

ABSTRACT
Cigarette smoke (CS)-induced accumulation of mitochondrial damage has been widely implicated in chronic obstructive pulmonary disease (COPD) pathogenesis. Mitophagy plays a crucial role in eliminating damaged mitochondria, and is governed by the PINK1 (PTEN induced putative protein kinase 1)-PRKN (parkin RBR E3 ubiquitin protein ligase) pathway. Although both increased PINK1 and reduced PRKN have been implicated in COPD pathogenesis in association with mitophagy, there are conflicting reports for the role of mitophagy in COPD progression. To clarify the involvement of PRKN-regulated mitophagy in COPD pathogenesis, prkn knockout (KO) mouse models were used. To illuminate how PINK1 and PRKN regulate mitophagy in relation to CS-induced mitochondrial damage and cellular senescence, overexpression and knockdown experiments were performed in airway epithelial cells (AEC). In comparison to wild-type mice, prkn KO mice demonstrated enhanced airway wall thickening with emphysematous changes following CS exposure. AEC in CS-exposed prkn KO mice showed accumulation of damaged mitochondria and increased oxidative modifications accompanied by accelerated cellular senescence. In vitro experiments showed PRKN overexpression was sufficient to induce mitophagy during CSE exposure even in the setting of reduced PINK1 protein levels, resulting in attenuation of mitochondrial ROS production and cellular senescence. Conversely PINK1 overexpression failed to recover impaired mitophagy caused by PRKN knockdown, indicating that PRKN protein levels can be the rate-limiting factor in PINK1-PRKN-mediated mitophagy during CSE exposure. These results suggest that PRKN levels may play a pivotal role in COPD pathogenesis by regulating mitophagy, suggesting that PRKN induction could mitigate the progression of COPD.
Abbreviations: AD: Alzheimer disease; AEC: airway epithelial cells; BALF: bronchoalveolar lavage fluid; AKT: AKT serine/threonine kinase; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CDKN1A: cyclin dependent kinase inhibitor 1A; CDKN2A: cyclin dependent kinase inhibitor 2A; COPD: chronic obstructive pulmonary disease; CS: cigarette smoke; CSE: CS extract; CXCL1: C-X-C motif chemokine ligand 1; CXCL8: C-X-C motif chemokine ligand 8; HBEC: human bronchial epithelial cells; 4-HNE: 4-hydroxynonenal; IL: interleukin; KO: knockout; LF: lung fibroblasts; LPS: lipopolysaccharide; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MTOR: mechanistic target of rapamycin kinase; 8-OHdG: 8-hydroxy-2ʹ-deoxyguanosine; OPTN: optineurin; PRKN: parkin RBR E3 ubiquitin protein ligase; PCD: programmed cell death; PFD: pirfenidone; PIK3C: phosphatidylinositol-4:5-bisphosphate 3-kinase catalytic subunit; PINK1: PTEN induced putative kinase 1; PTEN: phosphatase and tensin homolog; RA: rheumatoid arthritis; ROS: reactive oxygen species; SA-GLB1/β-Gal: senescence-associated-galactosidase, beta 1; SASP: senescence-associated secretory phenotype; SNP: single nucleotide polymorphism; TNF: tumor necrosis factor.
KEYWORDS: Cellular senescence, COPD, mitophagy, PINK1, PRKN
Introduction
Chronic obstructive pulmonary disease (COPD) characterized by progressive airflow limitation is mainly caused by the noxious effects of cigarette smoke (CS) exposure [1]. Recent studies have implicated impaired mitochondrial structural and functional integrity accompanied by excessive reactive oxygen species (ROS) production in COPD pathogenesis, resulting in enhanced programmed cell death (PCD) and cellular senescence during CS exposure [2–7]. Mitochondrial integrity is orchestrated by a complex layer of regulatory mechanisms, including biogenesis, both fusion and fission, and degradation. Mitochondria selective autophagy-lysosomal degradation, known as mitophagy, plays a crucial role in eliminating depolarized damaged mitochondria and is governed by post-translational modifications of PINK1 (PTEN-induced putative kinase 1) and PRKN (parkin RBR E3 ubiquitin protein ligase) [8].
Both PINK1 and PRKN gene mutations associated with mitochondrial dysfunction have been implicated in the development of hereditary recessive early-onset Parkinsonism [8–10]. A retrospective cohort study evaluating the clinical connection between Parkinson disease and COPD shows higher risk of developing Parkinson disease in COPD cases [11]. A recent paper demonstrates PRKN deficiency is related to increased inflammation and genomic instability, which can be involved in COPD pathogenesis [12]. We have reported a positive correlation between PRKN protein levels in lung homogenates and pulmonary function test in COPD cases [7]. Furthermore, a comprehensive validation study of single nucleotide polymorphism (SNP) variants of PRKN shows a potential implication of PRKN in developing COPD and lung cancer [12]. Accordingly, it is plausible that impairment of mitochondrial integrity caused by PRKN dysfunction can be a factor for both Parkinsonism and COPD development.
Phenotypic alterations shown by double-mutants of Pink1 and park/parkin, the Drosophila ortholog of mammalian PRKN, are identical to those observed in either mutant alone, indicating Pink1 and park are at least partially redundant [13]. Transgenic expression of park ameliorates phenotypic alterations caused by functional loss of Pink1, including mitochondrial dysfunction and apoptosis in Drosophila [13–15]. Conversely, PINK1 overexpression fails to rescue phenotypic changes evoked by PRKN mutations, indicating that PRKN functions downstream of PINK1. Furthermore, in contrast to PRKN overexpression, PINK1 overexpression demonstrates no effect on mitophagy in fibroblasts containing endogenous levels of both PINK1 and PRKN, suggesting that PRKN levels can be the rate-limiting factor in PINK1-PRKN-regulated mitophagy [16].
Although both PINK1 and PRKN have been widely implicated in COPD pathogenesis with respect to regulating mitophagy, there are conflicting reports regarding their effects on mitophagy status and their role in disease progression [4,6,7]. CS-induced excessive mitophagy activation caused by PINK1 accumulation is responsible for COPD progression via induction of programmed cell death (PCD), specifically necroptosis in human bronchial epithelial cells (HBEC) [6]. Conversely, we and others have reported that mitophagy activation is not sufficient to attenuate accelerated cellular senescence in COPD pathogenesis [4,7]. Insufficient mitophagy is attributed to reduced PRKN protein levels and impaired mitochondrial translocation of PRKN with concomitantly enhanced PINK1 accumulation [4,7]. PINK1 is targeted to the mitochondria but cleaved for proteasomal degradation during the steady state, resulting in very low expression levels. PINK1 is stabilized on the outer membrane in the setting of mitochondrial damage, indicating that high PINK1 protein levels may reflect accumulation of damaged mitochondria in COPD [17]. Hence, it remains unclear whether PINK1 accumulation or reduced PRKN is dominantly regulating mitophagy status and cell fate in terms of COPD pathogenesis.
In the present study, we have attempted to clarify the involvement of PRKN-regulated mitophagy and mitochondrial integrity in COPD pathogenesis by using prkn knockout mouse models. We have also attempted to further elucidate how PINK1 and PRKN are regulating mitophagy during CS-induced mitochondrial damage and cellular senescence by a combination of overexpression and knockdown experiments in HBEC.
Results
Aggravation of the CS-induced COPD phenotype in prkn KO mice
Our recent observation of the positive correlation between PRKN and the pulmonary function test indicates that PRKN protein levels can be implicated in the mechanism of COPD progression [7]. To clarify the role PRKN plays in determining COPD phenotype, prkn KO mice were used. First, lung parenchymal emphysema and airway wall thickening in response to CS exposure were examined in both wild-type and prkn KO mice. CS exposure for 6 months induced both lung emphysematous change and mild airway wall thickening in wild-type mice, which were significantly augmented in prkn KO mice (Figure 1(a,b)). Chronic inflammation in response to CS-exposure has been recognized to be a part of COPD pathogenesis. To evaluate inflammatory cell infiltrates in lungs, bronchoalveolar lavage fluid (BALF) was used for cell counting (Figure 1(c)). Only neutrophil counts were significantly increased in wild-type mice following CS exposure. Significant increase in total cell and macrophage counts were observed in CS-exposed prkn KO mice compared to CS-exposed wild-type mice. No significant difference was demonstrated in neutrophil and lymphocyte counts between the wild type and prkn KO.
Figure 1.
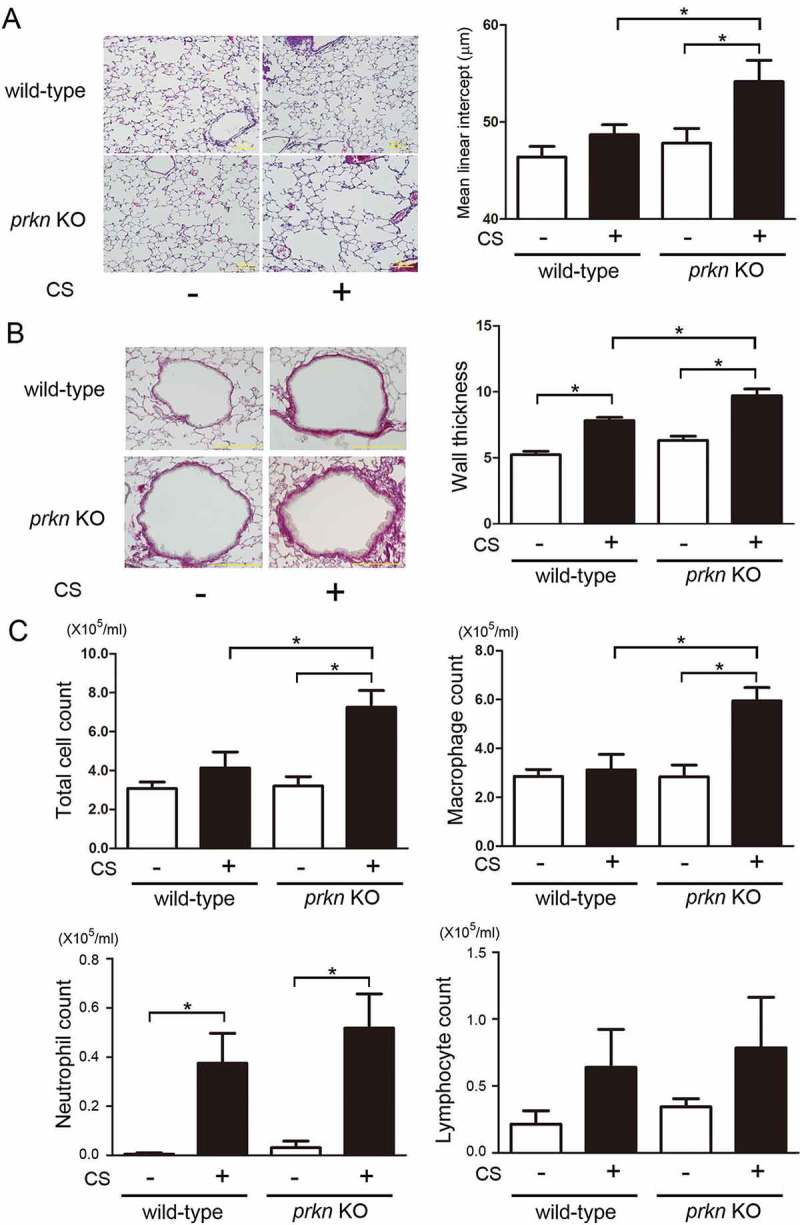
Effect of long-term cigarette smoke exposure on prkn knockout mice. (a) Photomicrographs of hematoxylin and eosin staining in control-air- and cigarette-smoke- (CS) exposed mouse lungs. Bar: 100 µm. Right panel shows the average (±SEM) of mean liner intercept by using Image J. Open bar is control-air- and filled bar is CS-exposed. Treatment groups were composed of control-air-exposed wild-type mice (n = 9), CS-exposed wild-type mice (n = 6), control-air-exposed prkn KO mice (n = 5), CS-exposed prkn KO mice (n = 7). All groups exposed for 6 months. *P < 0.05, by ANOVA and Tukey post-hoc test. (b) Photomicrographs of PSR staining in control-air- and CS-exposed mouse lungs. Bar: 100 µm. (c) Cell counts in bronchoalveolar lavage fluid (BALF). Treatment groups were composed of the same number of mice (n = 4). *P < 0.05, by ANOVA and Tukey post-hoc test.
Accelerated cellular senescence in prkn KO mice in response to CS-exposure
Accelerated cellular senescence has been widely implicated in COPD pathogenesis [18,19]. Our recent findings suggest that PRKN-mediated mitophagy has a key regulatory role in CS extract (CSE)-induced HBEC senescence through modulating mitochondrial integrity and ROS [7]. Hence, cellular senescence was evaluated in both wild-type and prkn KO mice after CS exposure for 6 months. Senescence-associated-GLB1/β-galactosidase staining showed clear positive staining of airway epithelial cells in CS-exposed prkn KO mice but few positive staining cells were detected in CS-exposed wild-type mice (Figure 2(a)). Consistent with GLB1 staining, acceleration of airway epithelial cell senescence in prkn KO mice was also shown by means of phospho-histone H2AFX (Ser139) staining of double-stranded DNA damage (Figure 2(b)).
Figure 2.
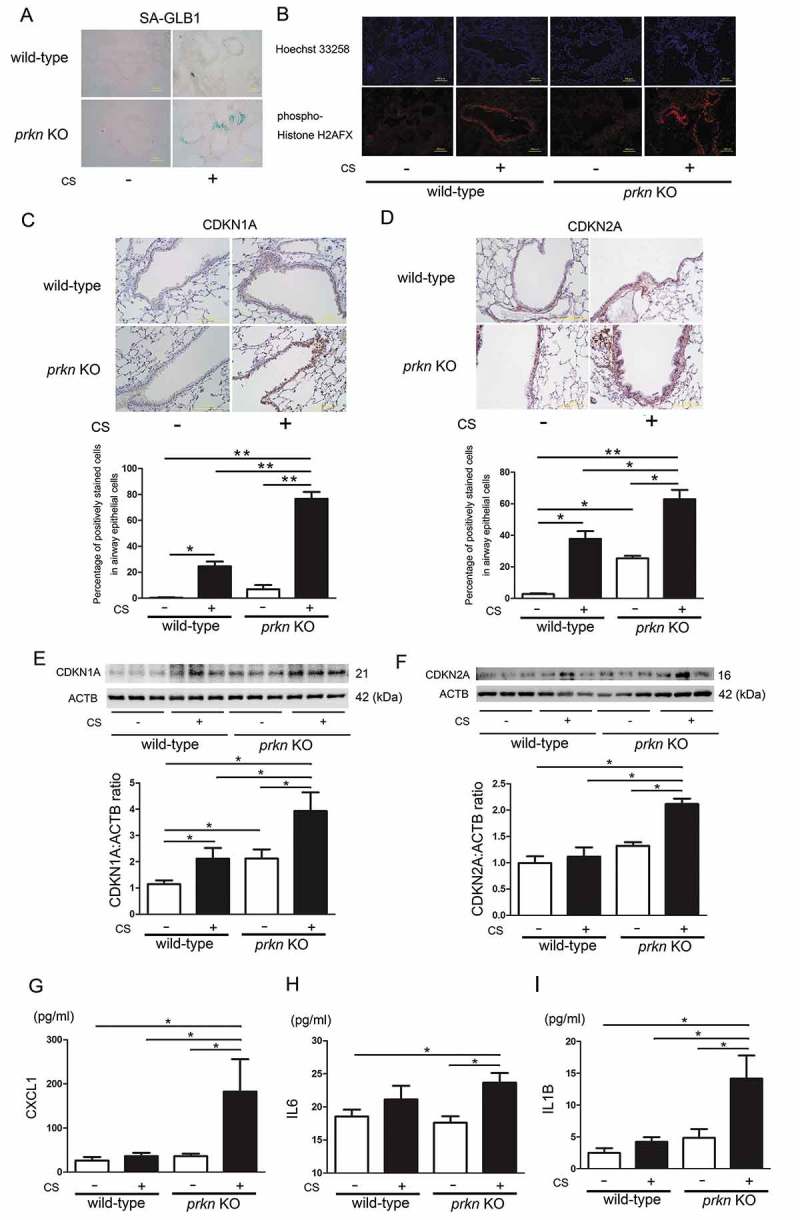
Long-term cigarette smoke exposure induces accelerated cellular senescence in prkn knockout mice. (a) Photomicrographs of GLB1 staining of control-air- and CS-exposed mouse lungs. (b) Photographs of immunofluorescent staining of phospho-Histone H2AFX (Ser139) in control-air- and CS-exposed mouse lungs. Bar: 100 µm. (c) Immunohistochemical staining of CDKN1A in control-air- and CS-exposed mouse lungs. Bar: 100 µm. The lower panel is the percentage (average ± SEM) of positively stained airway epithelial cells. *P < 0.05, **P < 0.001, by ANOVA and Tukey post-hoc test. (d) Immunohistochemical staining of CDKN2A in control-air- and CS-exposed mouse lungs. Bar: 100 μm. The lower panel is the percentage (average ± SEM) of positively stained airway epithelial cells. *P < 0.05, **P < 0.001, by ANOVA and Tukey post-hoc test. (e) WB using anti- CDKN1A and anti-ACTB antibodies, of lung homogenates from control-air- and CS-exposed mice for 6 months. The lower panel is the average (±SEM) taken from densitometric analysis of WB. Treatment groups were composed of control-air-exposed wild-type mice (n = 10), CS-exposed wild-type mice (n = 9), control-air-exposed prkn KO mice (n = 8), CS-exposed prkn KO mice (n = 9) *P < 0.05, by ANOVA and Tukey post-hoc test. All groups exposed for 6 months. (f) WB using anti- CDKN2A and anti-ACTB antibodies, of lung homogenates from control-air- and CS-exposed mice. The lower panel is the average (±SEM) taken from densitometric analysis of WB. Treatment groups were composed of control-air-exposed wild-type mice (n = 9), CS-exposed wild-type mice (n = 9), control-air-exposed prkn KO mice (n = 8), CS-exposed prkn KO mice (n = 9) *P < 0.05, by ANOVA and Tukey post-hoc test. (g) ELISA showing CXCL1 in lung homogenates from control-air- and CS-exposed mice. Shown is the average (±SEM). *P < 0.05, by ANOVA and Tukey post-hoc test. (h) ELISA showing IL6 in lung homogenates from control-air- and CS-exposed mice. Shown is the average (±SEM). *P < 0.05, by ANOVA and Tukey post-hoc test. (i) ELISA showing IL1B in lung homogenates from control air and CS-exposed mice. Shown is the average (±SEM). *P < 0.05, by ANOVA and Tukey post-hoc test.
To confirm airway epithelial cell senescence, immunohistochemical staining and western blotting of CDKN1A/p21 and CDKN2A/p16 (senescence-associated cyclin dependent kinase inhibitors) were performed. Both airway epithelial cells and alveolar epithelial cells in CS-exposed prkn KO mice demonstrated clear positive staining in both CDKN1A and CDKN2A (Figures 2(c,d) and S1)). Western blotting using lung homogenates further confirmed significant increases in CDKN1A and CDKN2A protein levels in lungs from CS-exposed prkn KO mice compared to CS-exposed wild-type mice (Figure 2(c,d)). Aberrant cytokine secretion of the senescence-associated secretory phenotype (SASP) is a characteristic feature of cellular senescence and has been proposed to be a part of COPD pathogenesis [20]. Thus we analyzed CXCL1 (chemokine [C-X-C] motif ligand 1), IL6 (interleukin 6), and IL1B (interleukin 1 beta) protein levels in lung homogenates, which are representative of SASP [21]. Consistent with the degree of cellular senescence, CXCL1 and IL1B protein levels were significantly increased in CS-exposed prkn KO mice compared to CS-exposed wild-type mice (Figure 2(e,g)). A mild but significant increase in IL6 was also observed in CS-exposed prkn KO mice (Figure 2(f)).
Accumulation of damaged mitochondria and oxidative modification in prkn KO mice in response to CS exposure
We have reported accumulation of fragmented and structurally distorted mitochondria in the airway epithelial cells of COPD lungs [5]. To elucidate the accumulation of damaged mitochondria reflecting insufficient mitophagy conferred by PRKN deficiency, electron microscopy evaluation was performed. No obvious structural alteration of mitochondria was observed in airway epithelial cells of wild-type mice nor prkn KO mice without CS exposure. Airway epithelial cells of CS-exposed wild-type mice showed accumulation of damaged mitochondria with abnormal swelling and crista disruption, which was significantly enhanced in prkn KO mice (Figure 3(a)). Total mitochondrial counts in airway epithelial cells (Figure 3(a)) and estimated mitochondrial mass shown by TOMM20 protein levels in lung homogenates were also significantly increased in lungs from CS-exposed prkn KO mice (Figure 3(b)). PINK1 protein levels were apparently increased in lungs from CS-exposed prkn KO mice, indicating that PINK1 levels may also reflect accumulation of damaged mitochondria (Figure 3(c)). Modest but significant reduction of PRKN protein levels was detected in CS-exposed wild-type mice (Figure 3(d)).
Figure 3.
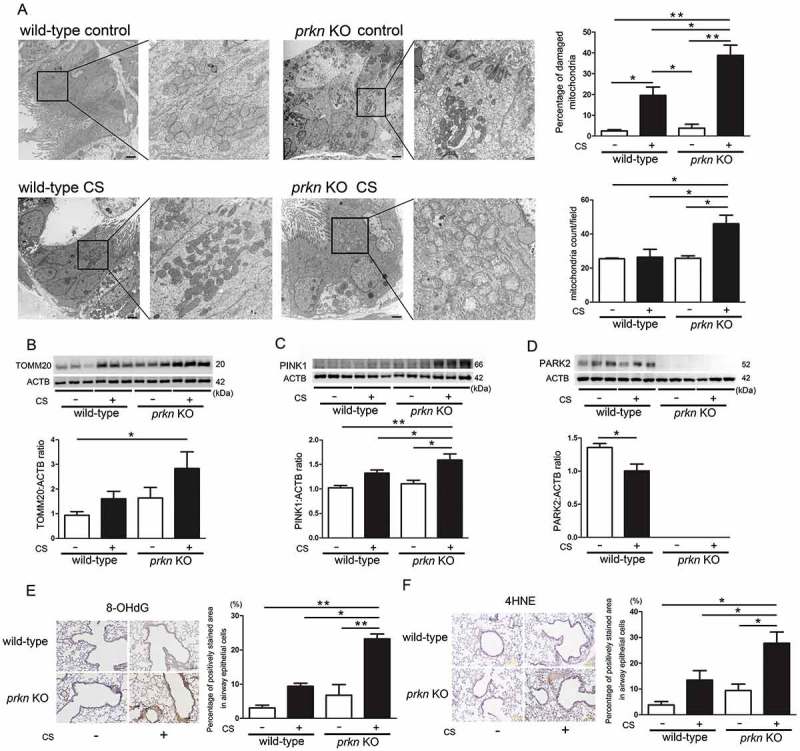
Alteration of mitochondrial structure, mitochondrial mass, and oxidative modifications in prkn knockout mice after long-term cigarette smoke exposure. (a) Electron microscopy detection of mitochondria in airway epithelial cells with cilia of control-air- and CS-exposed mouse lungs. Bar: 2 µm. Shown in upper panel is percentage of damaged mitochondria (average ± SEM) taken from 10 image fields (10,000 X) for each sample (n = 4 in each group). Shown in the lower panel is average (±SEM) of mitochondrial counts taken from 10 image fields (10,000 X) for each sample (n = 4 in each group). *P < 0.05, **P < 0.001, by ANOVA and Bonferroni post-hoc test. (b) WB using anti-TOMM20 and anti-ACTB antibodies, of lung homogenates from control-air- and CS-exposed mice for 6 months. The lower panel is the average (±SEM) taken from densitometric analysis of WB. Treatment groups were composed of control-air-exposed wild-type mice (n = 10), CS-exposed wild-type mice (n = 9), control-air-exposed prkn KO mice (n = 8), CS-exposed prkn KO mice (n = 9) *P < 0.05, by ANOVA and Tukey post-hoc test. (c) WB using anti-PINK1 and anti-ACTB antibodies, of lung homogenates from control-air- and CS-exposed mice for 6 months. The lower panel is the average (±SEM) taken from densitometric analysis of WB. *P < 0.05, by ANOVA and Tukey post-hoc test. (d) WB using anti-PRKN and anti-ACTB antibodies, of lung homogenates from control-air- and CS-exposed mice for 6 months. The lower panel is the average (±SEM) taken from densitometric analysis of WB. *P < 0.05, by unpaired Student t test. (e) Immunohistochemical staining of 8-hydroxy-2-deoxyguanosine (8-OHdG), oxidized derivative of deoxyguanosine in control-air- and CS-exposed mouse lungs. Bar: 100 µm. Shown in the right panel is percentage of positively stained areas in airway epithelial cells (average ± SEM) taken from 10 image fields (200X) for each sample (n = 4 in each group). *P < 0.05, **P < 0.001, by ANOVA and Bonferroni post-hoc test. (f) Immunohistochemical staining of 4-hydroxy-2-nonenal (4-HNE) of lipid peroxidation in control-air- and CS-exposed mouse lungs. Bar: 100 μm. Shown in the right panel is percentage of positively stained areas in airway epithelial cells (average ± SEM) taken from 10 image fields (200X) for each sample (n = 4 in each group). *P < 0.05, **P < 0.001, by ANOVA and Bonferroni post-hoc test.
CS exposure induced 8-hydroxy-2ʹ-deoxyguanosine (8-OHdG) staining, a major product of DNA oxidation, in wild-type mice, which was clearly enhanced in CS-exposed prkn KO mice. 4-Hydroxynonenal (4-HNE) staining, representing lipid peroxidation, was also markedly enhanced in CS-exposed prkn KO mice, suggesting the causal association between accumulation of damaged mitochondria and excessive mitochondrial ROS-mediated tissue damage in prkn KO mice during CS exposure (Figure 3(d,e)).
Increased PINK1 is associated with increased proteasomal degradation of PRKN
To further confirm PINK1 accumulation in association with reduced PRKN, PINK1 protein levels were evaluated during PRKN knockdown in HBEC. Consistent with PINK1 accumulation in CS-exposed prkn KO mice, PRKN knockdown significantly enhanced PINK1 in response to CSE exposure (Figure 4(a)), suggesting the involvement of PRKN in regulating PINK1 protein levels reflecting mitochondrial damage in HBEC. Conversely, PINK1 enhances not only PRKN translocation to mitochondria but also ubiquitination and degradation of PRKN through the ubiquitin-proteasome system (UPS) [16].
Figure 4.
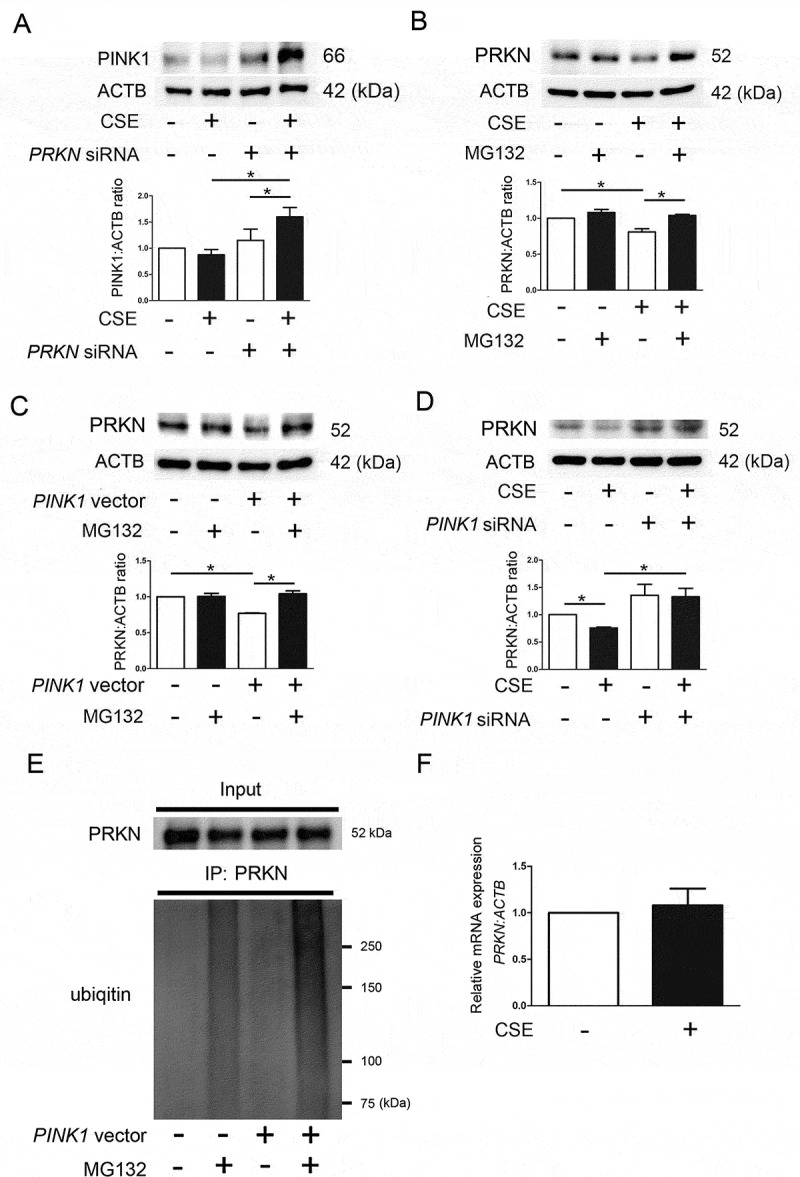
PINK1 regulates PRKN protein levels through proteasomal degradation in HBEC. (a) WB using anti-PINK1 and ACTB antibodies. HBEC were transfected with control siRNA or PRKN siRNA. CSE (1%) treatment was started 48 h post-transfection and protein samples were collected after 48-h treatment. Shown is a representative experiment of 4 showing similar results. The lower panel is the average (±SEM) relative increase in PINK1 normalized to ACTB, which are taken from densitometric analysis of WB from 4 independent experiments. *P < 0.05, by paired Student t test. (b) WB using anti-PRKN and ACTB antibodies. HBEC were treated with CSE (1%) for 24 h. MG132 (10 µM) treatment was started 6 h before collecting cell lysates. Shown is a representative experiment of 4 showing similar results. The lower panel is the average (±SEM) relative increase in PRKN normalized to ACTB, which are taken from densitometric analysis of WB from 4 independent experiments. *P < 0.05, by paired Student t test. (c) HBEC were transfected with control vector or PINK1 vector. MG132 treatment was started 48 h post-transfection and protein samples were collected after 6-h treatment. Shown is a representative experiment of 5 showing similar results. The lower panel is the average (±SEM) relative increase in PRKN normalized to ACTB, which are taken from densitometric analysis of WB from 5 independent experiments. *P < 0.05, by paired Student t test. (d) HBEC were transfected with control siRNA or PINK1 siRNA. CSE (1%) treatment was started 48 h post-transfection and protein samples were collected after 48-h treatment. Shown is a representative experiment of 4 showing similar results. The lower panel is the average (±SEM) relative increase in PRKN normalized to ACTB, which are taken from densitometric analysis of WB from 4 independent experiments. *P < 0.05, by paired Student t test. (e) Immunoprecipitation for detecting ubiquitinated PRKN. Cell lysates were collected HBEC treated with MG132 (10 ng/ml) for 6 h and treatment was started 48 h post-transfection with control or PINK1 vector. Immunoprecipitation was performed by using anti-PRKN and WB using an anti-ubiquitin antibody was performed. Shown is a representative experiment of 3 showing similar results. (f) HBEC were treated with CSE (1%) and mRNA samples were collected after treatment for 48 h. Real time-PCR was performed using primers to PRKN or ACTB, as a control. PRKN mRNA expression was normalized to ACTB. Shown is the fold increase (±SEM) relative to control treated cells (n = 3).
To gain a better understanding of the mechanisms for reduced PRKN protein levels in COPD with concomitant PINK1 accumulation, PRKN protein levels were examined in the setting of both PINK1 overexpression and knockdown in HBEC, respectively. Involvement of proteasomal degradation in PRKN reduction during CSE exposure was demonstrated by treatment with MG132, a proteasome inhibitor, which showed partial but significant recovery of PRKN protein levels (Figure 4(b)). Intriguingly, PINK1 overexpression alone reduced PRKN protein levels, which were significantly recovered by MG132 (Figure 4(c)). In contrast, PINK1 knockdown enhanced PRKN protein levels both in the absence and presence of CSE exposure (Figure 4(d)).
To clarify the involvement of mitophagy in PRKN reduction, protease inhibitors (E64d and pepstatin A) were used to prevent autolysosomal degradation. Protease inhibitors significantly inhibited PRKN reduction in response to CSE exposure (Figure S2). To further confirm proteasomal degradation of PRKN during PINK1 accumulation, PRKN ubiquitination was examined by means of immunoprecipitation assay. PINK1 overexpression with concomitant MG132 treatment significantly increased ubiquitinated PRKN protein levels (Figure 4(e)). No significant change was demonstrated in PRKN mRNA expression levels of HBEC in response to CSE exposure (Figure 4(f)). Taken together, it is likely that PRKN protein levels can be regulated by both the proteasome and by mitophagy, but proteasomal degradation modulated by PINK1 accumulation may have a pivotal role during insufficient mitophagy.
PRKN, relative to PINK1, is the dominant regulator of CSE-induced mitophagy
CS-exposed prkn KO mice demonstrated high PINK1 protein levels with concomitant accumulation of damaged mitochondria, reflecting insufficient mitophagic degradation, suggesting the pivotal role of PRKN in regulating mitophagy in COPD pathogenesis. To further elucidate the dominant role of PRKN compared with PINK1 in mitophagy regulation, combinations of overexpression and knockdown experiments were performed. Efficient knockdown and overexpression were confirmed by western blotting (Figure S3) and data not shown). Mitophagy was examined by means of colocalization analysis of confocal laser scanning microscopy images of TOMM20 (mitochondria) and MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta) (autophagosome) staining in BEAS-2B cells, an immortalized human bronchial epithelial cell line. CSE induced mitophagy shown by colocalization of TOMM20 and MAP1LC3B, which was further enhanced by PRKN overexpression but no alteration was demonstrated by PINK1 overexpression (Figure 5(a)). Consistent with our previous findings [7], CSE-induced mitophagy was apparently inhibited by PINK1 knockdown and PRKN knockdown, respectively (Figure 5(b)). Intriguingly PRKN overexpression clearly reversed PINK1 knockdown-mediated, (around 50% reduction of PINK1 protein levels was achieved: Figure S3), attenuation of CSE-induced mitophagy (Figure 5(b)). Conversely, PINK1 overexpression failed to reverse PRKN knockdown-mediated reduction of CSE-induced mitophagy (Figure 5(b)).
Figure 5.
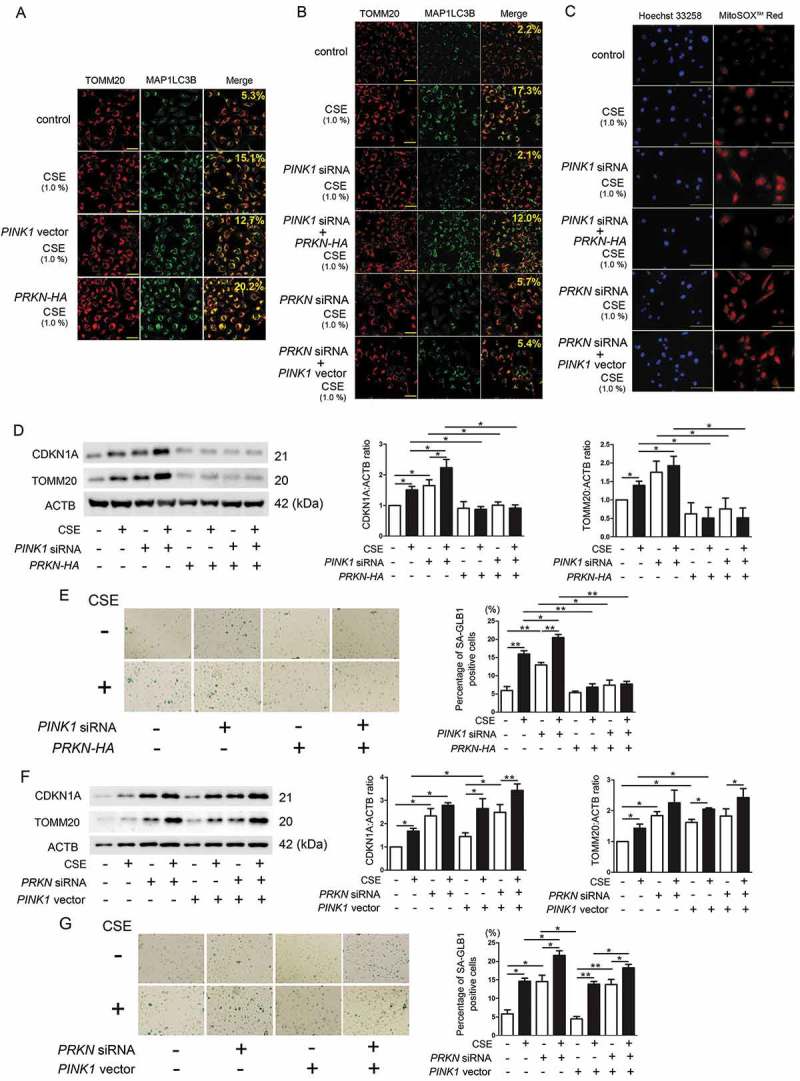
Dominant role of PRKN in the regulation of CSE-induced mitophagy. (a) Colocalization analysis of confocal laser scanning microscopy images of TOMM20 staining and MAP1LC3B staining. BEAS-2B cells were transfected with control vector, PINK1 vector, or PRKN-HA vector. CSE (1%) treatment was started 48 h post-transfection. BafA1 (20 nM) treatment was started 6 h before fixation and BEAS-2B cells were fixed after 24-h treatment with CSE. Bar: 100 μm. Shown percentage in merged image was calculated by dividing yellow intensity of mitophagy area by red intensity of mitochondrial area. (b) Colocalization analysis of TOMM20 staining and MAP1LC3B staining. BEAS-2B cells were transfected with the indicated combination of siRNA and expression vector, respectively. Bar: 100 µm. Shown percentage in merged image was calculated by dividing yellow intensity of mitophagy area by red intensity of mitochondrial area. (c) Photographs of Hoechst 33258 and MitoSOX Red fluorescence staining. HBEC were transfected with the indicated combination of siRNA and expression vector, respectively. CSE (1%) treatment was started 48 h post-transfection. HBEC were fixed after 24-h treatment with CSE. Bar: 100 µm. (d, f) WB using anti-CDKN1A, anti-TOMM20, and ACTB antibodies. HBEC were transfected with the indicated combination of siRNA and expression vector, respectively. CSE (1%) treatment was started 48 h post-transfection and protein samples were collected after 48-h treatment. Shown is a representative experiment of 4 showing similar results. The middle panel is the average (±SEM) relative increase in CDKN1A normalized to ACTB and the right panel is the average (± SEM) relative increase in TOMM20 normalized to ACTB, which are taken from densitometric analysis of WB from 4 independent experiments. *P < 0.05, by paired Student t test. (e, g) Photographs of senescence associated β-galactosidase (GLB1) staining in HBEC. Shown in right panel is the percentage (±SEM) of GLB1-positive cells from 5 independent experiments. *P < 0.05. **P < 0.001 by paired Student t test.
The dominant role of PRKN in mitophagy regulation in HBEC was also confirmed by means of colocalization analysis (Figure S4). In line with mitophagy status, increased mitochondrial ROS (MitoSOX Red) and mitochondrial mass (TOMM20), and accelerated cellular senescence (CDKN1A and GLB1 staining) by CS exposure, were enhanced by PINK1 and PRKN knockdown in HBEC, respectively (Figure 5(c-g)). CS-induced accumulation of mitochondrial ROS and mitochondrial mass, as well as accelerated cellular senescence enhanced by PINK1 knockdown, were clearly attenuated by PRKN overexpression. PINK1 overexpression failed to attenuate those alterations in the setting of PRKN knockdown (Figure 5(c-g)). The regulatory role of PRKN in CSE-induced HBEC senescence was further characterized by detecting CXCL8/IL8 (C-X-C motif chemokine ligand 8) secretion, a known SASP factor, from HBEC. CSE-induced CXCL8 secretion was apparently enhanced by PRKN knockdown, but was significantly reduced by PRKN overexpression (Figure S5). These findings support the notion that PRKN levels can be the rate-limiting factor in PINK1-PRKN-regulated mitophagy with respect to modulating cellular senescence during CSE exposure.
OPTN and CALCOCO2 are not involved in the mitophagy regulation in response to CSE exposure
A recent paper showed the existence of PINK1-mediated PRKN-independent mitophagy via recruitment of macroautophagy/autophagy receptors, CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) and OPTN (optineurin) [22]. To elucidate the involvement of CALCOCO2 and OPTN in mitophagy regulation during CSE exposure, knockdown experiments were performed in BEAS-2B cells. Although siRNA-mediated efficient knockdown of CALCOCO2 and OPTN were observed (Figure S3), no apparent attenuation in colocalization of TOMM20 and MAP1LC3B were demonstrated in response to CSE exposure (Figure 6(a)). Consistent with mitophagy status, mitochondrial ROS production (MitoSOX Red staining) was not affected by OPTN and CALCOCO2 knockdown (Figure 6(b)). Furthermore, in comparison to non-smoker lungs, no alteration in protein levels of CALCOCO2 and OPTN was detected in lung homogenates from COPD patients (Figure 6(c)). Patient characteristics are presented in Table 1. Consistent with our recent findings [7], significantly reduced PRKN but elevated PINK1 protein levels were demonstrated in COPD lungs (Figure 6(c)), suggesting that CALCOCO2 and OPTN-mediated PRKN-independent mitophagy was not involved major participant in the mechanisms for mitophagy regulation in COPD pathogenesis.
Figure 6.
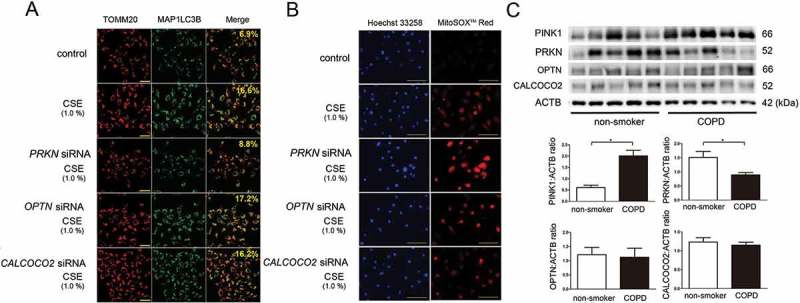
Effect of OPTN and CALCOCO2 knockdown on CSE-induced mitophagy. (a) Colocalization analysis of confocal laser scanning microscopy images of TOMM20 staining and MAP1LC3B staining. BEAS-2B cells were transfected with control siRNA, PRKN siRNA, OPTN (optineurin) siRNA, or CALCOCO2 (calcium binding and coiled-coil domain 2) siRNA. CSE (1%) treatment was started 48 h post-transfection. BafA1 (20 nM) treatment was started 6 h before fixation and BEAS-2B cells were fixed after 24-h treatment with CSE. Bar: 100 µm. Shown percentage in merged image was calculated by dividing yellow intensity of mitophagy area by red intensity of mitochondrial area. (b) Photographs of Hoechst 33258 and MitoSOX Red fluorescence staining. HBEC were transfected with the indicated siRNA, respectively. CSE (1%) treatment was started 48 h post-transfection. HBEC were fixed after 24-h treatment with CSE. Bar: 100 μm. (c) WB using anti-PINK1, anti-PRKN, anti-OPTN, anti-CALCOCO2, and anti-ACTB antibodies, of lung homogenates from non-smoker and COPD patients. The lower panel is the average (±SEM) taken from densitometric analysis of WB. Open bar is non-smoker (n = 5) and filled bar is COPD (n = 5). *P < 0.05, by Unpaired Student t test.
Table 1.
Patient characteristics (for lung homogenates).
| Non-smoker | COPD | ||
|---|---|---|---|
| (n = 5) | (n = 5) | P value | |
| Age (years) | 66.6 ± 5.4 | 62.8 ± 4.7 | NS* |
| Male (% of group) | 100.0 | 80.0 | NS** |
| SI (pack-years) | 0 | 44.5 ± 5.9 | <0.01* |
| FEV1/FVC (%) | 76.4 ± 4.9 | 66.7 ± 4.0 | <0.05* |
COPD, chronic obstructive pulmonary disease; FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; NS, not statistically significant; SI, Smoking index; Values are mean ± SD. *Mann-Whitney test, **Fisher exact test
Pirfenidone attenuates CSE-induced cellular senescence via inducing PRKN-mediated mitophagy
We have recently reported that pirfenidone (PFD), an anti-fibrotic modality of treatment for idiopathic pulmonary fibrosis, induces autophagy/mitophagy through the enhancement of PRKN expression in lung fibroblasts (LF) [23]. Hence, the role of PFD-mediated autophagy/mitophagy in the regulation of CSE-induced cellular senescence was examined in HBEC. PFD-induced autophagy activation was confirmed by detecting the conversion of MAP1LC3B from MAP1LC3B-I (free form) to MAP1LC3B-II (phosphatidylethanolamine-conjugated form) in the presence of protease inhibitors (E64d and pepstatin A), which was comparable to that observed by CSE treatment (Figure 7(a)). The combination of PFD and CSE further enhanced autophagy, suggesting that the mechanism for autophagy activation by PFD may not be identical to that by CSE (Figure 7(a)).
Figure 7.
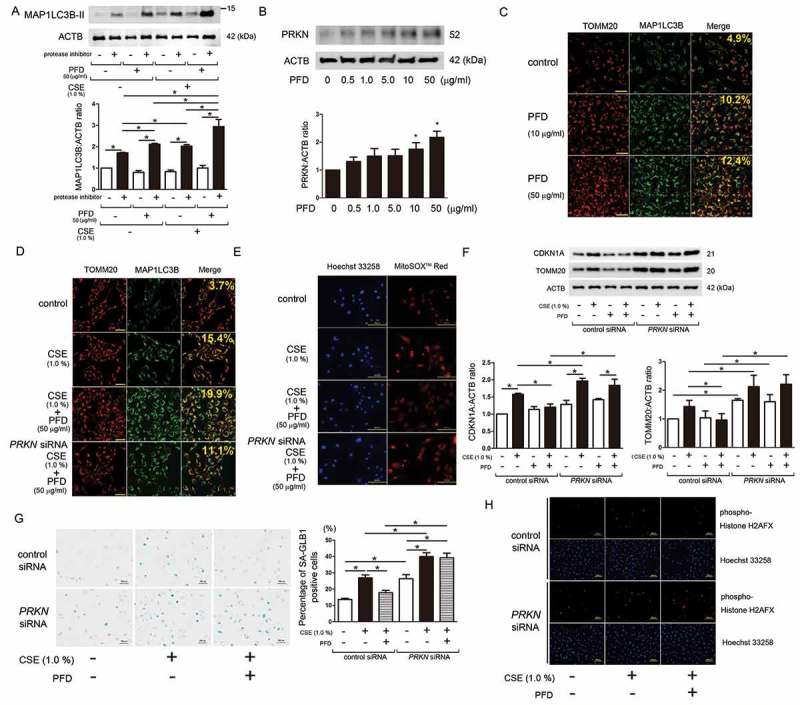
Effect of pirfenidone on autophagy/mitophagy and CSE-induced cellular senescence. (a) WB using anti- MAP1LC3B and anti-ACTB antibodies, of cell lysates from CSE treated HBEC in the presence or absence of pirfenidone (PFD) (50 µg/ml). Protein samples were collected after 24-h treatment with CSE and PFD. Protease inhibitor (E64d 10 μg/ml, pepstatin A 10 μg/ml) treatment was started 6 h before collecting cell lysates. In the lower panel is the average (±SEM) taken from 5 independent experiments shown as relative expression. *P < 0.05, by paired Student t test. (b) WB using anti-PRKN and anti-ACTB antibodies, of cell lysates from HBEC treated with the indicated concentration of PFD. Protein samples were collected after 2-h treatment with PFD. In the lower panel is the average (±SEM) taken from 7 independent experiments shown as relative expression. *P < 0.05, by paired Student t test. (c) Colocalization analysis of confocal laser scanning microscopy images of TOMM20 staining and MAP1LC3B staining. BEAS-2B cells were treated with the indicated concentration of PFD. BafA1 (20 nM) treatment was started 6 h before fixation and BEAS-2B cells were fixed after 24-h treatment with PFD. Bar: 100 µm Shown percentage in merged image was calculated by dividing yellow intensity of mitophagy area by red intensity of mitochondrial area. (d) Colocalization analysis of confocal laser scanning microscopy images of TOMM20 staining and MAP1LC3B staining. BEAS-2B cells were transfected with control siRNA or PRKN siRNA. CSE (1%) and PFD (50 µg/ml) treatment was started 48 h post-transfection. BafA1 (20 nM) treatment was started 6 h before fixation and BEAS-2B cells were fixed after 24-h treatment with CSE and PFD. Bar: 100 μm. Shown percentage in merged image was calculated by dividing yellow intensity of mitophagy area by red intensity of mitochondrial area. (e) Photographs of Hoechst 33258 and MitoSOX Red fluorescence staining. HBEC were transfected with control siRNA or PRKN siRNA. CSE (1%) and PFD (50 µg/ml) treatment was started 48 h post-transfection and HBEC were fixed after 24-h treatment. Bar: 100 µm (f) WB using anti-CDKN1A, anti-TOMM20, and ACTB antibodies. HBEC were transfected with control siRNA or PRKN siRNA. CSE (1%) and PFD (50 µg/ml) treatment was started 48 h post-transfection and protein samples were collected after 48-h treatment. Shown is a representative experiment of 3 showing similar results. In the lower panels are the average (±SEM) taken from 3 independent experiments shown as relative expression. *P < 0.05, by paired Student t test. (g) Photomicrographs of GLB1 staining of control siRNA or PRKN siRNA transfected HBEC. CSE (1%) and PFD (50 µg/ml) treatment was started 48 h post-transfection and HBEC were treated for 48 h. Bar: 500 µm. The right panel shows the percentage (±SEM) of GLB1-positive cells from 5 independent experiments. *P < 0.05, by paired Student t test. (h) Photographs of immunofluorescent staining of phospho-Histone H2AFX (Ser139) in control siRNA and PRKN siRNA transfected HBEC. CSE (1%) and PFD (50 µg/ml) treatment was started 48 h post-transfection and HBEC were fixed after 48-h treatment. Bar: 100 µm.
Next, changes in PRKN protein levels by PFD treatment were examined. PFD induced PRKN protein expression in a dose dependent manner, and significant induction was detected at the concentration of 10 μg/ml (Figure 7(b)). PFD-induced colocalization of TOMM20 and MAP1LC3B was demonstrated at the concentration of 10 μg/ml, but was clearly observed at the concentration of 50 μg/ml (Figure 7(c)). The effect of PFD on cellular senescence was also evaluated, and clear inhibition of CSE-induced CDKN1A expression was detected at the concentration of 50 μg/ml (Figure S6), thus 50 μg/ml of PFD was selected for further experiments to elucidate the anti-senescent role of mitophagy regulation by PFD.
CSE-induced mitophagy was enhanced by concomitant PFD treatment, and was clearly abrogated by PRKN knockdown (Figure 7(d)), indicating that PFD-mediated mitophagy was at least partly attributable to increased PRKN protein levels. Consistent with mitophagy regulation, CSE-induced mitochondrial ROS production was attenuated by PFD treatment, which was clearly abrogated by PRKN knockdown (Figure 7(e)). CSE treatment enhanced CDKN1A expression with concomitantly increased mitochondrial mass, which may reflect insufficient mitophagic degradation (Figure 7(f)). In line with mitophagy and mitochondrial ROS regulation, PFD significantly reduced cellular senescence and mitochondrial mass at the concentration of 50 μg/ml during CSE exposure (Figure 7(f)). However, these effects of PFD were clearly abrogated by PRKN knockdown (Figure 7(f)). PFD-mediated regulation of HBEC senescence via PRKN expression during CSE exposure was further confirmed by GLB1 staining (Figure 7(g)) and phospho-Histone H2AFX (Ser139) staining, respectively (Figure 7(h)). Taken together, these results suggest that PFD is a potential promising geroprotector, with the potential to inhibit cellular senescence caused by CSE exposure via inducing PRKN-mediated mitophagy.
Discussion
In comparison to wild-type mice, prkn KO mice demonstrate exaggerated airway wall thickening and emphysematous changes in response to long-term CS exposure. Airway epithelial cells in CS-exposed prkn KO mice show accumulation of damaged mitochondria and increased oxidative modifications accompanied by accelerated cellular senescence, suggesting the pivotal role of PRKN in regulating mitochondrial integrity and cellular senescence during CS exposure. In vitro experiments elucidate that PRKN overexpression is sufficient for inducing mitophagy during CSE exposure even in the setting of PINK1 knockdown, resulting in attenuation of mitochondrial ROS production and cellular senescence. Conversely PINK1 overexpression fails not only to enhance CSE-induced mitophagy but also to recover impaired mitophagy caused by PRKN knockdown, indicating that PRKN protein levels can be the rate-limiting factor in PINK1-PRKN-mediated mitophagy during CSE exposure in HBEC. Intriguingly, PINK1 overexpression reduces PRKN protein levels, which is partly recovered by proteasome inhibitors, suggesting that increased PINK1-mediated proteasomal degradation of PRKN can be involved in the mechanisms for decreased PRKN in COPD lungs. Knockdown experiments and protein expression levels in lung homogenates clarify that CALCOCO2 and OPTN are not involved in CSE-induced mitophagy and COPD pathogenesis. Consistent with recent reports including our findings [4,7], lung homogenates from human samples demonstrate decreased PRKN and increased PINK1 protein levels, further supporting the notion of the pivotal involvement of PRKN-mediated mitophagy in COPD pathogenesis. PFD induces PRKN protein expression and PRKN-mediated mitophagy, which prevents accumulation of mitochondrial ROS and cellular senescence during CSE exposure, suggesting that PFD can be a novel geroprotective modality of treatment for COPD with accelerated cellular senescence.
PINK1 is rapidly degraded in basal conditions but is stabilized on the outer membrane in response to mitochondrial depolarization, resulting in PINK1 accumulation following PRKN translocation to mitochondria. Although increased PINK1 protein levels may imply enhancement of mitophagy [6], increased PINK1 can also be recognized as reflecting accumulation of damaged mitochondria with stabilized PINK1 regardless of mitophagy status. Actually, PINK1 accumulation has been demonstrated during impaired mitophagy caused by reduced PRKN protein levels and subsequent malfunction of PINK1-PRKN crosstalk in skin fibroblasts from sporadic Alzheimer disease (AD) [24]. A previous paper shows upregulation of PINK1 in COPD lungs, which is considered as reflecting accumulation of mitochondrial damage [17], while another recent paper shows that PRKN translocation to mitochondria is hampered in CS-exposed mouse lungs and COPD lungs, resulting in impairment of mitophagy with enhanced PINK1 protein levels [4]. We believe that increased PINK1 expression levels are mainly attributed to accumulation of damaged mitochondria during insufficient mitophagy caused by PRKN reduction [7], which is strongly supported by our experimental results in the present study. However, it is still unclear whether PINK1 accumulation is a simple consequence of CS exposure or causally associated with COPD development. We have previously reported that PINK1 tended to be increased in COPD lungs but not in non-COPD smoker lungs [7], suggesting that increase in PINK1 is not only dependent on smoking status but also on aberrant accumulation of damaged mitochondria as a part of COPD pathogenesis.
Although PRKN expression levels appear to be a critical determinant of mitophagy during CS exposure, the precise mechanism for PRKN reduction in COPD lungs remains elusive. A recent comprehensive validation study shows the potential participation of a PRKN SNP in COPD and lung cancer development, but the role of this PRKN SNP in regulating protein levels remains to be determined [12]. PINK1 enhances ubiquitination and degradation of PRKN through UPS [16], resulting in a decreased cytosolic pool of PRKN. This suggests that increased PINK1 cannot only result from PRKN reduction during insufficient mitophagy, but is also itself a cause of subsequent PRKN reduction. Actually, we observed a significant decrease in PRKN protein levels with PINK1 overexpression and CSE exposure in HBEC, which was at least partly recovered by treatment with proteasome inhibitors (Figure 4(b,c)), suggesting the existence of a positive feedback-loop of PINK1 accumulation and PRKN reduction, resulting in a persistent state of decreased PRKN in COPD lungs (Figure 8). Although we observed no significant change in mRNA levels during in vitro CSE exposure models in HBEC (Figure 4(f)), significant reduction in mRNA levels was demonstrated in COPD lungs (Figure S7). Therefore, the mechanism for mRNA reduction remains unclear, regulation in both protein and mRNA levels can be responsible for PRKN reduction in COPD lungs caused by long-term CS exposure. Furthermore, post-mortem evaluation of Parkinson disease brain samples demonstrated that PRKN was inactivated by post-translational modifications, including oxidation and nitrosylation [25]. Those oxidative modifications are a prominent feature in COPD lungs [19], and were clearly demonstrated in mouse airway epithelial cells subjected to CS exposure (Figure 3(d,e)), suggesting that not only protein expression levels but also PRKN dysfunction conferred by oxidative modifications may have a role in insufficient mitophagy in COPD pathogenesis.
Figure 8.
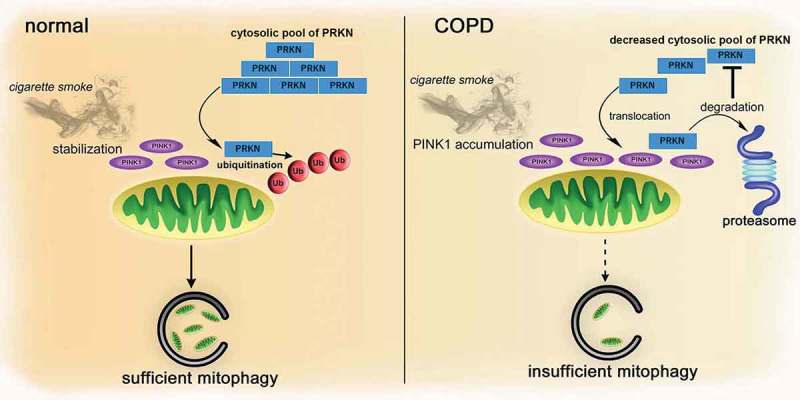
Hypothetical model of involvement of PRKN in the regulation of mitophagy during smoking stress in COPD pathogenesis. In normal conditions, CS-induced mitochondrial damage stabilizes PINK1 on the mitochondrial outer membrane, which recruits PRKN to the mitochondria from an abundant cytosolic pool, resulting in sufficient mitophagy, thus preventing accumulation of damaged mitochondria and accelerated cellular senescence. In contrast, in COPD, insufficient mitophagy triggers accumulation of damaged mitochondria with stabilized PINK1, which can be attributed to a decreased cytosolic PRKN pool, leading to further PRKN reduction by PINK1-mediated proteasomal degradation. We speculate the existence of a feedback loop of PINK1 accumulation and PRKN reduction linked to persistently decreased PRKN accompanied by insufficient mitophagy in COPD pathogenesis.
Accelerated cellular senescence in a variety of cell types has been implicated in COPD pathogenesis [18]. Although mitochondria are the major site of ATP production, dysfunctional mitochondria are the main source of intrinsic ROS, which has been widely implicated in the mechanisms for cellular senescence [26]. In line with our previous findings in COPD lungs [5], we demonstrated accumulation of damaged mitochondria, which was accompanied by enhanced oxidative modifications (8-OHdG and 4HNE) and accelerated cellular senescence in lung tissues of CS-exposed prkn KO mice (Figure 3). In contrast to mitochondria-localized PINK1, PRKN mediates its effects in multiple cellular compartments, including the cytosol, mitochondria, and nucleus [25]. Additionally, diverse roles of PRKN in maintaining mitochondrial function through regulating mitochondrial DNA integrity, biogenesis, dynamics, and intracellular transportation have been reported [25,27–29]. Accordingly, PRKN deficiency can be responsible for accumulation of damaged mitochondria and accelerated cellular senescence in CS-exposed prkn KO mice through not only mitophagy regulation, but other mechanisms for maintaining mitochondrial integrity. A recent paper shows that PRKN deficiency is linked to activation of class I phosphoinositide 3-kinase (PI3K)-AKT (AKT serine/t hreonine kinase)-MTOR (mechanistic target of rapamycin kinase) signaling through PTEN loss with respect to tumorigenesis [30]. Aberrant activation of PI3K-AKT-MTOR signaling has been widely implicated in COPD pathogenesis and recognized as a key regulator for accelerated cellular senescence [31]. Hence, it is plausible that reduced PRKN protein levels are associated with COPD pathogenesis via regulating mitochondrial function and cellular senescence through a variety of mechanisms, including insufficient mitophagy, impaired mitochondrial integrity, and aberrant MTOR activation.
Chronic inflammation has been widely shown in COPD pathogenesis [31], and PRKN may also have a direct role in controlling the inflammatory process. It has been reported that PRKN deficiency was associated with exacerbated inflammation induced by chronic systemic lipopolysaccharide (LPS) treatment [32]. Furthermore, PRKN-deficient mice demonstrate IL6 elevation in their serum, and increased IL1B and TNF (tumor necrosis factor) mRNA in primary bronchial epithelial cells [12], indicating intrinsic anti-inflammatory properties for PRKN. Conversely, a recent paper shows that PRKN may mediate endotoxin-induced acute inflammation in an acute lung injury model [33]. Furthermore, PRKN deficiency may inhibit inflammation via suppressing TP53/p53 degradation in rheumatoid arthritis (RA) [34], indicating that the role of PRKN in modulating the inflammatory process appears to be complicated and remains uncertain. We observed significantly increased inflammatory cytokines (CXCL1, IL6, and IL1B) in lung homogenates and macrophage cell counts in BALF from CS-exposed prkn KO mice, but only modest increase in cytokines and cell counts were demonstrated in prkn KO mice without CS exposure (Figure 3). Although PRKN-mediated direct regulation of inflammation cannot be completely excluded, we speculate that SASP attributed to accelerated cellular senescence is responsible for the inflammatory phenotype in CS-exposed prkn KO mice
PRKN overexpression was sufficient for inducing mitophagy and inhibiting cellular senescence in response to CSE exposure (Figure 5), indicating that PRKN induction is a potentially promising geroprotective modality of treatment for COPD with reduced PRKN and accelerated cellular senescence. We have recently reported that anti-fibrotic PFD is a candidate agent for inducing PRKN expression [23]. Although a significant increase in PRKN expression levels was detected at 10 μg/ml PDF in HBEC, which is comparable to the levels of maximum plasma concentration of PFD in a clinical setting, 5-fold higher concentration (50 μg/ml) was needed to demonstrate sufficient mitophagy with an anti-senescence property (Figures S6 and 7). Although PFD exerts a wide array of anti-fibrotic properties, including anti-inflammation, anti-pro-fibrotic growth factors, and anti-oxidative effects [35,36], its precise mechanism of action remains unknown. We speculate that mitophagy induction through increased PRKN expression plays a role in the anti-oxidative properties of PFD. Although higher concentrations of PFD than clinically achievable plasma levels is potentially necessary for the anti-senescence modality of treatment, because of the anatomical specificity, we speculate that inhalation therapy may be an interesting system to achieve sufficient local PFD concentration in lung epithelial cells. Hence, further studies are warranted for development of novel drug-delivery systems for PFD.
In summary, we demonstrated that PRKN plays a pivotal role in regulating mitophagy and thus mitochondrial integrity and cellular senescence during COPD pathogenesis. PINK1 is also essential for mitophagy, but PRKN protein levels are the limiting factor in PINK1-PRKN-regulated mitophagy during CS exposure in HBEC. Although efficient and clinically available PRKN inducing agents should be developed in future studies, PRKN induction by PFD can be a promising geroprotective modality via enhancement of mitophagy in the aging-associated pathology, COPD.
Materials and methods
Cell culture, antibodies, and reagents
Normal and COPD airways were obtained from 1st through 4th order bronchi from pneumonectomy and lobectomy specimens for primary lung cancer. Informed consent was obtained from all surgical participants as part of an approved ongoing research protocol by the ethics committee of Jikei University School of Medicine. HBEC were isolated with protease treatment and freshly isolated HBEC were plated onto rat-tail collagen type I (Sigma-Aldrich, C3867.)-coated (10 μg/ml) dishes, incubated overnight, and then the medium was changed to bronchial epithelial growth medium (Clonetics, CC-3170). Cultures were characterized immunohistochemically using anti-KRT/cytokeratin antibodies (Lu-5; BioCare Medical, 043) and anti-VIM/vimentin (Sigma-Aldrich, V6630), as previously described [1]. HBEC showed >95% positive staining with anti-KRT/cytokeratin and <5% positive staining with the anti-VIM antibody. HBEC were serially passaged and used for experiments until passage 3. HBEC were grown to 80% confluency before experiments. The experiments were performed with HBEC from non-COPD patients. The bronchial epithelial cell line BEAS-2B was cultured in RPMI1640 (Gibco Life Technologies, 11875–093) with 10% fetal calf serum (Gibco Life Technologies, 26140–079) and penicillin-streptomycin (Gibco Life Technologies, 15140–122). After growing to 80% confluency, BEAS-2B experiments were performed using media without FCS.
Antibodies used were rabbit anti-CDKN1A (Cell Signaling Technology, 2947), rabbit anti-CDKN2A (Abcam, ab54210), rabbit anti-PRKN (Cell Signaling Technology, 2132), rabbit anti-PINK1 (Cell Signaling Technology, 6946), rabbit anti-PINK1 (Abcam, ab23707), rabbit anti-PINK1 (Novus, BC100-494), rabbit anti-phospho-histone H2AFX/H2A.X (Ser139) (Cell Signaling Technology, 2577), rabbit anti-MAP1LC3B/LC3B (Novus, 600-1384), rabbit anti-MAP1LC3B/LC3B (Cell Signaling Technology, 3868), rabbit anti-CALCOCO2 (Cell Signaling Technology, 60732), anti-OPTN (LifeSpan BioSciences, LS-C331724), rabbit anti-4 hydroxynonenal (Abcam, ab46545), rabbit anti-8-OHdG/8 hydroxyguanosine (BIOSS, bs-1278R), mouse anti-TOMM20 (Santa Cruz Biotechnology, sc-17764), and mouse anti-ACTB/β-actin (Sigma-Aldrich, A5316). The following reagents were used: bafilomycin A1 (BafA1; Sigma-Aldrich, B1793), MG-132 (Enzo Life Sciences, BML-P102), CM-H2DCFDA (Life Technologies, C6827), Hoechst 33258 (Sigma-Aldrich, B2883), MitoSOX Red (Molecular Probes/Life Technologies, M36008), pepstatin A (Peptide Institute, 4397), E64d (Peptide Institute, 4321-v). Pirfenidone (PFD) was provided by Shionogi & Co., Ltd. (Osaka, Japan)
Plasmids, small interfering RNA and transfection
PRKN expression vector and PINK1 expression vector were obtained from Addgene (Addgene, 17613 and 13316; deposited by Ted Dawson and Mark Cookson). Small interfering RNA (siRNA) targeting PRKN (Applied Biosystems Life Technologies, s10043, s10044), PINK1 (Applied Biosystems Life Technologies, s35166, s35167), OPTN (Applied Biosystems Life Technologies, s19720), CALCOCO2/NDP52 (Applied Biosystems Life Technologies, s19995) and negative control siRNAs (Applied Biosystems Life Technologies, AM4635, AM4641) were purchased from Life Technologies. Specific knockdowns of PRKN and PINK1 were validated using 2 different siRNAs, respectively. Transfections of HBEC were performed using the Neon® Transfection System (Invitrogen Life Technologies, MPK5000), using matched optimized transfection kits (Invitrogen Life Technologies, MPK10096).
Preparation of cigarette smoke extract (CSE)
Cigarette smoke extract (CSE) was prepared as previously described with minor modifications [21]. The research reference cigarettes 3R4F were used for experiments (Louisville, KY, USA). Forty milliliters of cigarette smoke were drawn into a syringe and slowly bubbled into sterile serum-free cell culture media in 15-ml BD falcon tubes. One cigarette was used for the preparation of 10 mL of solution. CSE solution was filtered (0.22 μm; Merck Millipore, SLGS033SS) to remove insoluble particles and was designated as 100% CSE solution.
Western blotting
HBEC grown on 6-well culture plates were lysed in RIPA buffer (Thermo Fisher Scientific, 89900) containing a protease inhibitor cocktail (Roche Diagnostics, 11697498001) and 1 mM sodium orthovanadate (Wako, 13721–39-6), or lysed with Laemmli sample buffer. Western blotting was performed as previously described [37]. For each experiment, equal amounts of total protein were resolved by 7.5–15% SDS-PAGE. After SDS-PAGE, proteins were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, ISEQ00010), and incubation with specific primary antibody was performed for 2 h at 37°C, or 24 h at 4°C. After washing several times with PBST (Wako, 161–25521), the membrane was incubated with anti-rabbit IgG, HRP-linked secondary antibody (Cell Signaling Technology, 7074), anti-mouse IgG, HRP-linked secondary antibody, 7076) followed by chemiluminescence detection (Thermo scientific, 34080, and BIO-RAD, 1705061) with the ChemiDocTM Touch Imaging System (BIO-RAD, CA, USA). To compare the CDKN1A, CDKN2A, TOMM20, PINK1, PRKN, OPTN and CALCOCO2 expression levels in lung homogenates between different experimental sets, one homogenate sample from a normal patient or control mouse was selected as a loading control for all western blotting and used for further normalization of densitometric data analysis. Immunoprecipitation was performed as previously described by using anti-PRKN/PARK2 (Abcam, ab15964) and anti-ubiquitin (Enzo Life Science, BML-PW8810) [16].
Immunofluorescence staining
BEAS-2B cells were grown on 8-well culture slides. BEAS-2B cells transfected with a plasmid encoding PRKN expression vector and PINK1 expression vector were treated with CSE at 48 h post-transfection. BEAS-2B cells were transfected with control siRNA, PRKN siRNA, PINK1 siRNA, OPTN siRNA or CALCOCO2 siRNA and were treated with CSE at 48 h post-transfection. BEAS-2B cells were treated with BafA1 6 h before collection. BEAS-2B cells were fixed with 100% methanol (Wako, 137-01823). After blocking with 1% BSA (Sigma-Aldrich, A2153) for 60 min, the primary and secondary antibodies were applied according to the manufacturers’ instructions. Confocal laser scanning microscopy analysis of BEAS-2B cells was performed using mouse anti-TOMM20 and rabbit anti- MAP1LC3B (Cell Signaling Technology, 3868), and evaluated by fluorescence microscopy (Carl Zeiss LSM880, Tokyo, Japan). Quantitative measure of immunofluorescence staining to evaluate mitophagic activity was analyzed by dividing the yellow intensity of mitophagy areas by the red intensity of mitochondrial areas.
Fluorescence microscopy analysis of phospho-histone H2AFX and Hoechst 33258 staining were performed in lung frozen sections and HBEC, and evaluated by fluorescence microscopy (Olympus, BX60, Tokyo, Japan and Keyence, BZ-X700, Tokyo, Japan).
RNA isolation, polymerase chain reaction
RNA isolation, reverse transcription and Real-Time PCR were performed using the SYBR green method as previously described (Roche, LightCycler® 96 System, Tokyo, Japan) [23]. The primers used were PRKN sense primer, 5ʹ-AAATGCCCAGACAAGATGCC-3ʹ; PRKN antisense primer, 5ʹ-GGCCTCTCACGACTGAGTT-3ʹ; ACTB sense primer 5ʹ-CATGTACGTTGCTATCCAGGC-3ʹ ACTB antisense primer 5ʹ-CTCCTTAATGTCACGCACGAT-3ʹ. These primer sets yielded PCR products of 135 bp and 250 bp for PRKN and ACTB respectively. PCRs of PRKN were validated using 2 different primers. Primer sequences were from Primer Bank (http://pga.mgh.harvard.edu/primerbank).
Measurement of ROS production
HBEC were grown on 8-well culture slides. HBEC transfected with a plasmid encoding either a PRKN expression vector or PINK1 expression vector were treated with CSE at 48 h post-transfection. HBEC were transfected with control siRNA, PRKN siRNA, PINK1 siRNA, OPTN siRNA or CALCOCO2 siRNA and were treated with CSE at 48 h post-transfection. HBEC were fixed after 24-h treatment with CSE. Mitochondrial ROS production was analyzed by MitoSOX Red staining according to the manufacturer’s instructions, which was evaluated by fluorescence microscopy (Olympus, Tokyo, Japan and Keyence, BZ-X700).
GLB1 staining
Senescence associated GLB1/β-galactosidase staining was performed using lung frozen sections and HBEC grown on 6-well and 12-well culture plates according to the manufacturer’s instructions (Sigma-Aldrich, CS0030).
Mouse models and CS exposure protocol
C57BL/6J (CLEA Japan INC, Tokyo, Japan) and B6.129S4-Prkntm1Shn/J (Jackson Laboratories, Bar Harbor, ME) mice were purchased, and were maintained in the animal facility at the Jikei University School of Medicine. All experimental procedures were approved by the Jikei University School of Medicine Animal Care Committee. Six- to 8-week-old mice were exposed using a whole-body exposure system (SCIREQ ‘InExpose’) within a barrier facility. Mice were exposed at a total suspended particulates of 200 mg/m3 using 3R4F cigarettes for 5 days a week. After 6 months, immediately following sacrifice, the right lung was inflated with 10% buffered formalin at a pressure of 20 cm H2O. Fixed lungs were embedded in paraffin and cut into 4-µm sections for tissue staining. The sections stained with hematoxylin & eosin (H-E) and Picro-Sirius Red Stain (PSR) according to conventional protocols for histopathological evaluation. Immunohistochemistry was performed on the paraffin-embedded lung tissues as previously described with minor modifications [37]. The left lungs were removed and homogenized for protein evaluation or the frozen sections stained with GLB1 staining and immunofluorescence staining. Tracheotomy was performed to insert a tracheal tube for collecting bronchoalveolar lavage fluid (BALF) sample with PBS (Wako, 041-20211; total 3 ml). The cell numbers in BALF were counted using a hemocytometer. Differential cell counts in BALF were analyzed on 300 cells stained with Diff-Quick (Sysmex, 16920).
Airway morphometry
Airway morphometry was performed essentially as previously described [38]. Measurements of alveolar enlargement were estimated using Hematoxylin and Eosin (H&E) stained slides. Microtome sections from H&E stained sections of paraffin embedded mouse lungs were digitally imaged at 200X magnification (Olympus, Tokyo, Japan and Keyence, BZ-X700). 10 random fields were evaluated by a blinded investigator using an image analysis software, (Image J), and alveolar enlargement was estimated using the mean linear intercept (MLI) method as previously described [39]. Airway wall fibrosis was assessed by the presence of thick collagen bundles stained by the PSR Stain, which expresses wall thickness as a function of area of the airway wall/basement membrane length. Five random airways/each mouse were evaluated by a blinded investigator using image analysis software, (ImageJ).
CXCL1/KC, IL6, IL1B, CXCL8/IL-8 enzyme-linked immunosorbent assay
CXCL1, IL6, IL1B, and CXCL8 in lung homogenates and HBEC conditioned medium were measured using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Electron microscopy
Electron microscopy was performed as previously described [37]. Pieces from mouse lung were fixed with 2% glutaraldehyde, 0.1 M phosphate buffer (pH 7.4) and after 48 h of incubation were dehydrated with a graded series of ethanol. Fixed lung pieces were then embedded in epoxy resin (Epok812; Oken, 02–1001). Ultrathin sections were stained with uranyl acetate and lead citrate and observed with the Hitachi H-7500 transmission electron microscope (Hitachi, Tokyo, Japan). For quantitative evaluation of mitochondria in airway epithelial cells, 10 airway epithelial cells imaged at 10,000X magnification were selected for each sample and mitochondria were counted.
Statistics
Data are shown as the average (±SEM) taken from at least 3 independent experiments. Comparisons between 2 different groups were determined by Student t test for parametric data or Mann-Whitney test for nonparametric data. One-way analysis of variance was used for multiple comparisons and Tukey or Bonferroni post-hoc tests used to test for statistical significance. Significance was defined as P < 0.05. Statistical software used was Prism v.5 (GraphPad Software, Inc., San Diego, CA).
Funding Statement
This work was supported by grants from JSPS KAKENHI Grant Number JP15K09231 to J.A., 15K09233 to K. Nakayama, and JP15K09232 to K.Kuwano.
Acknowledgments
We wish to thank Stephanie Cambier (University of Washington, Seattle, USA), Emi Kikuchi and Dr. Toshiaki Tachibana (Jikei University School of Medicine, Tokyo, Japan) for technical support.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Araya J, Cambier S, Markovics JA, et al. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest. 2007. November;117(11):3551–3562. PubMed PMID: 17965775; PubMed Central PMCID: PMCPMC2040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].van der Toorn M, Slebos DJ, de Bruin HG, et al. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol. 2007. May;292(5):L1211–218. PubMed PMID: 17209140. [DOI] [PubMed] [Google Scholar]
- [3].van der Toorn M, Rezayat D, Kauffman HF, et al. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009. July;297(1):L109–114. PubMed PMID: 19411310; PubMed Central PMCID: PMCPMC2711811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ahmad T, Sundar IK, Lerner CA, et al. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J. 2015. July;29(7):2912–2929. PubMed PMID: 25792665; PubMed Central PMCID: PMCPMC4478793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hara H, Araya J, Ito S, et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am J Physiol Lung Cell Mol Physiol. 2013. November 15;305(10):L737–746. PubMed PMID: 24056969. [DOI] [PubMed] [Google Scholar]
- [6].Mizumura K, Cloonan SM, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014. September;124(9):3987–4003. PubMed PMID: 25083992; PubMed Central PMCID: PMCPMC4151233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ito S, Araya J, Kurita Y, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. 2015;11(3):547–559. PubMed PMID: 25714760; PubMed Central PMCID: PMCPMC4502689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Durcan TM, Fon EA.. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015. May 15;29(10):989–999. PubMed PMID: 25995186; PubMed Central PMCID: PMCPMC4441056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998. April 09;392(6676):605–608. PubMed PMID: 9560156. [DOI] [PubMed] [Google Scholar]
- [10].Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004. May 21;304(5674):1158–1160. PubMed PMID: 15087508. [DOI] [PubMed] [Google Scholar]
- [11].Li CH, Chen WC, Liao WC, et al. The association between chronic obstructive pulmonary disease and Parkinson’s disease: a nationwide population-based retrospective cohort study. Qjm. 2015. January;108(1):39–45. PubMed PMID: 25024356. [DOI] [PubMed] [Google Scholar]
- [12].Lee S, She J, Deng B, et al. Multiple-level validation identifies PARK2 in the development of lung cancer and chronic obstructive pulmonary disease. Oncotarget. 2016. July 12;7(28):44211–44223. PubMed PMID: 27329585; PubMed Central PMCID: PMCPMC5190090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Clark IE, Dodson MW, Jiang C, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006. June 29;441(7097):1162–1166. PubMed PMID: 16672981. [DOI] [PubMed] [Google Scholar]
- [14].Park J, Lee SB, Lee S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006. June 29;441(7097):1157–1161. PubMed PMID: 16672980. [DOI] [PubMed] [Google Scholar]
- [15].Yang Y, Gehrke S, Imai Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006. July 11;103(28):10793–10798. PubMed PMID: 16818890; PubMed Central PMCID: PMCPMC1502310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rakovic A, Shurkewitsch K, Seibler P, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem. 2013. January 25;288(4):2223–2237. PubMed PMID: 23212910; PubMed Central PMCID: PMCPMC3554895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hoffmann RF, Zarrintan S, Brandenburg SM, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013. October 02;14:97 PubMed PMID: 24088173; PubMed Central PMCID: PMCPMC3852998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aoshiba K, Nagai A. Senescence hypothesis for the pathogenetic mechanism of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2009. December 01;6(7):596–601. PubMed PMID: 19934355. [DOI] [PubMed] [Google Scholar]
- [19].Hara H, Araya J, Takasaka N, et al. Involvement of creatine kinase B in cigarette smoke-induced bronchial epithelial cell senescence. Am J Respir Cell Mol Biol. 2012. March;46(3):306–312. PubMed PMID: 21980054. [DOI] [PubMed] [Google Scholar]
- [20].Chilosi M, Carloni A, Rossi A, et al. Premature lung aging and cellular senescence in the pathogenesis of idiopathic pulmonary fibrosis and COPD/emphysema. Transl Res. 2013. September;162(3):156–173. PubMed PMID: 23831269. [DOI] [PubMed] [Google Scholar]
- [21].Fujii S, Hara H, Araya J, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012. August 01;1(5):630–641. PubMed PMID: 22934255; PubMed Central PMCID: PMCPMC3429567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015. August 20;524(7565):309–314. PubMed PMID: 26266977; PubMed Central PMCID: PMCPMC5018156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kurita Y, Araya J, Minagawa S, et al. Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir Res. 2017. June 02;18(1):114 PubMed PMID: 28577568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Martin-Maestro P, Gargini R, Perry G, et al. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum Mol Genet. 2016. February 15;25(4):792–806. PubMed PMID: 26721933; PubMed Central PMCID: PMCPMC4743695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Scarffe LA, Stevens DA, Dawson VL, et al. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014. June;37(6):315–324. PubMed PMID: 24735649; PubMed Central PMCID: PMCPMC4075431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013. June 06;153(6):1194–1217. PubMed PMID: 23746838; PubMed Central PMCID: PMCPMC3836174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vincow ES, Merrihew G, Thomas RE, et al. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A. 2013. April 16;110(16):6400–6405. PubMed PMID: 23509287; PubMed Central PMCID: PMCPMC3631677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuroda Y, Mitsui T, Kunishige M, et al. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet. 2006. March 15;15(6):883–895. PubMed PMID: 16449237. [DOI] [PubMed] [Google Scholar]
- [29].Rothfuss O, Fischer H, Hasegawa T, et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet. 2009. October 15;18(20):3832–3850. PubMed PMID: 19617636. [DOI] [PubMed] [Google Scholar]
- [30].Gupta A, Anjomani-Virmouni S, Koundouros N, et al. PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol Cell. 2017. March 16;65(6):999–1013 e7. PubMed PMID: 28306514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Barnes PJ. Senescence in COPD and its comorbidities. Annu Rev Physiol. 2017. February 10;79:517–539. [DOI] [PubMed] [Google Scholar]
- [32].Frank-Cannon TC, Tran T, Ruhn KA, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008. October 22;28(43):10825–10834. PubMed PMID: 18945890; PubMed Central PMCID: PMCPMC2603252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Letsiou E, Sammani S, Wang H, et al. Parkin regulates lipopolysaccharide-induced proinflammatory responses in acute lung injury. Transl Res. 2017. March;181:71–82. PubMed PMID: 27693468. [DOI] [PubMed] [Google Scholar]
- [34].Jung YY, Son DJ, Lee HL, et al. Loss of Parkin reduces inflammatory arthritis by inhibiting p53 degradation. Redox Biol. 2017. April 05;12:666–673. PubMed PMID: 28395174; PubMed Central PMCID: PMCPMC5388915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008. August 20;590(1–3):400–408. PubMed PMID: 18598692. [DOI] [PubMed] [Google Scholar]
- [36].Mitani Y, Sato K, Muramoto Y, et al. Superoxide scavenging activity of pirfenidone-iron complex. Biochem Biophys Res Commun. 2008. July 18;372(1):19–23. PubMed PMID: 18468515. [DOI] [PubMed] [Google Scholar]
- [37].Tsubouchi K, Araya J, Minagawa S, et al. Azithromycin attenuates myofibroblast differentiation and lung fibrosis development through proteasomal degradation of NOX4. Autophagy. 2017. June;14:1–15. PubMed PMID: 28613983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Minagawa S, Lou J, Seed RI, et al. Selective targeting of TGF-beta activation to treat fibroinflammatory airway disease. Sci Transl Med. 2014. June 18;6(241):241ra79 PubMed PMID: 24944194; PubMed Central PMCID: PMCPMC4341974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010. November 02;107(44):18880–18885. PubMed PMID: 20956295; PubMed Central PMCID: PMCPMC2973911. [DOI] [PMC free article] [PubMed] [Google Scholar]