

ABSTRACT
Macroautophagy/autophagy is an important intracellular mechanism for the maintenance of cellular homeostasis. Here we show that the CERKL (ceramide kinase like) gene, a retinal degeneration (RD) pathogenic gene, plays a critical role in regulating autophagy by stabilizing SIRT1. In vitro and in vivo, suppressing CERKL results in impaired autophagy. SIRT1 is one of the main regulators of acetylation/deacetylation in autophagy. In CERKL-depleted retinas and cells, SIRT1 is downregulated. ATG5 and ATG7, 2 essential components of autophagy, show a higher degree of acetylation in CERKL-depleted cells. Overexpression of SIRT1 rescues autophagy in CERKL-depleted cells, whereas CERKL loses its function of regulating autophagy in SIRT1-depleted cells, and overexpression of CERKL upregulates SIRT1. Finally, we show that CERKL directly interacts with SIRT1, and may regulate its phosphorylation at Ser27 to stabilize SIRT1. These results show that CERKL is an important regulator of autophagy and it plays this role by stabilizing the deacetylase SIRT1.
KEYWORDS: Autophagy, CERKL, deacetylation, retinitis pigmentosa, SIRT1, stabilization
Introduction
Macroautophagy (hereafter autophagy) is an important intracellular pathway, which plays a critical role in maintaining cellular homeostasis and regulating the response to environmental stressors [1–4]. When autophagy is activated, phagophores engulf the cytoplasmic material, and then mature into double-membrane autophagosomes; these subsequently fuse with lysosome, resulting in degradation of the delivered material [5–7]. Autophagy is classically described as a response that is immediately activated when cells are subjected to nutrient deprivation. However, increasing evidence shows that autophagy plays an important role in maintaining a stable intracellular environment by degrading toxic aggregates, misfolded proteins, and damaged organelles [2]. Dysregulation of autophagy is implicated in many diseases, including cancer, immune diseases and neurodegeneration [1,8–10].
In the eye, autophagy proteins are strongly expressed, and autophagic activity is increased under cellular stresses such as oxidative conditions, intense light and mitochondrial damage [11]. Therefore, autophagy is important to the health and function of the retinal pigment epithelium (RPE) and the retina itself [12,13]. In eyes, in addition to the function of maintaining the stable intracellular environment, a noncanonical autophagy pathway supports the phagocytosis of photoreceptor outer segments and maintains the retinoid levels in RPE [14]. Previous studies have shown that specific defects of autophagy in RPE result in alterations in autophagy flux and in vision defects [15], whereas photoreceptor-specific defects cause photoreceptor death and vision defects [12,13]. Alterations in autophagy flux are observed both in age-related macular degeneration (AMD) and in retinitis pigmentosa (RP) patients [16,17].
CERKL, one of the RP causative genes, was first identified in a RP Spanish family and later was associated with cone-rod dystrophy (CRD) [18–21]. Regarding CERKL location in the retina, immunofluorescence of mouse and zebrafish cryosections reveal a location in the ganglion cell layer (GCL), inner nuclear layer (INL), and the inner segments (IS) and outer segments (OS) of photoreceptors. Currently there is debate as to whether CERKL is located in RPE [22–27].
Previous studies of the function and disease mechanism of CERKL have shown that CERKL fails to show any kinase activity even though it shares an integral diacylglycerol kinase (DAGK) signature with CERK (ceramide kinase) [28–30]. Nonetheless, it has been demonstrated that CERKL protects cells from stress, especially oxidative stress [31]. In our previous study, we showed that CERKL interacts with TXN2 (thioredoxin 2) and acts as a novel player in the regulation of the mitochondrial PRDX (peroxiredoxin)-mediated antioxidant pathway [27]. These findings provide a new direction for research into CERKL, although further studies are still necessary. Recently, our team generated a stable cerkl knockout zebrafish model that displays rod-cone dystrophy and disturbance in photoreceptor OS phagocytosis; the molecular mechanism is still unclear [32]. As autophagy plays an important role in maintaining the stable intracellular environment, and the noncanonical pathway has a close connection with RPE phagocytosis, in this study we analyzed if and how CERKL regulated autophagy in vivo and in vitro.
In the present study, we demonstrated that in cerkl−/- zebrafish, the basal level of autophagy in photoreceptors and RPE cells was reduced prior to the onset of photoreceptor death. We also showed in vitro that CERKL played an important role in the formation of autophagosomes in APRE-19 cells. While CERKL did not directly affect the components of autophagosomes, it did affect post-transcriptional regulation of autophagy. CERKL affected the protein stability of SIRT1, the deacetylase of ATG5 and ATG7, by directly interacting with SIRT1 and regulating its phosphorylation. Our findings have suggested a novel molecular mechanism for the maintenance by CERKL of homeostasis and visual function in the retina.
Results
Autophagy is impaired in cerkl−/- zebrafish retinas
To understand the CERKL-mediated molecular mechanism in pathogenesis, we examined autophagy flux in cerkl−/- zebrafish retinas by determining the level of Lc3 and Sqstm1. LC3B-I is localized in the cytosol, and when it is conjugated with phosphatidylethanolamine and converted to LC3-II, the latter is present on phagophore membranes and autophagosomes and to a lesser extent in autolysosomes. Therefore, the assay of ‘LC3 puncta’ and the ratio of LC3-II to LC3-I provide a useful measure of autophagy initiation [33], while SQSTM1 is a well-characterized autophagy receptor and substrate that is elevated after autophagy inhibition [34,35]. Compared to the wild-type retinas, extracts of cerkl−/- retinas showed a significant decrease in the ratio of Lc3-II to Lc3-I, and a significant increase of Sqstm1 at 1-month old prior to the onset of retinal degeneration (RD) (Figures 1(a) and S1). A basal level of Lc3 puncta was present in control retinas, while cerkl−/- retinas showed fewer Lc3 puncta in IS, INL and RPE (Figure 1(b,c)). These results indicate that autophagy is impaired in cerkl−/- retinas.
Figure 1.
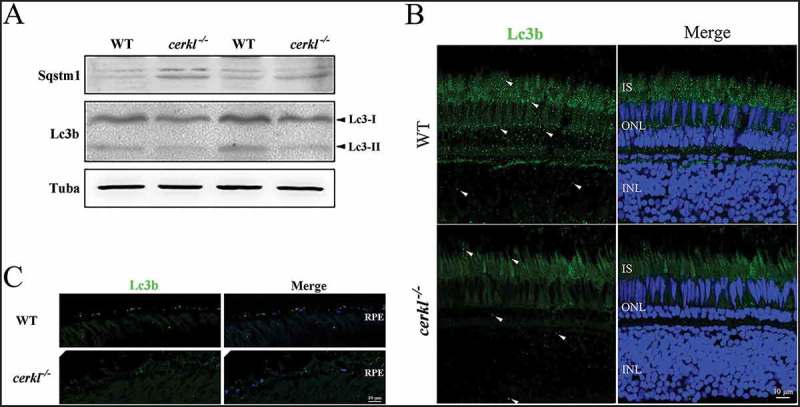
Autophagy flux was impaired in cerkl−/- retinas. (a) Immunoblotting of Lc3 and Sqstm1 in retinal extracts from wild-type (WT) and cerkl−/- zebrafish aged 1 month. (b) Immunostaining analysis of Lc3 in retinas of WT and cerkl−/- zebrafish aged 1 month. Scale bars: 10 μm. INL, inner nuclear layer; ONL, outer nuclear layer; IS, inner segments. (c) Immunostaining analysis of Lc3 in RPE of WT and cerkl−/- zebrafish aged 1 month. Scale bars: 10 μm.
Autophagy flux is impaired in CERKL-depleted ARPE-19 cells
To address the potential role of CERKL in regulating autophagy, we knocked down the expression of CERKL in ARPE-19 cells (Figure S2). As siCERKL1 was more effective than siCERKL2, we mainly used siCERKL1 (hereafter referred to as siCERKL) in subsequent experiments. Compared to the negative control (NC) ARPE-19 cells, the ratio of LC3-II to LC3-I was significantly decreased by 38% and SQSTM1 also increased by 23% in CERKL-depleted cells compared with the NC (Figures 2(a) and S3), indicating that basal autophagy was suppressed by knocking down CERKL.
Figure 2.
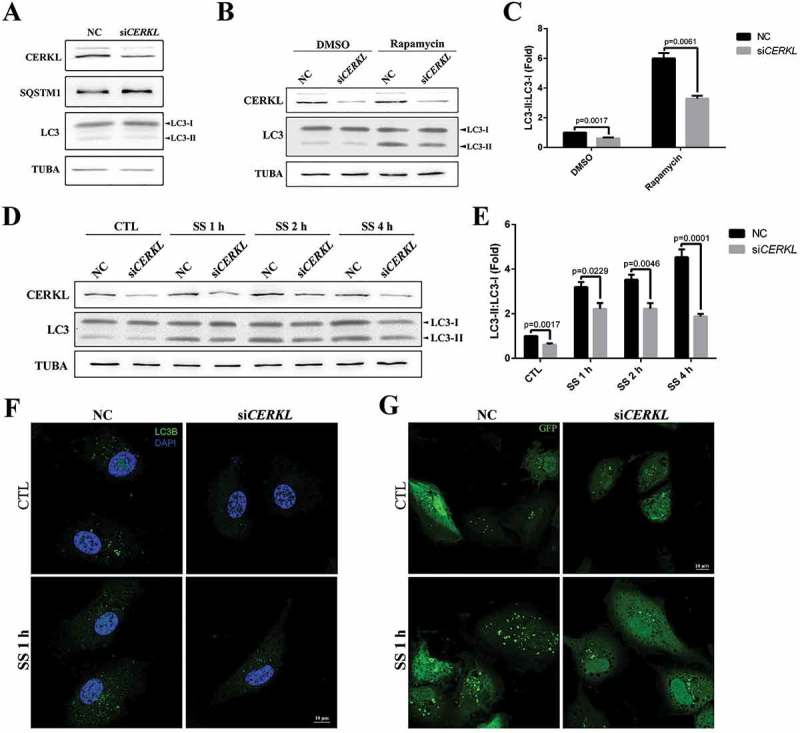
Autophagy was impaired in CERKL-depleted ARPE-19 cells. (a) Immunoblotting analysis of LC3 and SQSTM1 in negative control (NC) and CERKL-depleted ARPE-19 cells. (b and c) Immunoblotting and quantification of the levels of endogenous LC3-II:LC3-I ratio in NC and CERKL-depleted ARPE-19 cells post treatment with DMSO and 100 nM rapamycin for 2 h. Means± SEM of 5 repeats are shown. (d and e) Immunoblotting and quantification of the levels of endogenous LC3-II:LC3-I ratio in NC and CERKL-depleted ARPE-19 cells under normal and serum starvation (SS) condition for 1, 2 and 4 h. Means± SEM of 5 repeats are shown. (f) Immunostaining analysis of LC3 in NC and CERKL-depleted ARPE-19 cells induced by siRNA under normal and SS condition for 1 h. Scale bars: 10 μm. (g) Immunostaining of the distribution of exogenous GFP-LC3 in NC and CERKL-depleted ARPE-19 cells under normal and SS condition for 1 h. Scale bars: 10 μm.
In an effort to extend these results, autophagy was stimulated by rapamycin treatment or serum starvation in the NC and CERKL-depleted cells. As shown in Figure 2(b,c), treatment of NC cells with rapamycin caused a roughly 6-fold increase in autophagy, an effect that was suppressed in CERKL-depleted cells undergoing the same treatment. A similar result was observed when serum starvation was used to stimulate autophagy in CERKL-depleted cells: the level of autophagy increased with increased time of starvation in NC cells, whereas in CERKL-depleted cells the level of autophagy remained basically unchanged (Figure 2(d,e)).
To further confirm these results, the formation of endogenous LC3 puncta and exogenous GFP-LC3 puncta was analyzed in the NC and CERKL-depleted cells in both the normal and starved condition. As noted in Figure 2(f,g), the number of LC3 puncta decreased in CERKL-depleted cells in both conditions. We also quantified GFP-LC3 puncta formation. In CERKL-depleted cells, the number of GFP-LC3 puncta per cell decreased by 42.5% in normal conditions and decreased by 42.7% in the starved condition, compared with NC cells (Figure S4). Given that CERKL appears to be required for basal and starvation- and rapamycin-stimulated autophagy, we thought it possible that CERKL mainly affects the formation of autophagosomes.
In order to corroborate that autophagy flux was inhibited by the CERKL depletion, bafilomycin A1 was used to block the fusion of autophagosomes and lysosomes in NC and CERKL-depleted cells. As shown in Figure 3(a,b), the ratio of LC3-II to LC3-I increased approximately 4 times when treated with bafilomycin A1 in NC cells, whereas the ratio in CERKL-depleted cells increased approximately 3 times, indicating that CERKL-depletion did not block the autophagy flux. We also observed the colocalization of autophagosomes and lysosomes in NC and CERKL-depleted cells by transiently overexpressing GFP-LC3 and RFP-LAMP1 to detect the formation of autolysosomes. As noted in Figure 3(c), the decrease of autolysosomes in CERKL-depleted cells was mainly caused by the decreased formation of autophagosomes by CERKL depletion.
Figure 3.
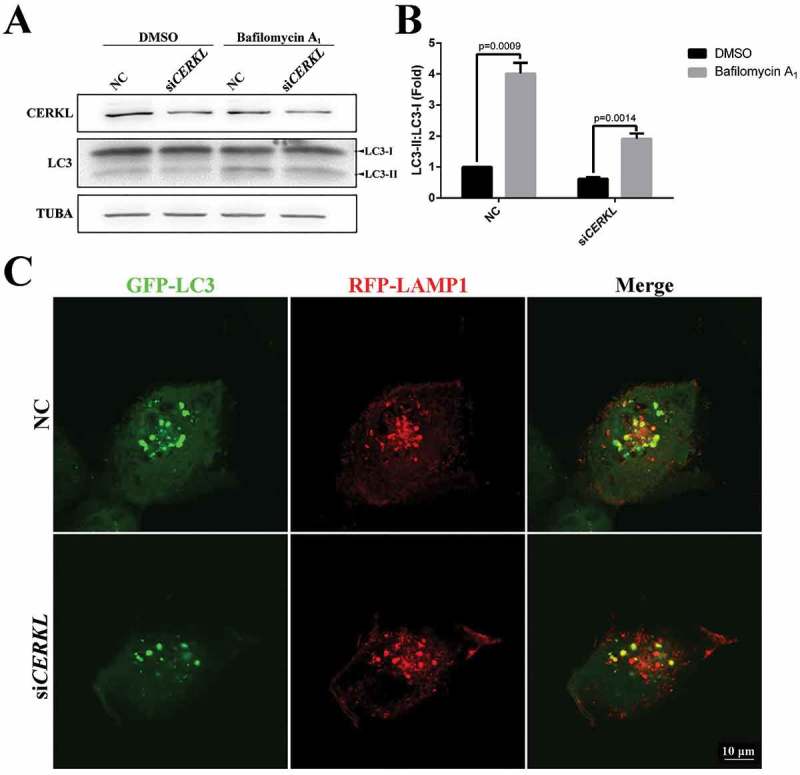
Autophagic flux was impaired in CERKL-depleted ARPE-19 cells. (a and b) Immunoblotting and quantification of the levels of endogenous LC3-II:LC3-I ratio in negative control (NC) and CERKL-depleted ARPE-19 cells under DMSO and 2.5 nM bafilomycin A1 condition for 3 h. Means± SEM of 5 repeats are shown. (c) Immunostaining analysis of the colocalization GFP-LC3 and RFP-LAMP1 in NC and CERKL-depleted ARPE-19 cells. Scale bars: 10 μm.
CERKL did not directly interact with the components of autophagosomes
Given that CERKL appears to be required for autophagy in ARPE-19 cells, we thought it possible that CERKL might directly influence the formation of autophagosomes. We therefore used immunofluorescence to investigate if CERKL might have a colocalization with autophagosomes. As seen in Figure 4(a), exogenous FLAG-CERKL shared no colocalization at all with LC3 puncta. Furthermore, there was no interaction between CERKL and ATG proteins (data not shown). We then considered if CERKL might participate in the process of autophagosome formation. As noted in Figure 4(b,d), the level of two of the major proteins of LC3 ubiquitin-like conjugation, ATG5 and ATG7, were assessed in NC or siCERKL ARPE-19 cells. No significant change was found. Based on these observations, we next investigated whether the upstream regulators of autophagy were impaired. The activation of the energy sensor AMP-activated protein kinase (AMPK) activates autophagy, and in atg5−/- mice, an autophagy-depleted model, there is increased activation of AMPK [36]. In CERKL-depleted cells, we also observed an increased activation of AMPK (Figure 4(c,d)), indicating that autophagy might be expected to be activated in CERKL-depleted cells, while in fact it was impaired.
Figure 4.
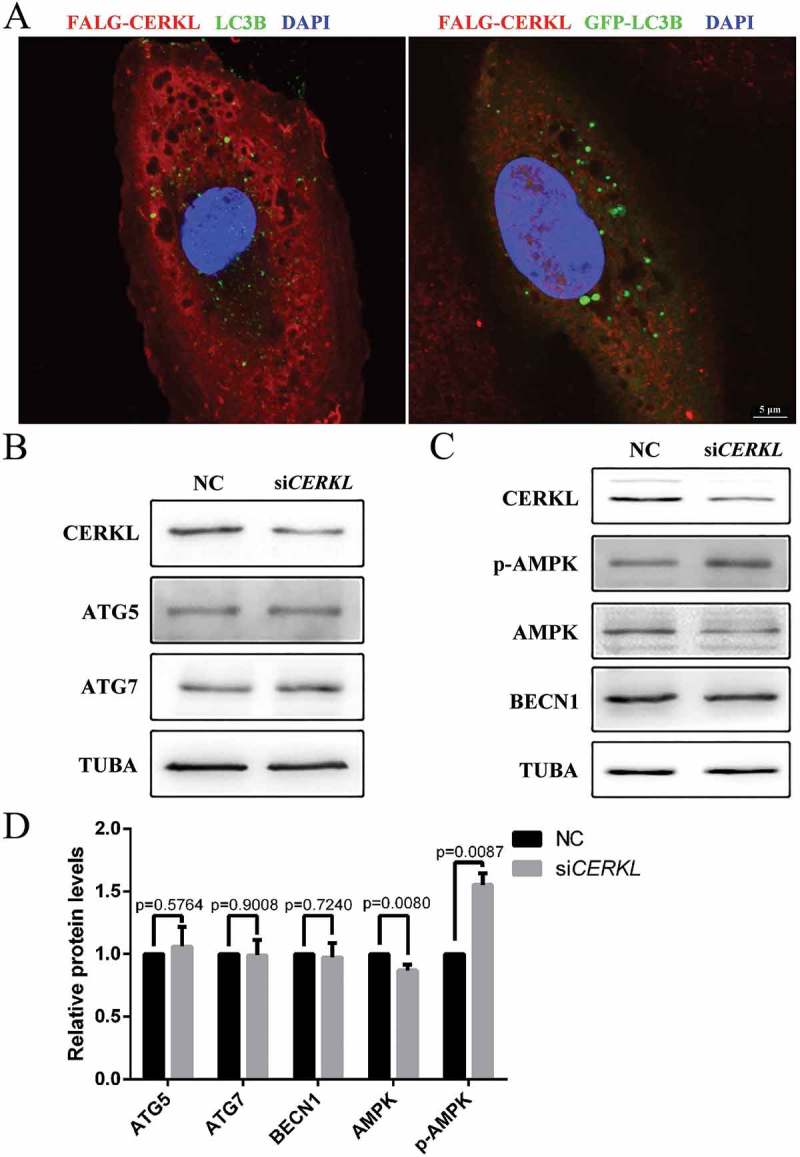
CERKL did not directly interact with the components of autophagosomes. (a) Immunostaining analysis of endogenous LC3B or exogenous GFP-LC3 and FLAG-CERKL in ARPE-19 cells. Scale bars: 5 μm. (b) Immunoblotting of the autophagy proteins ATG5 and ATG7 in NC and CERKL-depleted ARPE-19 cells. (c) Immunoblotting of the upstream regulator proteins of autophagy in NC and CERKL-depleted ARPE-19 cells. (d) Quantification of proteins presented in panels B and C. Relative expression of proteins in relation to TUBA. Means± SEM of 5 repeats are shown.
CERKL affected autophagy via SIRT1
A previous study suggests that autophagy is also regulated by protein post-translational modification [37]. Acetylation of autophagy proteins plays an important role in autophagic flux. SIRT1, a mammalian deacetylase, deacetylates several ATG proteins, such as ATG5, ATG7 and LC3, during starved conditions [38]. Thus, we examined the protein level of SIRT1 under a CERKL-depleted condition in vitro and in vivo. As seen in Figure 5(a-d), SIRT1 was significantly decreased by 52.2% in CERKL-depleted cells (siCERKL1) (Figure 5(a,b)), whereas in cerkl−/- zebrafish retina, Sirt1 levels decreased by approximately 70% (Figure 5(c,d)), indicating that CERKL affected autophagy via SIRT1. To confirm this hypothesis, we overexpressed SIRT1 in CERKL-depleted cells to rescue the impaired autophagy. As predicted, the ratio of LC3-II to LC3-I was increased after transient overexpression of SIRT1 in CERKL-depleted cells (Figure 5(e)). Given that CERKL affects autophagy via SIRT1, we assessed the level of acetylation in CERKL-depleted cells. As noted in Figure 6(a,b), the level of acetylated ATG5 and ATG7 both increased when CERKL was knocked down. Taken together, these data demonstrated that CERKL affected autophagy via the role of SIRT1 in regulating acetylation of ATG proteins.
Figure 5.
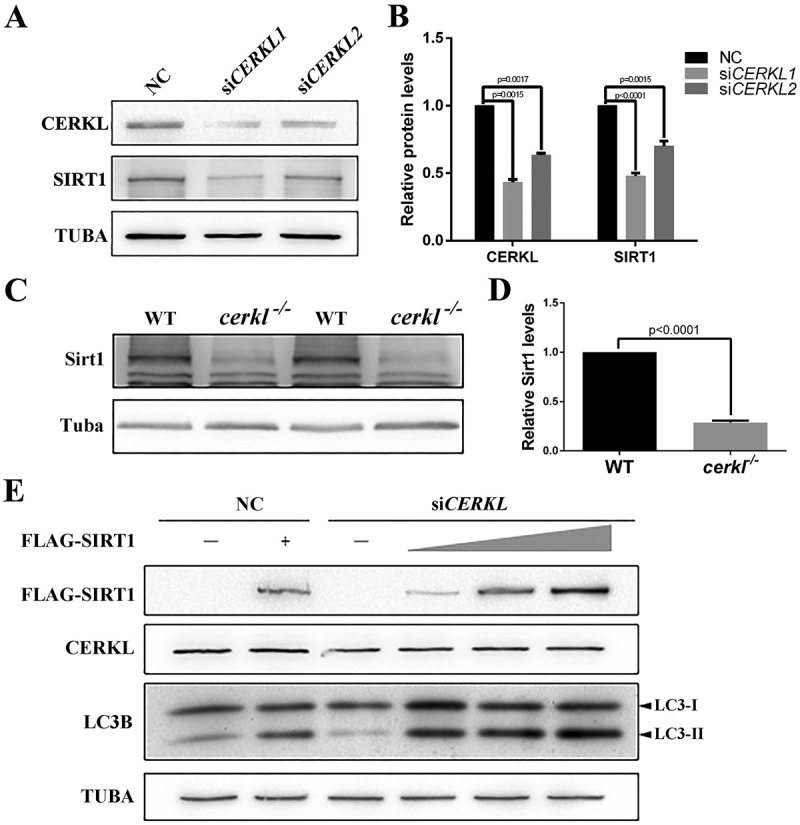
CERKL affected autophagy via SIRT1. (a and b) Immunostaining and quantification of the protein levels of SIRT1 in negative control (NC) and CERKL-depleted ARPE-19 cells. Relative expression of proteins in relation to TUBA. Means± SEM of 5 repeats are shown. (c and d) Immunostaining and quantification of the protein levels of Sirt1 in retinal extracts from wild-type (WT) and cerkl−/- zebrafish aged 1 month. Relative expression of Sirt1 in relation to Tuba. Means± SEM of 4 repeats are shown, each sample contain eyeballs of 4 zebrafish. (e) Immunoblotting of the levels of endogenous LC3-II:LC3-I ratio under a gradient expression of exogenous FLAG-SIRT1 in NC and CERKL-depleted ARPE-19 cells. Each well was transfected with 0.5 μg FLAG-SIRT1 plasmid in lane 2 and 4, 1 μg in lane 5, and 2 μg in lane 6.
Figure 6.
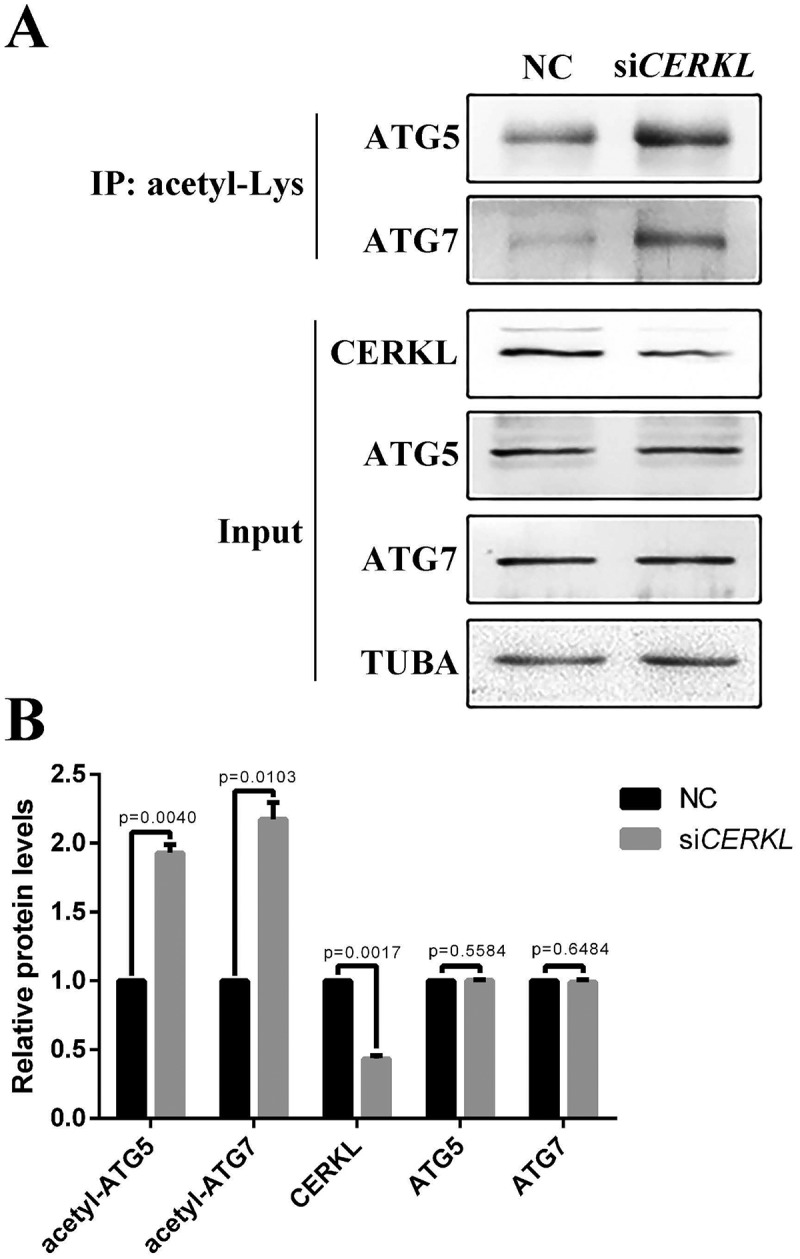
Acetylation analysis in CERKL-depleted ARPE-19 cells. (a) ARPE-19 cell extracts transfected with NC and siCERKL immunoprecipitated (IP) with pan acetyl-lysine antibody and analyzed with ATG5 and ATG7 antibodies. Input, whole cell extracts. (b) Quantification of proteins presented in panel A. Relative expression of proteins in relation to TUBA. Means± SEM of 5 repeats are shown.
CERKL regulated the stability of SIRT1
Given that knocking down CERKL reduced the protein level of SIRT1 (Figure 5(a,e)), both endogenous and exogenous, we then considered the possibility that CERKL might affect the protein stability of SIRT1. We therefore assayed the half-life of SIRT1 in CERKL-depleted cells. Protein levels of SIRT1 declined with a half-life of approximately 10 h by CERKL siRNA (Figure 7(a,b)). Previous work has suggested that the phosphorylation of SIRT1 at Ser27 (p-S27) increases the protein stability of SIRT1 [39], and that SIRT1 phosphorylation at Ser47 exhibits an opposite function, inducing a brief activation of SIRT1 function and degradation of SIRT1 thereafter by the proteasome [40]. Consequently, we investigated the level of SIRT1 p-S27 and SIRT1 p-S47 in CERKL-depleted cells. Interestingly the ratio of SIRT1 p-S27 and SIRT1 p-S47 to total SIRT1 were both reduced in CERKL-depleted cells (Figure 7(c,d)).
Figure 7.
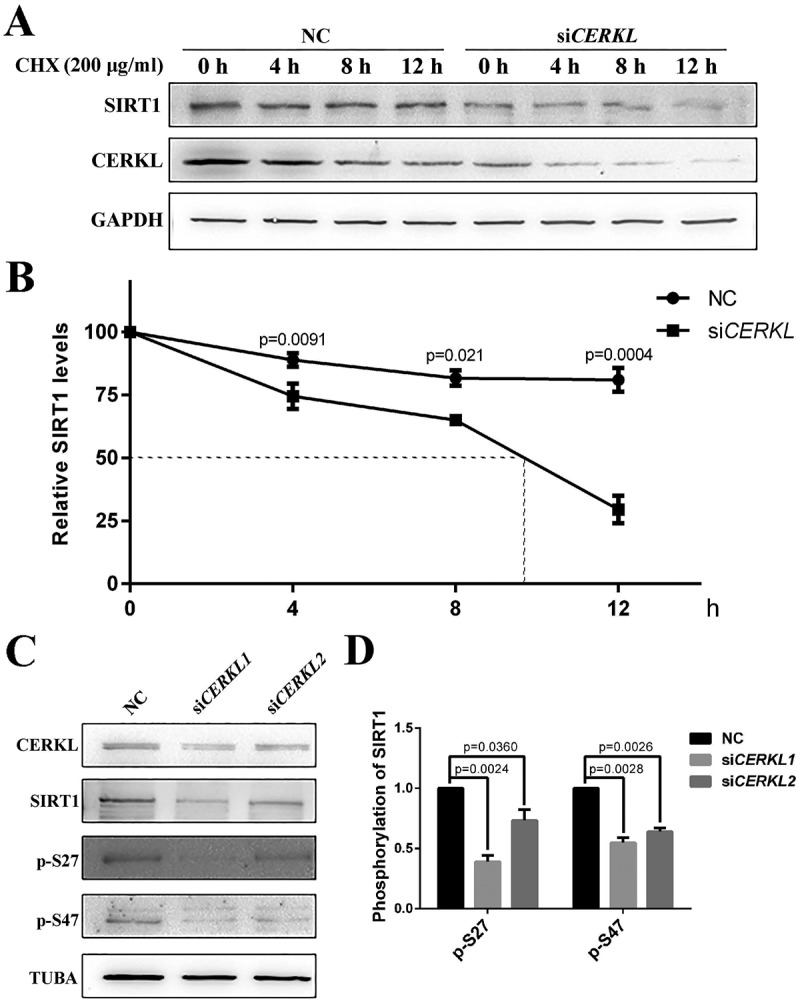
CERKL regulated the stability of SIRT1. (a) SIRT1 protein levels at indicated times post treatment with 200 μg/ml cycloheximide (CHX) in negative control (NC) and CERKL-depleted ARPE-19 cells. (b) Quantification of protein presented in panel A. Relative expression of SIRT1 in relation to GAPDH. Means± SEM of 3 repeats are shown. (c and d) Immunostaining and quantification of the protein levels of SIRT1 p-S27 and p-S47 in NC and CERKL-depleted ARPE-19 cells. Relative expression of proteins in relation to TUBA. Means± SEM of 5 repeats are shown.
To further confirm the results, we overexpressed CERKL in ARPE-19 cells. As noted in Figure 8(a,b), although endogenous SIRT1 did not show a significant upregulation in CERKL-overexpressing cells, the 2 phosphorylated forms of SIRT1, p-S27 and p-S47, increased significantly. As a result, the stability and activity of SIRT1 should both be upregulated. To confirm this inference, we then assessed the stability of SIRT1 and the level of autophagy in CERKL-overexpressing cells. As shown in Figure 8(c,d), SIRT1 displayed greater stability when CERKL was overexpressed. Autophagy detected through western blotting and immunofluorescence assay was also upregulated (Figures 8(e,f) and S6), along with the reduction of SQSTM1. In addition, we overexpressed CERKL in SIRT1-depleted cells and found that autophagy was not upregulated (Figure S7). These results further proved that the effect of CERKL on autophagy is dependent on SIRT1.
Figure 8.
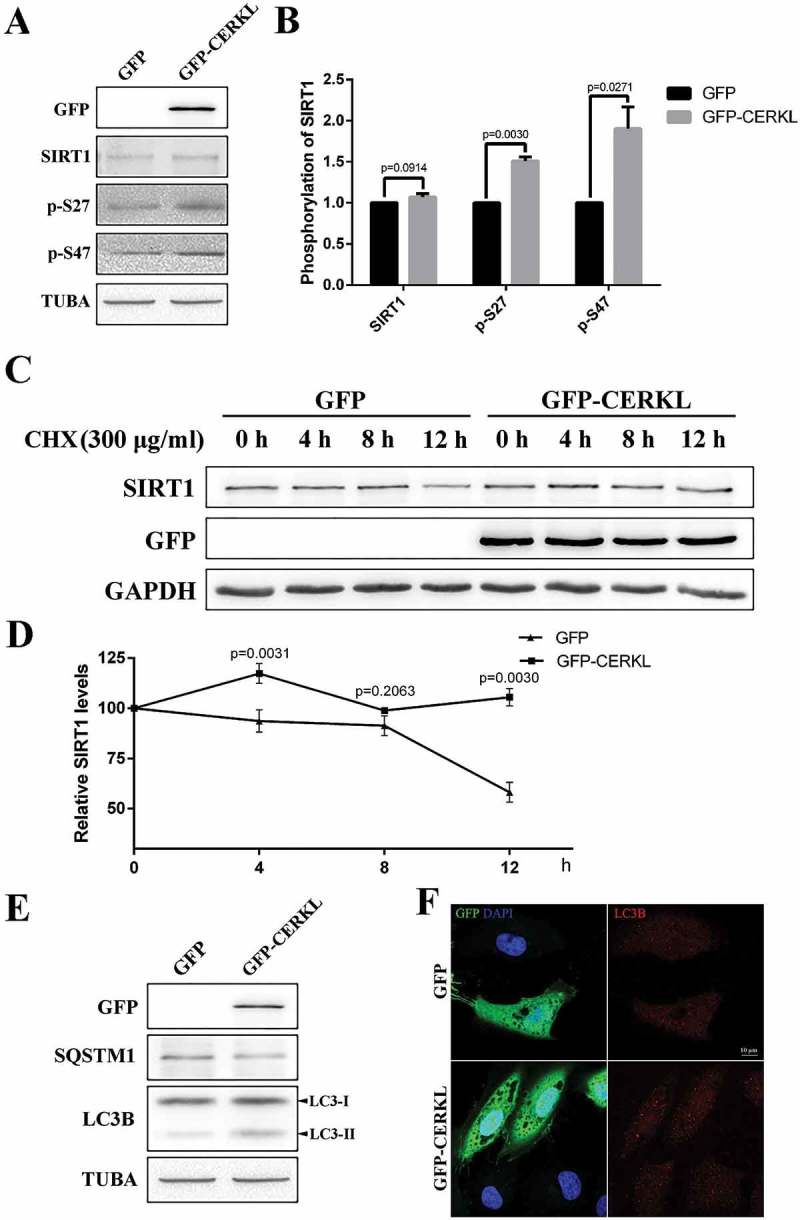
Overexpressing CERKL stabilized SIRT1. (a and b) Immunostaining and quantification of the protein levels of SIRT1 p-S27 and p-S47 in ARPE-19 cells overexpressing GFP and GFP-CERKL. Relative expression of proteins in relation to TUBA. Means± SEM of 5 repeats are shown. (c and d) SIRT1 protein levels at the indicated times post treatment with 300 μg/ml CHX in ARPE-19 cells overexpressing GFP and GFP-CERKL. Relative expression of SIRT1 in relation to GAPDH. Means± SEM of 3 repeats are shown. (e) Immunostaining analysis of LC3 and SQSTM1 in ARPE-19 cells overexpressing GFP and GFP-CERKL. (f) Immunostaining of the distribution of LC3 in ARPE-19 cells overexpressing GFP and GFP-CERKL. Scale bars: 10 μm.
CERKL directly interacted with SIRT1
Given that SIRT1 was stabilized by CERKL, we thought it possible that CERKL directly interacted with SIRT1 to affect its protein stability. Co-immunoprecipitation (co-IP) assays were performed to test this potential interaction. As shown in Figure 9(a,b), the anti-FLAG antibody (but not the control IgG) could immunoprecipitate GFP-CERKL, while the reverse co-IP with anti-GFP antibody also showed that GFP-CERKL interacted with FLAG-SIRT1. Moreover, immunofluorescence assays were done in an attempt to determine where the interaction occurs. Although GFP-CERKL exhibits dynamic cellular distributions, FLAG-SIRT1 and GFP-CERKL colocalized in both the cytoplasm and nucleus of ARPE-19 cells (Figure 9(c)). To investigate how CERKL affects the phosphorylation of SIRT1, we assayed the level of interaction between CERKL and phosphorylated SIRT1. As shown in Figure 9(d), SIRT1 p-S27 had less interaction with CERKL. We constructed vectors to simulate phosphorylated and unphosphorylated SIRT1, and measured their level of interaction with CERKL. Consistent with previous results, SIRT1S27D, the missense mutant simulating SIRT1 p-S27, had less interaction with CERKL compared with WT and with other mutants (Figure 9(e)).
Figure 9.
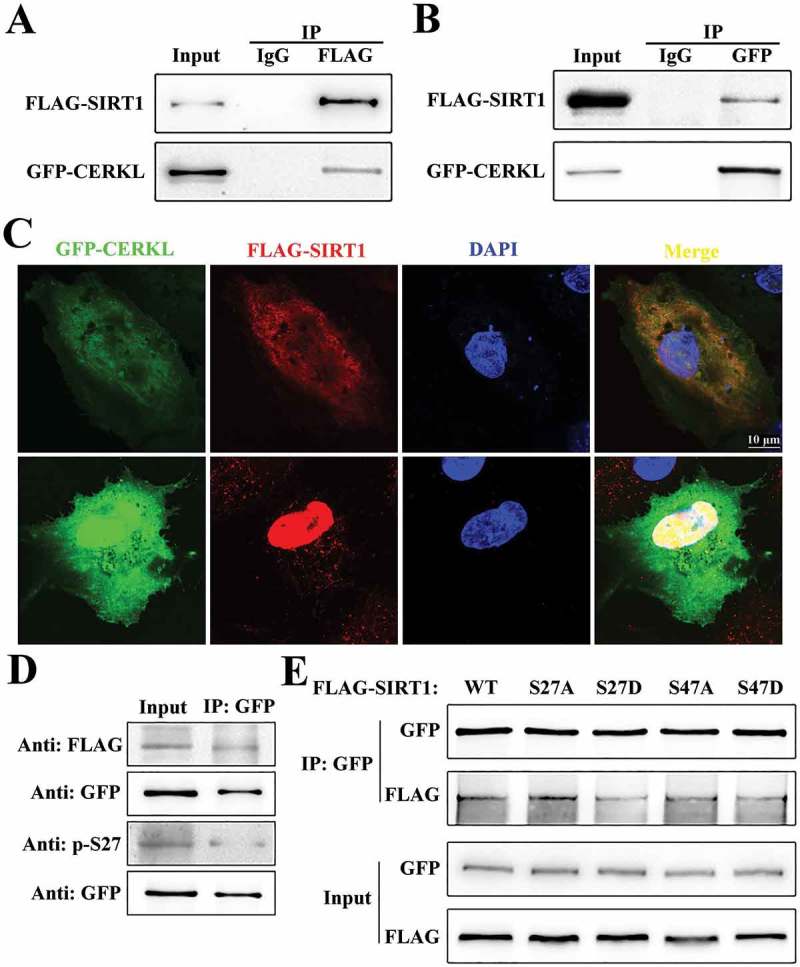
CERKL directly interacted with SIRT1. (a and b) Reciprocal co-immunoprecipitation assays. ARPE-19 cell extracts transfected with plasmids encoding GFP-CERKL and FLAG-SIRT1 were immunoprecipitated with one indicated tag antibody (FLAG in A, and GFP in B) and analyzed by immunoblotting analysis with the other antibody. (c) Immunostaining analysis of the colocalization of CERKL and SIRT1 in ARPE-19 cells transfected with plasmids encoding GFP-CERKL (green) and FLAG-SIRT1 (red). Scale bars: 10 μm. (d) ARPE-19 cell extracts from cells transfected with plasmids encoding GFP-CERKL and FLAG-SIRT1 were immunoprecipitated with GFP antibody and analyzed by immunoblotting analysis with FLAG antibody and SIRT1 p-S27 antibody. (e) ARPE-19 cell extracts from cells transfected with plasmids encoding GFP-CERKL and wild-type or mutant forms of FLAG-SIRT1 were immunoprecipitated with GFP and analyzed by immunoblotting analysis with FLAG antibody.
Discussion
CERKL, a gene causing autosomal recessive RP and CRD, plays a role in photoreceptor degeneration. Previous studies have shown that CERKL protects cells from stress, especially oxidative stress. Our previous data showed that CERKL modulates the redox state of TXN2 via protein-protein interaction to reduce reactive oxygen species. Here we demonstrate that CERKL plays a key role in autophagy in the retina by modulating the protein stability of an NAD-dependent deacetylase, SIRT1.
Autophagy is an intracellular process that allows for the degradation of proteins and organelles. Normally, autophagy involves the formation of double-membrane autophagosomes as a cellular response to stress and classically is induced by starvation. Other stressors, such as oxidative and endoplasmic reticulum (ER) stress, hypoxia, and infection can also trigger autophagy. Autophagy occurs at a basal level mainly to maintain homeostatic function during protein and organelle turnover. Suppression of basal autophagy in neural cells causes neurodegenerative disease. Here we found that autophagy in photoreceptor and RPE cells was reduced in cerkl−/- zebrafish compared with the wild type. As previous studies have shown that CERKL is located in photoreceptors and RPE [25,27], we speculated that CERKL could regulate autophagy in these cells.
In photoreceptors, protein aggregation, oxidative stress and ER stress increase as a result of autophagy downregulation [41]. The mouse model deleting Atg5 specifically in rods displays a reduced ONL from 2 months and photoreceptor death [13]. The Atg7ΔRod mouse shows no gross abnormalities, although the photoreceptors display sensitivity to light stress [11]. Atg5 deletion in cones results in a reduction in cone number from 4 months [12]. In RPE, LC3-associated phagocytosis, a noncanonical pathway that utilizes some of the autophagy machinery, controls the phagocytosis of daily shed photoreceptor outer segments during renewal of photoreceptors. The essential function of autophagy (to avoid lipofuscin and reactive oxygen species accumulation) also contributes to the antioxidant response of RPE cells. Atg5ΔRPE mice have decreased 11-cis retinal compared to normal mice and show loss of visual function [14]. Our study with cerkl−/- zebrafish displays phenotypes similar to those of the models above. In order to further prove that CERKL affected autophagy, we knocked down CERKL in ARPE-19 cells. In CERKL-depleted cells we observed the suppression of basal autophagy. In addition, by rapamycin treatment and starvation to stimulate autophagy, a reduction in autophagy was observed in CERKL-knockdown cells, which further indicated that CERKL mainly affects the formation of autophagosomes.
Conversely, the expression of the autophagy components ATG5 and ATG7 proteins was unchanged after knocking down CERKL. In addition, CERKL did not show any colocalization with the LC3 puncta, indicating that CERKL might not affect the assembly of autophagosomes. By contrast to the downregulation of autophagy in CERKL-depleted cells, the upstream activator AMPK was activated, indicating that when CERKL was knocked down in cells, the homeostasis was disrupted.
Given that the autophagy signaling pathway was not affected by knocking down CERKL, we turned our attention to the post-transcriptional regulation of autophagy. Post-transcriptional regulation is essential for modulating autophagy in order to adapt to different types of environmental stress, both with regard to amplitude of autophagy activity and its duration [37]. The post-translational modifications of autophagy can be classified into 3 categories: phosphorylation, ubiquitination and acetylation. SIRT1, a NAD-dependent deacetylase, is one of the main regulators of acetylation-deacetylation in autophagy [38]. Some components of the autophagy machinery, ATG5, ATG7, and LC3, are deacetylated by SIRT1 in response to starvation, an event necessary to induce autophagy. In CERKL-depleted cells, we found that the protein level of SIRT1 was reduced, and, as a consequence, the acetylation level of ATG5 and ATG7 increased. Therefore, we hypothesize that the role CERKL plays in autophagy is via the deacetylase activity of SIRT1. The results that SIRT1 could recover the suppressed autophagy in SIRT1-depleted cells and overexpressing CERKL in SIRT1-depleted cells could not upregulate autophagy further supported this hypothesis.
The SIRT1 mRNA was not downregulated by CERKL-depletion (Figure S5). However, the half-life of SIRT1 protein was markedly reduced following downregulation of CERKL. The protein stability of SIRT1 is regulated by post-translational phosphorylation at Ser27 and Ser47. SIRT1 is phosphorylated by MAPK8/JNK1 at Ser47, inducing a brief activation of SIRT1 function and degradation of SIRT1 thereafter by the proteasome [40]. The phosphorylation at Ser27 of SIRT1, induced by MAPK9/JNK2, increases SIRT1 protein stability. In the present study, we showed that the levels of SIRT1 p-S27 and p-S47, both regulated by CERKL, decreased with CERKL-depletion, and increased with CERKL overexpression. In addition, SIRT1 p-S27 showed less interaction with CERKL compared with total SIRT1. Therefore, we hypothesize that CERKL may stabilize SIRT1 by regulating the phosphorylation of SIRT1 at Ser27.
As the substrates of SIRT1 are not limited to ATG proteins, SIRT1 is able to deacetylate histones and initiate heterochromatin formation resulting in gene silencing. SIRT1 also deacetylates several non-histone target proteins, such as the tumor suppressor TP53/p53, members of the FOXO family, stress response proteins NFKB and XRCC6/Ku70, and the mitochondrial biogenesis regulator PPARGC1A/PGC-1α [42]. Given that SIRT1 plays a crucial role in the cellular stress response, this suggests that in cerkl−/- zebrafish, not only autophagy but other aspects of cell function such as metabolism, would also be affected. However, more experiments are needed to prove this hypothesis. Nevertheless, replenishment of autophagy or activation of SIRT1 may be a potential clinical treatment of retina degeneration caused by CERKL mutations.
Materials and methods
Zebrafish maintenance
All procedures of the animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the College of Life Science and Technology, Huazhong University of Science and Technology, and all experiments were conducted according to the relevant guidelines. Zebrafish larvae and adults were maintained at 26–28.5°C under a 14-h light/10-h dark cycle. Fertilized eggs were collected and maintained in E3 medium in an incubator (at ~ 28.5°C) for 72 h until the larvae hatched.
Plasmid constructs and RNA interference
The full-length CERKL and SIRT1 cDNAs were subcloned, respectively, into the pEGFP-C1 (Miaolingbio, P0134), p3xFLAG-CMV (Sigma, E7533) and pcDNA3.1 (Invitrogen, V79520) vectors. A FLAG tag was added to the end of the SIRT1 N terminus by PCR. Missense mutants were constructed by PCR based on the wild-type gene and verified by sequencing.
Small interfering RNAs (siRNAs) targeting different encoding regions of human CERKL were synthesized and purified by RiboBio (Guangzhou, China). CERKL siRNA 1 was used in the experiment. The targeting nucleotide sequences were as follows:
si-CERKL 1: 5′-GAATAATACTGGTGGATAT-3′,
si-CERKL 2: 5′-GGCAAATGATCCAGGGTCA-3′.
The siRNA duplexes with nonspecific sequences were used as siRNA negative control (si-NC).
Cell culture and co-immunoprecipitation
ARPE-19 cells (American Type Culture Collection, CRL-2302) were cultured at 37°C in DMEM/F12 (Gibco, 11330057) supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, 10378016).
Autophagy was induced by replacement of full growth medium with Earle’s buffered saline solution (EBSS; Sigma, E2888) for 1, 2 and 4 h, or by treating cells with 100 nM rapamycin (Sigma, 553210) in the presence of complete growth medium for the same period for 2 h.
Autophagy flux was blocked by treating cells with 2.5 nM bafilomycin A1 (Sigma, 196000) in the presence of complete growth medium for the same period for 3 h.
Cell transfection was performed with Lipofectamine™ 3000 (Invitrogen, L3000015). Plasmids encoding GFP-LC3 and RFP-LAMP1 were generously provided by Dr. Cui Xiukun (Henan University, Henan, China).
Cells were cotransfected with plasmids encoding GFP-CERKL and FLAG-SIRT1 or the corresponding mutant expression plasmids. After 24 h, cell lysates were harvested and immunoprecipitated with one antibody and protein G beads (Millipore, 16–266), and washed and analyzed by western blotting with the antibody for the other proteins.
Cells were transfected with siNC or siCERKL. After 72 h, cell lysates were harvested and immunoprecipitated with pan acetyl-lysine antibody (ABclonal, A2391) and protein G beads, and washed and analyzed by western blotting with the antibody.
For protein half-life experiments cells were treated with 200 μg/ml or 300 μg/ml cycloheximide (Amresco, 94271) 12 h following transfection with siRNAs or plasmids and subsequently sampled at the indicated times.
Antibodies
The list of antibodies used in the present study is provided in Table 1.
Table 1.
Table List of primary antibodies used in this study.
| Antibodies | Source | Recognize | Dilution |
|---|---|---|---|
| Anti-LC3B | Abcam Ab48394 | Zebrafish and human LC3B | 1:1000 for WB 1:200 for IF |
| Anti-CERKL | Sigma-Aldrich HPA035444 | Human CERKL | 1:1000 for WB |
| Anti-SQSTM1/p62 | ABclonal A7758 | Human SQSTM1 | 1:1000 for WB |
| Anti-ATG5 | ABclonal A7252 | Human ATG5 | 1:1000 for WB |
| Anti-ATG7 | ABclonal A0691 | Human ATG7 | 1:1000 for WB |
| Anti-SIRT1 | ABclonal A11267 | Zebrafish and human SIRT1 | 1:700 for WB |
| Proteintech 13161-1-AP | Zebrafish and human SIRT1 | 1:700 for WB | |
| Anti-phospho-SIRT1 (Ser27) | Cell Signaling Technology 2327 | Human SIRT1 p-S27 | 1:500 for WB |
| Anti-phospho-SIRT1 (Ser47) | Cell Signaling Technology 2314 | Human SIRT1 p-S47 | 1:200 for WB |
| Anti-pan acetyl-lysine | ABclonal A2391 | 1 mg per 1 ml cell lysate for IP | |
| Anti-AMPK | ABclonal A1229 | Human AMPKα | 1:1000 for WB |
| Anti-AMPK p-T172 | ABclonal AP0116 | Human phospho-PRKAA/AMPKα p-T172 | 1:1000 for WB |
| Anti-BECN1/BECLIN1 | Proteintech 11306-1-AP | Human BECN1 | 1:700 for WB |
| Anti-TUBA/α-TUBULIN | Proteintech 11224-1-AP | Zebrafish and human TUBA | 1:5000 for WB |
| Anti-GADPH | ABclonal AC027 | Human GAPDH | 1:3000 for WB |
| Anti-FLAG | MBL M185-3L | FLAG | 1:5000 for WB |
| Anti-GFP | Absmart | GFP | 1:5000 for WB |
| Proteintech 50430-2-AP | GFP | 1 mg per 1 ml cell lysate for IP |
Immunocytochemistry
Transfected cells were fixed in phosphate-buffered salien (PBS; Biosharp, 173665) containing 10% formaldehyde for 10 min, permeabilized with PBS containing 0.5% Triton X-100 (Sigma, X100) for 15 min and blocked with PBS containing 10% normal goat serum (Boster, AR1009) for 1 h at room temperature (RT). Cells were then incubated with the primary antibody (1:500–1000) solubilized in PBS containing 1% BSA (Biofroxx, 4240GR100) at 4°C overnight and Alexa Fluor 488 or 594 nm secondary antibody (1:1000; Invitrogen, A11005, A11008) for 1 h at 37°C. After staining with DAPI for 5 min, slides were mounted. Fluorescence images were captured using a confocal laser-scanning microscope (Fluo ViewTM FV1000 confocal microscope, Olympus Imaging).
Immunohistochemistry and histology
Zebrafish eyes were isolated and fixed with 4% paraformaldehyde in PBS for 12 h at 4°C, cryoprotected in 30% sucrose (Sinopharm, 10021418) overnight, and embedded in OCT compound (SAKURA, 4583). Cryostat sections (10- to 15-μm thick) containing the whole retina including the optic disk were rinsed with PDT (PBS solution containing 1% DMSO and 0.1% Triton X-100) for 10 min and blocked with blocking solution (PDT containing 1% BSA and 10% normal goat serum) for 1 h at RT. Primary antibodies (1:500–1000) were prepared in blocking solution containing 2% normal goat serum and slides were incubated overnight at 4°C. Slides were washed 3 times with PDT and incubated with Alexa Fluor 488 or 594 secondary antibody (1:1000) for 1 h at 37°C. DAPI was diluted with PBS to final 5 μg/mL and used to label the nucleus. The slides were washed 3 times with PBS and then mounted under glass coverslips. Fluorescence images were captured using a confocal laser-scanning microscope (Fluo ViewTM FV1000 confocal microscope, Olympus Imaging). The procedure details were conducted as described previously [43].
Western blotting
Cells and zebrafish eyes were collected and lysed in SDS lysis buffer (Beyotime, P0013G) with protease inhibitor cocktail (Roche, 04693159001). Protein concentration was determined using the BCA protein assay kit (Beyotime, P0010). Proteins were separated by 12% SDS-PAGE and transferred to nitrocellulose membranes (Millipore, HATF00010). The blots were incubated with primary antibodies (1:500–5000), followed by HRP-labeled secondary antibodies (1:20,000; ThermoFisher, 31430, 31460). ECL plus substrate (Pierce, 32132) was used for the detection of signals. The procedure was conducted as described previously [44].
RNA extraction and RT-qPCR
Total RNA of zebrafish was extracted using Trizol (Life, 15596018), and quantified by NanoDrop spectrometry (Thermo Scientific, Wilmington, DE, USA). cDNA was generated using MMLV reverse transcriptase (Invitrogen, 28025013). Real-time PCR was performed using AceQ® qPCR SYBR® Green Master Mix (Vazyme, Q141-02/03) according to the manufacturer’s instructions, and relative gene expression was quantified using the Step One PlusTM Real-Time PCR System (Life Technologies).
Statistical analysis
All data were presented as means± SEM. Data groups were compared by Student’s t-test (Prism 6.0 software; Graphpad Software, Inc., La Jolla, CA, USA). Differences between groups were considered statistically significant if p < 0.05.
Funding Statement
National Natural Science Foundation of China (No. 81870691, 31471194, 81371064, 81500762, 81670890, 81800870 and 31801041).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Lavandero S, Chiong M, Rothermel BA, et al. Autophagy in cardiovascular biology. J Clin Invest. 2015. January;125(1):55–64. PubMed PMID: 25654551; PubMed Central PMCID: PMCPMC4382263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rubinsztein DC, Marino G, Kroemer G.. Autophagy and aging. Cell. 2011. September 2;146(5):682–695. PubMed PMID: 21884931. [DOI] [PubMed] [Google Scholar]
- [3].Mizushima N, Komatsu M.. Autophagy: renovation of cells and tissues. Cell. 2011. November 11;147(4):728–741. PubMed PMID: 22078875. [DOI] [PubMed] [Google Scholar]
- [4].Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015. August;16(8):461–472. PubMed PMID: 26177004. [DOI] [PubMed] [Google Scholar]
- [5].Mizushima N. Autophagy: process and function. Genes Dev. 2007. November 15;21(22):2861–2873. PubMed PMID: 18006683. [DOI] [PubMed] [Google Scholar]
- [6].Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol. 2012. July;22(7):374–380. PubMed PMID: 22608991; PubMed Central PMCID: PMCPMC3383932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016. June 2;85:685–713. PubMed PMID: 26865532. [DOI] [PubMed] [Google Scholar]
- [8].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006. June 15;441(7095):885–889. PubMed PMID: 16625204. [DOI] [PubMed] [Google Scholar]
- [9].Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011. April 15;25(8):795–800. PubMed PMID: 21498569; PubMed Central PMCID: PMCPMC3078705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014. June;26:16–23. PubMed PMID: 24907664; PubMed Central PMCID: PMCPMC4253630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen Y, Sawada O, Kohno H, et al. Autophagy protects the retina from light-induced degeneration. J Biol Chem. 2013. March 15;288(11):7506–7518. PubMed PMID: 23341467; PubMed Central PMCID: PMCPMC3597791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhou Z, Vinberg F, Schottler F, et al. Autophagy supports color vision. Autophagy. 2015;11(10):1821–1832. PubMed PMID: 26292183; PubMed Central PMCID: PMCPMC4824586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou Z, Doggett TA, Sene A, et al. Autophagy supports survival and phototransduction protein levels in rod photoreceptors. Cell Death Differ. 2015. March;22(3):488–498. PubMed PMID: 25571975; PubMed Central PMCID: PMCPMC4326583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim JY, Zhao H, Martinez J, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013. July 18;154(2):365–376. PubMed PMID: 23870125; PubMed Central PMCID: PMCPMC3744125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yao J, Jia L, Khan N, et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy. 2015;11(6):939–953. PubMed PMID: 26075877; PubMed Central PMCID: PMCPMC4502815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bergmann M, Schutt F, Holz FG, et al. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004. March;18(3):562–564. PubMed PMID: 14715704. [DOI] [PubMed] [Google Scholar]
- [17].Mitter SK, Song C, Qi X, et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10(11):1989–2005. PubMed PMID: 25484094; PubMed Central PMCID: PMCPMC4502658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tuson M, Marfany G, Gonzalez-Duarte R. Mutation of CERKL, a novel human ceramide kinase gene, causes autosomal recessive retinitis pigmentosa (RP26). Am J Hum Genet. 2004. January;74(1):128–138. PubMed PMID: 14681825; PubMed Central PMCID: PMCPMC1181900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aleman TS, Soumittra N, Cideciyan AV, et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest Ophthalmol Vis Sci. 2009. December;50(12):5944–5954. PubMed PMID: 19578027. [DOI] [PubMed] [Google Scholar]
- [20].Tang Z, Wang Z, Wang Z, et al. Novel compound heterozygous mutations in CERKL cause autosomal recessive retinitis pigmentosa in a nonconsanguineous Chinese family. Arch Ophthalmol. 2009. August;127(8):1077–1078. PubMed PMID: 19667359. [DOI] [PubMed] [Google Scholar]
- [21].Littink KW, Koenekoop RK, van den Born LI, et al. Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and clinical characterizations. Invest Ophthalmol Vis Sci. 2010. November;51(11):5943–5951. PubMed PMID: 20554613; PubMed Central PMCID: PMCPMC3061516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vekslin S, Ben-Yosef T. Spatiotemporal expression pattern of ceramide kinase-like in the mouse retina. Mol Vis. 2010. December 03;16:2539–2549. PubMed PMID: 21151604; PubMed Central PMCID: PMCPMC3000240. [PMC free article] [PubMed] [Google Scholar]
- [23].Garanto A, Vicente-Tejedor J, Riera M, et al. Targeted knockdown of Cerkl, a retinal dystrophy gene, causes mild affectation of the retinal ganglion cell layer. Biochim Biophys Acta. 2012. August;1822(8):1258–1269. PubMed PMID: 22549043. [DOI] [PubMed] [Google Scholar]
- [24].Garanto A, Riera M, Pomares E, et al. High transcriptional complexity of the retinitis pigmentosa CERKL gene in human and mouse. Invest Ophthalmol Vis Sci. 2011. July 13;52(8):5202–5214. PubMed PMID: 21508105. [DOI] [PubMed] [Google Scholar]
- [25].Mandal NA, Tran JT, Saadi A, et al. Expression and localization of CERKL in the mammalian retina, its response to light-stress, and relationship with NeuroD1 gene. Exp Eye Res. 2013. January;106:24–33. PubMed PMID: 23142158; PubMed Central PMCID: PMCPMC3538916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Riera M, Burguera D, Garcia-Fernandez J, et al. CERKL knockdown causes retinal degeneration in zebrafish. PLoS One. 2013;8(5):e64048 PubMed PMID: 23671706; PubMed Central PMCID: PMCPMC3650063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li C, Wang L, Zhang J, et al. CERKL interacts with mitochondrial TRX2 and protects retinal cells from oxidative stress-induced apoptosis. Biochim Biophys Acta. 2014. July;1842(7):1121–1129. PubMed PMID: 24735978. [DOI] [PubMed] [Google Scholar]
- [28].Bornancin F, Mechtcheriakova D, Stora S, et al. Characterization of a ceramide kinase-like protein. Biochim Biophys Acta. 2005. February 21;1687(1–3):31–43. PubMed PMID: 15708351. [DOI] [PubMed] [Google Scholar]
- [29].Inagaki Y, Mitsutake S, Igarashi Y. Identification of a nuclear localization signal in the retinitis pigmentosa-mutated RP26 protein, ceramide kinase-like protein. Biochem Biophys Res Commun. 2006. May 12;343(3):982–987. PubMed PMID: 16581028. [DOI] [PubMed] [Google Scholar]
- [30].Graf C, Niwa S, Muller M, et al. Wild-type levels of ceramide and ceramide-1-phosphate in the retina of ceramide kinase-like-deficient mice. Biochem Biophys Res Commun. 2008. August 15;373(1):159–163. PubMed PMID: 18555012. [DOI] [PubMed] [Google Scholar]
- [31].Tuson M, Garanto A, Gonzalez-Duarte R, et al. Overexpression of CERKL, a gene responsible for retinitis pigmentosa in humans, protects cells from apoptosis induced by oxidative stress. Mol Vis. 2009;15:168–180. PubMed PMID: 19158957; PubMed Central PMCID: PMCPMC2628313. [PMC free article] [PubMed] [Google Scholar]
- [32].Yu S, Li C, Biswas L, et al. CERKL gene knockout disturbs photoreceptor outer segment phagocytosis and causes rod-cone dystrophy in zebrafish. Hum Mol Genet. 2017. June 15;26(12):2335–2345. PubMed PMID: 28398482. [DOI] [PubMed] [Google Scholar]
- [33].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. PubMed PMID: 26799652; PubMed Central PMCID: PMCPMC4835977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005. November 21;171(4):603–614. PubMed PMID: 16286508; PubMed Central PMCID: PMCPMC2171557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007. May;13(5):619–624. PubMed PMID: 17450150. [DOI] [PubMed] [Google Scholar]
- [36].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004. December 23;432(7020):1032–1036. PubMed PMID: 15525940. [DOI] [PubMed] [Google Scholar]
- [37].Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015. June;25(6):354–363. PubMed PMID: 25759175; PubMed Central PMCID: PMCPMC4441840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee IH, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA. 2008. March 4;105(9):3374–3379. PubMed PMID: 18296641; PubMed Central PMCID: PMCPMC2265142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ford J, Ahmed S, Allison S, et al. JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle. 2008. October;7(19):3091–3097. PubMed PMID: 18838864. [DOI] [PubMed] [Google Scholar]
- [40].Gao Z, Zhang J, Kheterpal I, et al. Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J Biol Chem. 2011. June 24;286(25):22227–22234. PubMed PMID: 21540183; PubMed Central PMCID: PMCPMC3121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Boya P, Esteban-Martinez L, Serrano-Puebla A, et al. Autophagy in the eye: development, degeneration, and aging. Prog Retin Eye Res. 2016. November;55:206–245. PubMed PMID: 27566190. [DOI] [PubMed] [Google Scholar]
- [42].Buler M, Andersson U, Hakkola J. Who watches the watchmen? Regulation of the expression and activity of sirtuins. FASEB J. 2016. December;30(12):3942–3960. PubMed PMID: 27591175. [DOI] [PubMed] [Google Scholar]
- [43].Liu F, Chen J, Yu S, et al. Knockout of RP2 decreases GRK1 and rod transducin subunits and leads to photoreceptor degeneration in zebrafish. Hum Mol Genet. 2015. August 15;24(16):4648–4659. PubMed PMID: 26034134. [DOI] [PubMed] [Google Scholar]
- [44].Cui X, Zhang J, Du R, et al. HSF4 is involved in DNA damage repair through regulation of Rad51. Biochim Biophys Acta. 2012. August;1822(8):1308–1315. PubMed PMID: 22587838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.