

Abstract
The function of the right ventricle determines the fate of patients with pulmonary hypertension. Since right heart failure is the consequence of increased afterload, a full physiological description of the cardiopulmonary unit consisting of both the right ventricle and pulmonary vascular system is required to interpret clinical data correctly. Here, we provide such a description of the unit and its components, including the functional interactions between the right ventricle and its load. This physiological description is used to provide a framework for the interpretation of right heart catheterisation data as well as imaging data of the right ventricle obtained by echocardiography or magnetic resonance imaging. Finally, an update is provided on the latest insights in the pathobiology of right ventricular failure, including key pathways of molecular adaptation of the pressure overloaded right ventricle. Based on these outcomes, future directions for research are proposed.
Short abstract
State of the art and research perspectives in pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension with theoretical and practical aspects http://ow.ly/18v830mgLiP
The cardiopulmonary unit: more than a sum of its parts
The function of the right ventricle is of great clinical importance in severe pulmonary hypertension (PH) since it determines the outcome of the disease [1, 2]. Given the fact that right heart failure in PH is the consequence of increased (arterial) afterload and not the mere consequence of a myocardial disease, a full description of the cardiopulmonary unit is required in the study of right heart failure (figure 1a and b). The cardiopulmonary unit is composed of two main functional subsystems, i.e. the right ventricle and the pulmonary vasculature, each having their own intrinsic characteristics (figure 1b). The ventricular pressure–volume loop analysis is central in understanding right ventricular physiology, while pressure–flow analysis is central in understanding pulmonary haemodynamics. For the right ventricle, intrinsic characteristics include contractility, chamber stiffness and, although perhaps less established, the time constant of ventricular relaxation (τ), which are all load independent; for the pulmonary vascular system, resistance and compliance provide intrinsic characteristics of the steady and pulsatile load.
FIGURE 1.

The cardiopulmonary system (CPS): a) function and b) characterisation. MVO2: myocardial oxygen consumption; RV: right ventricle; RA: right atrium; LA: left atrium; LV: left ventricle; Ees: end-systolic elastance; τ: time constant of ventricular relaxation; Eed: end-diastolic elastance; PVR: pulmonary vascular resistance; PAC: pulmonary arterial compliance; Ea: arterial elastance; PAP: pulmonary arterial pressure; PAWP: pulmonary arterial wedge pressure; EF: ejection fraction; CO: cardiac output. Subsystems (or units: heart, respectively its load) are characterised by their intrinsic function, which can be derived from the ventricular pressure–volume relationship and the pulmonary pressure–flow relationship. The system parameters result from cardiopulmonary interaction.
The interaction between the intrinsic ventricular characteristics and load results in global function, and is commonly described by cardiac output (CO) and ejection fraction (EF), on the one hand, and pressure (mean, systolic and diastolic pressure), on the other hand. The pressure gradient over the pulmonary circulation also falls into this category. Energy transfer of right ventricular to arterial load is a special form of interaction for which we reserve the term “coupling”. The concept of coupling is particularly important in physiologically describing the continuum of ventricular adaptation in pulmonary arterial hypertension (PAH): well-adapted right ventricles often have preserved ventriculo-arterial coupling, while maladapted right ventricles have varying degrees of altered ventriculo-arterial coupling (figure 2). These concepts will be discussed in more detail in the following section.
FIGURE 2.
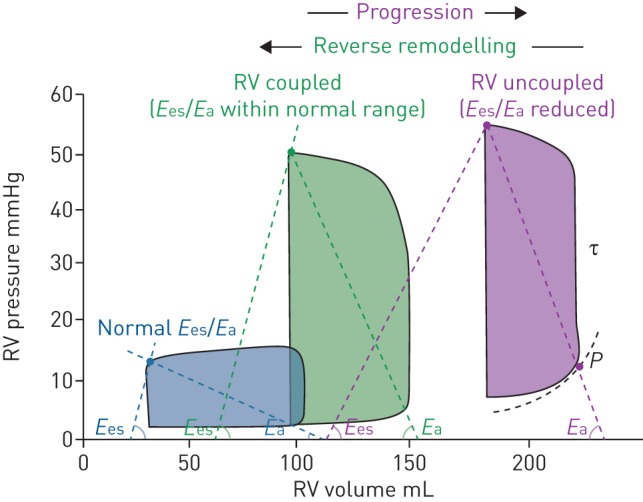
Right ventricular (RV) pressure–volume analysis. Pressure–volume loops at three different stages: normal (blue), pulmonary hypertension (green) and right ventricular failure (purple). Ees: end-systolic elastance; Ea: arterial elastance; τ: time constant of ventricular relaxation. P=α(eβV−1) describes the diastolic pressure–volume relation. Reproduced and modified from [11] with permission.
This distinction between intrinsic characteristic of a subsystem and global function or system characteristics has important physiological implications. For example, despite the decrease in right ventricular EF (RVEF) in patients with PAH, right ventricle contractility as measured by ventricular elastance (i.e. end-systolic elastance (Ees)) is usually increased and not decreased [3].
Definitions
The current section presents some definitions that may help standardise important concepts relevant to the field of right ventricular adaptation and right heart failure.
Right heart failure in PH can be defined as a clinical syndrome characterised by decreased right ventricular function that leads to insufficient blood flow and/or elevated filling pressures at rest or during physiologically demanding conditions, such as exercise, developmental growth or pregnancy. The cardinal symptoms of right heart failure include dyspnoea and fatigue as well as congestion. The severity of heart failure is often subcategorised according to New York Heart Association (NYHA) Functional Class and by the degree of congestion.
Right ventricular adaptation in PH represents a continuum with an adapted right ventricle at one end and a maladapted ventricle at the other end. An adapted right ventricle in PH is characterised by a slightly dilated right ventricle with preserved stroke volume (SV), systolic function and normal filling pressures, whereas a maladapted right ventricle is characterised by a dilated right ventricle with reduced SV, systolic function and increased filling pressures. As will be discussed in the following section, adapted ventricles usually have preserved ventriculo-arterial coupling, while maladapted ventricles usually have uncoupled ventricles (figure 2).
Subsystem function (intrinsic properties of the cardiac or pulmonary vascular system): description of the right ventricle in a load-independent manner (Ees and end-diastolic elastance (Eed)) or pulmonary vascular load in a manner independent of right ventricular function (pulmonary arterial vascular resistance (PVR), or arterial elastance (Ea), and pulmonary arterial compliance (PAC)).
System function results from the interaction of the two subsystems, i.e. the ventricular pump and its afterload. Interaction results in SV, CO, pressure and functional imaging parameters derived by echocardiography or cardiovascular magnetic resonance imaging (CMR). Descriptions of systolic function as RVEF or SV/right ventricular end-systolic volume (ESV) are also the result of functional interaction and not surrogates for coupling.
Coupling: the condition that occurs when right ventricular function is adapted to the pulmonary vascular load such that energy transfer is most efficient. This coupling is described by systolic and arterial elastance, Ees/Ea (figures 1b and 2).
Although this article focuses on the right ventricle, it is noteworthy that ventricular interdependency plays an important role in PH. Not only the right ventricle but also the left ventricle is involved in PH since the right ventricle and left ventricle have the septum in common, are encircled with common myocardial fibres and are within a (not acutely) distensible pericardium (figure 1a) [4, 5]. This ventricular interdependency becomes visible in PH as rapid leftward bowing of the septum during early left ventricle diastole. This typical septal motion abnormality has been shown to be a consequence of a prolonged contraction of the right ventricle free wall relative to that of the septum and the left ventricle free wall, causing interventricular relaxation dyssynchrony [6, 7]. As such, the septum acts as a whistleblower of a right–left ventricular tissue load imbalance, reflecting right ventricular tissue overload in the setting of PH. This rapid early-diastolic leftward motion of the septum is also associated with septal and left ventricular myocardial stretch during late right ventricular ejection [8], causing mechanical inefficiency of the right ventricle and contributing to left ventricular underfilling [9] and atrophy [10]. The septal curvature is a useful metric in PH, reflecting both the interventricular pressure gradient and relative interventricular size, as well as dyssynchrony of contraction [7].
Assessment of the right ventricle in a load-independent manner
Right ventricular function can be characterised by the pressure–volume relation (figure 2) [11], and measurement of right ventricular volumes and pressures are therefore useful for the assessment of the right ventricle in PH, particularly for physiological studies. A schematic overview of an approach to right ventricle phenotyping, integrating structural imaging, haemodynamics, molecular imaging and biomarkers is given in supplementary figure S1a.
Systole
Ees of the ventricle is a load-independent description of the right ventricle and the current reference measure of contractility; it depends on the contractile force of the myocyte and cardiac muscle mass (hypertrophy) [12, 13].
The derivation of Ees is based on the pressure–volume loop (figure 2). The best method to assess Ees is to decrease ventricular filling, e.g. by partial vena cava occlusion, and analyse the series of loops that result [14]. Connection of the end-systolic pressure (Pes)–volume points results in the Pes–volume relation and its slope is Ees. Since this approach is invasive and not widely available in the clinic, single-beat methods have been developed [13, 15]. Ees can be increased 5-fold in PAH patients, reflecting that the right ventricle is performing at a high contractile state [16]. In addition, the elastance response to pharmacological (usually dobutamine) or physiological stress (exercise) can be useful to assess contractile reserve. In the normal right ventricle, Ees usually increases significantly with exercise [3]. In the pressure overloaded right ventricle, although the baseline is higher than normal, the degree of increase is blunted with exercise or following dobutamine infusion [3, 17]. Contractile reserve at exercise is absent in more advanced disease states or might even decrease [3].
Diastole
In practice, right atrial pressure and central venous pressure are often used as surrogates for diastolic properties of the right ventricle; right atrial pressure has been consistently related to outcome in PAH starting with the original US National Institutes of Health registry study in 1991 [18]. Using load-independent metrics has the advantage of allowing a better understanding of the responsiveness to volume status or change in filling conditions. Eed of the ventricle is a load-independent representation of the diastolic function and is best described by a diastolic elastance curve determined by multiple pressure–volume loops obtained by decreased loading by partial vena cave occlusion. The relation is curvilinear and the most adequate description is obtained by fitting the relation with an exponential curve through the diastolic pressure–volume points, with the formula P=α(eβV−1), where α and β are curve-fitting constants (figure 2) [11, 19]. A single-beat method has been developed to determine Eed [20, 21]. Eed now can be calculated as Eed=αβeβVed, where Ved is the end-diastolic volume (EDV) [21]. The so-defined diastolic stiffness predicts outcome in PAH as well as the more complex β calculation [21].
Right ventricular diastolic function is closely associated with disease severity [20], but also with Ees [21, 22]. Right ventricle diastolic stiffness in severe PH is accompanied by fibrosis and specific biological alterations, such as reduced titin phosphorylation [20]. Although Eed is related to Ees, diastolic adaptability remains variable in patients with PH and whether it may serve as a biomarker of pending right ventricle failure is not completely resolved.
Assessment of the pulmonary circulation in a heart-independent manner
Pulmonary vascular resistance
When analysing the pulmonary circulation, it is useful to distinguish a steady and pulsatile component of the load. These main components of arterial load are PVR and total PAC. In the pulmonary system these two components are inversely related (see later). PVR is calculated as PVR=(mPAP−PAWP)/CO, where mPAP is the mean pulmonary arterial pressure and PAWP is the pulmonary arterial wedge pressure. A measure of total load can also be estimated from the pressure–volume loop (figure 2) as Ea=Pes/SV. It has been proposed to use Ea∼mPAP/SV when Pes is not available [23]; however, this approximation should be used with caution since, especially in higher pressures ranges, Pes is underestimated by mPAP [24].
Pulmonary capillary flow is a dynamic process. In basal states, flow through an individual capillary is intermittent. Capillary recruitment occurs when the probability of a given capillary carrying flow increases. Capillary recruitment and distension both play a role in the lung's accommodation of pulmonary arterial blood flow. At basal or moderately increased blood flows, capillary recruitment is predominant [25, 26]. At very high regional flows or pressures, after local full recruitment, capillary distension is predominant [26–29]. The local pattern of capillary recruitment/distension may vary regionally in the lung, with gravity, arterial and post-capillary venous pressures, airway pressures, and disease states all having effects [30–34].
Examining the pulmonary circulation as a whole, since in the (healthy) pulmonary circulation the arteries and veins are distensible, the assumption that the pulmonary vascular pressure difference–flow relationship is linear and crosses the origin is inaccurate. The simple formula then no longer holds and PVR can be described using two parameters: α (the pulmonary circulatory distensibility coefficient) and R0 (a reference resistance usually assumed to be the resistance at rest) [35, 36]. While α may be relevant for the detection of early pulmonary vascular remodelling and relates to exercise capacity, right ventricular function and outcome [37], it requires accurate measurement of pulmonary vascular pressure and flow at rest and at exercise. It therefore cannot be currently recommended as part of standard haemodynamic evaluation of patients referred for PH.
Pulmonary arterial compliance
The pulsatile load of the pulmonary circulation is most often evaluated using PAC. The best method to calculate PAC is based on a two-element Windkessel model with flow waveform and resistance as inputs to estimate the compliance value that best predicts systolic and diastolic pressures, the so-called “pulse pressure” (PP) method [38]. A simpler and accepted method to derive PAC is SV/PP [39]. This ratio assumes that the SV is buffered in the large elastic arteries in systole, without any peripheral outflow. However, there is a continuous flow toward the periphery, reducing the vascular volume increase during ejection, resulting in an overestimation of the true PAC [40]. It has been shown in intact experimental animals at various severities of induced PH that SV/PP overestimates PAC by 60–80% [40, 41]. This overestimation probably depends on patient status. CMR-determined proximal arterial compliance amounts to around 20% of PAC [42], showing the greater contribution of the smaller vessels compared with the systemic arterial vasculature.
Time constant of the pulmonary circulation
In the previous sections we described the steady and pulsatile components separately; however, as already mentioned, these are closely related by an inverse relationship. The PAP decay curve in diastole is determined by PVR and PAC. The combined effect can be formulated by the product of PVR and PAC. The unit of this product is time and, therefore, is called the arterial time constant (RC-time) [43].
The inverse relationship between PVR and PAC was first reported in 1971 [44]. A series of studies showed recently that PVR and PAC are inversely related, and RC-time is constant over a wide range of severities, aetiologies and treatments of PH (figure 3) [41, 45, 46]. It is noteworthy that RC-time is decreased when PAWP increases in patients with heart failure [47, 48]. The fact that PVR takes the wedge pressure into account, whereas compliance, calculated as SV/PP, does not, gives rise to an altered RC-time in post-capillary PH.
FIGURE 3.
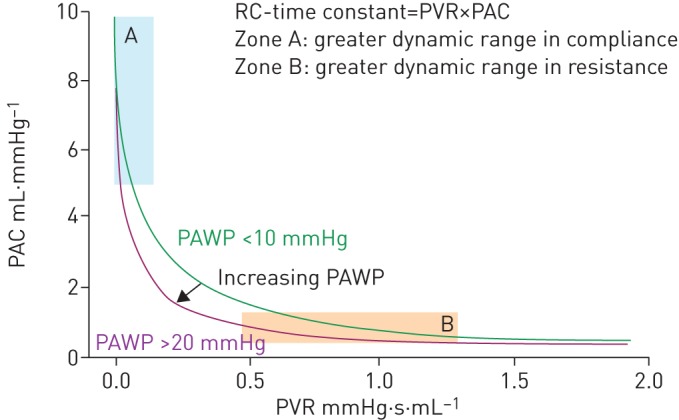
The resistance–compliance relationship: the relationship between pulmonary vascular resistance (PVR) and pulmonary arterial compliance (PAC) is characterised by a constant value of RC-time (i.e. the product of PVR×PAC). PAWP: pulmonary arterial wedge pressure. Reproduced and modified from [11] with permission.
Owing to the inverse relationship between PVR and PAC, the dynamic range in PAC will be greater in patients with mild pulmonary vascular disease (PVD) (zone A in figure 3), while the dynamic range in PVR will be greater in patients with more advanced PVD (zone B in figure 3). Therefore, PAC may be more sensitive to detect changes in PVD in the early phase of disease. Moreover, several studies using linear prediction models found that PAC is an independent predictor of outcome in PAH [49–52] over a wide range of PVR [53, 54]. Another implication of this remarkable structural characteristic of the pulmonary vasculature is that it dictates the reported tight correlation between systolic PAP (sPAP), diastolic PAP (dPAP) and mPAP in normal subjects and in patients with PH of all possible aetiologies [55, 56], independently of PAWP (figure 4) [57]:
FIGURE 4.

The mean, systolic and diastolic pulmonary arterial pressure (mPAP, sPAP and dPAP) relationships: relative values of pressures in the pulmonary circulation are tightly related. Reproduced and modified from [57] with permission.
and
The first relation implies that mPAP can be calculated from Doppler echocardiographic estimates of sPAP by the formula:
The mPAP calculated in this way provides an estimate of mPAP in patients referred for diagnostic work-up of PH [58]. In echocardiography-based studies, different thresholds for right ventricular systolic pressure (e.g. 30, 35 or 40 mmHg) have been used for defining PH. According to the formula, a right ventricular systolic pressure of 40 mmHg would best approximate a mPAP of 25 mmHg assuming a mild right ventricular outflow tract gradient and an insonation angle <15°.
Assessment of the cardiopulmonary unit
Functional interaction
In the clinic, the description of right ventricular function in relation to its load is of high prognostic value. Most imaging parameters as measured by echocardiography or CMR reflect system function of the interaction of the right ventricle and the vascular load. At present no recommendations on the most relevant measurements of right ventricular function can be made. Further clinical research is needed, preferably multicentre and prospective, with an a priori established list of variables of interest. It should not be overlooked that adequate measurements do not only qualify based on their prognostic capability or prediction of clinical worsening; they should be reproducible and easy to assess in PH centres of expertise.
However, there is ample evidence that direct or indirect measurements of right ventricular EDV and ESV, derivation of SV, and filling pressures contain the foremost prognostic information. This can be explained by the pressure–volume loops derived from the adapted and failing right ventricle. In the adapted right ventricle, increased afterload in PH leads to hypertrophy and an increased contractility with more or less preserved dimensions and SV [2, 4, 11, 59], whereas in progressive right ventricular failure, the right ventricle progressively dilates and decreases its SV.
It should be noted that SV/ESV is inversely related to RVEF, as indicated by the formula SV/ESV=EF/(1−EF). Indeed, Vanderpool et al. [22] showed this inverse relation exists in patients with PH [22]. Thus, both SV/ESV and RVEF contain similar prognostic information, and should be considered as parameters of functional interaction, not coupling. RVEF and SV/ESV have been shown to be equally predictive of outcome in patients with PAH [60]. Rigorously defined cut-off values for shortened survival are 0.35 for EF [60, 61] and 0.54 for SV/ESV [22, 60]. Although theoretically RVEF and SV/ESV should have similar predictive potential, due to the hyperbolic relationship between the two, the latter may be more sensitive to early changes [62]. Then again, it is more difficult to determine ESV in an accurate manner. A large-scale head-to-head comparison of these two parameters may resolve this matter.
Coupling
Coupling implies efficiency of energy transfer from the ventricle to the arterial load and can be calculated as the ratio Ees/Ea. The value will be between 1 and 2 if maximal energy transfer from ventricle to load occurs (figure 2).
Coupling has been reported in PAH patients, with single-beat calculation of the Ees/Ea ratio from CMR measurements of right ventricular volumes and right heart catheterisation (RHC) measurements of right ventricular pressures [63]. Single-beat determinations of Ees/Ea have been implemented in experimental animal studies to show, for example, that acutely administered prostacyclin has no intrinsic inotropic effect [64] and that β-blocker agents may either deteriorate (acutely) [11] or improve (chronically) [64] right ventriculo–arterial coupling.
Compared with controls, in PH the Ees/Ea was decreased, indicating insufficient contractility adaptation (“homeometric”) and impending right ventricular failure. Results have been confirmed in small cohorts of patients with either PAH or chronic thromboembolic PH, and in one case report of a patient with a systemic-like pressured right ventricle [3, 65–68]. In these studies, Ees/Ea was measured either by the single-beat method [3, 63, 65, 67] or using multiple pressure–volume loops obtained by decreasing venous return through a Valsalva manoeuvre [66, 68]. Ees/Ea was either maintained or decreased at rest, but consistently decreased at exercise. Decreased Ees/Ea at exercise was accompanied by an increase in right ventricular EDV [68].
Coupling is maintained at resting conditions for as long as the ventricle can adapt (figure 2). Only in the late stages of pressure overload does uncoupling occur [11, 21]. Therefore, coupling is not a sensitive parameter to identify an early disease state. Although coupling measured at exercise might contain more important clinical information, the complexity of these measurements will limit clinical use.
Practical assessment of pulmonary haemodynamics
Single variables such as PAPs, SV and CO are the result of the interaction of the right ventricle to its load. This implies that from a single variable, e.g. CO, no quantitative information can be obtained on either of the subsystems, i.e. the heart or the arterial load. For this reason, a change in PAP at exercise cannot be considered as a measure of right ventricular contractility.
Measuring the pressures correctly
Measurements of PAP and PAWP during rest and especially at exercise are technically challenging because of respiratory pressure swings. To avoid spurious increases in PAWP at end-expiration during exercise caused by dynamic hyperinflation and/or decrease in lung volume, it is preferable to average the reading of pulmonary vascular pressure curves over several respiratory cycles [69]. The 2015 European Society of Cardiology/European Respiratory Society PH guidelines recommend measurements at end-expiration at rest, as is standard procedure in most catheterisation laboratories, but allow for averaging over several respiratory cycles during exercise when respiration-related phasic changes become excessive [1, 70].
Provocation of the pulmonary circulation
Provocative testing of the pulmonary circulation with exercise or a fluid challenge has been used by some centres in clinical practice for decades, but has only recently been standardised.
Exercise
The upper limit of normal of mPAP during an incremental dynamic exercise challenge is now well established at 30 mmHg at CO <10 L·min−1 (figure 5), which corresponds to a total pulmonary resistance (TPR=mPAP/CO) of 3 WU [37, 71, 72]. It has been recently proposed that exercise-induced increases in mPAP >30 mmHg at CO <10 L·min−1 be called “exercise PH” [69].
FIGURE 5.

The mean pulmonary arterial pressure (mPAP)–cardiac output (CO) relationships: the ratio of ΔP/ΔCO in health and disease. PH: pulmonary hypertension; PAH: pulmonary arterial hypertension. Reproduced and modified from [71] with permission.
The cause of “exercise PH” is either an upstream transmission of increased PAWP, such as in heart failure, or an increase in PVR, such as in PVD, disturbed lung mechanics or hypoxia [37, 69, 71]. This differential diagnosis relies on a clinical probability and eventual invasive measurements of PAWP and left ventricular end-diastolic pressure. The upper limit of normal of PAWP during exercise is generally thought to be between 15 and 20 mmHg, but higher values can be recorded in athletes capable of very high CO and in elderly subjects [73]. Some consider 20 mmHg as a reasonable upper limit of normal [74]. However, a higher cut-off value of 25 mmHg has been proposed for the diagnosis of heart failure [75]. As for mPAP, a flow-corrected measure may be more appropriate, but there has been no study specifically addressing this. Since TPR normally decreases by up to 30% during exercise, the PAWP/CO slope should not exceed 2 mmHg·L−1·min−1. Mean PAWP/CO slopes around 1 mmHg·L−1·min−1 have been reported in control groups of studies on exercise testing in heart failure patients [71, 74, 75].
Fluid challenge
A fluid challenge will induce a rapid rise in PAWP in any condition associated with altered left ventricular diastolic compliance or mitral valvulopathy [76]. Fluid loading increases PAWP in healthy volunteers as a function of age, sex, amount infused and infusion rate [77]. There is an emerging consensus to infuse 500 mL of saline in 5–10 min as the best compromise between safety and stress efficacy, with 18 mmHg as the optimal PAWP cut-off to separate abnormal from normal [76–80]. This was recently underpinned by a report on 212 patients referred for PH who were challenged with 7 mL·kg−1 of saline given in <5 min (corresponding to 0.5 L for a 70-kg patient) [80]. To limit the number of healthy outliers [80], a cut-off value of 20 mmHg might be preferable. Both exercise and fluid loading increase systemic venous return, but exercise has additional effects, including sympathetic nervous system activation, intrathoracic pressure changes and mixed venous or arterial hypoxaemia. These differences probably impact on their respective efficacies for diagnosis of latent disease [81].
The emerging role of non-invasive assessment of right ventricular function
The role of right ventricular function estimated by non-invasive methods such as echocardiography (two-dimensional, three-dimensional, speckle tracking-derived echocardiography) or CMR is emerging rapidly in observational studies and has been reviewed elsewhere [82]. Speckle-derived strain technology allows measurements of regional strains by either method and demonstrates heterogeneity of right ventricular strain depending on the region (e.g. apical versus mid or basal region) and occasionally significant differences between disease aetiologies, such as idiopathic PAH (IPAH) and systemic sclerosis (SSc)-associated PAH. Recent larger studies in echocardiography have demonstrated that right ventricular longitudinal strain or indices of right ventricular end-systolic remodelling in combination with N-terminal pro-brain natriuretic peptide and NYHA Functional Class provide good discrimination of outcome in PAH [83, 84].
Imaging modalities may be incorporated into large multicentre clinical trials in PH using composite morbidity and mortality end-points, which would be ideal settings to validate potential right ventricle-based surrogate end-points. Since CMR has greater sensitivity and reproducibility than ultrasound, and can detect an efficacy signal with a small sample size in a relatively short period of time, validation of specific right ventricle surrogate CMR markers (e.g. RVEF, right ventricular mass, right ventricular mass index; left ventricle size and function) in the setting of smaller phase 2 trials would be useful to help identify potentially promising therapies. This could allow the creation of prediction scores constructed from a hierarchically organised series of identified independent predictors.
Recent insights in the pathobiology of right ventricular failure
Since the 5th World Symposium on Pulmonary Hypertension report in 2013 [2], there have been major advances in understanding the underlying pathobiology of right ventricular remodelling and failure. This knowledge has been derived both from animal models, reviewed elsewhere [85], and, importantly, from human tissue. Recent work has focused on several main themes and a thorough review of the complete breadth of right ventricular failure mechanisms is beyond the scope of the current article.
First, there has been an expansion of the understanding that genetic features may affect right ventricular stress responses. It is well recognised that patients with heritable forms of PAH (HPAH) have poorer survival than their IPAH counterparts, but recent work has demonstrated that this is related to more severe right ventricular failure in HPAH due to BMPR2 (bone morphogenetic protein receptor type 2) mutation despite similar levels of afterload, suggesting a genetic contribution to right ventricular failure [86]. In a different series of investigations, BMPR2 mutation was shown to promote lipotoxicity in the BMPR2 mutant right ventricle in rodents, in humans and in cultured cardiomyocytes with overexpression of mutant BMPR2 [87–89]. More recently, Potus et al. [90] demonstrated downregulation of miR-126 in human tissue and the monocrotaline model of right ventricular failure, with subsequent reduction in angiogenesis. Taken together, these data suggest a role for genetic regulation of right ventricular failure and may be used to develop new models of right ventricular failure.
The effect of sex hormones on the right ventricle has gained further attention. Male sex is known to be an independent risk factor for poor survival [91] and Jacobs et al. [92] found that despite a similar burden of PVD, male patients lack improvement in RVEF regardless of treatment, while their female counterparts improved RVEF. Frump et al. [93] demonstrated using the Sugen hypoxia models that oestrogen improves right ventricular function and bioenergetics, and reduces proapoptotic signalling and inflammatory cytokine expression, perhaps suggesting a protective role for oestrogen underlying the clinically recognised sex differences.
There is improved understanding of right ventricle metabolism with recent findings that right ventricular failure is characterised by reduced fatty acid oxidation in addition to increased glycolysis [87, 88, 94, 95]. Presently, it is unknown if enhanced fatty acid oxidation or improved glucose oxidation would most successfully improve right ventricular function. Finally, there is a new perception of the contribution of altered cytoskeletal function in the right ventricle. Pulmonary artery banding was used to induce right ventricular hypertrophy and dysfunction in rats, resulting in increased myocardial stiffness with increased fibrosis- and myofibril-mediated stiffness [96]. Using the monocrotaline model, Prins et al. [97] found abnormal t-tubule architecture associated with reduced junctophillin-2 expression. Colchicine ameliorated these findings, perhaps suggesting a new therapeutic avenue for the failing right ventricle. Taken together, these studies present new data on the role of the cytoskeleton and myocardial fibrosis in promoting right ventricular dysfunction.
Animal models of right ventricular failure related to group 2 PH should be developed and may require a “multiple-hit” rather than “single-hit” approach in order to more adequately reproduce the clinical syndrome [98].
Finally, insight into the pathophysiology of right ventricular function is also emerging from human right ventricle biopsies. Isolated cardiomyocytes thus obtained may allow for deep phenotyping and mechanistic investigations of specific pathways. Using this approach in patients with end-stage PAH undergoing heart–lung transplantation, and comparing with non-failing donors, Rain et al. [20] demonstrated increased fibrosis and stiffening of right ventricle sarcomeres in conjunction with decreased titin phosphorylation in PAH. A recent study by Hsu et al. [99], using right ventricle biopsies obtained during RHC under echocardiographic guidance, revealed that patients with SSc-PAH have depressed sarcomeric function, which is manifested by a significant decline in maximal force (Fmax) calcium dependence compared with non-PAH patients, while the opposite (i.e. increased calcium-activated Fmax) was demonstrated for patients with IPAH, in line with data obtained by Rain et al. [20] in (non-scleroderma) PAH patients. Collectively, these findings may explain the significant differences in outcomes and survival in these two groups of patients.
Future directions
A summary of future directions is given in supplementary figure S1b.
Our insights into the role of the right ventricle in PH have considerably improved over recent years. These insights allow for a full assessment of right ventricular function and pulmonary vascular load based on pressure and volume measurements in patients. Such an assessment permits us to test the differential effects of future drugs on the right ventricle and pulmonary vasculature. Use of CMR should be implemented in PAH clinical trials and clinical practice when possible. Molecular imaging should be used to translate from bench to bedside (and vice versa). Novel approaches are necessary to show the clinical value of echocardiography for predicting response and tailoring therapy.
Currently, the clinical relevance of an abnormal exercise pulmonary haemodynamic response is largely unknown and requires long-term follow-up studies. If PH is present, diuretics and atrioseptostomy are the only options available to treat the right ventricle, mainly by reducing wall tension [100].
Better characterisation of normal right ventricular function, early dysfunction and irreversible failure is needed. Growing insights into how to prevent right ventricular failure at a biological level need to be translated to the clinic via well-designed trials. If right ventricular failure is present, the role of inotropic agents is unknown. Based on the finding that contractile reserve of the right ventricle is absent in advanced disease, the effectiveness of inotropic drugs in end-stage right ventricular failure needs to be re-evaluated. Finally, the optimal medical care of patients in end-stage right heart failure needs to be defined.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figure S1 ERJ-01900-2018_Supplement (792.3KB, pdf)
Footnotes
Number 3 in the series “Proceedings of the 6th World Symposium on Pulmonary Hypertension” Edited by N. Galiè, V.V. McLaughlin, L.J. Rubin and G. Simonneau
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: A. Vonk Noordegraaf reports grants and speaker fees from Actelion, MSD and GSK, outside the submitted work.
Conflict of interest: K.M. Chin reports personal fees for consulting work on clinical trials from Actelion, grants (paid to institution) from Ironwood and Sonivie, personal fees for consulting work for a clinical registry from University of California San Diego, and research grants from the NIH, outside the submitted work.
Conflict of interest: F. Haddad has nothing to disclose.
Conflict of interest: P.M. Hassoun has nothing to disclose.
Conflict of interest: A.R. Hemnes reports personal fees from Actelion, Bayer, Complexa and United Therapeutics, and grants from the CMREF and NIH, outside the submitted work; and in addition has a patent issued: Annamometer (oral mechanism for detection of end-tidal CO2; not referenced in this work).
Conflict of interest: S.R. Hopkins is funded by the NIH via research grants to study the pulmonary circulation.
Conflict of interest: S.M. Kawut reports non-financial travel support from the ATS and Pulmonary Hypertension Association, grants from Actelion, United Therapeutics, Gilead, Lung Biotech, Bayer and Mallinkrodt, and grants and non-financial support from the CMREF, outside the submitted work and paid to his university; and has served in an advisory capacity (for grant review and other purposes) for United Therapeutics, Akros Pharmaceuticals, GSK and Complexa, Inc., without financial support or in-kind benefits.
Conflict of interest: D. Langleben reports grants, personal fees and non-financial support from Actelion and Bayer, personal fees from United Therapeutics and Merck, and grants from Northern Therapeutics, outside the submitted work.
Conflict of interest: J. Lumens has nothing to disclose.
Conflict of interest: R. Naeije has nothing to disclose.
References
- 1.Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37: 67–119. [DOI] [PubMed] [Google Scholar]
- 2.Vonk-Noordegraaf A, Haddad F, Chin KM, et al. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol 2013; 62: D22–D33. [DOI] [PubMed] [Google Scholar]
- 3.Spruijt OA, de Man FS, Groepenhoff H, et al. The effects of exercise on right ventricular contractility and right ventricular–arterial coupling in pulmonary hypertension. Am J Respir Crit Care Med 2015; 191: 1050–1057. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg MK, Redington AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation 2014; 129: 1033–1044. [DOI] [PubMed] [Google Scholar]
- 5.Naeije R, Badagliacca R. The overloaded right heart and ventricular interdependence. Cardiovasc Res 2017; 113: 1474–1485. [DOI] [PubMed] [Google Scholar]
- 6.Palau-Caballero G, Walmsley J, Van Empel V, et al. Why septal motion is a marker of right ventricular failure in pulmonary arterial hypertension: mechanistic analysis using a computer model. Am J Physiol Heart Circ Physiol 2017; 312: H691–H700. [DOI] [PubMed] [Google Scholar]
- 7.Haddad F, Guihaire J, Skhiri M, et al. Septal curvature is marker of hemodynamic, anatomical, and electromechanical ventricular interdependence in patients with pulmonary arterial hypertension. Echocardiography 2014; 31: 699–707. [DOI] [PubMed] [Google Scholar]
- 8.Lumens J, Arts T, Marcus JT, et al. Early-diastolic left ventricular lengthening implies pulmonary hypertension-induced right ventricular decompensation. Cardiovasc Res 2012; 96: 286–295. [DOI] [PubMed] [Google Scholar]
- 9.Marcus JT, Gan CT, Zwanenburg JJ, et al. Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol 2008; 51: 750–757. [DOI] [PubMed] [Google Scholar]
- 10.Manders E, Bogaard HJ, Handoko ML, et al. Contractile dysfunction of left ventricular cardiomyocytes in patients with pulmonary arterial hypertension. J Am Coll Cardiol 2014; 64: 28–37. [DOI] [PubMed] [Google Scholar]
- 11.Vonk Noordegraaf A, Westerhof BE, Westerhof N. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017; 69: 236–243. [DOI] [PubMed] [Google Scholar]
- 12.Suga H, Sagawa K, Shoukas AA. Load independence of the instantaneous pressure–volume ratio of the canine left ventricle and effects of epinephrine and heart rate on the ratio. Circ Res 1973; 32: 314–322. [DOI] [PubMed] [Google Scholar]
- 13.Brimioulle S, Wauthy P, Ewalenko P, et al. Single-beat estimation of right ventricular end-systolic pressure–volume relationship. Am J Physiol Heart Circ Physiol 2003; 284: H1625–H1630. [DOI] [PubMed] [Google Scholar]
- 14.Maughan WL, Shoukas AA, Sagawa K, et al. Instantaneous pressure–volume relationship of the canine right ventricle. Circ Res 1979; 44: 309–315. [DOI] [PubMed] [Google Scholar]
- 15.Westerhof N, Stergiopulos N, Noble MI, et al. Snapshots of Hemodynamics: An Aid for Clinical Research and Graduate Education. 3rd Edn. New York, Springer-Nature, 2018. [Google Scholar]
- 16.Spruijt OA, Bogaard HJ, Vonk-Noordegraaf A. Assessment of right ventricular responses to therapy in pulmonary hypertension. Drug Discov Today 2014; 19: 1246–1250. [DOI] [PubMed] [Google Scholar]
- 17.Guihaire J, Haddad F, Noly PE, et al. Right ventricular reserve in a piglet model of chronic pulmonary hypertension. Eur Respir J 2015; 45: 709–717. [DOI] [PubMed] [Google Scholar]
- 18.D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991; 115: 343–349. [DOI] [PubMed] [Google Scholar]
- 19.Vonk-Noordegraaf A, Westerhof N. Describing right ventricular function. Eur Respir J 2013; 41: 1419–1423. [DOI] [PubMed] [Google Scholar]
- 20.Rain S, Handoko ML, Trip P, et al. Right ventricular diastolic impairment in patients with pulmonary arterial hypertension. Circulation 2013; 128: 2016–2025. [DOI] [PubMed] [Google Scholar]
- 21.Trip P, Rain S, Handoko ML, et al. Clinical relevance of right ventricular diastolic stiffness in pulmonary hypertension. Eur Respir J 2015; 45: 1603–1612. [DOI] [PubMed] [Google Scholar]
- 22.Vanderpool RR, Pinsky MR, Naeije R, et al. RV–pulmonary arterial coupling predicts outcome in patients referred for pulmonary hypertension. Heart 2015; 101: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chemla D, Hebert JL, Coirault C, et al. Matching dicrotic notch and mean pulmonary artery pressures: implications for effective arterial elastance. Am J Physiol 1996; 271: H1287–H1295. [DOI] [PubMed] [Google Scholar]
- 24.Tello K, Richter MJ, Axmann J, et al. More on single-beat estimation of right ventriculo-arterial coupling in pulmonary arterial hypertension. Am J Respir Crit Care Med 2018; 2198: 816–818. [DOI] [PubMed] [Google Scholar]
- 25.Presson RG Jr, Baumgartner WA Jr, Peterson AJ, et al. Pulmonary capillaries are recruited during pulsatile flow. J Appl Physiol 2002; 92: 1183–1190. [DOI] [PubMed] [Google Scholar]
- 26.Orfanos SE, Ehrhart IC, Barman S, et al. Endothelial ectoenzyme assays estimate perfused capillary surface area in the dog lung. Microvasc Res 1997; 54: 145–155. [DOI] [PubMed] [Google Scholar]
- 27.Dupuis J, Goresky CA, Ryan JW, et al. Pulmonary angiotensin-converting enzyme substrate hydrolysis during exercise. J Appl Physiol 1992; 72: 1868–1886. [DOI] [PubMed] [Google Scholar]
- 28.Dupuis J, Goresky CA, Rouleau JL, et al. Kinetics of pulmonary uptake of serotonin during exercise in dogs. J Appl Physiol 1996; 80: 30–46. [DOI] [PubMed] [Google Scholar]
- 29.Toivonen HJ, Catravas JD. Effects of blood flow on lung ACE kinetics: evidence for microvascular recruitment. J Appl Physiol 1991; 71: 2244–2254. [DOI] [PubMed] [Google Scholar]
- 30.Wagner WW Jr, Latham LP, Hanson WL, et al. Vertical gradient of pulmonary capillary transit times. J Appl Physiol 1986; 61: 1270–1274. [DOI] [PubMed] [Google Scholar]
- 31.Langleben D, Orfanos SE, Giovinazzo M, et al. Pulmonary capillary endothelial metabolic dysfunction: severity in pulmonary arterial hypertension related to connective tissue disease versus idiopathic pulmonary arterial hypertension. Arthritis Rheum 2008; 58: 1156–1164. [DOI] [PubMed] [Google Scholar]
- 32.Asadi AK, Cronin MV, Sa RC, et al. Spatial-temporal dynamics of pulmonary blood flow in the healthy human lung in response to altered FIO2. J Appl Physiol 2013; 114: 107–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asadi AK, Sa RC, Kim NH, et al. Inhaled nitric oxide alters the distribution of blood flow in the healthy human lung, suggesting active hypoxic pulmonary vasoconstriction in normoxia. J Appl Physiol 2015; 118: 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langleben D, Orfanos SE, Giovinazzo M, et al. Acute vasodilator responsiveness and microvascular recruitment in idiopathic pulmonary arterial hypertension. Ann Intern Med 2015; 162: 154–156. [DOI] [PubMed] [Google Scholar]
- 35.Linehan JH, Haworth ST, Nelin LD, et al. A simple distensible vessel model for interpreting pulmonary vascular pressure–flow curves. J Appl Physiol 1992; 73: 987–994. [DOI] [PubMed] [Google Scholar]
- 36.Reeves JT, Linehan JH, Stenmark KR. Distensibility of the normal human lung circulation during exercise. Am J Physiol Lung Cell Mol Physiol 2005; 288: L419–L425. [DOI] [PubMed] [Google Scholar]
- 37.Naeije R, Vanderpool R, Dhakal BP, et al. Exercise-induced pulmonary hypertension: physiological basis and methodological concerns. Am J Respir Crit Care Med 2013; 187: 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stergiopulos N, Segers P, Westerhof N. Use of pulse pressure method for estimating total arterial compliance in vivo. Am J Physiol 1999; 276: H424–H428. [DOI] [PubMed] [Google Scholar]
- 39.Chemla D, Hebert JL, Coirault C, et al. Total arterial compliance estimated by stroke volume-to-aortic pulse pressure ratio in humans. Am J Physiol 1998; 274: H500–H505. [DOI] [PubMed] [Google Scholar]
- 40.Segers P, Brimioulle S, Stergiopulos N, et al. Pulmonary arterial compliance in dogs and pigs: the three-element windkessel model revisited. Am J Physiol 1999; 277: H725–H731. [DOI] [PubMed] [Google Scholar]
- 41.Lankhaar JW, Westerhof N, Faes TJ, et al. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol 2006; 291: H1731–H1737. [DOI] [PubMed] [Google Scholar]
- 42.Saouti N, Westerhof N, Helderman F, et al. RC time constant of single lung equals that of both lungs together: a study in chronic thromboembolic pulmonary hypertension. Am J Physiol Heart Circ Physiol 2009; 297: H2154–H2160. [DOI] [PubMed] [Google Scholar]
- 43.Saouti N, Westerhof N, Postmus PE, et al. The arterial load in pulmonary hypertension. Eur Respir Rev 2010; 19: 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reuben SR. Compliance of the human pulmonary arterial system in disease. Circ Res 1971; 29: 40–50. [DOI] [PubMed] [Google Scholar]
- 45.Lankhaar JW, Westerhof N, Faes TJ, et al. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J 2008; 29: 1688–1695. [DOI] [PubMed] [Google Scholar]
- 46.Bonderman D, Martischnig AM, Vonbank K, et al. Right ventricular load at exercise is a cause of persistent exercise limitation in patients with normal resting pulmonary vascular resistance after pulmonary endarterectomy. Chest 2011; 139: 122–127. [DOI] [PubMed] [Google Scholar]
- 47.Metkus TS, Mullin CJ, Grandin EW, et al. Heart rate dependence of the pulmonary resistance × compliance (RC) time and impact on right ventricular load. PLoS One 2016; 11: e0166463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tedford RJ, Hassoun PM, Mathai SC, et al. Pulmonary capillary wedge pressure augments right ventricular pulsatile loading. Circulation 2012; 125: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghio S, D'Alto M, Badagliacca R, et al. Prognostic relevance of pulmonary arterial compliance after therapy initiation or escalation in patients with pulmonary arterial hypertension. Int J Cardiol 2017; 230: 53–58. [DOI] [PubMed] [Google Scholar]
- 50.Mahapatra S, Nishimura RA, Oh JK, et al. The prognostic value of pulmonary vascular capacitance determined by Doppler echocardiography in patients with pulmonary arterial hypertension. J Am Soc Echocardiogr 2006; 19: 1045–1050. [DOI] [PubMed] [Google Scholar]
- 51.Douwes JM, Roofthooft MT, Bartelds B, et al. Pulsatile haemodynamic parameters are predictors of survival in paediatric pulmonary arterial hypertension. Int J Cardiol 2013; 168: 1370–1377. [DOI] [PubMed] [Google Scholar]
- 52.Campo A, Mathai SC, Le Pavec J, et al. Hemodynamic predictors of survival in scleroderma-related pulmonary arterial hypertension. Am J Respir Crit Care Med 2010; 182: 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pellegrini P, Rossi A, Pasotti M, et al. Prognostic relevance of pulmonary arterial compliance in patients with chronic heart failure. Chest 2014; 145: 1064–1070. [DOI] [PubMed] [Google Scholar]
- 54.Al-Naamani N, Preston IR, Paulus JK, et al. Pulmonary arterial capacitance is an important predictor of mortality in heart failure with a preserved ejection fraction. JACC Heart Fail 2015; 3: 467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chemla D, Castelain V, Humbert M, et al. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. Chest 2004; 126: 1313–1317. [DOI] [PubMed] [Google Scholar]
- 56.Amsallem M, Sternbach JM, Adigopula S, et al. Addressing the controversy of estimating pulmonary arterial pressure by echocardiography. J Am Soc Echocardiogr 2016; 29: 93–102. [DOI] [PubMed] [Google Scholar]
- 57.Handoko ML, De Man FS, Oosterveer FP, et al. A critical appraisal of transpulmonary and diastolic pressure gradients. Physiol Rep 2016; 4: e12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D'Alto M, Romeo E, Argiento P, et al. Accuracy and precision of echocardiography versus right heart catheterization for the assessment of pulmonary hypertension. Int J Cardiol 2013; 168: 4058–4062. [DOI] [PubMed] [Google Scholar]
- 59.Galiè N, Palazzini M, Manes A. Pulmonary arterial hypertension: from the kingdom of the near-dead to multiple clinical trial meta-analyses. Eur Heart J 2010; 31: 2080–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brewis MJ, Bellofiore A, Vanderpool RR, et al. Imaging right ventricular function to predict outcome in pulmonary arterial hypertension. Int J Cardiol 2016; 218: 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van de Veerdonk MC, Kind T, Marcus JT, et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011; 58: 2511–2519. [DOI] [PubMed] [Google Scholar]
- 62.Vanderpool RR, Rischard F, Naeije R, et al. Simple functional imaging of the right ventricle in pulmonary hypertension: can right ventricular ejection fraction be improved? Int J Cardiol 2016; 223: 93–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuehne T, Yilmaz S, Steendijk P, et al. Magnetic resonance imaging analysis of right ventricular pressure–volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation 2004; 110: 2010–2016. [DOI] [PubMed] [Google Scholar]
- 64.de Man FS, Handoko ML, van Ballegoij JJ, et al. Bisoprolol delays progression towards right heart failure in experimental pulmonary hypertension. Circ Heart Fail 2012; 5: 97–105. [DOI] [PubMed] [Google Scholar]
- 65.Wauthy P, Naeije R, Brimioulle S. Left and right ventriculo-arterial coupling in a patient with congenitally corrected transposition. Cardiol Young 2005; 15: 647–649. [DOI] [PubMed] [Google Scholar]
- 66.Tedford RJ, Mudd JO, Girgis RE, et al. Right ventricular dysfunction in systemic sclerosis-associated pulmonary arterial hypertension. Circ Heart Fail 2013; 6: 953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCabe C, White PA, Hoole SP, et al. Right ventricular dysfunction in chronic thromboembolic obstruction of the pulmonary artery: a pressure–volume study using the conductance catheter. J Appl Physiol 2014; 116: 355–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hsu S, Houston BA, Tampakakis E, et al. Right ventricular functional reserve in pulmonary arterial hypertension. Circulation 2016; 133: 2413–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kovacs G, Herve P, Barbera JA, et al. An official European Respiratory Society statement: pulmonary haemodynamics during exercise. Eur Respir J 2017; 50: 1700578. [DOI] [PubMed] [Google Scholar]
- 70.Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2015; 46: 903–975. [DOI] [PubMed] [Google Scholar]
- 71.Lewis GD, Bossone E, Naeije R, et al. Pulmonary vascular hemodynamic response to exercise in cardiopulmonary diseases. Circulation 2013; 128: 1470–1479. [DOI] [PubMed] [Google Scholar]
- 72.Herve P, Lau EM, Sitbon O, et al. Criteria for diagnosis of exercise pulmonary hypertension. Eur Respir J 2015; 46: 728–737. [DOI] [PubMed] [Google Scholar]
- 73.Oliveira RK, Agarwal M, Tracy JA, et al. Age-related upper limits of normal for maximum upright exercise pulmonary haemodynamics. Eur Respir J 2016; 47: 1179–1188. [DOI] [PubMed] [Google Scholar]
- 74.Borlaug BA, Nishimura RA, Sorajja P, et al. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail 2010; 3: 588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andersen MJ, Ersboll M, Bro-Jeppesen J, et al. Exercise hemodynamics in patients with and without diastolic dysfunction and preserved ejection fraction after myocardial infarction. Circ Heart Fail 2012; 5: 444–451. [DOI] [PubMed] [Google Scholar]
- 76.Borlaug BA. Invasive assessment of pulmonary hypertension: time for a more fluid approach? Circ Heart Fail 2014; 7: 2–4. [DOI] [PubMed] [Google Scholar]
- 77.Fujimoto N, Borlaug BA, Lewis GD, et al. Hemodynamic responses to rapid saline loading: the impact of age, sex, and heart failure. Circulation 2013; 127: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fox BD, Shimony A, Langleben D, et al. High prevalence of occult left heart disease in scleroderma-pulmonary hypertension. Eur Respir J 2013; 42: 1083–1091. [DOI] [PubMed] [Google Scholar]
- 79.Robbins IM, Hemnes AR, Pugh ME, et al. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circ Heart Fail 2014; 7: 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.D'Alto M, Romeo E, Argiento P, et al. Clinical relevance of fluid challenge in patients evaluated for pulmonary hypertension. Chest 2017; 151: 119–126. [DOI] [PubMed] [Google Scholar]
- 81.Andersen MJ, Olson TP, Melenovsky V, et al. Differential hemodynamic effects of exercise and volume expansion in people with and without heart failure. Circ Heart Fail 2015; 8: 41–48. [DOI] [PubMed] [Google Scholar]
- 82.Vonk Noordegraaf A, Haddad F, Bogaard HJ, et al. Noninvasive imaging in the assessment of the cardiopulmonary vascular unit. Circulation 2015; 131: 899–913. [DOI] [PubMed] [Google Scholar]
- 83.Fine NM, Chen L, Bastiansen PM, et al. Outcome prediction by quantitative right ventricular function assessment in 575 subjects evaluated for pulmonary hypertension. Circ Cardiovasc Imaging 2013; 6: 711–721. [DOI] [PubMed] [Google Scholar]
- 84.Amsallem M, Sweatt AJ, Aymami MC, et al. Right heart end-systolic remodeling index strongly predicts outcomes in pulmonary arterial hypertension: comparison with validated models. Circ Cardiovasc Imaging 2017; 10: e005771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guihaire J, Bogaard HJ, Flecher E, et al. Experimental models of right heart failure: a window for translational research in pulmonary hypertension. Semin Respir Crit Care Med 2013; 34: 689–699. [DOI] [PubMed] [Google Scholar]
- 86.van der Bruggen CE, Happe CM, Dorfmuller P, et al. Bone morphogenetic protein receptor type 2 mutation in pulmonary arterial hypertension: a view on the right ventricle. Circulation 2016; 133: 1747–1760. [DOI] [PubMed] [Google Scholar]
- 87.Brittain EL, Talati M, Fessel JP, et al. Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension. Circulation 2016; 133: 1936–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Talati MH, Brittain EL, Fessel JP, et al. Mechanisms of lipid accumulation in the bone morphogenetic protein receptor type 2 mutant right ventricle. Am J Respir Crit Care Med 2016; 194: 719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hemnes AR, Brittain EL, Trammell AW, et al. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 2014; 189: 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Potus F, Ruffenach G, Dahou A, et al. Downregulation of microRNA-126 contributes to the failing right ventricle in pulmonary arterial hypertension. Circulation 2015; 132: 932–943. [DOI] [PubMed] [Google Scholar]
- 91.Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010; 122: 164–172. [DOI] [PubMed] [Google Scholar]
- 92.Jacobs W, van de Veerdonk MC, Trip P, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014; 145: 1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frump AL, Goss KN, Vayl A, et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension: effects of endogenous and exogenous sex hormones. Am J Physiol Lung Cell Mol Physiol 2015; 308: L873–L890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Graham BB, Kumar R, Mickael C, et al. Severe pulmonary hypertension is associated with altered right ventricle metabolic substrate uptake. Am J Physiol Lung Cell Mol Physiol 2015; 309: L435–L440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Archer SL, Fang YH, Ryan JJ, et al. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ 2013; 3: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rain S, Andersen S, Najafi A, et al. Right ventricular myocardial stiffness in experimental pulmonary arterial hypertension: relative contribution of fibrosis and myofibril stiffness. Circ Heart Fail 2016; 9: e002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Prins KW, Tian L, Wu D, et al. Colchicine depolymerizes microtubules, increases junctophilin-2, and improves right ventricular function in experimental pulmonary arterial hypertension. J Am Heart Assoc 2017; 6: e006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Meng Q, Lai YC, Kelly NJ, et al. Development of a mouse model of metabolic syndrome, pulmonary hypertension, and heart failure with preserved ejection fraction. Am J Respir Cell Mol Biol 2017; 56: 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hsu S, Kokkonen-Simon KM, Kirk JA, et al. Right ventricular myofilament functional differences in humans with systemic sclerosis-associated versus idiopathic pulmonary arterial hypertension. Circulation 2018; 137: 2360–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Westerhof BE, Saouti N, van der Laarse WJ, et al. Treatment strategies for the right heart in pulmonary hypertension. Cardiovasc Res 2017; 113: 1465–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary figure S1 ERJ-01900-2018_Supplement (792.3KB, pdf)