

Abstract
Increasing evidence indicates that pattern recognition receptors (PRRs) are involved in neuropathic pain after peripheral nerve injury (PNI). While a significant number of studies support an association between neuropathic pain and the innate immune response mediated through Toll-like receptors, a family of PRRs, the roles of other types of PRRs are largely unknown. In this study, we have focused on the macrophage-inducible C-type lectin (Mincle), a PRR allocated to the C-type lectin receptor family. Here, we show that Mincle is involved in neuropathic pain after PNI. Mincle-deficient mice showed impaired PNI-induced mechanical allodynia. After PNI, expression of Mincle mRNA was rapidly increased in the injured spinal nerve. Most Mincle-expressing cells were identified as infiltrating leucocytes, although the migration of leucocytes was also observed in Mincle-deficient mice. Furthermore, Mincle-deficiency affected the induction of genes, which are reported to contribute to neuropathic pain after PNI in the dorsal root ganglia and spinal dorsal horn. These results suggest that Mincle is involved in triggering sequential processes that lead to the pathogenesis of neuropathic pain.
Introduction
Neuropathic pain results from damage to the peripheral nervous system (PNS) and/or the central nervous system (CNS), and is characterized by allodynia (pain evoked by a normally innocuous stimulus) and hyperalgesia (enhanced pain evoked by a noxious stimulus). Neuropathic pain remains a persistent clinical problem because of an incomplete understanding of the complex pathogenesis. Among such complex mechanisms underlying neuropathic pain, the neuroimmune interface has been reported to play an important role in inducing these hypersensitivities1–3. After peripheral nerve injury (PNI), various mediators such as cytokines, chemokines, and neurotransmitters, released from the damaged nerves, alter the responsiveness of various immune cells in the PNS and CNS4. Various immune cells are recruited to the injury site. These cells release numerous immune molecules such as cytokines and chemokines that can directly alter the nociceptive information4. Recently, the pattern recognition receptors (PRRs) have been receiving attention as molecules that induce pathological changes in chronic pain5–7. PRRs were originally classified as the types of receptors that sense exogenous pathogen-associated molecular patterns (PAMPs) and induce an innate immune response8. Now it is evident that endogenous molecules released from dying or damaged cells, referred to as damage-associated molecular patterns (DAMPs), also activate the PRRs during tissue injuries9. The ability of PRRs to be activated by DAMPs is particularly interesting because cells expressing PRRs can be activated in injured tissues and then can trigger reactions that lead to neuropathic pain under sterile conditions. Toll-like receptors (TLRs) are a well-studied family of PRRs and have been reported to play a significant role in innate immunity8. Notably, TLRs are expressed by numerous cell types in the CNS and PNS including both neuronal and non-neuronal cells such as the microglia (the resident macrophages in the CNS)10,11 and contribute to the induction and maintenance of neuropathic pain conditions6,7. While evidence regarding the roles of TLRs in neuropathic pain is accumulating, those of another type of PRRs, consisting of the C-type lectin receptors (CLRs), have not yet been investigated. Macrophage-inducible C-type lectin (Mincle), one of the CLRs, is expressed mainly in the myeloid cells and is induced after exposure to various stimuli and stress12. Mincle is known to recognize not only the PAMPs derived from mycobacterium and fungus13,14, but also the DAMPs derived from dead cells15,16, to induce inflammatory responses. In rodent models of cerebrovascular disorders, Mincle is reported to be expressed in microglia-like cells within damaged brain areas, and contributes to innate immune responses17,18. Therefore, we hypothesized that Mincle activation by DAMPs in damaged nerves could trigger the innate immune response and the following inflammatory events that would result in induction of neuropathic pain after PNI. Here, we show for the first time that Mincle is involved in pathological processes in neuropathic pain. Mincle-deficient mice showed impaired development of mechanical allodynia after PNI. Expression of Mincle mRNA was rapidly increased in the injured spinal nerve (SN) after PNI and most Mincle-expressing cells were identified as infiltrating neutrophils but not the dorsal root ganglion (DRG) neurons. Furthermore, the increase in Mincle mRNA expression and neutrophil migration in injured SN after PNI was found to depend on myeloid differentiation primary response gene 88 (MyD88), one of the major adaptor proteins of the TLR signalling pathways. Our results suggest that Mincle in the injured nerve induces neuropathic pain by triggering sequential processes that lead to tactile allodynia in the DRG and spinal dorsal horn.
Results
Mincle contributes to the mechanical hypersensitivity after peripheral nerve injury
Mice lacking the Mincle gene (Mincle−/−) appeared healthy and showed no obvious abnormality on visual inspection14. Before assessing pain behaviour, we examined the locomotor activity and anxiety of Mincle−/− mice because these factors are known to influence the reliability of assessment of pain behaviours. The open-field test was used to assess anxiety-like behaviour and locomotor activity (Supplementary Fig. S1a,b). The locomotor activity was also evaluated using rotarod (Supplementary Fig. S1c). There were no significant differences between wild-type (WT) and Mincle−/− mice in any of the parameters, based on data obtained from the open-field and rotarod tests. We concluded that Mincle−/− mice did not possess considerable dysfunction in locomotor activity and anxiety-like behaviours.
Next, we investigated whether Mincle contributes to the mechanical hypersensitivity after PNI. Basal mechanical sensitivity of Mincle−/− mice was not different from that of WT mice (Fig. 1a,b). PNI induced mechanical allodynia in the ipsilateral hind paw of WT mice as previously reported19. However, Mincle−/− mice exhibited attenuation of mechanical allodynia in the ipsilateral hind paw, and the paw withdrawal thresholds (PWTs) of these mice were significantly higher than those of WT mice (Fig. 1a). The contralateral PWTs after PNI remained unchanged in Mincle−/− as well as WT mice (Fig. 1b). We also evaluated the pain behaviour of Mincle−/− mice by using the formalin test, a different type of pain model. Pain responses (licking and biting) in both the first and second phases after formalin injection were indistinguishable between WT mice and Mincle−/− mice (Fig. 1c,d). These results indicate that Mincle is required for the development of tactile allodynia after PNI but not pain responses induced by the injection of formalin.
Figure 1.

Mincle deficiency attenuates mechanical allodynia after peripheral nerve injury. Fifty percent paw withdrawal threshold (PWT) in ipsi- (a) or contra- (b) lateral side of WT and Mincle−/− mice before (pre) and after PNI (n = 7 in each group, repeated ANOVA followed by Bonferroni post hoc test, *P < 0.05, **P < 0.01, ***P < 0.001 vs. the ipsilateral side of WT mice). (c) The duration (sec) of nociceptive behaviours after formalin injection (n = 7 in each group, MANOVA). (d) Total durations (sec) of nociceptive behaviours for the first phase (0–10 min) and for the second phase (10–60 min) in the formalin test (n = 7 in each group, Student’s t test).
PNI induces expression of Mincle mRNA in injured spinal nerves
Next, we tried to identify tissue(s) and/or cell type(s) expressing Mincle after PNI. First, we assumed that Mincle would be expressed in spinal microglia because: (1) it has been well established that spinal microglia are activated and show massive proliferation after PNI, and these changes are important in the pathogenesis of nerve injury-induced pain hypersensitivity20–24; (2) in these reports, TLRs expressed in the microglia contribute to neuropathic pain pathology1,5,6,20. Then, we tested the expression of Mincle mRNA in the spinal dorsal horn of WT mice before and after PNI using real-time PCR. However, Mincle mRNA was found to be too low to be detected and remained at undetectable levels even after PNI, unexpectedly. As a positive control of microglial gene expression, mRNA encoding ionized calcium-binding adaptor molecule-1 (Iba1), a microglial marker, was also examined by the same method. The expression of Iba1 mRNA was detected to be substantial even before PNI and increased after PNI, as expected (Supplementary Fig. S2a). These results indicate that Mincle expression in spinal microglia seems to be largely limited and not to be induced after PNI.
Next, we examined Mincle mRNA expression in the DRG tissue, as a number of studies have reported that the expression levels of various genes are changed after PNI in the DRG and that the changes are thought to markedly contribute to the generation of mechanical allodynia3,25–27. The DRG tissue that we used for the initial experiments included the (injured) SN (Fig. 2a). The expression of Mincle mRNA was barely detectable in the naïve mice. However, once the SN was transected, a rapid and marked increase of Mincle mRNA was observed in the ipsilateral but not the contralateral DRG tissue (Fig. 2b). To identify the part of the tissue containing Mincle mRNA, we then divided the DRG tissue into the DRG itself and SN (Fig. 2a). A remarkable increase in Mincle mRNA was observed in the SNs (Fig. 2c) with virtually the same time course as observed in the DRG tissues (Fig. 2b), while only a slight increase was observed in the DRGs. These results indicate that Mincle is expressed in cells in the injured SN, including immune cells, such as the neutrophils, which are known to infiltrate the injured SN after PNI, or the SN resident cells such as the Schwann cells.
Figure 2.
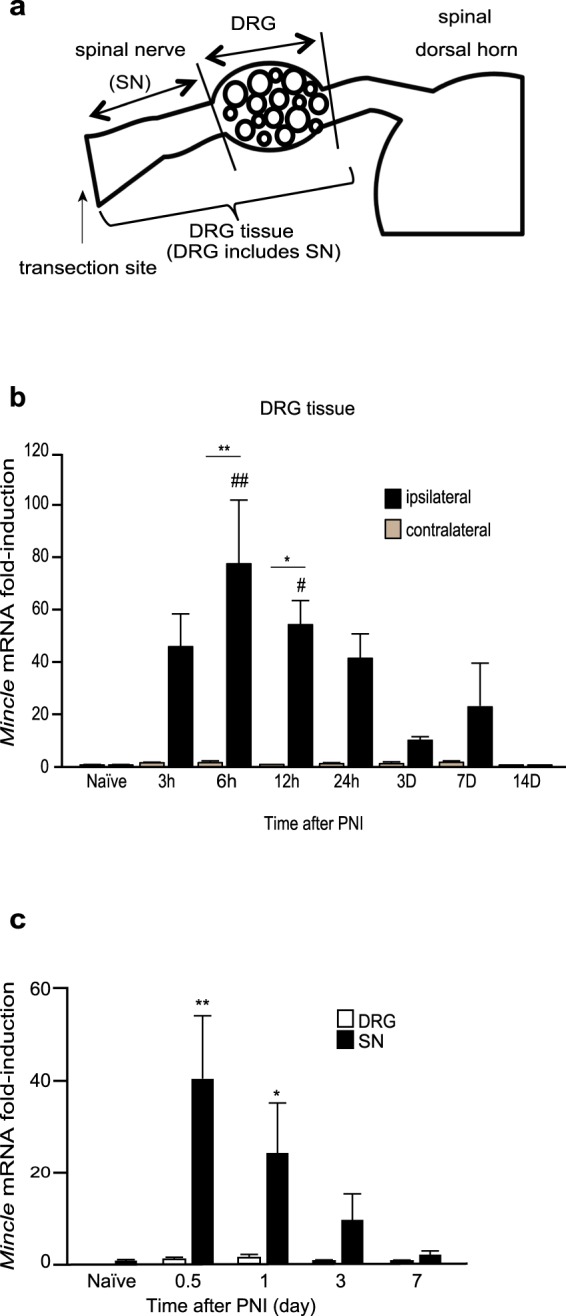
PNI induces expression of Mincle mRNA in injured spinal nerve. (a) Schematic representation of the tissue used for real-time PCR analysis. (b,c) The time course of Mincle mRNA expression before (naïve) and after PNI in WT mice. The expression level of Mincle gene was normalized to that of Gapdh gene. (b) Total RNA was obtained from the DRG tissue, which includes the SN (a). Data are presented as fold-induction compared to naïve contralateral side (n = 4–5 in each group, two-way ANOVA followed by Tukey’s post hoc test, #P < 0.05, ##P < 0.01 vs. naïve, *P < 0.05, **P < 0.01 vs. contralateral side). (c) Total RNA was obtained from the SN or DRG (a). Bar graphs show fold-change relative to the naïve SN (n = 3–7 in each group, one-way ANOVA followed by Dunnett post hoc test, *P < 0.05, **P < 0.01 vs. naïve controls). Values are mean ± SEM.
Mincle mRNA is expressed in the cells infiltrating the injured nerve
Previous studies have shown that neutrophils are the predominant immune cells migrating to the injured tissue within hours of injury, reaching a peak at 24 h3,28. The recruitment and activation of resident macrophages and invasion of monocytes from the peripheral blood follow neutrophil migration29,30. Haematoxylin-stained sections from the injured DRG and SN were observed to assess whether infiltrating cells were found in these tissues. Consistent with a previous report28, a large number of infiltrating cells were found in the ipsilateral SN 12 h after PNI (Supplementary Fig. S3a). Most of these cells seemed to be neutrophils because they had a lobular nucleus. Because Mincle is reported to be expressed in activated phagocytes and neutrophils12,14,15, we assumed that the possible source of induced Mincle mRNA expression observed in the injured SN was the neutrophils. To test this assumption, a mixture of Ly6G- (neutrophil marker) and/or F4/80- (monocytes/macrophages marker) positive cells were depleted from SN samples collected 12 h after nerve injury by using the columns designed for depletion of target cells (Fig. 3a,b). The real-time PCR analysis in each sample (contralateral, ipsilateral total, ipsilateral dpleted fraction, and trapped fraction) revealed that Mincle mRNA was virtually eliminated in the depleted fraction as expected, while it was preserved in the trapped fraction (Fig. 3c). The result indicated that the major source of Mincle mRNA 12 h after PNI was Ly6G- and/or F4/80-positive cells.
Figure 3.

Elimination of Mincle mRNA by depletion of neutrophils and macrophages/monocytes from injured SN cells. (a) Representative flow cytometry dot plots of Ly6G+ and/or F4/80+ cells and (b) the proportions of these cells in samples of contralateral total, ipsilateral total, ipsilateral depleted fraction, and ipsilateral trapped fraction cells 12 h after PNI. (a) Lower-right (red frame) and upper-left (blue frame) quadrant of the panels representing neutrophils (Ly6G+) and macrophages/monocytes (F4/80+), respectively. Note that neutrophils and macrophages/monocytes were adequately removed from the ipsilateral depleted fraction (lower-left panel in a and lower panels in b). (c) The Mincle mRNA expression in each sample. The expression level of Mincle gene was normalized to that of Gapdh gene. PCR amplifications were done in triplicate. Data are representative of two independent experiments. Note that Mincle mRNA expression in the ipsilateral depleted fraction was virtually eliminated.
The localization of Mincle mRNA in the DRG tissue (including the SN) was further examined using in situ hybridization histochemistry (ISHH). A large number of Mincle mRNA-expressing cells were observed in the ipsilateral SN, especially at the transection site, but not in the contralateral SN 12 h after PNI (Fig. 4a,b). Three days after PNI, these cells were still observed in the ipsilateral SN (Fig. 4c), although they were decreased in number. Unlike the ipsilateral SN, Mincle mRNA-expressing cells were scarcely found in the ipsilateral DRG at any of the time points observed. These results were consistent with our results obtained using real-time PCR. To determine the cell types expressing Mincle after PNI, we combined ISHH for Mincle mRNA with immunohistochemistry (IHC) using the cell-type-specific markers. Some of the Mincle mRNA signals were found to be colocalised with Ly6G (Fig. 4d) or F4/80 (Fig. 4e) 12 h or 3 days after PNI, respectively. Since Mincle mRNA-expressing cells were preferentially located in the epineurium (Supplementary Fig. S4), densities of these cells were quantified separately in the epineurium or nerve. The cell density of Mincle mRNA-expressing cells in the epineurium was much higher (7.38/104 μm2) than that in the nerve (0.47/104 μm2) 12 h after PNI. Majority (70.1%) of these in the epineurium and more than half (54.8%) of these in the nerve showed Ly6G-immunoreactivity. In total, 67.4% of these cells were identified as neutrophils (Supplementary Table S1). The cell density of Mincle mRNA-expressing cells in the epineurium was still higher (1.46/104 μm2) than that in the nerve (0.76/104 μm2) 3 days after PNI, however, it was much lower than that 12 h after PNI. Among these cells, 31.4% of these in the epineurium and 45.4% of these in nerve showed F4/80-immunoreactivity. In total, 41.8% of these cells were identified as monocytes/macrophages (Supplementary Table S2). These results indicate that the neutrophils and monocytes/macrophages express Mincle mRNA after PNI. In addition, Mincle mRNA-expressing cells that showed neither Ly6G nor F4/80 immunoreactivities were also found. Although we did not perform further analysis, these cells may be Schwann cells as judged from their morphology.
Figure 4.

PNI up-regulates Mincle mRNA at the transection site. (a–c) Dark field and bright field images of the same area of ISHH revealed mRNA distribution of Mincle mRNA after PNI in the L4 DRG and SN, 12 h after PNI, contralateral (a), ipsilateral (b), and 3 days after PNI, ipsilateral site (c). Arrowheads indicate positive cells. Scale bar: Dark field and bright field images, 400 μm, transection site, 80 μm. Counterstained with haematoxylin. (d,e) Bright-field photomicrographs of combined ISHH for Mincle mRNA with immunostaining with Ly6G or F4/80 in the ipsilateral SN. Arrowheads indicate cells single-labelled with ISHH (aggregation of grains). Arrows indicate cells double-labelled with ISHH (aggregation of grains) and IHC (brown staining). Counterstained with haematoxylin. Scale bar: 20 μm.
Leucocyte infiltration in injured SN is comparable between WT and Mincle−/− mice
As described above, Mincle was expressed in leucocytes, which infiltrated the injured SN after PNI. Because Mincle is reported to trigger neutrophil infiltration into the injured tissue15, we investigated whether Mincle-deficiency would affect migration of immune cells to the injured SN. First, we attempted to analyse the infiltrating cells in a haematoxylin-stained section of the injured DRG tissue (that included the SN) to compare between WT and Mincle−/− mice. Although a large number of infiltrating cells were observed in the injured SN (Supplementary Fig. S3a,b), we found it difficult to quantify the number of infiltrating cells by this method, because haematoxylin staining posed limitations with cell identification and it was difficult to obtain sections of uniform size. To solve these problems, we employed the flow cytometry as a rapid and reliable screening method for the characterization and quantification of migrating leucocytes to the injured tissue after PNI. We focused on the neutrophils on day 1 and monocytes/macrophages on day 3 after PNI, based on the observation of these cells in our ISHH experiment. Consistent with previous reports28–30, migration of neutrophils (CD11b+Ly6G+) or monocytes/macrophages (CD11b+F4/80+) were observed in the injured but not uninjured SN of WT mice 1 day and 3 days after PNI, respectively (Fig. 5a,c). Although the proportions were smaller than in the SN, these cells were also observed to migrate to the ipsilateral but not contralateral DRG (Fig. 5b,d). Unlike in a previous report15, infiltrating cells were also observed in the ipsilateral SN and DRG of Mincle−/− mice, and the proportions of leucocytes were comparable between WT and Mincle−/− mice. These results indicate that Mincle is not involved in leucocyte migration to the injured tissue in this model.
Figure 5.

Normal leucocyte infiltration at the injured site of Mincle−/− mice. Representative flow cytometry dot plot of CD11b+Ly6G+ cells in the SN (a) and DRG (b) 1 day after PNI. Upper-right quadrant (red frame) of the left panels representing infiltrating neutrophils (CD11b+Ly6G+). The bar graph (right) summarises the proportions of neutrophils in the ipsilateral or contralateral tissues of WT or Mincle−/− mice (n = 5 in each group, one-way ANOVA followed by Tukey’s post hoc test, ***P < 0.001 vs. contralateral side). Representative flow cytometry dot plot of CD11b+F4/80+ cells in the SN (c) and DRG (d) 3 days after PNI. Upper-right quadrant (red frame) of the left panels representing infiltrating monocytes/macrophages (CD11b+F4/80+). The bar graph (right) summarises the proportions of monocytes/macrophages in the ipsilateral or contralateral tissues of WT or Mincle−/− mice (n = 4 in each group, one-way ANOVA followed by Tukey’s post hoc test, **P < 0.01 vs. contralateral side). Values are mean ± SEM.
Mincle affects the induction of neuropathic pain-related genes in the DRG and spinal dorsal horn after PNI
Previous reports have shown that inflammatory cytokines (IL-1β, IL-6, and TNF-α) are released from activated immune cells, and are involved in the pathogenesis of neuropathic pain2,26,27,31. In the DRG, neurons, satellite cells, and resident macrophages are reported to be activated and to modulate neuronal activity after PNI3,25. Therefore, we investigated the temporal expression of these cytokines in the DRG and SN after PNI using real-time PCR. In the SNs, the expression of these cytokines in Mincle−/− mice was comparable to that in WT mice (Fig. 6a). In the DRGs, however, induction of Tnfa mRNA was significantly suppressed in Mincle−/− mice at day 1 after PNI (Fig. 6b). These results suggest that after PNI, reactions induced by Mincle activation in the SN affect the expression of TNF-α in the DRG.
Figure 6.

PNI-induced Tnfa expression in the injured DRG is reduced in Mincle−/− mice. Total RNA was isolated from the SN (a) and DRG (b) from WT and Mincle−/− mice before (naïve) and after PNI. Gene expression was measured using real-time PCR and presented as fold-induction compared to naïve WT mice (n = 3–6 in each group, two-way ANOVA followed by Tukey’s post hoc test, **P < 0.01 vs. WT mice, #P < 0.05, ##P < 0.01, ###P < 0.001 vs. naïve controls). Values are mean ± SEM.
As described above, the importance of microglial proliferation and activation in neuropathic pain after PNI is well established20–22. Because PNI failed to induce mechanical allodynia in Mincle−/− mice, we tested the possibility that Mincle-deficiency might impair microglial proliferation in the ipsilateral dorsal horn after PNI. As reported previously21,23, Iba1-immuno-reactivity revealed massive proliferation of spinal microglia in the ipsilateral dorsal horn of WT mice 7 days after PNI (Fig. 7a). Surprisingly, proliferation of spinal microglia was observed to a similar degree in Mincle−/− mice (Fig. 7a,b). This discrepancy between microglial proliferation and reduction of mechanical allodynia has been also described in previous reports of neuropathic pain models in mice lacking P2Y12R22, interferon regulatory factor-5 (IRF5)23, and DNAX-activating protein of 12 kDa (DAP12)24,32. Therefore, we assumed that Mincle-deficiency might negatively affect complete activation of spinal microglia. To test this idea, we quantified the expression of inflammatory cytokines (Il1b, Il6, and Tnfa) using real-time PCR, since these cytokines have been reported to be expressed by activated microglia1,20,23. Unexpectedly, expression of these inflammatory cytokines in Mincle−/− mice was comparable to that in WT mice (Fig. 7c). We also examined the expression of Irf5, because this gene was reported to be expressed in microglia and to be involved in neuropathic pain21,23,33. Interestingly, real-time PCR analysis revealed that the induction of Irf5 mRNA expression was significantly suppressed in Mincle−/− mice 3 days after PNI (Fig. 7d). These results suggest that after PNI, reactions induced by Mincle activation in the SN affect the expression of IRF5 in the spinal dorsal horn.
Figure 7.

Mincle deficiency inhibits complete activation of spinal microglia after PNI. (a) Representative images showing Iba1-immunoreactivity in the L4 spinal dorsal horn of WT and Mincle−/− mice 7 days after PNI (Scale bar: 100 μm). (b) Number of Iba1-immunoreactive cells counted in the ipsilateral and contralateral dorsal horn of WT and Mincle−/− mice (one-way ANOVA followed by Tukey’s post hoc test, ##P < 0.01 vs. contralateral side). Real-time PCR analysis of mRNA of proinflammatory cytokines (c) and neuropathic pain-related gene (d) in the ipsilateral spinal dorsal horn of WT and Mincle−/− mice before (naïve) and after PNI. Bar graphs show fold-change compared to naïve WT mice (n = 3–7 in each group, one-way ANOVA followed by Tukey’s post hoc test. **P < 0.01 vs. WT mice, #P < 0.05, ##P < 0.01 vs. naïve controls). Values are mean ± SEM.
Increase of Mincle mRNA in injured SN depends on MyD88 in neuropathic pain model
It has been reported that the TLR signalling pathways are known to contribute to Mincle expression12,34. The MyD88 protein is one of the major adaptor proteins that mediate the TLR signalling pathways8, and it is also known to mediate Mincle expression35. On the other hand, previous studies have reported that MyD88−/− mice showed reduced mechanical allodynia in the neuropathic pain model36 and MyD88 in nociceptors contributes to the infiltration of innate and adaptive immune cells into injured DRG after PNI37,38. Therefore, we examined whether the increase of Mincle mRNA in the injured DRG and SN were cancelled in MyD88−/− mice 12 h after PNI. Real-time PCR analysis revealed that the nerve injury-induced increase of Mincle mRNA in the injured SN was significantly attenuated in MyD88−/− mice compared to that in WT mice (Fig. 8a). Next, we investigated whether MyD88-deficiency affects the migration of leucocytes into the SN and DRG. Flow cytometry analysis revealed that the proportion of neutrophils (CD11b+Ly6G+) in the injured SN 1 day after PNI was significantly decreased in MyD88−/− mice compared to that in WT mice (Fig. 8b). Based on these results, it is suggested that the migration of neutrophils appears to be MyD88-dependent in this model, and that the reduction of Mincle mRNA in the SN of MyD88−/− mice might be due, at least in part, to the reduced infiltration of neutrophils.
Figure 8.
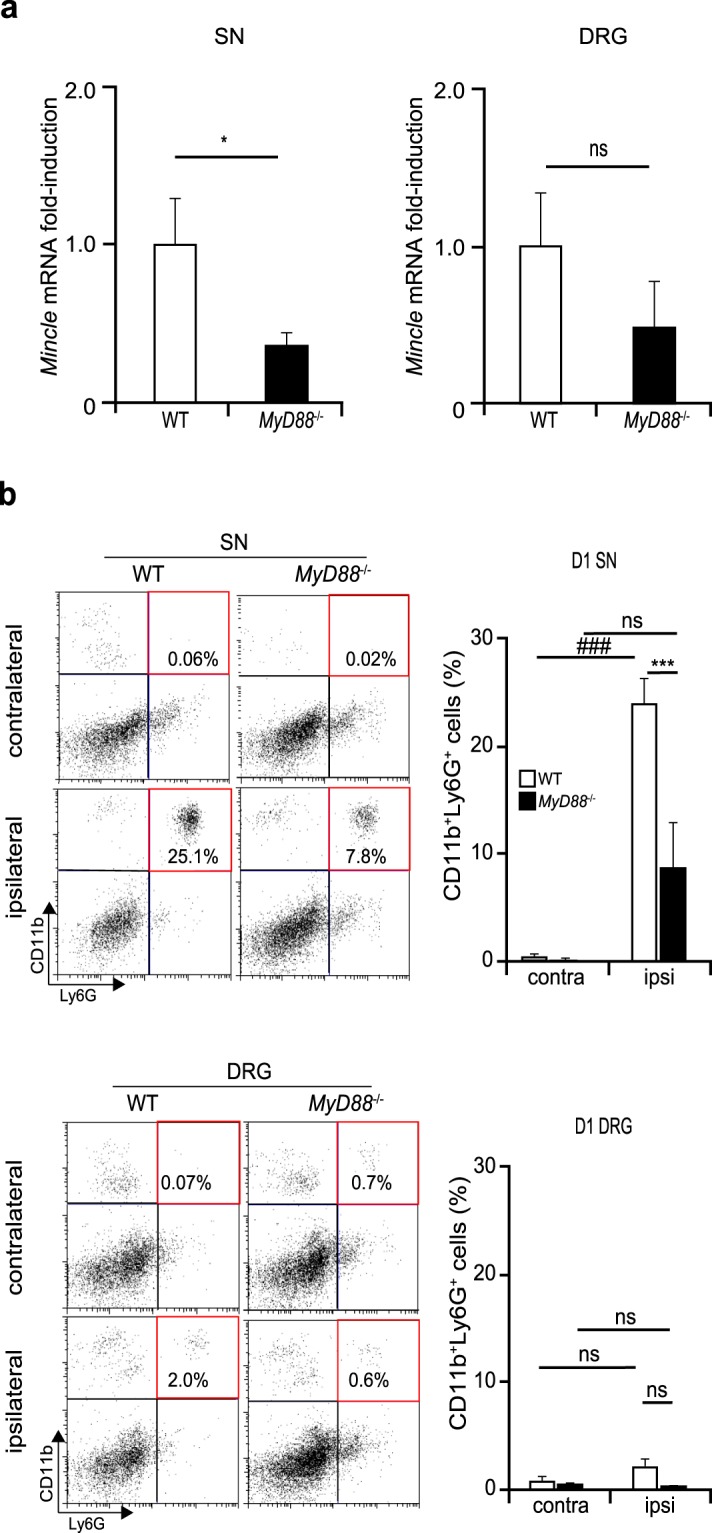
Increase of Mincle mRNA in injured SN depends on MyD88 in neuropathic pain model. (a) Real-time PCR analysis of Mincle mRNA in the injured SN and DRG 12 h after PNI in WT and MyD88−/− mice. Bar graphs show fold-change compared to naïve WT mice (n = 3 in each group, Student’s t test. *P < 0.05 vs. WT mice). (b) Representative flow cytometry dot plot of CD11b+Ly6G+ cells (left) in the SN (upper) and DRG (lower) 1 day after PNI. Upper-right quadrant (red frame) representing infiltrating neutrophils (CD11b+Ly6G+). The bar graph (right) summarises the proportions of neutrophils in the ipsilateral or contralateral tissues of WT or MyD88−/− mice (n = 3 in each group, one-way ANOVA followed by Tukey’s post hoc test, ***P < 0.001 vs. WT mice, ###P < 0.001 vs. contralateral side). Values are mean ± SEM.
Discussion
The present study provides the first evidence of the contribution of Mincle to pathological changes in a mouse model of neuropathic pain. The major findings of this study are as follows: (1) mechanical allodynia induced by PNI was attenuated in Mincle−/− mice; (2) Mincle mRNA was markedly induced in the injured SN, and migrating immune cells, especially neutrophils, were considered to be the major source of Mincle mRNA; (3) Mincle-deficiency affected the expression of certain genes, which are reported to contribute to neuropathic pain after PNI.
Recent studies on neuropathic pain have revealed that the TLRs, a family of PRRs, are involved in neuropathic pain after PNI6,7. It has been reported that when a peripheral nerve is injured, a variety of immune cells are recruited to the site of injury and release numerous cytokines and chemokines, which coordinate the cellular response to nerve injury and directly alter the sensory transduction properties and ongoing activity of primary afferents1,3,4.
These immune molecules, especially the proinflammatory cytokines, are also known to be upregulated in the DRG and spinal dorsal horn3,26,27. While a significant number of studies support a link between this disease and the TLRs, the current data demonstrate that Mincle also plays a role in triggering such sequential reactions.
Because PNI failed to induce mechanical allodynia in Mincle−/− mice, we compared the expression profile of cytokines and pain-related molecules in the DRG and spinal dorsal horn between Mincle−/− and WT mice before and after PNI. Induction of Tnfa in the DRG and Irf5 in the spinal dorsal horn were found to be significantly suppressed in Mincle−/− mice compared to those in WT controls. This result suggested that Mincle-mediated signals (and the subsequent reactions) in the injured SN play a role in the upregulation of Tnfa and Irf5 mRNA in the DRG and spinal dorsal horn, respectively. A growing body of literature demonstrates that TNF-α plays an important role in initiating inflammatory reactions of the innate immune processes and stimulation of inflammatory cells in injured nerves, and contributes to the generation of pain27,31,39,40. The direct application of TNF-α is known to induce ectopic firing in Aδ-, Aβ-, and C-fibers39,40, and also induce thermal hyperalgesia and mechanical allodynia41–43. TNF-α is also reported to enhance the sensitivity of nociceptors through the phosphorylation of Nav1.8 and Nav1.9 sodium channels44. Furthermore, a previous study has shown that TNF-α can increase excitatory synaptic transmission directly at the central terminal of primary afferents, and glutamatergic currents in the dorsal horn neurons1,45. After nerve injury, TNF-α is synthesized and probably released by various cell types in the DRG, such as the satellite glial cells, and even in sensory neurons46,47. Ogawa et al. demonstrated that silencing of TNF-α in the injured DRG reduces mechanical allodynia after PNI46. Together with these studies, our results indicate that TNF-α produced in the DRG by Mincle-dependent mechanisms might play an important role in the pathogenesis of neuropathic pain. It is not clear whether TNF-α produced in DRG neurons can be released from the central terminals of primary afferents, while some of the cytokines and chemokines such as CCL2, CCL21, and colony-stimulating factor1 (CSF1) are known to be released1,2,21,24.
In the spinal dorsal horn where the central axons of DRG neurons terminate, microglia have been reported to proliferate, exhibit marked changes in cell surface protein expression, and release various chemical mediators after PNI4,20. These responses are reported to be involved in the pathology of neuropathic pain. Because mechanical allodynia was ameliorated in Mincle−/− mice after PNI, we expected that the proliferation of spinal microglia would be attenuated. However, the number of these cells in Mincle−/− mice was found to be comparable to that in WT mice after PNI. This discrepancy between microglial proliferation and the ameliorated mechanical allodynia has been also reported in mice lacking P2Y12R22, IRF523, and DAP1224,32. Recently, CSF1, which is synthesized in DRG neurons and released from their central terminals to the spinal dorsal horn after PNI, has been reported to promote the proliferation of spinal microglia24. Consistent with this, Csf1 mRNA was found to be also induced in the DRG of Mincle−/− mice (Supplementary Fig. S2b). Together with other studies, the discrepancy mentioned here shows that induction of mechanical allodynia after PNI required not only microglial proliferation but also change(s) in microglial profile(s). IRF5 expressed in spinal microglia is reported to contribute to neuropathic pain after PNI23. In the current experiments, we found that upregulation of this Irf5 in the spinal dorsal horn after PNI was significantly suppressed in Mincle−/− mice. This result is consistent with our data from behavioural experiments. Although IRF5 is reported to drive P2X4R+-reactive microglia after PNI23, we found the expression of P2rx4 in Mincle−/− mice to be similar to that in WT mice (data not shown). A previous study also reported that IRF5 is critical for expression of M1 macrophage-related genes including proinflammatory cytokines (Il1b, Il6, and Tnfa)48; however, no differences were detected in the expression of these genes between Mincle−/− and WT mice in our experiments. Unidentified downstream pathways from IRF5 might also be involved in the induction of neuropathic pain after PNI.
After PNI, Mincle mRNA was induced only in the ipsilateral DRG tissue that included the SN, but not in the spinal dorsal horn. Since previous studies have shown that TLRs are expressed in the microglia5–7 and recognize DAMPs as their agonists directly6, we expected that Mincle would be also expressed in spinal microglia after PNI. However, Mincle mRNA in the ipsilateral dorsal horn remained at undetectable levels even after PNI while microglial proliferation (Fig. 7a,b) and upregulation of Iba1 mRNA (microglial marker) were observed (supplementary Fig. S2a). Thus, Mincle expression in spinal microglia appeared to be largely limited and was not induced after PNI. In rodent models of cerebrovascular disorders (CVD), Mincle is reported to be expressed in microglia-like cells, neurons, and endothelial cells17,18. In the CVD model, the blood-brain-barrier is broken down and blood cells gain access into the cerebral parenchyma. In contrast, no infiltrating microglia-like cells are observed in the PNI model49. Thus, Mincle-expressing microglia-like cells in the CVD model might be from blood and this could be a reason for the undetectable levels of Mincle mRNA in the spinal dorsal horn even after PNI in our model.
In the DRG tissue containing the SN, we further dissected the region of Mincle mRNA expression, and remarkable Mincle mRNA induction was observed in the injured SN, but not in the DRG. These results indicate a possibility that the immune cells that migrated to the injured SN may be the source of Mincle mRNA. This possibility was strengthen by the results of depletion experiments in which Mincle mRNA was virtually eliminated after depletion of neutrophils and macrophages/monocytes from the cells in injured SN. The presence of Mincle mRNA-expressing cells in injured SN was also confirmed by ISHH. Furthermore, ISHH with IHC also revealed the Mincle mRNA-expressing cells 12 h after PNI were mostly the migrating neutrophils, while those 3 days after PNI were monocytes/macrophages and some other unidentified cells. However, none of DRG neurons, including nociceptors, were found to express Mincle mRNA. Since systemic depletion of the neutrophils or macrophages reduce mechanical hypersensitivity after PNI28,50, it is suggested that these infiltrating cells expressing Mincle are required to generate neuropathic pain.
In some cases, Mincle mRNA signals were also detected in a population of cells that were neither neutrophils nor monocytes/macrophages. These cells may be other types of migrating immune cells or resident cells in the SN such as the Schwann cells. It has been reported that Schwann cells also proliferate and release inflammatory mediators after PNI and could play a key role in the development of neuropathic pain states4,51. Taken together, current results suggest that the Mincle-expressing cells for induction of neuropathic pain likely include neutrophils and macrophages, however, it would be possible that some unidentified cell types, being assumed to include Schwann cells, may also contribute to neuropathic pain.
When a peripheral nerve is injured, various immune cells are recruited to the site of injury. Mincle is known to promote neutrophil infiltration into the necrotic thymus tissue after whole body irradiation15. Thus, we assumed that migration of leucocytes to the injured nerve would be reduced in Mincle−/− mice. However, the migration of leucocytes into the injured nerve was comparable between WT and Mincle−/− mice. In addition, mRNA expression of the Ccl2 chemokine, which is known to regulate the migration of leucocytes, also showed similar induction in the injured SN and DRG between WT and Mincle−/− mice after PNI (Supplementary Fig. S2c). The TLRs are also known to be required for the induction of leucocyte migration into the injured nerve51. Numerous studies have shown the TLRs and their signalling protein MyD88 are involved in neuropathic pain and deletion of these genes has resulted in reduced tactile allodynia in the rodent model of neuropathic pain6,36–38. The following mechanisms have been proposed: (1) MyD88-deficiency results in less DRG reactivity to nerve injury36 and (2) MyD88 is required for microglial proliferation in the ipsilateral dorsal horn36. In the experiments using MyD88 conditional knockout mice, neuronal MyD88 is reported to be required for peripheral nerve injury-induced CCL2 up-regulation and macrophage infiltration into the DRG, and microglial proliferation37. In addition, MyD88 in nociceptive neurons is reported to be necessary for driving innate and adaptive immunity in DRGs38. Consistent with these observations, infiltration of neutrophils and monocytes/macrophages in the injured SN of MyD88−/− mice were impaired in our experiments. In these MyD88−/− mice, impairment of the increased Mincle mRNA in injured SN was also observed. These results strengthen the idea that the main source of Mincle mRNA in the injured SN was the migrating neutrophils and monocytes/macrophages. Alternatively, the impaired increase of Mincle mRNA in MyD88−/− mice may be due to the lack of TLR signalling because TLR ligands, such as lipopolysaccharides are known to induce Mincle mRNA expression12,52. Taken together with our results, it was suggested that Mincle may be involved at least in part in the MyD88-dependent mechanical allodynia observed in previous studies.
In this study, we demonstrated the contribution of Mincle to neuropathic pain pathology. Mincle mRNA was induced in migrating immune cells and unidentified cells in the injured SN. Recently, β-glucosylceramide (GlcCer), a glycolipid, was reported as a newly identified endogenous ligand for Mincle16. Glycolipids including β-GlcCer have been reported to be abundant in the nervous system including the brain53,54. Thus, β-GlcCer may be a possible candidate endogenous ligand recognized by Mincle in the neuropathic pain model. After PNI, β-GlcCer may be released or exposed in the injured region within the SN as DAMPs and Mincle-expressing cells may recognize it and activate subsequent reactions in the DRG and spinal dorsal horn, leading to neuropathic pain. It has been reported that TNF-α can induce Irf5 mRNA in primary cultures of healthy human epidermal keratinocytes55. Together with our result, it is conceivable that TNF-α induced in the DRG by Mincle dependent reaction(s) in the SN would be released in the spinal dorsal horn and induce IRF5 expression in the microglia which is known to result in neuropathic pain. Although further investigation is required, our results showed that Mincle may be involved in the pathology of neuropathic pain, and blockade of Mincle dependent pathways may provide new therapeutic strategies for neuropathic pain.
Methods
Animals
All experiments were approved by the Saga University Animal Care and Ethical Use Committee (#27-064-0, #28-063-0, and #28-027-0) and animals were treated in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions’ (Ministry of Education, Culture, Sports, Science and Technology of Japan) and the ethical guidelines of Saga University. All animal experiments were performed in male mice aged from 10 to 20 weeks at the initiation of the experiments.
Mincle-deficient (Mincle−/−) mice that had been backcrossed to C57BL/6 mice for at least eight generations14 were used, and C57BL/6 (SLC, Japan) were used as the wild type (WT) controls. MyD88-deficient (MyD88−/−) mice that had been backcrossed to C57BL/6 were also used.
Mice were housed in groups of 1-5 per cage at a temperature of 23.0 ± 2 °C and humidity of 55 ± 5% with a 12-h light-dark cycle and were fed food and water ad libitum.
Neuropathic pain model
We used the spinal nerve injury model56 with some modifications19. Under isoflurane (2%) anaesthesia, a small incision was made at L3-S1, and the paraspinal muscle and fat were removed from the L5 transverse process. The part of the transverse process was removed, and then the left L4 spinal nerve was carefully isolated and cut. The muscles and skin were closed in two layers with a 6–0 silk suture.
Assessment of mechanical sensitivity
To assess mechanical sensitivity, mice were placed individually in a transparent glass cylinder and separated from each other by an opaque divider, which was placed on a wire mesh, and habituated to the test apparatus for 1 h prior to data collection. After that, calibrated von Frey filaments (0.02~2.0 g, Stoelting, Wood Dale, IL) were applied to the mid plantar surface of the hind paws of mice with or without PNI, until they withdrew their paws in response to the mechanical stimuli. The 50% paw withdrawal threshold was determined using the up-down method57.
Formalin test
Mice were placed in individual boxes and allowed to habituate to the new environment for 15 min. Subsequently, formalin (5%, 20 μL) was subcutaneously injected into the dorsum of the hind paw. The duration of licking and biting responses to the injected hind paw was recorded as a nociceptive response, for 60 min at 5 min intervals after the injection19. An initial acute phase [first phase, during the first 10 min after the formalin injection] was followed by a relatively short quiescent period and then by a prolonged tonic response [second phase, beginning 10 min after the formalin injection]. The first phase is presumed to be a result of a direct activation of the primary afferent nociceptors, and the second phase involves peripheral inflammatory events and central sensitization58. The cumulative time of nociceptive behaviours during the first and second phases were analysed separately.
Real-time PCR
Mice were deeply anaesthetized with sodium pentobarbital (50 mg/kg) and then sacrificed by transcardial perfusion with 20 mL of RNA-later (Thermo Fisher Scientific, Waltham, MA) and tissues were removed immediately. RNA extraction was carried out using RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. After removal of the genomic DNA by treatment with DNase (Wako Nippon Gene, Osaka, Japan), complementary DNA synthesis was performed using the ReverTra Ace qPCR RT Master Mix (Toyobo, Osaka, Japan). Real-time PCR was performed using the Thunderbird SYBR qPCR Mix (Toyobo) with a StepOnePlus (Applied Biosystems, Foster City, CA) and the data were analysed using StepOne Software version 2.3 (Applied Biosystems). PCR amplifications were performed at 95 °C for 60 s, and 95 °C for 5 s, 60 °C for 30 s for 40 cycles. Ct values for genes of interest were normalized to correspond to those for a reference gene encoding glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and represented as a fold-change to a control, which was calculated using the 2−ΔΔCt method59. The sequences of the primers used in this study are presented below.
Gapdh (GU214026.1): 5′-AACTTTGGCATTGTGGAAGG-3′ (forward), 5′-ACACATTGGGGGTAGGAACA-3′ (reverse); Mincle (NM_019948.2): 5′-CAGTGGCAATGGGTGGATGATAC-3′ (forward), 5′-AGTCCCTTATGGTGGCACAGTC-3′ (reverse); Il1b (NM_008361): 5′-AGATGAAGGGCTGCTTCCAAAC-3′ (forward), 5′-GTTGATGTGCTGCTGCGACAT-3′ (reverse); Tnfa (NM_013693.3): 5′-AAGCCTGTAGCCCACGTCGTA-3′ (forward), 5′-GGCACCACTAGTTGGTTGTCTTTG-3′ (reverse); Il6 (NM_031168.1): 5′-CCACTTCACAAGTCGGAGGCTTA-3′ (forward), 5′-GCAAGTGCATCATCGTTGTTCATAC-3′ (reverse); Irf5 (NM_001252382.1): 5′-TGGGGACAACACCATCTTCA-3′ (forward), 5′-CTGGAAGTCACGGCTTTTGT-3′ (reverse).
Immunohistochemistry
Mice were deeply anaesthetized with sodium pentobarbital (50 mg/kg) and then sacrificed by transcardial perfusion with 20 mL of 4% paraformaldehyde in 0.1 M sodium phosphate buffer (PB; pH 7.4) before or 7 days after PNI. The L4 segment of the spinal cord was removed, post-fixed in the same fixative at 4 °C overnight and placed in 30% sucrose solution for 24 h at 4 °C. Transverse L4 spinal cord sections (30 μm) were cut, rinsed three times for 10 min in PBS and preincubated with 1% normal donkey serum containing 1% bovine serum albumin and 0.1% triton X-100 in PBS for 30 min. Sections were then incubated overnight at 4 °C with the primary antibody. Rabbit anti-Iba1 (ionized calcium-binding adapter molecule-1, 1:300, Wako Pure Chemical Industries, Osaka, Japan, RRID: AB_839504) was used. After rinsing three times for 10 min in PBS, these sections were incubated with the secondary antibody. Donkey anti-rabbit IgG conjugated Alexa Fluor 488 (1:400, Thermo Fisher Scientific, RRID: AB_141708) was used. TO-PRO-3 (1:200, Molecular Probes, Eugene, OR) was also used for nuclear staining together with the secondary antibody. Three to five sections of the L4 spinal cord segments of each mouse were randomly selected and analysed using a LSM5 Exciter-Zen 2009 (Carl Zeiss, Oberkochen, Germany). Iba1-immunopositive profiles in the dorsal horn were counted if the profile contained TO-PRO-3 positive nucleus.
Flow cytometry
Mice were deeply anaesthetized using sodium pentobarbital (50 mg/kg) and then sacrificed by transcardial perfusion with 20 mL of cold PBS at 1 day or 3 days after PNI. The L4 DRG and spinal nerve, ipsilateral and contralateral to PNI, were removed immediately and placed in ice-cold PBS. Tissues were treated with collagenase D (1 mg/mL in RPRI 1640 containing 2% FCS, Roche, Mannheim, Germany) for 1 h at 37 °C with gently shaking. The tissues were homogenized by passing through a mesh. Cells were washed with RPRI 1640 (2% FCS) 2 times. The cells were immunostained with rat anti mouse CD11b-APC (0.4 μg/mL, clone: M1/70, Tonbo Biosciences, San Diego, CA, RRID: AB_2621556), rat anti mouse Ly6G-PE (2 μg/mL, clone: 1A8, BD Biosciences, Franklin Lakes, NJ, RRID: AB_394208), and rat anti mouse F4/80-PE (2 μg/mL, clone: BM8.1, Tonbo Biosciences, RRID: AB_2621795). Neutrophils were defined as CD11b+ and Ly6G+, while monocytes/macrophages were CD11b+ and F4/80+. The total number of neutrophils and monocytes/macrophages was analysed using a FACS Calibur (BD Biosciences) and FlowJo software (BD Biosciences).
Depletion of Ly6G+ and/or F4/80+ cells from injured SNs
The injured and uninjured L4 spinal nerves were removed at 12 h after PNI as described above. To obtain a sufficient number of cells, a sample was collected from 3 animals, and treated together with collagenase D (1 mg/mL in RPRI 1640 containing 2% FCS, Roche) for 1 h at 37 °C with gently shaking. The tissues were homogenized by passing through a mesh. Cells were washed with RPRI 1640 (2% FCS) 2 times. Two-thirds of the cells obtained from injured SNs were incubated with rat anti mouse Ly6G-FITC (2 μg/mL, clone: 1A8, Tonbo Biosciences, RRID_AB_2621704) and rat anti mouse F4/80-PE (2 μg/mL, clone: BM8.1, Tonbo Biosciences) for 30 min on ice, washed with MACS Running buffer (Miltenyi Biotec, Bergisch Gladbach, Germany), and then incubated with anti-FITC and anti-PE antibody-coated immunomagnetic microbeads (Miltenyi Biotec, RRID: AB_244371, AB_244373, respectively) for 15 min on ice. After cells were washed with MACS Running buffer, unlabeled cells were collected (depleted fraction) from the total cells by using LD Columns (Miltenyi Biotec) which were designed for stringent depletion of labelled cells. Cells remaining in the columns (trapped fraction) were also collected. Four SN samples (contralateral total, ipsilateral total, ipsilateral depleted fraction, and ipsilateral trapped fraction) were further analysed. After confirming successful depletion by flow cytometry, RNA extraction from each sample was carried out using RNeasy Micro Kit (Qiagen), and the expression of Mincle mRNA was evaluated by real-time PCR in triplicate.
In situ hybridization
Mice were deeply anaesthetized using sodium pentobarbital (50 mg/kg) and then sacrificed by transcardial perfusion with 20 mL of 4% paraformaldehyde in 0.1 M PB 12 h or 3 days after PNI. The L4 whole DRGs were removed, post-fixed in the same fixative at room temperature for 3 h, and the samples were placed in 20% sucrose solution at 4 °C overnight. Then, the samples were rapidly frozen with powdered dry ice, and cut on a cryostat (HM-500-O, Thermo Fisher Scientific) at 8 μm thickness. The sections were mounted onto MAS-coated glass slides (Matsunami, Osaka, Japan). Using the enzyme-digested clones, 35 S UTP-labelled mouse Mincle antisense and sense cRNA probes (cDNA nucleotides 142–673, accession number NM_019948.2; in pCRII TOPO vector) were synthesized. The protocol for ISHH has been described in detail previously60. In order to examine the distribution of Mincle mRNA, we combined IHC with ISHH. The following antibodies were used: rat anti mouse F4/80 monoclonal antibody (1:1,000, Clone Cl: A3-1, AbD Serotec, RRID:AB_323806), rat anti mouse Ly6G monoclonal antibody (1:1,000, Clone RB6-8C5, AbD Serotec, RRID:AB_2137489). The treatment of sections and methods of double labelling with IHC and ISHH have been described in a previous paper60.
Statistical analysis
Data are represented as mean ± SEM. Statistical significances were determined using repeated analyses of variance (ANOVA) followed by Bonferroni post hoc test (Fig. 1a,b), multivariate analysis of variance (MANOVA) (Fig. 1c), two-way ANOVA followed by Tukey’s post hoc test (Figs 2b, 6a,b and 7c,d), one-way ANOVA followed by Dunnett’s post hoc test (Fig. 2c), Tukey’s post hoc test (Figs 5a–d, 7b and 8b), or unpaired Student’s t test (Figs 1d, 8a), using the JMP® Pro 12 software (SAS Institute Inc., Cary, NC). Differences were considered to be statistically significant at P < 0.05.
Supplementary information
Acknowledgements
Mice used were maintained at Division of Biological Resources and Development, Analytical Research Center for Experimental Sciences, Saga University. Real-time PCR, flow cytometry and confocal microscopy were conducted at Division of Instrumental Analysis in the research center. We thank all the staff members of the research center. Financial support from the JSPS KAKENHI Grant numbers 24390150 (TY), 15H04766 (TY) and 17K15791 (AI) is gratefully acknowledged.
Author Contributions
A.I., T.Y., N.H., H.H., S.Y. and H.Y. designed the experiments; A.I., T.Y., Y. Murata., Y. Miyake., E.I., S.I. and K.K. conducted the experiments and analysed the data; A.I. and T.Y. drafted the manuscript. All authors reviewed the manuscript.
Data Availability
The datasets generated during and/or analysed during the current study are available with the corresponding author, and can be accessed on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-37318-8.
References
- 1.Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014;14:217–231. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinho-Ribeiro FA, Verri WA, Jr., Chiu IM. Nociceptor sensory neuron-immune interactions in pain and inflammation. Trends Immunol. 2017;38:5–19. doi: 10.1016/j.it.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- 4.Calvo M, Dawes JM, Bennett DL. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012;11:629–642. doi: 10.1016/S1474-4422(12)70134-5. [DOI] [PubMed] [Google Scholar]
- 5.Kato J, Agalave NM, Svensson CI. Pattern recognition receptors in chronic pain: mechanisms and therapeutic implications. Eur. J. Pharmacol. 2016;788:261–273. doi: 10.1016/j.ejphar.2016.06.039. [DOI] [PubMed] [Google Scholar]
- 6.Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci. Bull. 2012;28:131–144. doi: 10.1007/s12264-012-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicotra L, Loram LC, Watkins LR, Hutchinson MR. Toll-like receptors in chronic pain. Exp. Neurol. 2012;234:316–329. doi: 10.1016/j.expneurol.2011.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–281. doi: 10.1016/j.tins.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto M, et al. A novel LPS-Inducible C-type lectin is a transcriptional target of NF-IL6 in macrophages. J. Immunol. 1999;163:5039–5048. [PubMed] [Google Scholar]
- 13.Ishikawa E, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 2009;206:2879–2888. doi: 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamasaki S, et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc. Natl. Acad. Sci. USA. 2009;106:1897–1902. doi: 10.1073/pnas.0805177106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamasaki S, et al. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 2008;9:1179–1188. doi: 10.1038/ni.1651. [DOI] [PubMed] [Google Scholar]
- 16.Nagata, M. et al. Intracellular metabolite beta-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl. Acad. Sci. USA114 (2017). [DOI] [PMC free article] [PubMed]
- 17.Suzuki Y, et al. Involvement of Mincle and Syk in the changes to innate immunity after ischemic stroke. Sci. Rep. 2013;3:3177. doi: 10.1038/srep03177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie Y, et al. Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav. Immun. 2017;60:346–360. doi: 10.1016/j.bbi.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 19.Tsuda M, et al. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol. Pain. 2009;5:28. doi: 10.1186/1744-8069-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 21.Beggs S, Trang T, Salter MW. P2X4R + microglia drive neuropathic pain. Nat. Neurosci. 2012;15:1068–1073. doi: 10.1038/nn.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tozaki-Saitoh H, et al. P2Y12 receptors in spinal microglia are required for neuropathic pain after peripheral nerve injury. J. Neurosci. 2008;28:4949–4956. doi: 10.1523/JNEUROSCI.0323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masuda T, et al. Transcription factor IRF5 drives P2X4R + -reactive microglia gating neuropathic pain. Nat. Commun. 2014;5:3771. doi: 10.1038/ncomms4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guan Z, et al. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 2016;19:94–101. doi: 10.1038/nn.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu P, Bembrick AL, Keay KA, McLachlan EM. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav. Immun. 2007;21:599–616. doi: 10.1016/j.bbi.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 26.Clark AK, Old EA, Malcangio M. Neuropathic pain and cytokines: current perspectives. J. Pain. Res. 2013;6:803–814. doi: 10.2147/JPR.S53660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sommer C, Kress M. Recent findings on how proinflammatory cytokines cause pain: peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci. Lett. 2004;361:184–187. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Perkins NM, Tracey DJ. Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience. 2000;101:745–757. doi: 10.1016/S0306-4522(00)00396-1. [DOI] [PubMed] [Google Scholar]
- 29.Mueller M, et al. Rapid response of identified resident endoneurial macrophages to nerve injury. Am. J. Pathol. 2001;159:2187–2197. doi: 10.1016/S0002-9440(10)63070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrin FE, Lacroix S, Aviles-Trigueros M, David S. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain. 2005;128:854–866. doi: 10.1093/brain/awh407. [DOI] [PubMed] [Google Scholar]
- 31.Leung L, Cahill CM. TNF-alpha and neuropathic pain–a review. J. Neuroinflammation. 2010;7:27. doi: 10.1186/1742-2094-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobayashi M, Konishi H, Sayo A, Takai T, Kiyama H. TREM2/DAP12 Signal Elicits Proinflammatory Response in Microglia and Exacerbates Neuropathic Pain. J. Neurosci. 2016;36:11138–11150. doi: 10.1523/JNEUROSCI.1238-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsuda M, et al. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 34.Miyake Y, Masatsugu OH, Yamasaki S. C-type lectin receptor MCL facilitates Mincle expression and signaling through complex formation. J. Immunol. 2015;194:5366–5374. doi: 10.4049/jimmunol.1402429. [DOI] [PubMed] [Google Scholar]
- 35.Lee WB, et al. Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the Mincle pathway. PLoS Pathog. 2012;8:e1002614. doi: 10.1371/journal.ppat.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stokes JA, Cheung J, Eddinger K, Corr M, Yaksh TL. Toll-like receptor signaling adapter proteins govern spread of neuropathic pain and recovery following nerve injury in male mice. J. Neuroinflammation. 2013;10:148. doi: 10.1186/1742-2094-10-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu XJ, et al. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci. Rep. 2016;6:28188. doi: 10.1038/srep28188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu XJ, et al. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. 2014;24:1374–1377. doi: 10.1038/cr.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang JM, Li H, Liu B, Brull SJ. Acute topical application of tumor necrosis factor alpha evokes protein kinase A-dependent responses in rat sensory neurons. J. Neurophysiol. 2002;88:1387–1392. doi: 10.1152/jn.2002.88.3.1387. [DOI] [PubMed] [Google Scholar]
- 40.Sorkin LS, Xiao WH, Wagner R, Myers RR. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience. 1997;81:255–262. doi: 10.1016/S0306-4522(97)00147-4. [DOI] [PubMed] [Google Scholar]
- 41.Murata Y, et al. Changes in pain behavior and histologic changes caused by application of tumor necrosis factor-alpha to the dorsal root ganglion in rats. Spine. 2006;31:530–535. doi: 10.1097/01.brs.0000201260.10082.23. [DOI] [PubMed] [Google Scholar]
- 42.Homma Y, Brull SJ, Zhang JM. A comparison of chronic pain behavior following local application of tumor necrosis factor alpha to the normal and mechanically compressed lumbar ganglia in the rat. Pain. 2002;95:239–246. doi: 10.1016/S0304-3959(01)00404-3. [DOI] [PubMed] [Google Scholar]
- 43.Wagner R, Myers RR. Endoneurial injection of TNF-alpha produces neuropathic pain behaviors. Neuroreport. 1996;7:2897–2901. doi: 10.1097/00001756-199611250-00018. [DOI] [PubMed] [Google Scholar]
- 44.Jin X, Gereau RW. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J. Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogawa N, et al. Gene therapy for neuropathic pain by silencing of TNF-alpha expression with lentiviral vectors targeting the dorsal root ganglion in mice. PLoS One. 2014;9:e92073. doi: 10.1371/journal.pone.0092073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu YL, et al. Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007;52:708–715. doi: 10.1016/j.neuropharm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 48.Krausgruber T, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011;12:231–238. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 49.Tashima R, et al. Bone marrow-derived cells in the population of spinal microglia after peripheral nerve injury. Sci. Rep. 2016;6:23701. doi: 10.1038/srep23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu T, van Rooijen N, Tracey DJ. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain. 2000;86:25–32. doi: 10.1016/S0304-3959(99)00306-1. [DOI] [PubMed] [Google Scholar]
- 51.Boivin A, et al. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J. Neurosci. 2007;27:12565–12576. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lang R. Recognition of the mycobacterial cord factor by Mincle: relevance for granuloma formation and resistance to tuberculosis. Front. Immunol. 2013;4:5. doi: 10.3389/fimmu.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Farfel-Becker T, et al. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 2014;23:843–854. doi: 10.1093/hmg/ddt468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu RK, Nakatani Y, Yanagisawa M. The role of glycosphingolipid metabolism in the developing brain. J Lipid Res. 2009;50:S440–S445. doi: 10.1194/jlr.R800028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banno T, Gazel A, Blumenberg M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 2004;279:32633–32642. doi: 10.1074/jbc.M400642200. [DOI] [PubMed] [Google Scholar]
- 56.Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- 57.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 58.Tjolsen A, Berge OG, Hunskaar S, Rosland JH, Hole K. The formalin test: an evaluation of the method. Pain. 1992;51:5–17. doi: 10.1016/0304-3959(92)90003-T. [DOI] [PubMed] [Google Scholar]
- 59.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 60.Kobayashi K, et al. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J. Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available with the corresponding author, and can be accessed on reasonable request.