

Summary
DNA replication is a fundamental biological process that is tightly regulated in all cells. In bacteria, DnaA controls when and where replication begins by building a step-wise complex that loads the replicative helicase onto chromosomal DNA. In many low-GC Gram-positive species, DnaA recruits the DnaD and DnaB proteins to function as adaptors to assist in helicase loading. How DnaA, its adaptors, and the helicase form a complex at the origin is unclear. We addressed this question by using the bacterial two-hybrid assay to determine how the initiation proteins from Bacillus subtilis interact with each other. We show that cryptic interaction sites play a key role in this process and we map these regions for the entire pathway. In addition, we found that the SirA regulator that blocks initiation in sporulating cells binds to a surface on DnaA that overlaps with DnaD. The interaction between DnaA and DnaD was also mapped to the same DnaA surface in the human pathogen Staphylococcus aureus, demonstrating the broad conservation of this surface. Therefore, our study has unveiled key protein interactions essential for initiation and our approach is widely applicable for mapping interactions in other signaling pathways that are governed by cryptic binding surfaces.
Keywords: DNA replication, Replicative Helicase, Bacillus subtilis, Staphylococcus aureus, Two-Hybrid Assay, Protein Interaction Domains
Abbreviated Summary
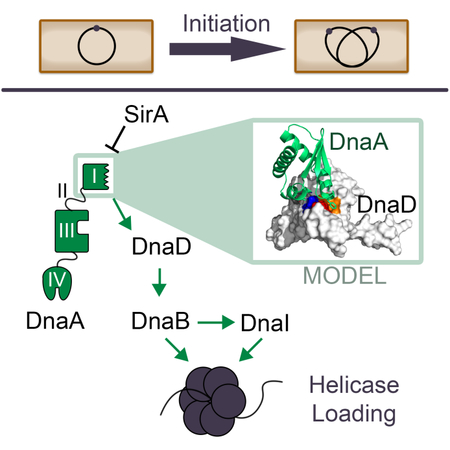
In bacteria, DnaA determines when and where DNA replication begins by initiating a protein interaction cascade. In this work we identified cryptic binding sites that regulate the ordered assembly of the initiation complex from DnaA to the replicative helicase in Bacillus subtilis (green arrows). The first interaction in the cascade was found to be mediated by a surface on DnaA Domain I that is widely used to interact with regulators in many bacterial species.
Introduction
All cells use a dedicated replicative helicase loading pathway to ensure DNA replication begins at the right time and place. Bacteria use the master regulator DnaA to recognize the origin of replication (oriC) and induce melting of the origin DNA (Mott & Berger, 2007). DnaA then recruits the replicative helicase with its associated loader, representing the first step in replication fork assembly. DnaA is widely conserved, but importantly the way it recruits the replicative helicase differs significantly between Gram-positive and Gram-negative species. In Escherichia coli, DnaA recruits the replicative helicase through a direct interaction (Marszalek & Kaguni, 1994; Sutton, Carr, Vicente, & Kaguni, 1998). In contrast, the Gram-positive model organism B. subtilis requires the essential proteins DnaD and DnaB (not to be confused with the DnaB helicase from E. coli) to allow for helicase loading. Specifically, DnaA recruits DnaD to the origin first; DnaD then recruits DnaB; and, finally, DnaB recruits and helps load the helicase (Smits, Goranov, & Grossman, 2010). DnaD and DnaB may also play additional roles in vivo, including contributions to DNA remodeling, gene expression and DNA repair (Bonilla & Grossman, 2012; Carneiro et al., 2006; Collier, Machon, Briggs, Smits, & Soultanas, 2012; Grainger, Machon, Scott, & Soultanas, 2010; Schneider, Zhang, Soultanas, & Paoli, 2008; Scholefield & Murray, 2013; Smits, Merrikh, Bonilla, & Grossman, 2011; Zhang, Allen, Roberts, & Soultanas, 2006; Zhang et al., 2005, 2008). Because DnaD and DnaB link DnaA to the replicative helicase we refer to both proteins as “adaptors” in regards to their function in replication initiation and helicase loading.
Interactions involving the DnaD and DnaB adaptors are tightly regulated and therefore add a layer of control over origin firing that is not present in Gram-negative bacteria. For example, B. subtilis is able to sporulate as a means of surviving nutrient limitation and DNA replication initiation must be prevented during sporulation. Inhibition of replication initiation is due to a small protein called SirA that is only produced once the cell is committed to sporulate (Rahn-Lee, Gorbatyuk, Skovgaard, & Losick, 2009; Wagner, Marquis, & Rudner, 2009). SirA directly binds to the DnaA initiator and acts as a potent inhibitor of its activity (Duan, Huey, & Herman, 2016; Jameson et al., 2014; Rahn-Lee, Merrikh, Grossman, & Losick, 2011). Though the mechanism of action is unknown, SirA prevents DnaA from accumulating at the origin and recruiting the DnaD and DnaB adaptors (Rahn-Lee et al., 2011; Wagner et al., 2009).
DnaD and DnaB share similar protein architectures and are conserved among Gram-positive bacteria with low GC-content, which includes many human pathogens such as Staphylococcus aureus, Listeria monocytogenes, Streptococcus pneumoniae, and Bacillus anthracis (Marston et al., 2010). In addition to DnaA, other types of initiators also use DnaD and DnaB to load the replicative helicase. For example, PriA uses DnaD and DnaB to restart stalled replication forks and some plasmid initiators use DnaD and DnaB during plasmid duplication (Bruand, Farache, McGovern, Ehrlich, & Polard, 2001; Hassan et al., 1997; Marsin, McGovern, Ehrlich, Bruand, & Polard, 2001). Multi-drug resistance plasmids and phage genomes can also express their own versions of DnaD- and DnaB-like adaptor proteins when replicating in low GC-content Gram-positive hosts (Rokop, Auchtung, & Grossman, 2004; Schumacher et al., 2014). Therefore, determining how DnaD and DnaB function as adaptors in replication initiation is critical for understanding the regulation of DNA replication in multiple contexts. To this end, we sought to determine if and how interactions mediated by DnaD and DnaB are regulated.
DnaD and DnaB form large aggregates which has largely prevented an in vitro approach to studying the interactions with their binding partners (Carneiro et al., 2006; Schneider et al., 2008; Zhang et al., 2005, 2008). In addition, the helicase loading proteins are membrane associated in B. subtilis (Grainger et al., 2010; Meile, Wu, Ehrlich, Errington, & Noirot, 2006; Rokop et al., 2004), which makes reconstituting an in vitro system very difficult. Therefore, the most productive attempts have used yeast two-hybrid based screens to circumvent these in vitro issues yielding mixed results. For example, the DnaA and DnaD interaction gives a positive signal in yeast two-hybrid assays only under certain experimental conditions (Ishigo-oka, Ogasawara, & Moriya, 2001). In contrast, DnaD and DnaB do not generate any signals with each other in yeast two-hybrid assays (Ishigo-oka et al., 2001; Noirot-Gros et al., 2002; Rokop et al., 2004). There is some evidence that DnaD and DnaB may weakly interact in vitro (Bruand et al., 2005; Marsin et al., 2001), however the strongest support for a direct interaction is a gain-of-function mutation in DnaB that generates a robust interaction with DnaD and increases the frequency of initiation in vivo (Bruand et al., 2005; Rokop et al., 2004). This gain-of-function mutation (S371P) has been mapped to the C-terminus of DnaB and likely induces a conformational change, though it is unclear if this mimics a physiological process. Consequently, there is a pressing need to establish a method for detecting interactions with the wild type DnaD and DnaB adaptors.
To investigate how DnaD and DnaB control replication initiation, we used a bacterial two-hybrid (B2H) assay to map their interaction surfaces. While the full-length initiation proteins did not interact, specific combinations of isolated domains from DnaA, DnaD, DnaB, and the replicative helicase produced robust signals. Using this approach, the interaction between DnaA and DnaD was mapped to the same hot spot used by the SirA inhibitor. In addition, we found that DnaD used its N-terminal winged helix domain to mediate all of its interactions, while DnaB instead favored its C-terminal domain to bind its partners. Together, these results indicate that protein interactions at the B. subtilis origin are controlled through conformational changes that expose cryptic interaction surfaces to build a step-wise initiation complex.
Results
Identification of interaction sites in initiation proteins
A B2H assay was used to detect interactions between the B. subtilis initiation proteins (Fig. 1A). Positive interactions generate cyclic AMP within a cya- E. coli host cell, which can be detected as blue colonies in the presence of X-Gal or growth using maltose as the only carbon source (Karimova, Pidoux, Ullmann, & Ladant, 1998). Both DnaD and DnaB homo-oligomerize (Marsin et al., 2001) and these self-interactions could be readily detected in the assay (Fig. 1B). In contrast, DnaD and DnaB did not interact with each other or with DnaA under the same conditions (Fig. 1C). As a further control, a DnaB variant with the S371P gain-of-function mutation was also used (Bruand et al., 2005; Rokop et al., 2004), which generated a signal within the B2H assay as expected (Fig. 1C).
Figure 1. Full-length DnaA, DnaD and DnaB do not interact in a B2H assay.

(A) A schematic of the helicase loading pathway in B. subtilis. Black arrows indicate potential protein-protein interactions while blue curved arrows indicate self-interactions. (B) B2H of T18- with T25-tagged DnaD (top) or DnaB (bottom) to demonstrate self-interactions mediated by the adaptor proteins. (C) B2H of T18-tagged DnaD co-expressed with either T25-tagged DnaA, wild type DnaB (DnaBWT), or the gain-of-function DnaB variant (DnaBS371P).
Because we could not detect interactions between the full-length proteins, we chose an alternative strategy to map their interaction surfaces. DnaA, DnaD, and DnaB were separated into their constituent domains (Fig. 2A) and tested against each other in all possible pairwise combinations using both X-Gal and minimal media containing maltose to determine positive interactions. This approach reveals potential interaction surfaces on individual domains that may be occluded in the conformation favored by the full-length proteins.
Figure 2. Mapping Interacting Domains Involved in DNA Replication Initiation.

(A) Schematic of DnaA, DnaD, DnaB, DnaC, and DnaI divided into their individual domains. Amino acid boundaries of each protein fragment are indicated. WHD = Winged Helix Domain; CTD = C-terminal Domain; NTD = N-terminal Domain; ZBD = Zinc Binding Domain. (B) SDS-polyacrylamide gel showing purified domains from DnaD and DnaB. (C) B2H of T25-tagged DnaA domains co-expressed with T18-tagged full-length YabA (YabAFL), full-length SirA (SirAFL), or the domains from DnaD. (D) B2H of T25-tagged DnaB domains co-expressed with T18-tagged DnaD domains. (E) B2H of T25-tagged DnaI or DnaB domains co-expressed with T18-tagged DnaC domains. (F) B2H of T25-tagged DnaB domains co-expressed with T18-tagged DnaI domains.
To experimentally determine if the domain boundaries were correct, we purified each individual domain from the DnaD and DnaB adaptors and found they could fold as stable products (Fig. 2B). For DnaA, we tested its domains with known binding partners in the B2H. DnaA domain I (DnaADI) was found to interact with the SirA inhibitor (Jameson et al., 2014; Rahn-Lee et al., 2011) while DnaA domain III (DnaADIII) interacted with YabA as expected (Cho, Ogasawara, & Ishikawa, 2008) (Fig. 2C). Therefore, the individual domains appear to be suitable for screening in the B2H assay as they are stable and exhibit other known functions based on prior reports.
For the DnaA-DnaD interaction, we found that the winged helix domain from DnaD (DnaDWHD) interacted with DnaADI (Fig. 2C). DnaDWHD also interacted with its downstream partner, DnaB, but in this case the signal was split between the DnaB winged helix domain (DnaBWHD) and its second C-terminal domain (DnaBCTD2) (Fig. 2D). These results were also corroborated when using growth on minimal media containing maltose (Fig. S1). The winged helix domain of DnaD mediates a dimer-tetramer equilibrium; however, expressing DnaDWHD on its own shifts the equilibrium to favor the tetrameric form (Schneider et al., 2008). We entertained the possibility that the tetrameric form of DnaDWHD specifically interacts with its protein partners. To test this idea directly, DnaDWHD was truncated to prevent its tetramerization as determined by size exclusion chromatography (Fig. S2A and S2B). Our results show that preventing DnaDWHD tetramerization did not disturb any of the B2H signals (Fig. S2C). Therefore, we find it unlikely that the oligomeric state of DnaD dictates its interaction status. Instead, our results suggest that full-length DnaA, DnaD and DnaB may undergo conformational changes to expose cryptic binding sites and form stable complexes with each other during initiation.
The same B2H method was also used to map the interaction between the DnaB adaptor and the replicative helicase (called DnaC in B. subtilis; not to be confused with the DnaC loader from E. coli). The major signal came from DnaBCTD2 interacting with the C-terminal end of the helicase (DnaCCTD), although weak signals with DnaCNTD were also apparent (Fig. 2E). The zinc binding domain of the B. subtilis ATPase loader (called DnaI) is also known to interact with the C-terminal end of the DnaC helicase, but interactions between DnaI and DnaCNTD have not been investigated (Ioannou, Schaeffer, Dixon, & Soultanas, 2006; Loscha et al., 2009; Noirot-Gros et al., 2002; P. Soultanas, 2002). Therefore, the DnaI domains (Fig. 2A) were screened against the DnaC helicase domains. We found that the DnaI zinc binding domain (DnaIZBD) can interact with both the N- and C-terminal ends of the helicase (Fig. 2E). These results were also reproduced using growth on minimal media supplemented with maltose (Fig. S3). Therefore, we suggest that DnaI may have multiple interaction modes with the replicative helicase during loading.
Studies have indicated that the DnaB adaptor and DnaI loader act together to load the helicase and consequently we also investigated whether these two proteins can interact with each other (Velten et al., 2003). We found that the C-terminal domains from DnaB (DnaBCTD2) and DnaI (DnaICTD) could interact in the B2H assay (Fig. 2F), however the signal was weak. We could also detect this interaction using minimal media supplemented with maltose (Fig. S3C). Therefore, DnaB, DnaI, and DnaC may form a ternary complex during helicase loading.
DnaA can recruit the replicative helicase directly in E. coli or indirectly through its ATPase loader in Aquifex aeolicus (Marszalek et al., 1996; Mott, Erzberger, Coons, & Berger, 2008; Panos Soultanas, 2012; Sutton et al., 1998). Although we attempted to detect interactions between B. subtilis DnaA and either the DnaC helicase or DnaI loader, we were unable to find any positive signals (Fig. S4). With these results we suggest that DnaA may have to rely on the DnaD and DnaB adaptors for helicase loading in B. subtilis.
DnaD and DnaB adopt very similar structures, although they mediate different interactions during initiation. Therefore, as an added control, we attempted to swap DnaD and DnaB in our B2H assays to ensure the interactions we detected are specific. Our results indicate that despite sharing similar structures, DnaD and DnaB could not substitute for each other in our assays as expected (Fig. S5).
The DnaD wing interacts with the DnaA initiator
After identifying all of the interacting domains, we chose to focus on the interaction between DnaA and DnaD as this is likely the most regulated step of the helicase loading pathway. The ConSurf server was used to map conserved residues on the DnaDWHD crystal structure [PDB 2V79 (Schneider et al., 2008)] revealing potential interaction sites (Ashkenazy, Erez, Martz, Pupko, & Ben-Tal, 2010; Celniker et al., 2013; Glaser et al., 2003; Landau et al., 2005). A conserved patch was found in a cleft formed by the β-strands of the wing in DnaDWHD (Fig. 3A). When the conserved hydrophobic cleft residues F51 and I83 were mutated to alanines the interaction with DnaADI was lost (Fig. 3B). Furthermore, reducing the length of the loop (Δloop) in the DnaDWHD wing (Schneider et al., 2008) also disrupted binding to DnaADI (Fig. 3B). This indicates that the DnaD wing region is necessary for forming a complex with DnaA.
Figure 3. The DnaDWHD Wing Forms a Binding Cleft for DnaA.

(A) The crystal structure of DnaDWHD (PDB 2V79) is shown as a surface representation and colored according to sequence conservation as indicated in the legend (top). The boxed inset shows a semi-transparent surface with F51, I83 and E95 represented as sticks. (B) B2H of T18-tagged DnaDWHD variants co-expressed with either T25-tagged DnaADI, DnaBFL or DnaDWHD. (C) SDS-polyacrylamide gel stained with coomassie blue showing the glutaraldehyde crosslinking of wild type DnaDWHD (WT) or the F51A and I83A variants to reveal self-interactions. The various oligomeric forms are symbolized by dots at the right-hand side of the gel, with each dot representing one DnaDWHD protomer. The molecular weight marker is labeled on the left-hand side in kDa.
To ensure the F51A and I83A DnaDWHD mutations did not cause structural defects, we purified both variants and verified that they could tetramerize using size exclusion chromatography (data not shown) and chemical crosslinking (Fig. 3C). Tetramerization uses surfaces distal to the wing and is therefore an excellent test for wide-ranging structural defects (Schneider et al., 2008). We also found that the DnaDWHD wing variants could still interact with DnaB (Fig. 3B), which further indicates these variants are structurally sound. Note that the DnaDWHD Δloop variant was previously found to be stable and therefore was not tested (Schneider et al., 2008).
To test if the wing region is also critical in full-length DnaD, we used a temperature sensitive B. subtilis strain (dnaD23) where the native DnaD protein is not functional at 48°C (Bruand, Sorokin, Serror, & Ehrlich, 1995). Ectopically expressing the F51A or I83A full-length DnaD variants from the amyE locus did not rescue this strain at the restrictive temperature (Fig. 4A). Therefore, the DnaD wing is also required for function in vivo.
Figure 4. The DnaDWHD Binding Cleft is Essential.

(A) dnaD23 strains ectopically expressing WT DnaD or its winged helix variants (F51A or I83A) were incubated at the permissive temperature (30°C) or the restrictive temperature (48°C). The dnaD23 parental strain was also included as a control (labeled “None” on the plates). (B) The same strains described in (A) were also incubated at the permissive temperature without (0%) or with (1%) xylose induction. Small colonies indicate a growth defect upon DnaD over-expression.
Overexpressing DnaD in B. subtilis causes a growth defect that manifests in small colony sizes. This phenotype depends on DNA binding by DnaD but is independent of its interaction with DnaA (Marston et al., 2010). Consequently, we predicted that if the F51A and I83A DnaD variants were only defective in binding DnaA, but not in other functions, they should also induce a small colony phenotype when overexpressed. The dnaD23 strains used in Fig 4A was therefore incubated with high levels of xylose at the permissive temperature. Under these conditions, both F51A and I83A DnaD produced small colonies (Fig. 4B). With these results we suggest that functions outside of DnaA binding, which may include DNA remodeling activities, are still preserved.
DnaD and SirA bind the same interaction hot spot in DnaA
As SirA and DnaD both bind to DnaADI, we asked whether they recognize the same binding site. SirA binds to a surface on DnaADI that is considered an interaction hot spot because it is recognized by diverse binding partners in different bacterial species (Zawilak-Pawlik, Nowaczyk, & Zakrzewska-Czerwinska, 2017). Intriguingly, this surface cannot tolerate mutations in B. subtilis despite the fact that the interaction with SirA is not essential (Jameson et al., 2014). We hypothesized that DnaD may also bind to this DnaADI hot spot and that mutating this region is lethal because it knocks out the interaction with the DnaD adaptor.
To test our hypothesis, six different DnaADI variants were used: three with mutations on the hot spot that are lethal in B. subtilis (T26A, W27A, and F49A) (Jameson et al., 2014) and three with mutations on the same surface that are still viable (N47A, F49Y, and A50V) (Duan et al., 2016; Rahn-Lee et al., 2011). In the B2H assay, only the mutations that are lethal in B. subtilis knocked out the interaction with DnaD (Fig. 5A). As a control, the DnaA variants were also tested against SirA where all variants ablated the interaction (Fig. 5A), with the exception of T26A which is consistent with previous studies (Jameson et al., 2014). Therefore, we conclude that DnaD and SirA bind to overlapping surfaces on DnaADI.
Figure 5. DnaDWHD Binds to the DnaADI hot spot.

(A) B2H of T25-tagged DnaADI variants co-expressed with T18-tagged SirA or DnaDWHD. “Viable” and “non-viable” refer to the effect these DnaA variants have when introduced into B. subtilis. (B) Model of the DnaDWHD and DnaADI complex. DnaADI is represented as a green ribbon diagram with α2 and α3 labeled, while DnaDWHD is shown as a white surface. The conserved residues that line the binding cleft in DnaDWHD are colored with F51 in orange, I83 in blue, and E95 in red. The boxed inset shows a zoomed view of the interaction interface with the DnaDWHD F51, I83 and E95 sidechains shown as spheres. DnaDWHD I76 is also shown and colored in grey. The DnaADI interacting residues (T26, W27 and F49) are shown as green sticks with oxygen atoms colored red and nitrogen atoms colored blue. (C) B2H of T25-tagged DnaADI co-expressed with T18-tagged DnaDWHD variants. (D) SDS-polyacrylamide gel stained with coomassie blue showing the glutaraldehyde crosslinking of the wild type DnaD winged helix domain (WHDWT) and DnaA domain I (DIWT). The dots at the right hand side of the gel show the oligomeric state of each band with the grey dots symbolizing DnaD and the green dots symbolizing DnaA. The band representing the complex is further distinguished with an asterisk. (E) SDS-polyacrylamide gel stained with coomassie blue showing the glutaraldehyde crosslinking of a variant of the DnaD winged helix domain (WHDF51A) or DnaA domain I (DIF49A) that cannot interact in a B2H assay. The band representing the complex is marked by an asterisk.
As crystal structures of both partners are available, we also modeled the complex between DnaDWHD and DnaADI using the Rosetta docking algorithm (Chaudhury et al., 2011; Lyskov et al., 2013; Lyskov & Gray, 2008). This model predicted a hydrophobic interaction surface on DnaD consisting of I83, F51 and the aliphatic portion of the E95 sidechain (Fig. 5B). DnaADI docks on this hydrophobic surface using T26, W27 and F49 (Fig. 5B). Therefore, the residues identified through our B2H assays are predicted to be in direct contact within the complex.
The DnaD crystal structure indicated that I83 may serve a structural role by packing against I76 and stabilizing the DnaD wing (Fig. 5B). We entertained the possibility that mutating I83 increases the flexibility of the wing region by destroying this anchoring point; however, this is unlikely to be the reason the interaction with DnaA was lost because mutating the other isoleucine involved (I76A) did not prevent DnaA binding (Fig. 5C). Therefore, we conclude that I83 is directly important for complex formation.
To ensure we are detecting a direct interaction in our B2H assay, we also tested if purified DnaADI and DnaDWHD could form a complex in vitro. When treated with a chemical crosslinker, a unique band consistent with the size of a DnaADI-DnaDWHD complex could be detected (Fig. 5D) and we confirmed this band consisted of both binding partners through mass spectrometry (Fig. S6). Critically, the DnaDWHD F51A and DnaADI F49A variants could not be crosslinked (Fig. 5E), which supports the B2H results that show these substitutions destroy the interaction (Fig. 3B and Fig. 5A). Further, this result demonstrates the in vitro crosslinking is capturing a specific interaction that depends on the same residues as determined in the B2H assay.
S. aureus DnaA binds its cognate DnaD adaptor using the domain I hot spot
SirA and DnaD bind to overlapping surfaces on the DnaA domain I hot spot, however the majority of species that rely on the DnaD adaptor do not have a SirA homolog. This led us to ask if the DnaA hot spot also recruits DnaD to the origin in species that lack SirA, or whether these organisms instead evolved a different interaction surface (Fig. 6A).
Figure 6. DnaD Binds to the DnaA domain I hot spot in S. aureus.

(A) Schematic of DnaA demonstrating three different possibilities in bacteria: species that have both the DnaD adaptor and SirA regulator, species that have DnaD but lack SirA, and species that lack DnaD and SirA. (B) B2H of T18-tagged SaDnaDWHD co-expressed with T25-tagged variants of SaDnaADI. (C) Sequence alignment of the DnaA hot spot from B. subtilis, S. aureus, and E. coli. The three hot spot residues mutated in the B2H assays are highlighted in yellow, with residues critical for the DnaD interaction in B. subtilis indicated by blue arrow heads. Residues critical for the direct interaction between E. coli DnaA and the replicative helicase are indicated by red arrow heads. The numbering at the top of the alignment refers to the B. subtilis sequence. (D) B2H of T18-tagged BsDnaDWHD variants co-expressed with T25-tagged BsDnaADI. (E) B2H of T25-tagged DnaADI from B. subtilis (Bs), S. aureus (Sa) or E. coli (Ec) co-expressed with T18-tagged BsDnaDWHD or SaDnaDWHD.
To test this possibility, we used the human pathogen S. aureus because it lacks a SirA homolog. DnaA and DnaD from S. aureus (herein referred to as SaDnaA and SaDnaD) interacted in a B2H using domain I from SaDnaA and the winged helix domain from SaDnaD (Fig. 6B). The hot spot residues equivalent to T26, W27 and F49 in B. subtilis DnaA (T25, F26, and F48 in SaDnaA) were then mutated to alanines (Fig. 6C). Mutating the two aromatic residues on the interaction hot spot (SaDnaA F26A and F48A) decreased the signal in the B2H assay, while mutating the threonine (SaDnaA T25A) had only a modest effect (Fig. 6B). Therefore, the hot spot on DnaA domain I still interacts with its cognate adaptor in species that lack SirA, but the molecular contacts differ slightly from the interaction site we identified in B. subtilis.
Out of the three residues on the DnaA hot spot investigated to this point, the contribution of the conserved threonine (T26 in BsDnaA; T25 in SaDnaA) was still unclear. Our model predicted that the interaction between DnaA and DnaD is primarily hydrophobic in B. subtilis, with the bulky methyl group from the threonine contributing directly to the interaction interface (Fig. 5B). To test this further, T26 in BsDnaA was mutated to a serine which would effectively remove only the methyl group in question. Indeed, we found that mutation to serine was sufficient for preventing the interaction with DnaD in a B2H (Fig. 6D). To determine if other hydrophobic residues could substitute for T26 in the B. subtilis DnaA-DnaD complex, we also tested a T26M mutation and found the interaction with DnaD was maintained (Fig. 6D). These results strongly support the model that the DnaA hot spot is providing a hydrophobic surface that docks onto the DnaD wing. Therefore, it appears that the conserved threonine contributes to this hydrophobic hot spot in B. subtilis DnaA, but is largely dispensable in S. aureus.
Due to the differences in the molecular contacts between BsDnaD and SaDnaD with the DnaA hot spot, we next tested whether these DnaD proteins could be swapped in our B2H assay. We found that both BsDnaA and SaDnaA could only recognize their cognate adaptors, while E. coli DnaA could not recognize either DnaD homolog as expected (Fig. 6E). Therefore, the interaction between DnaA and DnaD is highly specific despite the fact that it relies on a conserved interaction hot spot in domain I of DnaA.
Discussion
We have identified the protein interaction domains required to recruit and load the replicative helicase during initiation. Our results indicate that unlike in E. coli and A. aeolicus, B. subtilis DnaA recruits the helicase through the accessory proteins DnaD and DnaB instead of through direct contacts or the ATPase loader (Fig. 7A and 7B). Additionally, most of the B. subtilis initiation proteins tend to interact with the chromosome using their C-terminal ends and with their protein partners using their N-terminal ends (Fig. 7C). This is with the notable exception of the DnaB adaptor, which can bind both DNA and its protein partners with its C-terminal domain (Fig. 2D–F). It was previously noted that truncating the C-terminal end of DnaB is lethal and prevents it from being recruited to the origin (Grainger et al., 2010), underscoring the functional importance of this region.
Figure 7. Model of Protein and DNA Interacting Domains Used for Initiation.

(A) Schematic of DnaA interactions from Gram-negative bacterial systems. A direct interaction between DnaA and the replicative helicase representing the E. coli system is shown with a blue arrow, while the interaction between DnaA and the ATPase loader representing A. aeolicus is shown with a red arrow. (B) Schematic of interactions involved in initiation for B. subtilis with interacting proteins connected by blue arrows. (C) Schematic of interacting domains for the initiation proteins in B. subtilis. Double-headed arrows connect domains that interact with each other. DNA binding domains are highlighted in yellow. Note that the order of interaction between DnaB, DnaI and the DnaC helicase are not known. The line connecting DnaB and DnaI is dotted to represent a weak interaction.
We have also mapped the interaction surfaces in the DnaA-DnaD complex using a B2H assay. This interaction required a hydrophobic cleft in the DnaD winged helix domain (Fig. 3A), which is often used to bind DNA in winged helix folds (Harami, Gyimesi, & Kovacs, 2013). Interestingly, a DnaD-like plasmid initiator from S. aureus does use this cleft to bind DNA; however, in this case the cleft is positively charged and adopts a different conformation than the same site in B. subtilis DnaD (Schumacher et al., 2014). Therefore, the winged helix cleft is critical for DnaD-like proteins, but can be adapted for different functions. These results strongly echo licensing factors that load the replicative helicase in Eukaryotes and Archaea, which often contain winged helix domains that can bind to either DNA, protein, or both (Khayrutdinov et al., 2009).
DnaD binds to an interaction hot spot on DnaA domain I and, critically, mutations on this surface that are not tolerated in vivo also prevent the interaction with DnaD (Fig. 5A). Therefore, we propose that this surface is essential in vivo because it recruits DnaD to the origin. Considering that this DnaA surface also binds to SirA, it is possible that SirA competes with DnaD as part of its inhibitory function. This may prevent DnaD recruitment by DnaA and thereby ensure that the origin cannot fire during sporulation. SirA also destabilizes the interaction between DnaA and oriC in vivo (Rahn-Lee et al., 2011; Wagner et al., 2009), which could be a consequence of displacing or impairing the DnaA interaction with DnaD.
The interaction hot spot on DnaA domain I is widely conserved in bacteria and likely plays critical roles in mediating interactions required for replicative helicase loading even in species that lack DnaD and/or SirA (Zawilak-Pawlik et al., 2017). For example, the domain I hot spot in E. coli DnaA binds directly to the replicative helicase using residue F46 (equivalent to F49 in B. subtilis DnaA) (Abe et al., 2007; Seitz, Weigel, & Messer, 2000; Sutton et al., 1998). In addition, the E. coli hot spot also contacts the protein DiaA which further promotes initiation (Ishida et al., 2004; Keyamura, Abe, Higashi, Ueda, & Katayama, 2009; Keyamura et al., 2007; Su’etsugu et al., 2013). In H. pylori, the domain I hot spot instead interacts with the HobA protein, which adopts a similar structure to DiaA (Natrajan, Hall, Thompson, Gutsche, & Terradot, 2007; Natrajan, Noirot-Gros, Zawilak-Pawlik, Kapp, & Terradot, 2009; Zawilak-Pawlik et al., 2011). Therefore, it appears that this hot spot is capable of mediating species-specific interactions with various positive (DnaD, DiaA, HobA) or negative (SirA) regulators.
The interactions between DnaB, DnaI and the replicative helicase in B. subtilis were also investigated. We found that the C-terminal domain of DnaB (DnaBCTD2) could bind the ATPase domains from both DnaI and the helicase, though the interaction with the helicase produced a much higher signal (Fig. 2E and 2F). These interactions appeared to be specific because the structurally similar C-terminal domain from DnaD could not interact under the same conditions, nor could DnaBCTD2 interact with the DnaA ATPase domain (Fig. S5). It was previously shown that DnaI could also interact with the ATPase domain from the replicative helicase (Ioannou et al., 2006; Loscha et al., 2009; Noirot-Gros et al., 2002; P. Soultanas, 2002), though we also detected an interaction with the N-terminal end of the helicase (Fig. 2E). It is interesting to note that structural studies have found DnaI associates only with the C-terminal (ATPase) end of the helicase when it is preassembled as a hexameric ring (Lin, Naveen, & Hsiao, 2016; Liu, Eliason, & Steitz, 2013). However, in B. subtilis the helicase is loaded using a “ring maker” mechanism (Davey & O’Donnell, 2003; Velten et al., 2003). That is, DnaI instead interacts with individual helicase subunits and assembles them as a ring around the single-stranded DNA exposed at the origin. Therefore, it will be interesting to investigate in the future whether additional contacts, perhaps involving the N-terminal end of the helicase, are required for DnaI to interact with the helicase when it is in its disassembled form prior to loading.
It is intriguing that the full-length DnaA, DnaD, and DnaB proteins did not interact in the B2H (Fig. 1C), yet their isolated domains could readily associate (Fig. 2C and 2D). We propose that these complexes are regulated through conformational changes that expose cryptic protein interaction sites, which are occluded in the full-length proteins but revealed in the isolated domains. For both DnaA and DnaD, the interacting surfaces necessary for the B2H signal were also required in vivo [Fig. 4A and (Jameson et al., 2014)], demonstrating these surfaces are indeed functional in the context of the full-length proteins. Intriguingly, for the DnaI loader it was previously shown that the full-length protein readily interacts with its protein partner (DnaC helicase), but its DNA binding activity is cryptic (Ioannou et al., 2006). Regardless of which activity is masked, our results suggest that factors which lead to conformational changes may play a critical role in regulating initiation frequency in low GC Gram-positive bacteria.
Experimental Procedures
Plasmid Construction
All plasmids were constructed using the Gibson assembly method (Gibson et al., 2009) with the primers described in Supporting Information Table S1. The assembled plasmids were isolated from single colonies and sent to the University of Michigan Sequencing Core for verification (https://seqcore.brcf.med.umich.edu/).
Plasmids used for the B2H assays are described in Supporting Information Table S2 including a list of all the primers used for Gibson assembly. The vectors were amplified using the following primers: pUT18C (oLM140 and oLM141), pUT18 (oLM218 and oLM217), pKT25 (oLM146 and oLM147), and pKNT25 (oLM212 and oLM211).
Plasmids used for integrating dnaD variants at the amyE locus are described in Supporting Information Table S3 including a list of all the primers used for Gibson assembly. All dnaD gene variants were cloned into pJS103 (Liao, Schroeder, Gao, Simmons, & Biteen, 2015) which has a xylose inducible promoter driving expression of dnaD linked to an erythromycin resistance cassette. These regions are flanked by sequences homologous to amyE which allows for the plasmid to integrate at this locus through a double-crossover recombination event. The pJS103 vector was amplified by oLM197 and oLM198 in preparation for Gibson assembly.
Plasmids used for protein expression are described in Supporting Information Table S4 including a list of all the primers used for Gibson assembly. The gene fragment encoding the DnaDWHD (amino acids 1–128) and its variants as well as DnaBWHD (amino acids 1–153) were cloned into pET28a encoding an N-terminal HIS6-MBP-SUMO tag (Burby & Simmons, 2017b). The vector was amplified with oJS638 and oJS639 in preparation for Gibson assembly. The gene fragments encoding DnaDCTD (amino acids 129–232), DnaBCTD1 (amino acids 185–300), DnaBCTD2 (amino acids 303–472), and DnaADI (amino acids 1–82) were cloned into pE-SUMO. The pE-SUMO vector was amplified using oLM1 and oLM2.
All site-directed mutagenesis was conducted using the primer extension method (Reikofski & Tao, 1992). This required the use of at least four primers: two flanking primers that amplify the desired region and two partially complementary primers that encode the desired mutation. Each flanking primer was paired with the appropriate mutagenic primer to amplify the gene in two pieces. These amplification products were then gel purified, mixed in equal molar amounts, and used as the template in a second PCR with the flanking primers only.
Building the dnaD23 strain
The dnaD23 strain was created using CRISPR/Cas9 gene-editing to introduce an A166T mutation in dnaD using a method we developed previously (Burby & Simmons, 2017a, 2017b). A detailed description of the process can be viewed in the supporting information section.
Purification of DnaDWHD and DnaBWHD
BL21 (DE3) cells were transformed with the relevant expression plasmid (Supporting Information Table S4) and incubated at 37°C until reaching an OD600 of 0.7. Protein expression was induced using a final concentration of 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) and the cells were incubated at 37°C for 3 hours. The cells were then harvested, washed once with 1X PBS to remove residual media, and resuspended in 50 mL buffer A (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 5% glycerol, 1 mM DTT) with a protease inhibitor tablet (Complete EDTA-free, Roche). Sonication was used to lyse the sample, after which the soluble fraction was loaded onto a 4 mL amylose gravity column. The column was washed four times with 10 mL of buffer A, followed by eluting any bound protein using buffer A supplemented with 10 mM D-maltose while collecting 1 mL fractions.
Fractions containing the desired protein were pooled and adjusted to 100 mM NaCl and incubated with Ulp1 protease for two hours at room temperature. The digested sample was filtered and loaded onto a 5 mL HiTrap™ Q FF column (GE life sciences) equilibrated with buffer B (20 mM Tris-HCl pH 8.0, 1 mM DTT, 1 mM EDTA, 5% glycerol) supplemented with 100 mM NaCl. The tagless protein was then eluted with a 90 minute gradient from 100 mM to 500 mM NaCl at 1 mL min−1 and further purified using a HiPrep™ 16/60 Sephacryl™ S-200 HR column (GE life sciences) equilibrated in buffer B containing 150 mM NaCl at 0.75 mL min−1. The final eluted sample was concentrated using an Amicon centrifugal filter (Amicon Ultra-15 with Ultracel-10 membrane). The sample was supplemented with 25% glycerol and flash frozen for storage at −80°C.
Purification of DnaDCTD, DnaBCTD1, and DnaBCTD2
BL21 (DE3) cells were transformed with the relevant expression plasmid (Supporting Information Table S4) and incubated at 37°C until reaching an OD600 of 0.7. Protein expression was induced using a final concentration of 0.5 mM IPTG and the cells were incubated at 12°C for 16 hours. The cells were then harvested, washed once with 1X PBS to remove residual media, and resuspended in 50 mL buffer C (20 mM Tris-HCl pH 8.0, 400 mM NaCl, and 5% glycerol) with a protease inhibitor tablet (Complete EDTA-free, Roche). Sonication was used to lyse the sample, after which the soluble fraction was loaded onto a 5 mL HiTrap™ IMAC FF column (GE life sciences) equilibrated with Buffer C. The column was washed with 40 mL of Buffer C supplemented with 2 M NaCl followed by 30 mL of Buffer C with 15 mM imidazole. The bound protein was then eluted using Buffer C supplemented with 300 mM imidazole while collecting 1 mL fractions. Fractions containing the desired protein were pooled and added dropwise to 50 mL of Buffer D (20 mM Tris-HCl pH 8.0, 1 mM DTT, 5% glycerol, and 150 mM NaCl). The tag was digested using Ulp1 protease for 2 hours at room temperature.
The digested sample was dialyzed overnight against Buffer C at 4°C. The sample was then filtered and loaded onto a 5 mL HiTrap™ IMAC FF column equilibrated with Buffer C. The column was washed with 15 mL of Buffer C followed by 15 mL of Buffer C supplemented with 15 mM imidazole. The washes that contained the tagless protein were pooled and concentrated using an Amicon centrifugal filter (Amicon Ultra-15 with Ultracel-10 membrane). The concentrated protein was purified further using a HiPrep™ 16/60 Sephacryl™ S-200 HR column (GE life sciences) using buffer E (20 mM Tris-HCl pH 8.0, 400 mM NaCl, 1 mM DTT, 1 mM EDTA, and 5% glycerol) at 0.75 mL min−1. The final eluted sample was concentrated using an Amicon centrifugal filter (Amicon Ultra-15 with Ultracel-10 membrane), supplemented with 25% glycerol, and flash frozen for storage at −80°C.
Purification of DnaA Domain I
BL21 (DE3) cells were transformed with the relevant expression plasmid (Supporting Information Table S4) and incubated at 37°C until reaching an OD600 of 0.7. Protein expression was induced using a final concentration of 0.5 mM IPTG and the cells were incubated for 3 hours at 37°C. The cells were then harvested and resuspended in 50 mL of Buffer C with a protease inhibitor tablet (Complete EDTA-free, Roche). Sonciation was used to lyse the cells and the soluble fraction was then loaded onto a 5 mL HiTrap™ IMAC FF column (GE life sciences) equilibrated with Buffer C. The column was washed with 8 column volumes of Buffer C supplemented with 15 mM imidazole and the protein was eluted with Buffer C supplemented with 300 mM imidazole. Fractions containing the desired protein were pooled, the salt was adjusted to 100 mM NaCl, and DTT was added to a final concentration of 1 mM. Ulp1 protease was then added and incubated for 2 hours at room temperature. Dialysis was performed to remove the imidazole and DTT followed by reloading the sample onto a 5 mL HiTrap™ IMAC FF column (GE life sciences) equilibrated with Buffer C. The tagless protein flowed through the column and this sample was collected and further purified using a HiPrep™ 16/60 Sephacryl™ S-200 HR column (GE life sciences) using buffer B supplemented with 150 mM NaCl at 0.75 mL min−1. The final eluted sample was concentrated using an Amicon centrifugal filter (Amicon Ultra-15 with Ultracel-10 membrane), adjusted to 25% glycerol, and flash frozen for storage at −80°C.
Bacterial two-hybrid assays
BTH101 cells were co-transformed with a plasmid encoding a T18 fusion of interest and a plasmid encoding a T25 fusion of interest (details of the specific plasmids can be viewed in Supporting Information Table S2). Co-transformants were grown in 3 mL of LB media (supplemented with 100 μg mL−1 ampicillin and 25 μg mL−1 kanamycin) at 37°C until an OD600 of between 0.5 and 1.0 was reached. The cultures were adjusted to an OD600 of 0.5, diluted 1/1000 in LB, and spotted (5 μL per spot) onto LB agar plates containing 40 μg mL−1 of X-Gal (5-bromo-4-chloro-3-indoxyl-β-D-galactopyranoside), 0.5 mM IPTG, 100 μg mL−1 ampicillin, and 25 μg mL−1 kanamycin. The plates were incubated for two days at 30°C followed by an additional 24 hours at room temperature while being protected from light. All two-hybrid experiments were performed a minimum of three times working from fresh co-transformations.
Chemical Crosslinking
To test for self-interactions, purified DnaDWHD and its variants (16 μM final concentration) were mixed with increasing amounts of glutaraldehyde (0.03%, 0.06%, 0.12%) in 10 μL reactions containing 20 mM HEPES-KOH pH 7.7, 150 mM NaCl, 1 mM DTT, 1 mM EDTA, and 5% glycerol. Each reaction was incubated for 15 minutes at room temperature, followed by quenching with 1 μL of 1 M Tris-HCl pH 8.0 and incubating for an additional 10 minutes. SDS-loading dye (62.5 mM Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 100 mM DTT, and 0.01% bromophenol blue) was then added to the samples, which were loaded onto a 4–20% gradient SDS-polyacrylamide gel (Bio-Rad), electrophoresed in 1X TAE running buffer for 30 minutes at 200 V, and stained with coomassie blue.
To detect the DnaADI-DnaDWHD complex, the proteins were incubated together for 1 hour at room temperature at a final concentration of 16 μM each. The procedure used to crosslink DnaD self-interactions (see above) was then employed to capture the complex. The band that was thought to represent the DnaADI-DnaDWHD complex was excised from the gel and sent for mass spectrometry analysis (msbioworks, Ann Arbor, MI, www.msbioworks.com) to confirm it contained both proteins.
Complementation of dnaD23ts by dnaD Variants
The relevant strains (Supporting Information Table S5) were streaked onto LB agar containing 0.001% D-xylose and incubated at 30°C (permissive temperature) or 48°C (restrictive temperature) overnight. To test for the small colony phenotype, strains were instead plated on LB agar with (1%) or without (0%) D-xylose and incubated overnight at 30°C.
Modeling the DnaA-DnaD Complex
The structures of DnaADI (PDB 4TPS) and DnaDWHD (PDB 2V79) were used to model the interaction between the DnaADI α2-α>3 surface and the DnaDWHD winged helix cleft (Jameson et al., 2014; Schneider et al., 2008). The final model was generated using the Rosetta Docking program available on ROSIE (Rosetta Online Server that Includes Everyone) (Chaudhury et al., 2011; Lyskov et al., 2013; Lyskov & Gray, 2008). Figures were prepared using the PyMOL Molecular Graphics System (v1.8.0.3), Schrödinger, LLC.
Supplementary Material
Acknowledgments
This work was supported by R01GM107312 to L.A.S. We wish to thank Jeremy Schroeder for contributing the pJS103 plasmid and Peter Burby for contributing the pPB41 plasmid used for CRISPR/Cas9 gene editing in B. subtilis.
References
- Abe Y, Jo T, Matsuda Y, Matsunaga C, Katayama T, & Ueda T (2007). Structure and function of DnaA N-terminal domains: specific sites and mechanisms in inter-DnaA interaction and in DnaB helicase loading on oriC. The Journal of Biological Chemistry, 282(24), 17816–17827. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Erez E, Martz E, Pupko T, & Ben-Tal N (2010). ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Research, 38(Web Server issue), W529–W533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilla CY, & Grossman AD (2012). The primosomal protein DnaD inhibits cooperative DNA binding by the replication initiator DnaA in Bacillus subtilis. Journal of Bacteriology, 194(18), 5110–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruand C, Farache M, McGovern S, Ehrlich SD, & Polard P (2001). DnaB, DnaD and DnaI proteins are components of the Bacillus subtilis replication restart primosome. Molecular Microbiology, 42(1), 245–255. [DOI] [PubMed] [Google Scholar]
- Bruand C, Sorokin A, Serror P, & Ehrlich SD (1995). Nucleotide sequence of the Bacillus subtilis dnaD gene. Microbiology, 141(Pt 2), 321–322. [DOI] [PubMed] [Google Scholar]
- Bruand C, Velten M, McGovern S, Marsin S, Sérèna C, Ehrlich SD, & Polard P (2005). Functional interplay between the Bacillus subtilis DnaD and DnaB proteins essential for initiation and re-initiation of DNA replication. Molecular Microbiology, 55(4), 1138–1150. [DOI] [PubMed] [Google Scholar]
- Burby PE, & Simmons LA (2017a). CRISPR/Cas9 Editing of the Bacillus subtilis Genome. Bio-Protocol, 7(8), e2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burby PE, & Simmons LA (2017b). MutS2 Promotes Homologous Recombination in Bacillus subtilis. Journal of Bacteriology, 199(2), e00682–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro MJVM, Zhang W, Ioannou C, Scott DJ, Allen S, Roberts CJ, & Soultanas P (2006). The DNA-remodelling activity of DnaD is the sum of oligomerization and DNA-binding activities on separate domains. Molecular Microbiology, 60(4), 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker G, Nimrod G, Ashkenazy H, Glaser F, Martz E, Mayrose I, Pupko T, Ben-Tal N (2013). ConSurf: Using Evolutionary Data to Raise Testable Hypotheses about Protein Function. Israel Journal of Chemistry, 53(3–4), 199–206. [Google Scholar]
- Chaudhury S, Berrondo M, Weitzner BD, Muthu P, Bergman H, & Gray JJ (2011). Benchmarking and analysis of protein docking performance in Rosetta v3.2. PloS One, 6(8), e22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E, Ogasawara N, & Ishikawa S (2008). The functional analysis of YabA, which interacts with DnaA and regulates initiation of chromosome replication in Bacillus subtilis. Genes & Genetic Systems, 83(2), 111–125. [DOI] [PubMed] [Google Scholar]
- Collier C, Machon C, Briggs GS, Smits WK, & Soultanas P (2012). Untwisting of the DNA helix stimulates the endonuclease activity of Bacillus subtilis Nth at AP sites. Nucleic Acids Research, 40(2), 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey MJ, & O’Donnell M (2003). Replicative helicase loaders: ring breakers and ring makers. Current Biology: CB, 13(15), R594–596. [DOI] [PubMed] [Google Scholar]
- Duan Y, Huey JD, & Herman JK (2016). The DnaA inhibitor SirA acts in the same pathway as Soj (ParA) to facilitate oriC segregation during Bacillus subtilis sporulation: oriC segregation during sporulation. Molecular Microbiology, 102(3), 530–544. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA 3rd, & Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods, 6(5), 343–345. [DOI] [PubMed] [Google Scholar]
- Glaser F, Pupko T, Paz I, Bell RE, Bechor-Shental D, Martz E, & Ben-Tal N (2003). ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics, 19(1), 163–164. [DOI] [PubMed] [Google Scholar]
- Grainger WH, Machon C, Scott DJ, & Soultanas P (2010). DnaB proteolysis in vivo regulates oligomerization and its localization at oriC in Bacillus subtilis. Nucleic Acids Research, 38(9), 2851–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harami GM, Gyimesi M, & Kovacs M (2013). From keys to bulldozers: expanding roles for winged helix domains in nucleic-acid-binding proteins. Trends in Biochemical Sciences, 38(7), 364–371. [DOI] [PubMed] [Google Scholar]
- Hassan AK, Moriya S, Ogura M, Tanaka T, Kawamura F, & Ogasawara N (1997). Suppression of initiation defects of chromosome replication in Bacillus subtilis dnaA and oriC-deleted mutants by integration of a plasmid replicon into the chromosomes. Journal of Bacteriology, 179(8), 2494–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou C, Schaeffer PM, Dixon NE, & Soultanas P (2006). Helicase binding to DnaI exposes a cryptic DNA-binding site during helicase loading in Bacillus subtilis. Nucleic Acids Research, 34(18), 5247–5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T, Akimitsu N, Kashioka T, Hatano M, Kubota T, Ogata Y, … Katayama T (2004). DiaA, a novel DnaA-binding protein, ensures the timely initiation of Escherichia coli chromosome replication. The Journal of Biological Chemistry, 279(44), 45546–45555. [DOI] [PubMed] [Google Scholar]
- Ishigo-oka D, Ogasawara N, & Moriya S (2001). DnaD Protein of Bacillus subtilis Interacts with DnaA, the Initiator Protein of Replication. Journal of Bacteriology, 183(6), 2148–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson KH, Rostami N, Fogg MJ, Turkenburg JP, Grahl A, Murray H, & Wilkinson AJ (2014). Structure and interactions of the Bacillus subtilis sporulation inhibitor of DNA replication, SirA, with domain I of DnaA. Molecular Microbiology, 93(5), 975–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, & Ladant D (1998). A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proceedings of the National Academy of Sciences of the United States of America, 95(10), 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyamura K, Abe Y, Higashi M, Ueda T, & Katayama T (2009). DiaA dynamics are coupled with changes in initial origin complexes leading to helicase loading. The Journal of Biological Chemistry, 284(37), 25038–25050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyamura K, Fujikawa N, Ishida T, Ozaki S, Su’etsugu M, Fujimitsu K, … Katayama T (2007). The interaction of DiaA and DnaA regulates the replication cycle in E. coli by directly promoting ATP DnaA-specific initiation complexes. Genes & Development, 21(16), 2083–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayrutdinov BI, Bae WJ, Yun YM, Lee JH, Tsuyama T, Kim JJ, Hwang E, Ryu KS, Cheong HK, Cheong C, Ko JS, Enomoto T, Karplus PA, Guntert P, Tada S, Jeon YH, & Cho Y (2009). Structure of the Cdt1 C-terminal domain: conservation of the winged helix fold in replication licensing factors. Protein Science, 18(11), 2252–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, & Ben-Tal N (2005). ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Research, 33(Web Server issue), W299–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Schroeder JW, Gao B, Simmons LA, & Biteen JS (2015). Single-molecule motions and interactions in live cells reveal target search dynamics in mismatch repair. Proceedings of the National Academy of Sciences of the United States of America, 112(50), E6898–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-C, Naveen V, & Hsiao C-D (2016). EM structure of a helicase-loader complex depicting a 6:2 binding sub-stoichiometry from Geobacillus kaustophilus HTA426. Biochemical and Biophysical Research Communications, 473(1), 243–248. [DOI] [PubMed] [Google Scholar]
- Liu B, Eliason WK, & Steitz TA (2013). Structure of a helicase-helicase loader complex reveals insights into the mechanism of bacterial primosome assembly. Nature Communications, 4, 2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscha KV, Jaudzems K, Ioannou C, Su X-C, Hill FR, Otting G, Dixon NE, & Liepinsh E (2009). A novel zinc-binding fold in the helicase interaction domain of the Bacillus subtilis DnaI helicase loader. Nucleic Acids Research, 37(7), 2395–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S, Chou F-C, Conchuir SO, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P, Borgo B, Havranek JJ, Kuhlman B, Kortemme T, Bonneau R, Gray JJ, & Das R (2013). Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). PloS One, 8(5), e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S, & Gray JJ (2008). The RosettaDock server for local protein-protein docking. Nucleic Acids Research, 36(Web Server issue), W233–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsin S, McGovern S, Ehrlich SD, Bruand C, & Polard P (2001). Early steps of Bacillus subtilis primosome assembly. The Journal of Biological Chemistry, 276(49), 45818–45825. [DOI] [PubMed] [Google Scholar]
- Marston FY, Grainger WH, Smits WK, Hopcroft NH, Green M, Hounslow AM, Grossman AD, Craven CJ, & Soultanas P (2010). When simple sequence comparison fails: the cryptic case of the shared domains of the bacterial replication initiation proteins DnaB and DnaD. Nucleic Acids Research, 38(20), 6930–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek J, & Kaguni JM (1994). DnaA protein directs the binding of DnaB protein in initiation of DNA replication in Escherichia coli. The Journal of Biological Chemistry, 269(7), 4883–4890. [PubMed] [Google Scholar]
- Marszalek J, Zhang W, Hupp TR, Margulies C, Carr KM, Cherry S, & Kaguni JM (1996). Domains of DnaA protein involved in interaction with DnaB protein, and in unwinding the Escherichia coli chromosomal origin. The Journal of Biological Chemistry, 271(31), 18535–18542. [DOI] [PubMed] [Google Scholar]
- Meile J-C, Wu LJ, Ehrlich SD, Errington J, & Noirot P (2006). Systematic localisation of proteins fused to the green fluorescent protein in Bacillus subtilis: identification of new proteins at the DNA replication factory. Proteomics, 6(7), 2135–2146. [DOI] [PubMed] [Google Scholar]
- Mott ML, & Berger JM (2007). DNA replication initiation: mechanisms and regulation in bacteria. Nature Reviews. Microbiology, 5(5), 343–354. [DOI] [PubMed] [Google Scholar]
- Mott ML, Erzberger JP, Coons MM, & Berger JM (2008). Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell, 135(4), 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natrajan G, Hall DR, Thompson AC, Gutsche I, & Terradot L (2007). Structural similarity between the DnaA-binding proteins HobA (HP1230) from Helicobacter pylori and DiaA from Escherichia coli. Molecular Microbiology, 65(4), 995–1005. [DOI] [PubMed] [Google Scholar]
- Natrajan G, Noirot-Gros MF, Zawilak-Pawlik A, Kapp U, & Terradot L (2009). The structure of a DnaA/HobA complex from Helicobacter pylori provides insight into regulation of DNA replication in bacteria. Proceedings of the National Academy of Sciences of the United States of America, 106(50), 21115–21120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noirot-Gros M-F, Dervyn E, Wu LJ, Mervelet P, Errington J, Ehrlich SD, & Noirot P (2002). An expanded view of bacterial DNA replication. Proceedings of the National Academy of Sciences, 99(12), 8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn-Lee L, Gorbatyuk B, Skovgaard O, & Losick R (2009). The Conserved Sporulation Protein YneE Inhibits DNA Replication in Bacillus subtilis. Journal of Bacteriology, 191(11), 3736–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn-Lee L, Merrikh H, Grossman AD, & Losick R (2011). The Sporulation Protein SirA Inhibits the Binding of DnaA to the Origin of Replication by Contacting a Patch of Clustered Amino Acids. Journal of Bacteriology, 193(6), 1302–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reikofski J, & Tao BY (1992). Polymerase chain reaction (PCR) techniques for site-directed mutagenesis. Biotechnology Advances, 10(4), 535–547. [DOI] [PubMed] [Google Scholar]
- Rokop ME, Auchtung JM, & Grossman AD (2004). Control of DNA replication initiation by recruitment of an essential initiation protein to the membrane of Bacillus subtilis. Molecular Microbiology, 52(6), 1757–1767. [DOI] [PubMed] [Google Scholar]
- Schneider S, Zhang W, Soultanas P, & Paoli M (2008). Structure of the N-terminal oligomerization domain of DnaD reveals a unique tetramerization motif and provides insights into scaffold formation. Journal of Molecular Biology, 376(5), 1237–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholefield G, & Murray H (2013). YabA and DnaD inhibit helix assembly of the DNA replication initiation protein DnaA: Regulation of DnaA by YabA and DnaD. Molecular Microbiology, 90, 147–159. [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Tonthat NK, Kwong SM, Chinnam NB, Liu MA, Skurray RA, & Firth N (2014). Mechanism of staphylococcal multiresistance plasmid replication origin assembly by the RepA protein. Proceedings of the National Academy of Sciences of the United States of America, 111(25), 9121–9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Weigel C, & Messer W (2000). The interaction domains of the DnaA and DnaB replication proteins of Escherichia coli. Molecular Microbiology, 37(5), 1270–1279. [DOI] [PubMed] [Google Scholar]
- Smits WK, Goranov AI, & Grossman AD (2010). Ordered association of helicase loader proteins with the Bacillus subtilis origin of replication in vivo. Molecular Microbiology, 75(2), 452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits WK, Merrikh H, Bonilla CY, & Grossman AD (2011). Primosomal Proteins DnaD and DnaB Are Recruited to Chromosomal Regions Bound by DnaA in Bacillus subtilis. Journal of Bacteriology, 193(3), 640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas P (2002). A functional interaction between the putative primosomal protein DnaI and the main replicative DNA helicase DnaB in Bacillus. Nucleic Acids Research, 30(4), 966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas Panos. (2012). Loading mechanisms of ring helicases at replication origins. Molecular Microbiology, 84(1), 6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su’etsugu M, Harada Y, Keyamura K, Matsunaga C, Kasho K, Abe Y, … Katayama T (2013). The DnaA N-terminal domain interacts with Hda to facilitate replicase clamp-mediated inactivation of DnaA. Environmental Microbiology, 15(12), 3183–3195. [DOI] [PubMed] [Google Scholar]
- Sutton MD, Carr KM, Vicente M, & Kaguni JM (1998). Escherichia coli DnaA protein. The N-terminal domain and loading of DnaB helicase at the E. coli chromosomal origin. The Journal of Biological Chemistry, 273(51), 34255–34262. [DOI] [PubMed] [Google Scholar]
- Velten M, McGovern S, Marsin S, Ehrlich SD, Noirot P, & Polard P (2003). A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Molecular Cell, 11(4), 1009–1020. [DOI] [PubMed] [Google Scholar]
- Wagner JK, Marquis KA, & Rudner DZ (2009). SirA enforces diploidy by inhibiting the replication initiator DnaA during spore formation in Bacillus subtilis. Molecular Microbiology, 73(5), 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawilak-Pawlik A, Donczew R, Szafranski S, Mackiewicz P, Terradot L, & Zakrzewska-Czerwinska J (2011). DiaA/HobA and DnaA: a pair of proteins co-evolved to cooperate during bacterial orisome assembly. Journal of Molecular Biology, 408(2), 238–251. [DOI] [PubMed] [Google Scholar]
- Zawilak-Pawlik A, Nowaczyk M, & Zakrzewska-Czerwinska J (2017). The Role of the N-Terminal Domains of Bacterial Initiator DnaA in the Assembly and Regulation of the Bacterial Replication Initiation Complex. Genes, 8(5), 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Allen S, Roberts CJ, & Soultanas P (2006). The Bacillus subtilis primosomal protein DnaD untwists supercoiled DNA. Journal of Bacteriology, 188(15), 5487–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Carneiro MJVM, Turner IJ, Allen S, Roberts CJ, & Soultanas P (2005). The Bacillus subtilis DnaD and DnaB proteins exhibit different DNA remodelling activities. Journal of Molecular Biology, 351(1), 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Machon C, Orta A, Phillips N, Roberts CJ, Allen S, & Soultanas P (2008). Single-molecule atomic force spectroscopy reveals that DnaD forms scaffolds and enhances duplex melting. Journal of Molecular Biology, 377(3), 706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.