hindlimb. AJP Endocrinol Metab. 2013;304:E229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kelleher AR, Gordon BS, Kimball SR, et al. Changes in REDD1, REDD2, and atrogene mRNA expression are prevented in skeletal muscle fixed in a stretched position during hindlimb immobilization. Physiol Rep. 2014;2:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kelleher AR, Pereira SL, Jefferson LS, et al. REDD2 expression in rat skeletal muscle correlates with nutrient-induced activation of mTORC1: responses to aging, immobilization, and remobilization. Am J Physiol Endocrinol Metab. 2015;308:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gingras AC, Kennedy SG, O’Leary MA, et al. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12:502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Haghighat A, Mader S, Pause A, et al. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. Embo J. 1995;14:5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Suzuki C, Garces RG, Edmonds KA, et al. PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc Natl Acad Sci U S A. 2008;105:3274–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dennis MD, Shenberger JS, Stanley BA, et al. Hyperglycemia mediates a shift from cap-dependent to cap-independent translation via a 4E-BP1-dependent mechanism. Diabetes. 2013;62:2204–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rommel C, Bodine SC, Clarke BA, et al. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–1013. [DOI] [PubMed] [Google Scholar]
- [40].Binsch C, Jelenik T, Pfitzer A, et al. Absence of the kinase S6k1 mimics the effect of chronic endurance exercise on glucose tolerance and muscle oxidative stress. Mol Metab. 2017;6:1443–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ohanna M, Sobering AK, Lapointe T, et al. Atrophy of S6K1 −/− skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol. 2005;7:286–294. [DOI] [PubMed] [Google Scholar]
- [42].Marabita M, Baraldo M, Solagna F, et al. S6K1 is required for increasing skeletal muscle force during hypertrophy. Cell Rep. 2016;17:501–513. [DOI] [PubMed] [Google Scholar]
- [43].Steiner JL, Pruznak AM, Deiter G, et al. Disruption of genes encoding eIF4E binding proteins-1 and −2 does not alter basal or sepsis-induced changes in skeletal muscle protein synthesis in male or female mice. PLoS One. 2014;9:99582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Merkin J, Russell C, Chen P, et al. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science. 2012;338(6114):1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Apponi LH, Corbett AH, Pavlath GK. RNA-binding proteins and gene regulation in myogenesis. Trends Pharmacol Sci. 2011;32:652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bland CS, Wang ET, Vu A, et al. Global regulation of alternative splicing during myogenic differentiation. Nucleic Acids Res. 2010;38:7651–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brinegar AE, Xia Z, Loehr JA, et al. Extensive alternative splicing transitions during postnatal skeletal muscle development are required for Ca 2+ handling. Elife. 2017;6:e27192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Prokic I, Cowling BS, Laporte J. Amphiphysin 2 (BIN1) in physiology and diseases. J Mol Med. 2014;92:453–463. [DOI] [PubMed] [Google Scholar]
- [49].Fugier C, Klein AF, Hammer C, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17:720–725. [DOI] [PubMed] [Google Scholar]
- [50].Tjondrokoesoemo A, Park KH, Ferrante C, et al. Disrupted membrane structure and intracellular Ca 2+ signaling in adult skeletal muscle with acute knockdown of Bin1. PLoS One. 2011;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee E, Marcucci M, Daniell L, et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297:1193–1196. [DOI] [PubMed] [Google Scholar]
- [52].Pineda-Lucena A, Ho CSW, Mao DYL, et al. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J Mol Biol. 2005;351:182–194. [DOI] [PubMed] [Google Scholar]
- [53].Ribeiro I, Yuan L, Tanentzapf G, et al. Phosphoinositide regulation of integrin trafficking required for muscle attachment and maintenance. PLoS Genet. 2011;7:e1001295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. [DOI] [PubMed] [Google Scholar]
- [55].Böhm J, Vasli N, Maurer M, et al. Altered splicing of the BIN1 muscle-specific exon in humans and dogs with highly progressive centronuclear myopathy. PLoS Genet. 2013;9:e1003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tuluc P, Molenda N, Schlick B, et al. A Cav1.1 Ca2+ channel splice variant with high conductance and voltage-sensitivity alters EC coupling in developing skeletal muscle. Biophys J. 2009;96:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sultana N, Dienes B, Benedetti A, et al. Restricting calcium currents is required for correct fiber type specification in skeletal muscle. Development. 2016;143:1547–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Tang ZZ, Yarotskyy V, Wei L, et al. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Cav1.1 calcium channel. Hum Mol Genet. 2012;21:1312–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Santoro M, Masciullo M, Bonvissuto D, et al. Alternative splicing of human insulin receptor gene (INSR) in type i and type II skeletal muscle fibers of patients with myotonic dystrophy type 1 and type 2. Mol Cell Biochem. 2013;380:259–265. [DOI] [PubMed] [Google Scholar]
- [60].Mosthaf L, Grako K, Dull TJ, et al. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. Embo J. 1990;9:2409–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Seino S, Bell G. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun. 1989;159:312–316. [DOI] [PubMed] [Google Scholar]
- [62].Belfiore A, Frasca F, Pandini G, et al. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586–623. [DOI] [PubMed] [Google Scholar]
- [63].Talukdar I, Sen S, Urbano R, et al. HnRNP A1 and hnRNP F modulate the alternative splicing of exon 11 of the insulin receptor gene. PLoS One. 2011;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Echeverria GV, Cooper TA. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res. 2014;42:1893–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Sen S, Talukdar I, Webster NJG. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol Cell Biol. 2009;29:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bodine SC, Stitt TN, Gonzalez M, et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–1019. [DOI] [PubMed] [Google Scholar]
- [67].Savkur RS, Philips AV, Cooper TA, et al. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am J Hum Genet. 2004;74:1309–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Michel RN, Dunn SE, Chin ER. Calcineurin and skeletal muscle growth. Proc Nutr Soc. 2004;63:341–349. [DOI] [PubMed] [Google Scholar]
- [69].Kimura T, Pace SM, Wei L, et al. A variably spliced region in the type 1 ryanodine receptor may participate in an inter-domain interaction. Biochem J. 2007;401:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Delling U, Tureckova J, Lim HW, et al. A calcineurin-NFATc3-dependent pathway regulates skeletal muscle differentiation and slow myosin heavy-chain expression. Mol Cell Biol. 2000;20:6600–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Liu Y, Cseresnyés Z, Randall WR, et al. Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers. J Cell Biol. 2001;155:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Chin ER, Olson EN, Richardson JA, et al. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998;12:2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Giudice J, Loehr JA, Rodney GG, et al. Alternative splicing of four trafficking genes regulates myofiber structure and skeletal muscle physiology. Cell Rep. 2016;17:1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lee KM, Tarn WY. Coupling pre-mRNA processing to transcription on the RNA factory assembly line. RNA Biol. 2013;10:380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2016;18:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Chen W, Jia Q, Song Y, et al. Alternative polyadenylation: methods, findings, and impacts. Genomics Proteomics Bioinformatics. 2017;15:287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Blazie SM, Babb C, Wilky H, et al. Comparative RNA-Seq analysis reveals pervasive tissue-specific alternative polyadenylation in Caenorhabditis elegans intestine and muscles. BMC Biol. 2015;13:4 DOI: 10.1186/s12915-015-0116-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Boutet SC, Cheung TH, Quach NL, et al. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell. 2012;10:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Hirai H, Verma M, Watanabe S, et al. MyoD regulates apoptosis of myoblasts through microRNA-mediated down-regulation of Pax3. J Cell Biol. 2010;10:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Crist CG, Montarras D, Pallafacchina G, et al. Muscle stem cell behavior is modified by microRNA-27 regulation of Pax3 expression. Proc Natl Acad Sci U S A. 2009;11:13383–13387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Fredericks AM, Cygan KJ, Brown BA, et al. RNA-binding proteins: splicing factors and disease. Biomolecules. 2015;5:893–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Hasan A, Cotobal C, Duncan CDS, et al. Systematic analysis of the role of RNA-binding proteins in the regulation of RNA stability. PLoS Genet. 2014;10:e1004684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Batra R, Charizanis K, Manchanda M, et al. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell. 2014;56:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Martinez-Contreras R, Cloutier P, Shkreta L, et al. Alternative splicing in the postgenomic era. New York (NY): Springer Verlag New York; 2007. [Google Scholar]
- [86].Martinez-Contreras R, Fisette JF, Nasim FUH, et al. Intronic binding sites for hnRNP A/B and hnRNP F/H proteins stimulate pre-mRNA splicing. PLoS Biol. 2006;4:172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sun S, Zhang ZUO, Fregoso O, et al. Mechanisms of activation and repression by the alternative splicing factors RBFOX1/2. Rna. 2012;18:274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Paul S, Dansithong W, Kim D, et al. Interaction of musleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. Embo J. 2006;25:4271–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Li Q, Zheng S, Han A, et al. The splicing regulator PTBP2 controls a program of embryonic splicing required for neuronal maturation. Elife. 2014;3:e01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wagner EJ, Garcia-Blanco MA. RNAi-mediated PTB depletion leads to enhanced exon definition. Mol Cell. 2002;10:943–949. [DOI] [PubMed] [Google Scholar]
- [91].Xue Y, Zhou Y, Wu T, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Llorian M, Schwartz S, Clark TA, et al. Position-dependent alternative splicing activity revealed by global profiling of alternative splicing events regulated by PTB. Nat Struct Mol Biol. 2010;17:1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Keppetipola N, Sharma S, Li Q, et al. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol. 2012;47:360–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Tang ZZ, Sharma S, Zheng S, et al. Regulation of the mutually exclusive exons 8a and 8 in the CaV1.2 calcium channel transcript by polypyrimidine tract-binding protein. J Biol Chem. 2011;286:10007–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hall MP, Nagel RJ, Fagg WS, et al. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. Rna. 2013;19:627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Galarneau A, Richard S. Target RNA motif and target mRNAs of the quaking STAR protein. Nat Struct Mol Biol. 2005;12:691–698. [DOI] [PubMed] [Google Scholar]
- [97].Dasgupta T, Ladd A. The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA binding proteins. Wiley Interdiscip Rev RNA. 2012;3:104–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Vlasova IA, Tahoe NM, Fan D, et al. Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol Cell. 2008;29:263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kress C, Gautier-Courteille C, Osborne HB, et al. Inactivation of CUG-BP1/CELF1 causes growth, viability, and spermatogenesis defects in mice. Mol Cell Biol. 2007;27:1146–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Giudice J, Xia Z, Li W, et al. Neonatal cardiac dysfunction and transcriptome changes caused by the absence of Celf1. Sci Rep. 2016;6:35550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wang ET, Ward AJ, Cherone JM, et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015;25:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wang ET, Cody NAL, Jog S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kühn U, Gündel M, Knoth A, et al. Poly(A) tail length is controlled by the nuclear Poly(A)-binding protein regulating the interaction between Poly(A) polymerase and the cleavage and polyadenylation specificity factor. J Biol Chem. 2009;284:22803–22814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Jenal M, Elkon R, Loayza-Puch F, et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell. 2012;149:538–553. [DOI] [PubMed] [Google Scholar]
- [105].Abbassi-Daloii T, Yousefi S, de Klerk E, et al. An alanine expanded PABPN1 causes increased utilization of intronic polyadenylation sites. Aging Mech Dis. 2017;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Goers ES, Purcell J, Voelker RB, et al. MBNL1 binds GC motifs embedded in pyrimidines to regulate alternative splicing. Nucleic Acids Res. 2010;38:2467–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. [DOI] [PubMed] [Google Scholar]
- [108].Suenaga K, Lee KY, Nakamori M, et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PLoS One. 2012;7:e33218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Singh RK, Kolonin AM, Fiorotto ML, et al. Rbfox-splicing factors maintain skeletal muscle mass by regulating calpain3 and proteostasis. Cell Rep. 2018;24:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Pedrotti S, Giudice J, Dagnino-Acosta A, et al. The RNA-binding protein Rbfox1 regulates splicing required for skeletal muscle structure and function. Hum Mol Genet. 2015;24:2360–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Singh RK, Xia Z, Bland CS, et al. Rbfox2-coordinated alternative splicing of Mef2d and Rock2 controls myoblast fusion during myogenesis. Mol Cell. 2014;55:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Lovci MT, Ghanem D, Marr H, et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol. 2013;20:1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Sellier C, Cerro-Herreros E, Blatter M, et al. RbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat Commun. 2018;9:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Runfola V, Sebastian S, Dilworth FJ, et al. Rbfox proteins regulate tissue-specific alternative splicing of Mef2D required for muscle differentiation. J Cell Sci. 2015;128:631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Penn BH, Bergstrom DA, Dilworth FJ, et al. A MyoD -generated feed-forward circuit temporally patterns gene expression dining skeletal muscle differentiation. Genes Dev. 2004;18:2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Mal A, Sturniolo M, Schiltz RL, et al. A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: inhibition of the myogenic program. Embo J. 2001;20:1739–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Mal A, Harter ML. MyoD is functionally linked to the silencing of a muscle-specific regulatory gene prior to skeletal myogenesis. Proc Natl Acad Sci USA. 2003;100:1735–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Puri PL, Sartorelli V, Yang XJ, et al. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell. 1997;1:35–45. [DOI] [PubMed] [Google Scholar]
- [119].Asp P, Blum R, Vethantham V, et al. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci USA. 2011;108:E149–E158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Gómez-Del Arco P, Perdiguero E, Yunes-Leites PS, et al. The chromatin remodeling complex Chd4/NuRD controls striated muscle identity and metabolic homeostasis. Cell Metab. 2016;23:881–892. [DOI] [PubMed] [Google Scholar]
- [121].Schor IE, Rascovan N, Pelisch F, et al. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc Natl Acad Sci USA. 2009;106:43225–43230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Schor IE, Fiszbein A, Petrillo E, et al. Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. Embo J. 2013;32:2264–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Sims RJ, Millhouse S, Chen CF, et al. Recognition of trimethylated histone H3 Lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Saint-André V, Batsché E, Rachez C, et al. Histone H3 lysine 9 trimethylation and HP1γ favor inclusion of alternative exons. Nat Struct Mol Biol. 2011;18:337–344. [DOI] [PubMed] [Google Scholar]
- [125].Luco RF, Pan Q, Tominaga K, et al. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Yong-Eun K, Chungoo P, Kyoon Eon K, et al. Histone and RNA-binding protein interaction creates crosstalk network for regulation of alternative splicing. Biochem Biophys Res Commun. 2018;499:30–36. [DOI] [PubMed] [Google Scholar]
- [127].Jiang H, Mankodi A, Swanson MS, et al. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. [DOI] [PubMed] [Google Scholar]
- [128].Jauvin D, Chrétien J, Pandey SK, et al. Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol Ther Nucleic Acids. 2017;7:465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Nakamori M, Sobczak K, Puwanant A, et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol. 2014;74:862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Thomas JD, Sznajder ŁJ, Bardhi O, et al. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017;31:1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Fu YH, Pizzuti A, Fenwick RG Jr., et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. [DOI] [PubMed] [Google Scholar]
- [132].Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3 ′ untranslated region of the gene. Science. 1992;255:1253–1255. [DOI] [PubMed] [Google Scholar]
- [133].Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron I of ZNF9. Science. 2001;293:864–867. [DOI] [PubMed] [Google Scholar]
- [134].Yuan Y, Compton SA, Sobczak K, et al. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35:5474–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Thomas JD, Oliveira R, Sznajder ŁJ, et al. Myotonic dystrophy and developmental regulation of RNA processing. Compr Physiol. 2018;8:509–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. [DOI] [PubMed] [Google Scholar]
- [137].Kuyumcu-Martinez NM, Wang G-S, Cooper T. Increased steady state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyper-phosphorylation. Mol Cell. 2007;28:68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Wheeler TM, Leger AJ, Pandey SK, et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2014;488:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016;17:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Nakamori M, Hamanaka K, Thomas JD, et al. Aberrant myokine signaling in congenital myotonic dystrophy. Cell Rep. 2017;21:1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Pelosi M, De Rossi M, Barberi L, et al. IL-6 impairs myogenic differentiation by downmodulation of p90RSK/eEF2 and mTOR/p70S6K axes, without affecting AKT activity. Biomed Res Int. 2014;2014:206026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Tuffery-Giraud S, Miro J, Koenig M, et al. Normal and altered pre-mRNA processing in the DMD gene. Hum Genet. 2017;136:1155–1172. [DOI] [PubMed] [Google Scholar]
- [143].Lim KRQ, Maruyama R, Yokota T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther. 2017;11:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Echigoya Y, Lim KRQ, Trieu N, et al. Quantitative antisense screening and optimization for exon 51 skipping in duchenne muscular dystrophy. Mol Ther. 2017;25:2561–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Echevarría L, Aupy P, Goyenvalle A. Exon-skipping advances for Duchenne muscular dystrophy. Hum Mol Genet. 2018;27:163–172. [DOI] [PubMed] [Google Scholar]
- [146].Kendall GC, Mokhonova EI, Moran M, et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci Transl Med. 2012;4:164ra160. [DOI] [PubMed] [Google Scholar]
- [147].Kapeli K, Martinez FJ, Yeo GW. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet. 2017;136:1193–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Polymenidou M, Lagier-Tourenne C, Hutt KR, et al. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012;1462:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: A cross-sectional study. Lancet Neurol. 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Barker HV, Niblock M, Lee Y-B, et al. RNA misprocessing in C9orf72-linked neurodegeneration. Front Cell Neurosci. 2017;11:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Mori K, Weng SM, Arzberger T, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. [DOI] [PubMed] [Google Scholar]
- [153].Lee YB, Chen HJ, Peres JN, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA Foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. [DOI] [PubMed] [Google Scholar]
- [155].Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- [156].Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Xiao S, Sanelli T, Dib S, et al. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci. 2011;47:167–180. [DOI] [PubMed] [Google Scholar]
- [158].Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Tank EM, Figueroa-Romero C, Hinder LM, et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat Commun. 2018;9:2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Tada M, Doi H, Koyano S, et al. Matrin 3 is a component of neuronal cytoplasmic inclusions of motor neurons in sporadic amyotrophic lateral sclerosis. Am J Pathol. 2018;188:507–514. [DOI] [PubMed] [Google Scholar]
- [161].Farrawell NE, Lambert-Smith IA, Warraich ST, et al. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci Rep. 2015;5:13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Johnson JO, Pioro EP, Boehringer A, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17:664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Uemura Y, Oshima T, Yamamoto M, et al. Matrin3 binds directly to intronic pyrimidine-rich sequences and controls alternative splicing. Genes Cells. 2017;22:785–798. [DOI] [PubMed] [Google Scholar]
- [164].Banerjee A, Vest KE, Pavlath GK, et al. Nuclear poly(A) binding protein 1 (PABPN1) and matrin3 interact in muscle cells and regulate RNA processing. Nucleic Acids Res. 2017;45:10706–10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Zhang ZC, Chook YM. Structural and energetic basis of ALS-causing mutations in the atypical proline–tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc Natl Acad Sci U S A. 2012;109:12017–12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Deng HX, Zhai H, Bigio EH, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. 2002;4:20–26. [DOI] [PubMed] [Google Scholar]
- [168].Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Mendell JR, Al-Zaidy S, Shell R, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713–1722. [DOI] [PubMed] [Google Scholar]
- [170].Passini MA, Bu J, Richards AM, et al. Antisense oligonucleotides ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Azzouz M, Le T, Ralph G. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest. 2004;114:1726–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Naryshkin NA, Weetall M, Dakka A, et al. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 2014;345:688–693. [DOI] [PubMed] [Google Scholar]