

Abstract
Emerging evidence suggests that the signalling of the Receptor for Advanced Glycation End products (RAGE) is critical for skeletal muscle physiology controlling both the activity of muscle precursors during skeletal muscle development and the correct time of muscle regeneration after acute injury. On the other hand, the aberrant re‐expression/activity of RAGE in adult skeletal muscle is a hallmark of muscle wasting that occurs in response to ageing, genetic disorders, inflammatory conditions, cancer, and metabolic alterations. In this review, we discuss the mechanisms of action and the ligands of RAGE involved in myoblast differentiation, muscle regeneration, and muscle pathological conditions. We highlight potential therapeutic strategies for targeting RAGE to improve skeletal muscle function.
Keywords: RAGE, AGEs, S100B, HMGB1, myogenesis, muscle wasting
Introduction
Skeletal muscle is the most abundant tissue and endocrine organ of the human body1, 2 playing vital roles in breathing, posture maintenance, locomotion, whole‐body metabolism, and reservoir of glucose and amino acids that can support energy demand during extreme metabolic perturbation. Skeletal muscle is a highly plastic tissue that can adapt its mass and functionality in response to different conditions, including ageing, diabetes, chronic and metabolic disorders, immobilization, inflammatory myopathies, and various genetic muscle disorders, such as muscular dystrophy.3, 4 Due to its essential role, skeletal muscle wasting accelerates disability and mortality in various disease conditions.
RAGE (Receptor for Advanced Glycation End products) was purified from bovine lung homogenates in 1992, thanks to its ability to bind and mediate the effects of Advanced Glycation End products (AGEs), which are produced by the non‐enzymatic glycation of lipids or proteins upon exposure to reducing sugars.5 RAGE is a multiligand receptor considered a key mediator of several physiological (e.g. tissue differentiation and regeneration/repair and resolution of inflammation) and pathological (e.g. inflammation, diabetes, cardiovascular diseases, neurodegeneration, and cancer) processes through the activation of multiple cellular signalling cascades, depending on the cell type, the ligands involved, and the density of the receptor on the cell surface.6, 7
The intracellular signalling cascades activated by RAGE change in response to environmental cues (i.e. the presence and concentration of RAGE ligands) in many systems, including skeletal muscle tissue. Indeed, an appropriate recruitment of RAGE concurs to skeletal muscle development and restoration of muscle homeostasis in physiological conditions and upon acute muscle injury, respectively.8, 9 On the other hand, over‐stimulation of RAGE concurs to muscle damage and altered muscle metabolism in ageing and pathological conditions, such as metabolic perturbations and myopathies, all characterized by chronic inflammation and increased production of reactive oxygen species (ROS). Therefore, the fate (i.e. proliferation, differentiation, or death) of muscle precursor cells, myofibre trophism, and the success of muscle regeneration appears to be strongly dependent on the extent of RAGE activity and the availability of specific ligands. In the current review, we summarize information about the role of RAGE as foe or friend in muscle tissue and raise questions about RAGE as a potential target in the prevention and treatment of muscle wasting.
The receptor RAGE
RAGE structure
RAGE is a member of the immunoglobulin (Ig) superfamily, which includes Igs, cell surface receptors, and adhesion molecules.5, 10 In humans and mice, RAGE is encoded by AGER and Ager, respectively, which localizes in both species next to the HLA (human leukocyte antigen) locus on chromosome 6, in close proximity of the MHC class III complex.11, 12 RAGE is highly conserved across different mammalian species at both DNA and protein level.13 Despite its name, however, RAGE is a multiligand receptor recognizing high mobility group B1 (HMGB1), certain S100 proteins, and other compounds besides a number of AGEs (see below). In humans, RAGE is a 55‐kDa cell surface receptor whose mature form consists of three regions: an extracellular region of 321 amino acids, a single hydrophobic transmembrane‐spanning domain of 19 amino acids, and a short cytosolic tail of 41 amino acids.5 The extracellular region is divided into three Ig‐like domains: one N‐terminal V‐type (variable) and two C‐type (constant, C1 and C2) domains14 (Figure 1 ). V and C1 domains form a structural and functional unit (VC1) connected with C2 domain by a short (seven amino acid long) and flexible linker region. Although the VC1 unit binds directly the majority of RAGE ligands, the C2 domain is important for the stabilization of ligands interaction.15, 16
Figure 1.
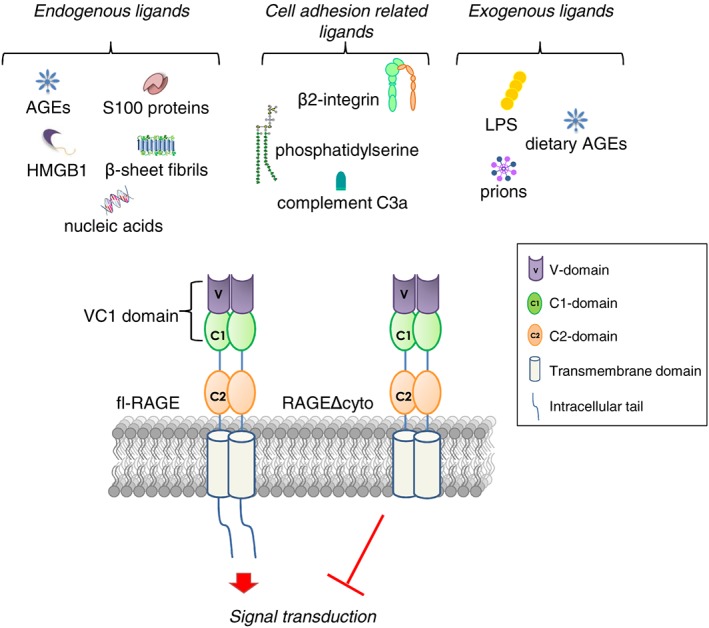
Schematic representation of fl‐RAGE structure and its of ligands. The dominat negative isoform of RAGE (RAGEΔcyto) lacking of cytoplasmatic tail is reported.
RAGE has two potential N‐glycosylation sites located in the V domain.17 Carboxylation of these N‐glycans and their surface exposure may influence ligand binding directly, for example, by enhancing HMGB1 and AGE binding and subsequent signal transduction.18 The transmembrane region has a role in anchoring RAGE to the cellular membrane, while the C‐terminal tail lacks known signalling motifs (e.g. kinase domains or phosphorylation sites) but is essential for RAGE‐mediated intracellular signalling.5, 19 A truncated form of RAGE (RAGEΔcyto), in which the cytosolic tail is deleted, recognizes and binds RAGE ligands without activating downstream signalling20 (Figure 1 , see Box 1).
Box 1. RAGE isoforms.
Twenty‐two different variants occurring at mRNA and protein level have been identified in 18 different tissues by real‐time PCR assays, 15 of which were named HsapRAGEv1 to v15.186 The most frequent splice variant, transcript variant 1 (Tv1‐RAGE), is the membrane‐bound form of RAGE consisting of the three classical domains (fl‐RAGE). The other variants are characterized by different exon/intron structure, causing changes in the sequence/function of the corresponding proteins, including changes in amino acid sequence of the extracellular ligand‐binding V domain (RAGE_v4 and RAGE_v5); the presence of a potential starting codon at the end of exon 7 producing two short variants lacking a large part of the extracellular domain of RAGE (RAGE_v11 and RAGE_v12); the presence of potential initiating codon in exon 3 producing a transmembrane protein lacking the V domain (RAGE_v2); the inclusion of the intron 1, 4, 6, or 9 translating into RAGE_v2, _v6, _v14, and _v1 variants, respectively; and the deletion of the entire or part of exon 8 resulting in loss of the transmembrane or cytosolic domain (RAGE_v3 and RAGE_v7, respectively).11 In human brain, a slightly truncated version of fl‐RAGE characterized by a partial deletion of exons 10 and 11, resulting in a predicted deletion of 16 amino acids, was also identified. This unique form of RAGE, named RAGEΔ, possesses an identical extracellular domain to fl‐RAGE but lacks a significant amount of the intracellular signal transduction domain.187 Truncation of the cytoplasmic tail confers a dominant negative effect on RAGE function, impairing its activation by blocking key signalling pathways downstream of RAGE.7 Several RAGE splice variants are potential targets of the nonsense‐mediated mRNA decay pathway and thus are likely to be degraded before protein expression. On the other hand, the mRNA isoforms lacking the signal sequence on exon 1 might be able to undergo protein expression, but the expressed protein could be subject to premature degradation.188 The only human variants that have been detected at the protein level in vivo are fl‐RAGE, sRAGE, and esRAGE.11
Human AGER is composed of 11 exons and 10 introns of variable length and a 3′UTR (untranslated) region. Several splice variants of RAGE have been identified (see Box 1). Those variants that lack both the transmembrane‐spanning domain and the intracellular tail likely generate circulating soluble isoforms of RAGE, collectively known as soluble RAGE (sRAGE). Alternative mRNA splicing and proteolytic cleavage of full‐length RAGE (fl‐RAGE) are the two primary mechanisms giving rise to sRAGE variants, including endogenous secretory RAGE (esRAGE) and cleaved RAGE (cRAGE). esRAGE (or RAGE_v1) is the C‐truncated form of RAGE originating from alternative splicing at exon 9 that causes inclusion of part of intron 9 and deletion of exon 10, whereas cRAGE arises from proteolytic cleavage by the membrane metalloproteinase, ADAM10, of fl‐RAGE at the cell surface21 (see Box 1).
RAGE ectodomain cleavage (shedding) occurs constitutively, but it can be induced by ligand binding, cell stimulation with phorbol 12‐myristate 13‐acetate, ionomycin, or cholesterol depletion.21 sRAGE can act as a decoy receptor that intercepts RAGE ligands, thereby blocking them. sRAGE can be detected in human blood at picomolar levels, and a strong correlation exists between circulating sRAGE levels and several pathological conditions, such as cardiovascular disease, diabetes, hypertension, and dementia/Alzheimer's disease (AD)11 (see Box 1).
RAGE ligands
RAGE mediates several physiological and pathological processes through the interaction with a large variety of ligands (Figure 1 ). AGEs are a group of non‐enzymatic adducts of proteins, lipids, and nucleic acids, which form in a time‐dependent manner under conditions of enhanced oxidative stress, especially when target molecules turnover slowly and aldose (such as glucose and ribose) levels are elevated.6 Over 20 different AGE modifications have been characterized including Nɛ‐carboxymethyl‐lysine (CML), pentosidine, Nɛ‐carboxyethyl‐lysine (CEL) adducts, methylglyoxal‐derived AGEs, argpyrimidine, and others, which makes AGEs a heterogeneous class of compounds. Formation of AGEs, which occurs naturally during ageing or in conditions of hyperglycaemia and oxidative stress, leads to the activation of different signalling pathways mediated by a series of cell surface receptors, the most studied of which is RAGE.22 As AGEs are heterogeneous, it is likely that not all of them interact with RAGE. Although CML is recognized as a major ligand of RAGE, other AGE compounds were reported to be RAGE ligands. Indeed, CEL (Nε‐carboxy‐ethyl‐lysine), argypirimidine, imidazoles, pentosidine, and methylglyoxal (MGL)‐derived AGEs have been reported to interact with RAGE and to activate intracellular signal transduction cascades suggesting that they might play a role in the complications of diabetes, cancer, and chronic inflammation23, 24, 25, 26, 27 (see Boxes 2 and 3).
Box 2. RAGE and inflammation.
An increase in RAGE expression has been documented in several acute and chronic diseases, such as diabetes, rheumatoid arthritis, osteoarthritis, arteriosclerosis, chronic renal disease, sepsis, and vasculitis. RAGE activity plays a role in the regulation of the immune system response and inflammatory processes.42, 189 Indeed, RAGE is expressed in neutrophils, T and B lymphocytes, monocytes, macrophages, and dendritic cells,188 and RAGE has been found in endothelial cells during inflammation or upon injury. By interacting directly with the adhesion molecule β2 integrin Mac‐1 present on leukocytes, RAGE stimulates leukocyte recruitment in case of acute inflammation.44
Most RAGE ligands (e.g. HMGB1, S100 proteins, and AGEs) are involved in acute and chronic inflammatory events, and many signalling cascades triggered by RAGE engagement determine the intracellular activation of the pro‐inflammatory NF‐κB (Figure 1 ).190 The ‘alarmin’ HMGB1 is a recognized DAMP, since it is secreted in case of inflammation, tissue injury, or infectious events, and it is released by necrotic cells, macrophages, NK cells, and dendritic cells. By interacting with RAGE, HMGB1 causes the activation of NF‐κB, thereby inducing the production of several cytokines, including interleukin (IL)‐6, IL‐1α, and TNF‐α (tumour necrosis factor). Macrophages from Ager −/− mice produce lower amounts of pro‐inflammatory cytokines after the stimulation with HMGB1, confirming the important role of RAGE is this process.191
S100–RAGE interactions might have important consequences in inflammatory and degenerative events, since they determine the activation of endothelial and vascular smooth muscle cells, neurons, astrocytes, microglia/macrophages, and other cell types, inducing the activation of different signalling pathways leading to production of pro‐inflammatory adhesion molecules and cytokines. In general, S100 proteins have to be present at high levels to generate these effects, except for S100B, which activates RAGE in neurons at low and high concentrations with trophic and toxic effects, respectively,192, 193, 194 and in macrophages with anti‐inflammatory and pro‐inflammatory effects at low and high concentrations, respectively.9 By impinging on NF‐κB, AGE–RAGE interactions also are involved in inflammatory processes.188 RAGE stimulation by AGEs increases levels of the pro‐inflammatory (M1) macrophage markers, iNOS and CD86, and the pro‐inflammatory cytokines, IL‐6 and TNF‐α.195 A positive feedback regulation of the RAGE promoter following activation of NF‐κB causes increase in RAGE expression levels, thus amplifying and prolonging the inflammatory process.20, 30
Box 3. RAGE and cancer.
RAGE plays a key role in the onset and progression of several cancer types, where it has been implicated in cell proliferation and survival, autophagy, invasion and metastasis, and angiogenesis.189, 196, 197 RAGE expression is found changed in cancer cells of a variety of human tumours, where RAGE is also expressed in cells of the tumour micro‐environment, such as endothelial and smooth muscle cells, fibroblasts, tumour‐associated macrophages, and leukocytes.198 Although RAGE results to be a potent inducer of tumour growth and malignant conversion, migration, and invasion in many cancer types (e.g. glioma, melanoma, breast, liver, bladder, lung, gastric, pancreatic, and colorectal and prostate cancers),197, 199, 200 a RAGE tumour‐suppressive function has been reported in other tumour types (e.g. oral and esophageal squamous cell carcinomas, non‐small cell lung carcinoma, and rhabdomyosarcomas).169, 170, 175, 201 Most types of solid tumours overexpress RAGE ligands, further implicating RAGE activity in tumour biology. Tissue‐specific differences in the expression levels of RAGE, its splice variants, and its ligands allow to hypothesize that RAGE might exhibit tumour‐suppressive functions in tissues characterized by constitutive RAGE expression, while it may act as a tumour‐promoter in tissues characterized by low or absent RAGE expression.189
In addition to AGEs, RAGE is a signal transduction receptor for amyloid‐β peptide (Aβ) and β‐sheets fibrils28; β2‐integrin Mac/CD11b; complement C3a; lipopolysaccharides (LPS); phosphatidylserine on the surface of apoptotic cells29, 30; S100/calgranulins, a family of closely related calcium‐binding proteins that accumulate extracellularly at sites of chronic inflammation20; and HMGB1 (amphoterin), a DNA‐binding protein released by cells undergoing necrosis that mediates inflammatory reactions and is involved in the pathophysiology of several diseases.19, 31 Besides binding ligands actively participating in chronic inflammatory and immune responses, RAGE interacts with molecules on the surface of leukocytes and bacteria,32, 33 nucleic acids, and prions34, 35 (Figure 1 ). Yet RAGE activity is not restricted to the promotion of inflammatory responses, having a role in tissue regeneration/repair as well36 (see Box 2).
Since RAGE can interact with pathogen associated molecular patterns (PAMPs) such as LPS, and damage‐associated molecular patterns (DAMPs) such as HMGB1, it can be considered as a pattern recognition receptor (PRR), like toll‐like receptors (TLR), pointing to cooperation between RAGE and TLRs in immune response.37, 38 In contrast to TLRs, which are expressed on the cells of the innate immune system and are involved in the recognition of microbial‐associated antigens, RAGE ligands are also of endogenous origin and typically accumulate in tissues during ageing, inflammation, or in response to other tissue stresses39, 40 (see Boxes 2 and 3).
Ligand domains interacting with RAGE display a negative charge on the surface. This acidic character of the RAGE ligands is complementary to the highly basic, electropositive surface of the RAGE ligand‐binding domain. Thus, a strong electrostatic interaction drives the formation of a tight receptor‐ligand complex, leading to sustained activation of the downstream signalling pathways.28, 41 Binding of ligands to RAGE does not accelerate its clearance or degradation but rather begins a sustained period of cellular activation mediated by receptor‐dependent signalling. Moreover, the enhanced surface expression of RAGE in environments rich in its ligands explains how upregulation of this receptor can contribute to an ascending spiral of RAGE‐dependent cellular perturbation. Taken together, these features of RAGE allow the receptor to propagate cellular dysfunction in a number of pathophysiologically relevant situations, most often dictated by the presence and persistence of RAGE ligands in tissues.14, 42
RAGE expression
RAGE is constitutively or inducibly expressed depending on the cell type and the developmental stage. RAGE is extensively expressed in a constitutive manner during embryonic and early post‐natal life, especially in the brain, but its expression level decreases after the accomplishment of development. This feature suggests that RAGE might play a role during tissue development, even if Ager −/−mice develop normally and do not show any overt phenotype and reproduce normally.43 This is probably because in Ager −/− mice, compensatory mechanisms are induced to replace RAGE activities. During the adult life, and in physiological conditions, RAGE is expressed at low levels in a wide range of differentiated cells, such as vascular endothelial cells, smooth muscle cells, almost all cells of the central nervous system, several cells of the immune system (such as monocytes, macrophages, neutrophils, dendritic cells, leukocytes, and CD4+ and CD8+ T cells), and cardiomyocytes.44 However, RAGE is upregulated under pathological conditions in which its ligands accumulate and/or transcription factors regulating RAGE are activated, such as cancer, retinal disease, atherosclerosis, cardiovascular disease, AD, respiratory disorders, liver diseases, and diabetes6, 44 (see Boxes 2 and 3). Known exceptions are represented by the skin and lung, which express constitutively RAGE at high levels throughout life.39
RAGE not only localizes in the cytoplasm and to plasma membrane but also is found in mitochondria and the nucleus of specific cells, where it might have different functions from that of surface RAGE. In pancreatic tumour cells, released HMGB1 induces mitochondrial translocation of RAGE by promoting its ERK1/2 (mitogen‐activated protein kinases)‐dependent phosphorylation. Mitochondrial RAGE is suggested to promote the first step in the process of ATP production to sustain the increased metabolic needs and growth of tumour cells. Although localization of RAGE in mitochondria has been observed in normal mouse pancreas, lung, heart, and liver cells,45 the physiological relevance of mitochondrial RAGE is unknown. Recently, nuclear accumulation of RAGE has been shown in pathological settings that challenge the genomic integrity such as Helicobacter pylori infection,46 diabetes,47 cancer, and under severe oxidative stress that affects the nuclear integrity.48, 49 RAGE, which accumulates in the nucleus after induction of DNA damage in an ATM serine/threonine kinase‐dependent manner,50 has been identified as a homeostatic regulator of DNA repair, suggesting an interesting role of the receptor in the prevention of cellular senescence, inflammation, pulmonary fibrosis, and cancer.
RAGE signalling
Homodimerization is a crucial event for signal transduction in many classes of receptors. Dimerization or oligomerization through self‐association of V–V or C1–C1 domains is an important step for RAGE signalling after binding its ligands (Figures 1 and 2 ), especially if these are in an oligomeric configuration (such as S100B and AGEs).51, 52 RAGE C‐terminal region is not homologous to any known kinase domain, and receptor dimerization and its enhancement by ligand binding affect the association of RAGE cytosolic tail with its interacting adaptor proteins to trigger signal transduction.53 Also, upon binding by certain ligands, the phosphorylation of RAGE at Ser391 in the cytoplasmic domain by PKCζ (protein kinase C‐zeta) promoted the recruitment of the adaptor protein, TIRAP (toll‐interleukin 1 receptor adaptor protein) and RAGE's downstream signalling54 (Figure 2 ).
Figure 2.
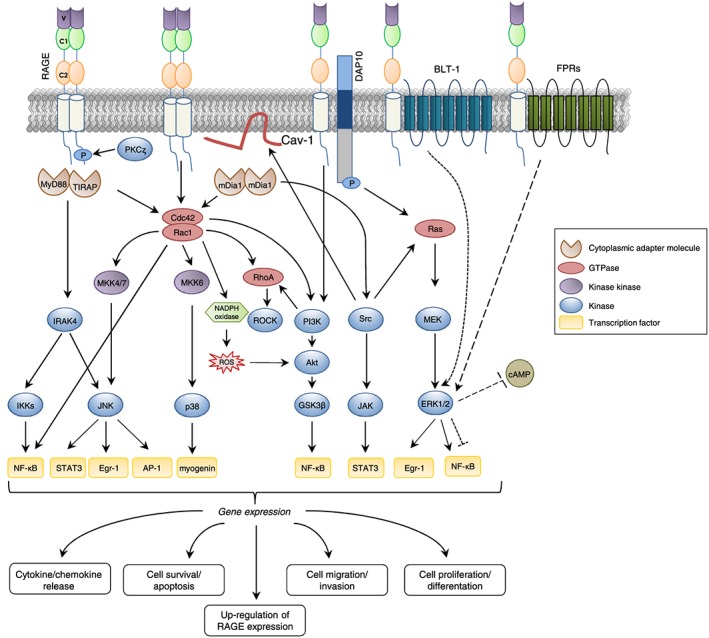
Activation of RAGE signalling. Preassembly of RAGE into dimers and multimers is necessary for RAGE to bind ligands and activate downstream signalling pathways. Upon ligand binding, the RAGE's intracellular domain changes its conformation allowing the interaction with intracellular partners. RAGE signalling involves GTPases and kinases that activate transcription factors. RAGE activation leads to changes in gene expression and altered cellular function including migration, survival, cytokine production, inflammation, proliferation, differentation, and upregulation of RAGE expression itself.
Since the cytosolic domain of RAGE lacks intrinsic tyrosine kinase activity, over the past years, several studies have addressed the question how RAGE can activate signal transduction. Some cytoplasmic or transmembrane adaptor proteins have been identified whose interaction with the cytoplasmic domain of RAGE translates into the activation of specific pathways leading to sometimes opposite cellular events (such as growth, migration, invasion, apoptosis, differentiation, and/or inflammatory cytokine and chemokine secretion)55 (see Boxes 2 and 3).
Most signalling cascades activated by RAGE, such as cdc42/Rac1 (cell division control protein 42/Ras‐related C3 botulinum toxin substrate 1), Src (Sarcoma family of proteins), Egr‐1 (early growth response protein‐1), PI3K/Akt (phosphoinositol‐3‐kinase/protein kinase B), and GSK3β (glycogen synthase kinase 3β), are regulated by the adaptor molecule mDia‐1 (diaphanous related formin 1), a member of the GEF (guanine nucleotide exchange factor) family. Recruitment of Dia‐1 by RAGE results in increased cell motility and/or chemokine secretion through activation of the Rac1/Cdc42 pathway.7, 56, 57 In U‐87MG human glioma cells, DOCK7 (dedicator of cytokinesis 7), another member of the GEF family, is recruited to the cytoplasmic tail of RAGE to activate Rac1/Cdc42, resulting in an enhancement of cell migration.58 Moreover, RAGE cytosolic domain is connected to the tyrosine kinase protein, Src, which is related to several downstream signal factors, such as caveolin‐1, ERK1/2, p38 MAPK, JNK (c‐Jun N‐terminal kinase), AP‐1 (activating protein 1), and NF‐κB (nuclear factor kappa B) and JAK/STAT3 (janus kinase/signal transducer and activator of transcription)7, 57 (Figure 2 ).
Two additional adaptor proteins, TIRAP and MyD88 (myeloid differentiation primary response gene 88), which are involved in TLR2/4 signalling, have been shown to be connected with the phosphorylated form of RAGE. Recruitment of these two proteins translates into the activation of Akt, IRAK4 (interleukin‐1 receptor‐associated kinase 4), p38 MAPK, JNK, and the IKKs (IκB kinases), leading to regulation of cell survival, apoptosis, and secretion of inflammatory cytokines.7, 54 Recently, DAP10 (dnax‐activating protein 10) has been identified as a transmembrane adaptor for RAGE necessary to regulate cell survival and proliferation via activation of the PI3K‐Akt and Ras‐ERK1/2 pathways in human keratinocytes.59
In addition, RAGE interacts with novel co‐receptors, FPRs (Formyl Peptide Receptors), and BLT1 (Leukotriene B4 Receptor 1), which are members of the GPCR (G‐protein‐coupled receptor) family. In glial cells, RAGE association with FPRs leads to ERK1/2 activation and accelerated consumption of intracellular cAMP resulting in increased inflammatory response in the neurodegenerative AD.55, 60 The RAGE/BLT1 interaction in neutrophils results in ERK1/2‐mediated reduction of NF‐κB activity and increased chemotaxis, whereas in the absence of RAGE, BLT1 activates ERK1/2 and NF‐κB signalling, inducing inflammatory cytokine production.61 Moreover, stimulation of RAGE in smooth muscle cells, tubular epithelial myofibroblasts, myoblasts, osteoblasts, and monocytic cells translates into the activation of ERK1/2, whereas p38 and SAPK/JNK MAPKs are activated in monocytes/macrophages and tumour cells.6, 62, 63 Other pathways connected to RAGE are rho‐GTPases, PI3K, and the JAK/STAT3 pathways. Activation of PI3K and ERK1/2 signalling cascades has also been observed in microglial cell lines.44, 57
RAGE and skeletal muscle physiology
The process of skeletal muscle formation during embryonic development, known as myogenesis, is a highly regulated process that involves the determination of multipotential mesodermal cells to myoblasts, the exit of myoblasts from the cell cycle, and their differentiation and fusion into muscle fibres.64 During myogenesis, a distinct subpopulation of myoblasts do not differentiate and remain closely associated with myofibres as satellite cells (SCs) in a quiescent state.65 In case of muscle damage consequent to trauma, intense physical exercise, chronic inflammation, cancer, or muscular dystrophies, quiescent SCs activate to repair muscle tissue. Muscle regeneration is also known as adult myogenesis, since it reproduces the events occurring during embryonic development, with the involvement of the same transcription factors as those controlling SCs proliferation and differentiation.66
Myogenesis is regulated by the sequential expression of PAX3 and PAX7 (paired‐domain transcription factors) that operate upstream of basic helix–loop–helix transcription factors known as MRFs (myogenic regulatory factors). PAX7 is expressed in and marks quiescent SCs and is essential for the proliferation and survival of myogenic progenitors, preventing their precocious differentiation. The MRFs (namely, Myf5, MyoD, myogenin, and MRF4) are timely activated in myoblasts to regulate downstream targets such as MyHC (myosin heavy chain), MCK (muscle creatine kinase), and troponin T, necessary for terminal differentiation and generation of functional myofibres.67, 68 PAX7 becomes repressed in differentiating myoblasts/myocytes, which are characterized by the expression of the terminal differentiation marker myogenin.67, 68, 69, 70 Indeed, the expression of PAX7 and myogenin are mutually exclusive in myoblasts, and downregulation of PAX7 is required for myoblast terminal differentiation to occur.71, 72 In myoblasts committed to differentiation, PAX7 expression is downregulated post‐transcriptionally by microRNA‐1, ‐206, and ‐486,73, 74 and post‐translationally by MyoD and myogenin.75 PAX7 is also essential for SC self‐renewal.72, 76 In fact, during muscle regeneration, some myoblasts reacquire a quiescent state through asymmetric division, thus reconstituting the SC reserve pool.66 Many steps of myogenesis can be recapitulated in vitro by culturing primary myogenic cells or myoblast cell lines in low serum conditions, in which myoblasts can form multinucleated myotubes.64 An essential feature of skeletal muscle formation is selective apoptosis that eliminates differentiation‐incompetent myoblasts during myogenesis.77
In skeletal muscle tissue, RAGE expression is developmentally regulated. RAGE can be detected in immature, nearly mature, and some mature myofibres up to 11 days after birth in rodents, with RAGE expression being restricted to the sarcolemma.63 However, RAGE is absent in adult muscle tissue. The presence of both positive and negative muscle fibres in 11‐day‐old rats suggests that repression of RAGE expression occurs around this time.63 This pattern of expression is typical of RAGE, which is expressed during development in several cell types and is repressed in their adult counterparts,36 suggesting that RAGE might play a role during muscle development. Accordingly, proliferating and differentiating myoblasts express nearly constant levels of RAGE mRNA, whereas protein levels significantly decline during late phases of differentiation in vitro.63, 78
RAGE engagement by serum factors, such as HMGB1, promotes and sustains myoblast differentiation and myotube formation.63, 79 The density of RAGE molecules on their surface and the relative concentration of HMGB1 and, likely, other RAGE ligands appear to determine myoblasts' final fate. HMGB1–RAGE interaction in myoblasts results in activation of p38 MAPK via Cdc42–Rac1–MKK6, triggering myogenin and MyHC expression and MCK induction63 (Figure 3 ). Besides promoting differentiation, RAGE engagement by HMGB1 in myoblasts induces cell proliferation arrest through p38 MAPK‐mediated inhibition of the Raf‐MEK‐ERK1/2 pathway with ensuing downregulation of cyclin D1 expression and Rb (retinoblastoma suppressor protein) phosphorylation and upregulation of the proliferation inhibitor, p21WAF1, and JNK inactivation.8, 80 Moreover, HMGB1–RAGE interaction in myoblasts promotes apoptosis via reduction of the anti‐apoptotic factor Bcl‐2 (B‐cell lymphoma 2) expression8 (Figure 3 ).
Figure 3.
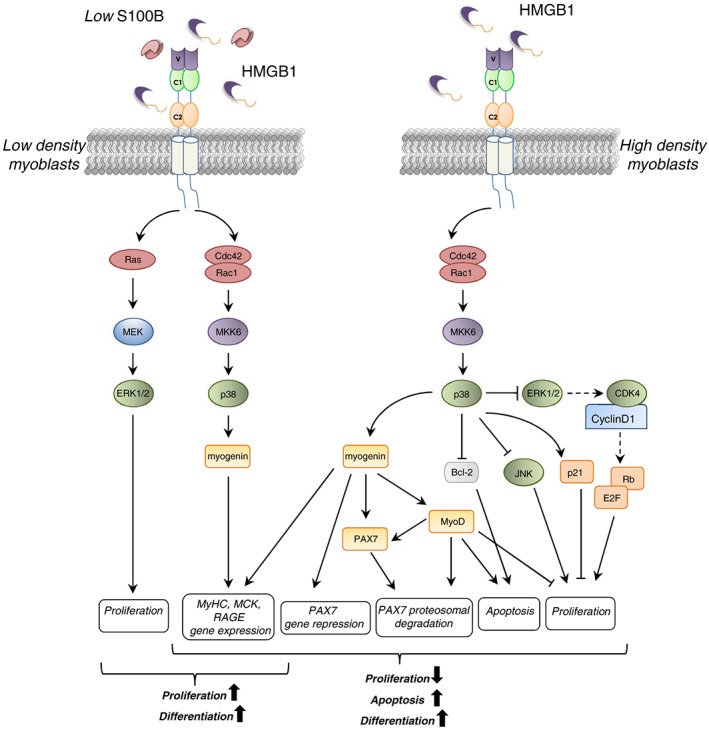
Schematics of S100B/RAGE and HMGB1/RAGE interactions in low‐density (LD) and high‐density (HD) myoblasts. In LD myoblast cultures, interaction of S100B or HMBG1 with RAGE results in the simultaneous stimulation of proliferation and activation of the myogenic programme (left). In HD myoblast cultures, HMGB1‐activated RAGE promotes myoblast differentiation, apoptosis, and myocyte fusion into myofibres (right). HMGB1‐dependent RAGE activation induces p38 MAPK‐dependent expression of myogenin, which upregulates MyoD and represses PAX7 expression, leading to the reduction of proliferation and terminal differentiation. In HD condition, S100B binds bFGF, and the S100B/bFGF complex crosslinks RAGE and FGFR1 on apposed cells, blocking RAGE signalling and enhancing FGFR1 signalling.
RAGE signalling also modifies myoblast morphology and motility. Functional inactivation of RAGE in myoblasts results in decreased cell size along with a less organized cytoskeletal F‐actin and increased filopodia formation, hallmarks of proliferating, migrating, and poorly adherent cells.8 On the other hand, fl‐RAGE‐expressing myoblasts exhibit high levels of adhesion molecules, such as β1‐integrin, NCAM, VCAM, and caveolin‐3, which play an important role in myoblast adhesiveness and differentiation.8 Thus, RAGE activity contributes to the increased adhesion properties and reduced motility of differentiating myoblasts. Further, RAGE activation and signalling in myoblasts result in reduced activity of matrix metalloproteinases (MMPs) 1 and 2,8 important for myoblast migration and invasiveness during early phases of embryonic skeletal muscle formation, when proliferating myoblasts have to reach the place of muscle formation, and during muscle regeneration, when activated SCs migrate towards damaged myofibres.81
However, different RAGE ligands elicit different effects by interacting with the receptor on the myoblast surface. Indeed, S100B can bind RAGE without activating it and block the RAGE promyogenic signalling by preventing its interaction with HMGB1.79, 82 S100B is able to differentially activate RAGE or bFGF (basic fibroblast growth factor)/FGFR1 (fibroblast growth factor receptor 1) depending on its own concentration, the presence or absence of bFGF, and myoblast density.79, 82 In low‐density myoblast cultures, which better reflect the beginning of the regeneration process in vivo, low S100B engages RAGE, thereby simultaneously stimulating cell proliferation via MEK‐ERK1/2 and activating the myogenic programme via p38 MAPK79 (Figure 3 ). S100B oligomers present in the extracellular space bind to RAGE V domain and promote RAGE oligomerization and stabilization, resulting in its activation.83 At high S100B concentrations, higher‐order S100B oligomers might additionally interact with FGFR1‐bound bFGF resulting in the formation of a RAGE/S100B/bFGF/FGFR1 complex on the same cell.79 Here, RAGE cannot efficiently oligomerize and signal, while bFGF‐FGFR1 signalling is enhanced. However, in the absence of bFGF, either low or high S100B activates RAGE in low‐density myoblast cultures, resulting in the simultaneous stimulation of proliferation and the myogenic programme.79 Similar to RAGE, other cell surface receptors belonging to the Ig superfamily such as NCAM (neural cell adhesion molecule), CDO (cell adhesion associated oncogene), and BOC (brother of CDO) are expressed during embryonal skeletal muscle development and sustain myoblast differentiation.84, 85
Although RAGE is not expressed in healthy adult skeletal muscles,63 activated/proliferating SCs and differentiating myoblasts re‐express RAGE upon muscle injury, and RAGE expression is repressed at the completion of the regeneration process.11 ROS produced following muscle damage induce NF‐κB (p65)‐dependent expression of RAGE in activated SCs. Further accumulation of RAGE in differentiating myoblasts is dependent on myogenin.78 During muscle regeneration, RAGE activity is driven by the presence of the RAGE ligands, S100B and HMGB1, which are released with different kinetics upon injury. S100B released by skeletal muscles early after injury stimulates RAGE signalling and enhances RAGE expression in activated SCs, inducing myoblast proliferation and activating the myogenic programme.9, 79 However, during the proliferation phase, S100B and bFGF are present at the same time in the extracellular space, and S100B promotes myoblast expansion by enhancing bFGF/FGFR1 signalling. At this time, RAGE is recruited into a RAGE/S100B/bFGF/FGFR1 tetracomplex that impairs RAGE signalling.79 HMGB1 is passively released from muscle tissue following damage and progressively accumulates during the regeneration process.78, 79, 86 By binding RAGE on the myoblast surface, HMGB1 activates p38 MAPK, thus upregulating the expression of myogenin, which in turn upregulates RAGE expression. This positive feedback loop leads to myogenin accumulation culminating in the repression of PAX7 expression through binding of myogenin to specific consensus sequences in the PAX7 gene promoter.78 This mechanism strongly contributes to the mutually exclusive presence of PAX7 and myogenin in muscle precursor cells. Besides translating into reduced myoblast proliferation rate, the downregulation of PAX7 subsequent to HMGB1–RAGE interaction promotes symmetric division in activated SCs, thereby limiting SC self‐renewal (Figure 4 ). Therefore, RAGE might accelerate myogenic differentiation and the regeneration process and optimize myofibre size and the SC number.78 Consistent with these results, acutely injured skeletal muscles of Ager −/− mice are characterized by excess myoblast proliferation and elevated myoblast asymmetric division leading to delayed regeneration.11 Also, during early regeneration phases, the number of infiltrating macrophages in Ager −/− injured muscles is smaller than that of controls, in accordance with the role of RAGE in macrophage migration,29 thus conditioning the timing of muscle regeneration78 (Figure 4 ).
Figure 4.
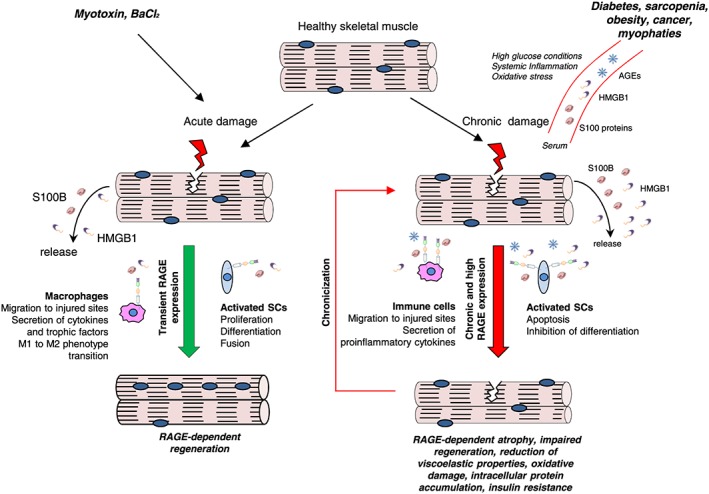
Role of RAGE in acute or chronic muscle injury. Upon acute skeletal muscle injury, re‐expressed RAGE in activated satellite cells results in their proliferation and differentiation ultimately leading to regeneration. Under the action of S100B, RAGE signalling promotes proliferation and simultaneously activates the myogenic programme. RAGE activated by HMGB1 causes proliferation arrest and stimulates terminal differentiation of myoblasts into myocytes that build up new myofibres and/or repair damaged myofibres. RAGE‐mediated signalling in infiltrating macrophages modulates the inflammatory response in injured muscles, contributing significantly to accelerate regeneration. S100B/RAGE axis also promotes macrophage infiltration of acutely damaged skeletal muscle and macrophage polarization to M2 phenotype (left). On the contrary, in the course of muscle diseases characterized by chronic inflammation, high glucose conditions, or oxidative stress, an accumulation of RAGE ligands occurs in the serum and damaged myofibres. The overexpressed and overstimulated RAGE amplifies the inflammatory response by stimulating proinflammatory cytokines secretion, induces apoptosis of myocytes, and causes lethal accumulation of oxidative stress contributing to chronic myofibre damage (right).
Infiltrating M1 (classically activated) macrophages contribute significantly to muscle regeneration by removing cell debris and providing important factors for the activation, migration, and proliferation of SCs.87 The transition of macrophages from a pro‐inflammatory (M1) to an anti‐inflammatory (M2) phenotype is crucial for efficient tissue repair,88 since M2 (alternatively activated) macrophages dampen the inflammatory response and promote muscle regeneration.29, 88 RAGE‐mediated signalling in infiltrating macrophages modulates the inflammatory response in injured muscles, contributing significantly to accelerate regeneration. Indeed, absence of RAGE results in a delayed and amplified inflammatory response9, 78 (Figure 4 ).
As mentioned earlier, complement C3a interacts with RAGE,29 and complement C3a has been recently shown to promote muscle regeneration following acute injury by activation of the so‐called alternative complement's pathway required for initiating recruitment of circulating monocytes into injured muscle.89 It can be hypothesized that RAGE might participate in the muscle regeneration process by transducing complement C3a effects. Besides, the S100B/RAGE axis promotes macrophage infiltration of acutely damaged skeletal muscle and macrophage polarization to M2 (anti‐inflammatory and pro‐regenerative) phenotype.9
Thus, in acutely injured muscles, RAGE affects muscle regeneration by a dual mechanism, promoting macrophage infiltration and polarization to M2 phenotype and regulating SC/myoblast activity, thus allowing proper myoblast proliferation and differentiation phases (Figure 4 ). However, excess RAGE signalling consequent to accumulation of S100B (e.g. in muscular dystrophy) results in excess macrophage infiltration and prolongation of the inflammatory phase, ultimately leading to disturbed regeneration9 (Figure 4 ).
HMGB1 levels increased in muscles after ischaemia induced by femoral artery ligation, and blockade of endogenous HMGB1 by administration of its truncated form, BoxA, resulted in reduction of vessel density and delayed regeneration, the opposite being observed with intramuscular administration of HMGB1.90 The ability of HMGB1 to sustain skeletal muscle regeneration was attributed to a direct effect on myoblasts, enhancing their recruitment to the site of injury, as well as to the arteriogenic action of HMGB1. Although myoblasts and endothelial cells were found to express the HMGB1 receptors, RAGE and TLR4, the authors did not establish which receptor mediates the HMGB1's effects in these conditions. However, others reported that, despite HMGB1–RAGE interaction is important for the recovery of the skeletal muscle following acute injury consequent to hind limb ischaemia, RAGE was proposed not to play a significant role in muscle regeneration.91 In these conditions, neutralization of HMGB1 resulted in prolonged muscle necrosis, suggesting that HMGB1 released in the setting of ischaemia is critical for promoting muscle recovery. However, while TRL4 KO mice showed impaired regeneration, RAGE KO mice exhibited muscle histology that was indistinguishable from control animals, suggesting that HMGB1 effects are mediated by TLR4 rather than RAGE in these experimental conditions.91 It is possible that strain differences of RAGE and TRL4 KO mice affect muscle regeneration after ischaemia. Another possibility stands on the fact that HMGB1 can exist into two redox forms, an oxidized one and a reduced one, that are able to differentially activate RAGE and TRL4. The HMGB1's ability to activate RAGE is restricted to the oxidized form of the protein.92, 93 Further studies are required to determine the exact role of RAGE and TLR4 in the HMGB1's effects in muscle tissue after ischaemic injury.
RAGE and skeletal muscle atrophy
Skeletal muscle wasting (atrophy) is a complex and highly regulated process, characterized by a substantial decrease in muscle mass, myofibre area, protein content, regenerative capacity, and muscle strength, together with increased myocyte apoptosis and fatigability.4, 94 In addition to cancer, muscle wasting is associated with ageing, disuse, diabetes, denervation, and many chronic diseases characterized by a variable degree of systemic chronic inflammation, in which muscle atrophy contributes to morbidity and mortality.3 In healthy young individuals, skeletal muscle maintains mass and function through the balance of intricate signalling networks that regulate muscle protein synthesis (hypertrophy) and degradation (atrophy),95, 96 leading to physiological levels of muscle proteins. In muscle atrophy, this balance shifts towards a catabolic state resulting in the breakdown of myofibrillary proteins due to activation of the two major intracellular proteolytic systems (i.e. ubiquitin‐proteasome [UPP] and autophagy) and decrease in protein synthesis.3, 95, 96 Myogenic precursors, including SCs, also might contribute to the maintenance of skeletal mass in health and diseases,97 and recent work has shown that atrophy factors inhibit their regenerative and fusion potential.98, 99
Systemic atrophy factors affect muscle mass by interfering with catabolic and anabolic pathways. In particular, inflammatory cytokines deactivate the PI3K/AKT/mTOR (mammalian target of rapamycin) pathway, leading to the reduction of protein synthesis and activation of the FOXO3 (forkhead box O3) transcription factor. FOXO3 is sufficient to induce the expression of the muscle‐specific ubiquitin ligases atrogin‐1/MAFbx (muscle atrophy F‐box protein) and MuRF‐1 (muscle RING finger‐1), collectively called atrogenes, and at the same time autophagy‐associated proteins (Atgs), leading to selective proteolysis of MyHC in skeletal muscle.100, 101, 102 On the other side, inflammatory cytokines activate NF‐κB and p38 MAPK, resulting in a direct induction of MuRF‐1 and atrogin‐1, respectively.3, 96, 103 Many other pathways such as AMPK (AMP‐activated kinase), ERK, and JNK are involved in the activation of UPP system by regulation of FOXO3 activity and atrogene expression.104, 105
RAGE in diabetes‐induced atrophy
Diabetic myopathy is characterized by impaired muscle growth and development, loss of muscle mass, a switch to a glycolytic myofibre phenotype, muscle weakness, and a reduced physical functional capacity that influence the rate of co‐morbidity development.106, 107 Also, the ability of SCs to repair damaged myofibres is adversely affected by diabetes environment probably due to prolonged exposure to high glucose.97, 108, 109
Early evidences showed that CML accumulates in myofibres of the plantaris muscle of diabetic rats and that the sites where CML accumulates are associated with fibre‐type transformation, which in turn is associated with muscular endurance.110, 111 Increased blood levels of different AGEs, including CML, and increased levels of unspecified muscle AGEs and RAGE were found in diabetic patients and mice.112 Recently, the role of AGEs and RAGE as molecular players in diabetic myopathy has been examined in some depth using different approaches, that is, diabetic mice, cultured muscle cells, and patient samples.112, 113 In a cell culture model mimicking diabetic conditions, high glucose levels promote the production and accumulation of AGE–BSA, which in turn are responsible for the membrane repair defect. In particular, AGE–BSA, even in the absence of high glucose, might cause repair defects by RAGE engagement, resulting in cell death and loss of muscle mass.113 Consistently, RAGE‐deficient cells are resistant to glucose‐induced repair defects.113
It is intriguing that the alagebrium chloride, an AGE crosslink breaker with chelating proprieties, used as a therapeutic compound in diabetic complications in various experimental animal models,114 is a potential therapeutic candidate to improve diabetic myopathy, including muscle wasting, muscle atrophy, and muscle regenerative capacity, by reducing plasma and muscle AGE levels in diabetic mice.112 Accordingly, AGE–BSA induced atrophy in myotube cultures and strongly inhibited myoblast differentiation through a RAGE‐mediated mechanism. In particular, AGE–BSA activated AMPK, a negative mediator of muscle mass, and downregulated Akt signalling, leading to activation of the atrophy marker Atrogin‐1 and reduction of myotube size. Moreover, AGE–BSA treatment showed detrimental effects in myoblast differentiation and fusion by reducing myogenin and MHC expression via phosphorylation of AMPK and dephosphorylation of Akt.112 The detailed mechanism acting in the negative regulation of muscle SC/stem cell homeostasis by AGE/RAGE in vivo remains to be elucidated.
A decrease in insulin action on target tissues, defined as insulin resistance (IR), is characteristic of type 2 diabetes.115, 116 Abnormal accumulation of MGL, a highly reactive AGE precursor, occurs in diabetic patients,117, 118 and MGL‐derived AGEs were shown to activate RAGE signalling.26, 119, 120, 121, 122, 123 When MGL was reduced in type 2 diabetic patients, IR was improved.124 Recently, an interesting study proposed a novel MGL‐derived AGE inhibitor, MK‐I81, as a potential therapeutic compound to alleviate AGE‐mediated downregulation of insulin signal transduction and insulin action in skeletal muscle cells restoring insulin sensitivity.125
In addition to those formed endogenously, AGEs are abundant in exogenous sources such as food prepared under elevated temperatures. Dietary AGEs have been shown to induce IR in mice also in absence of hyperglycaemia.126 Accordingly, reduced intake of AGEs has been shown to decrease the incidence of type I diabetes in non‐obese diabetic mice127 and improved insulin sensitivity in db/db mice.128, 129 Also, similar to exposure to glycated albumin, chronic exposure of L6 myoblasts to dietary AGEs induces the formation of a complex including RAGE‐PKCα‐Src that leads to phosphorylation of IRS (insulin receptor substrate) and inhibition of insulin action.126, 130 Lastly, muscle tissue from weight gainers and elderly subjects show a marked increase in immunostaining intensity for CML and RAGE in association with immunostaining of markers of oxidative stress and inflammation.131 Future studies would hopefully provide a complete picture of the role of RAGE and its ligands in the pathophysiology of skeletal muscle disorders in diabetes (Figure 4 ).
RAGE in sarcopenia
Sarcopenia defines an age‐related, continuous decline in muscle mass, quality, and strength.132, 133 Sarcopenia is characterized by an overall decrease in size and number of skeletal muscle fibres, mostly the type 2 or fast‐twitch muscle fibres, and a marked infiltration of skeletal muscle with fibrous and adipose tissue.4, 134 Many factors contribute to sarcopenia, including changes in skeletal muscle metabolism, hormonal changes, compromised regeneration, elevated levels of inflammatory cytokines, changes in vasculature, inactivity, and oxidative stress. Besides the activation of autophagy/lysosome systems leading to protein degradation, there is consensus regarding an age‐related decline in the performance of SCs as a primary cause of sarcopenia. In this regard, both extrinsic factors of the extracellular environment (the so‐called SC ‘niche’) and intrinsic properties of the SCs have been proposed to contribute to sarcopenia.135, 136, 137, 138
Interestingly, early work139 showed that compared with their young counterparts, SCs isolated from aged (>72 years old) human subjects (i) exhibit higher intracellular levels of the RAGE ligand, S100B, known to inhibit myoblast differentiation via NF‐κB‐dependent repression of MyoD (and myogenin) expression140; (ii) release lower amounts of mitogenic factors such as S100B and bFGF; and (iii) express a truncated form of RAGE in plasma membrane. The altered levels of S100B and RAGE correlated positively with the defective proliferation and differentiation potential exhibited by aged SCs, a notion reinforced by the observation that either transient transfection with fl‐RAGE or knockdown of S100B switched aged SCs to a young phenotype and conversely, functional inactivation of RAGE or transient transfection with S100B switched young SCs to an aged phenotype.139 Lastly, treatment of aged SCs with S100B, bFGF, or young SC‐conditioned medium enhanced aged SC proliferation and their subsequent differentiation. Also interestingly, chronic oxidative conditions lead to accumulation of S100B in an NF‐κB‐dependent manner in myoblasts and S100B‐stimulated conversion of myoblasts into brown adipocytes via an NF‐κB/YY1/miR‐133 axis and NF‐κB/YY1/BMP‐7 axis, events prevented by S100B knockdown.141 In this same experimental setting, interstitial cells and, unexpectedly, a subpopulation of myofibres in muscles of geriatric but not young mice were found to co‐express S100B and the brown adipocyte marker, uncoupling protein‐1. However, RAGE expression and activity were not studied in this experimental setting, thus whether RAGE has a role in S100B‐induced myoblast‐brown adipocyte conversion and/or RAGE transduces effects of chronic oxidative stress in myofibres remain elusive.
AGE‐modified proteins are known to become resistant to degradation by the UPP machinery, and the accumulation of modified proteins in several tissues, including skeletal muscle, has been considered as one of several potential mechanisms producing age‐related functional decline of diverse organs.142 Several evidences point to a correlation between the decline of muscular function in healthy aged subjects and circulating or muscle CML, CEL, and pentosidine. In this respect, the AGE product, pentosidine, is increased by more than 200% in skeletal muscle of older adults compared with younger adults, suggesting that glycated‐related cross‐linking of intramuscular connective tissue might cause the reduction of viscoelastic properties of muscle, thus contributing to the decline in muscle function with ageing.143, 144 CML may also play a role in sarcopenia through upregulation of inflammation and endothelial dysfunction in the microcirculation of skeletal muscle via RAGE by inducing the loss of myocytes and loss of muscle mass and strength145 (see Figure 4 ).
RAGE and skeletal muscle diseases
Myopathies are a large group of clinical disorder of skeletal muscles characterized by progressive weakness due to abnormalities of myofibre structure and metabolism, which ultimately leads to severe disability of the patient, pain, and death. Based on aetiology, myopathies can be divided into two main categories: inherited (i.e. muscular dystrophies, congenital myopathies, mitochondrial myopathies, and metabolic myopathies) and acquired (i.e. myositis or idiopathic inflammatory myopathies, toxic myopathies, and myopathies associated with systemic conditions).146, 147
RAGE in inherited myopathies
Muscular dystrophies (MDs) are a group of disorders that share clinical and pathological features, characterized by progressive muscle degeneration, affecting principally limb, axial, and facial muscles. There are different forms of MDs depending on the severity, age of onset, rate of progression, consequent implications, and prognosis.148 The actual classification of MDs into seven major groups is based on the links between protein mutations, their localization and functions, and the final clinical phenotype and include Duchenne muscular dystrophy (DMD), limb girdle muscular dystrophies (LGMDs), and facioscapulohumeral muscular dystrophy (FSHD).148
RAGE is detectable and co‐localizes with CML and NF‐κB in regenerating myofibres and less frequently in degenerating or necrotic myofibres of patients with LGMDs, even if with a lower staining intensity than in inflammatory myopathies.149 Moreover, RAGE is overexpressed in FSHD muscles when compared with normal and DMD muscles150 likely consequent to increased production of AGEs due to elevated ROS levels. AGE/RAGE interaction is known to increase ROS production via NF‐κB activation,150, 151 thus perpetuating the oxidative cellular damage involved in the FSHD pathogenesis.152
RAGE in acquired myopathies
Myositis is a heterogeneous group of disorders that are characterized clinically by chronic muscle weakness, fatigue, and damaged myofibres due to infiltration of inflammatory cell in muscle tissue. Based on differences in clinical and histopathological features, myositis can be subdivided into polymyositis (PM) and dermatomyositis (DM), which are chronic autoimmune diseases, and inclusion body myositis (IBM), the most common degenerative muscle disease in adults over the age of 50.153
In PM and DM, an overexpression of RAGE and its ligands, HMGB1 and CML, has been demonstrated in immune cells as well as in regenerating muscle fibres. Treatment with glucocorticoids, conventionally used in patients with PM and DM, was associated with a reduced expression of HMGB1, suggesting a clinical relevance of HMGB1–RAGE signalling to initiate and sustain muscle inflammation.149, 154
The hallmark of IBM is interfibre protein accumulation (inclusions) among which is β‐amyloid, besides muscle infiltration with inflammatory cells.155 Interestingly, HMGB1 and RAGE levels increase in skeletal muscle of patients with IBM,156 and the HMGB1–RAGE axis induces intracellular protein accumulation in IBM as well as cell death. Thus, similar to AD where the β‐amyloid–RAGE interaction is crucial for the cellular damage and cognitive neuronal dysfunction,157 RAGE may be a relevant mediator in the degenerative mechanism of muscle in IBM. Furthermore, there is evidence of significantly elevated serum levels of pentosidine and CML and accumulation of CML in the muscle tissue of patients with fibromyalgia (FM), a frequent rheumatic disorder of unknown aetiology and pathogenesis.158 In this same study, a more intensive staining for RAGE, NF‐κB, and CD68‐positive monocytes/macrophages was found in the interstitial connective tissue of muscle of FM patients suggesting that AGEs' binding to RAGE might prolong NF‐κB activity leading to the secretion of pro‐inflammatory cytokines, which in turn might contribute to pain generation and perpetuation of inflammation.
Myasthenia gravis (MG) is an autoimmune disease caused by the production of antibodies against receptors at the neuromuscular junction such as nicotinic acetylcholine receptor and is clinically characterized by weakness of facial, ocular, and limb muscles.159 An analysis of a broad inflammatory circulating protein and cytokine profile in patients affected by different MG subtypes revealed the presence of EN‐RAGE (also known as S100A12 protein) in the sera of MG patients.160, 161 Besides being a potential biomarker in MG, EN‐RAGE/S100A12 might contribute to MG pathogenesis by binding to RAGE to activate NF‐κB and induce secretion of pro‐inflammatory cytokines, a property shared with several other S100 proteins.20, 162
RAGE and skeletal muscle metabolism
Skeletal muscle can be considered as the largest metabolic organ in the human body, and metabolic dysfunctions occurring in skeletal muscle in diabetes, obesity, sarcopenia, cancer cachexia, and muscular dystrophies are characterized by a ‘metabolic reprogramming’ and can affect the whole‐body nutrient homeostasis.1, 2 Recently, various authors have advanced the idea that the presence of tumour promotes metabolic reprogramming of the skeletal muscle tissue in order to counter the high energy demand of tumour tissue and sustain proliferation and cell‐death resistance of tumour cells. In this scenario, the dramatic muscle wasting occurring in cancer, known as cancer cachexia, may be considered as a part of the metabolic reprogramming in cancer patients. A metabolic crosstalk has been identified in colorectal cancers between tumour masses and skeletal muscle. During the development and progression of colorectal cancer, expression of the autophagy‐inducing stress protein HMGB1 increased in the muscle of tumour‐bearing animals.163 This effect was associated with decreased expression and activity of the pyruvate kinase PKM1, which mediates the linker reaction between glycolysis and the TCA cycle in normal tissues muscle via RAGE. Colon cancer cells release HMGB1 in the serum, which decreases mTOR levels in muscle through RAGE‐dependent p38 phosphorylation, thus increasing autophagy‐associated proteins (beclin‐1 and LC3II). This results in increased plasma glutamate and free amino acids that supply energy to cancer cells.163
RAGE and rhabdomyosarcoma
Rhabdomyosarcomas (RMSs) are highly malignant tumours of skeletal muscle origin accounting for more than 50% of soft tissue sarcomas in childhood. It is widely accepted that RMSs represent aberrant stages of normal muscle development: like normal muscle precursor cells, RMS cells express PAX3, PAX7, and MRFs such as myogenin, all of which are hallmarks of RMS.164, 165 Alveolar RMSs (ARMSs) and embryonal RMSs (ERMSs) are the most frequent variants, with ERMSs accounting for 60–70% of RMSs. ERMSs show the phenotypic and biological features of embryonic muscles, suggesting that they may arise from muscle progenitor cells, such as activated SCs or poorly differentiated myoblasts165, 166 The tumour properties of ARMSs and ERMSs have been associated to PAX genes because of the transcriptional hyperactivity of PAX3/FOXO1A (forkhead box protein O1) or PAX7/FOXO1A fusion transcripts in ARMSs and the high level of PAX7 expression in ERMSs.167, 168 As mentioned earlier, the persistence of PAX7 in myoblasts results in prolonged proliferation and defective differentiation,74, 75 implying that conditions leading to accumulation of PAX7 in myoblasts can potentially promote ERMS formation. Moreover, PAX activity regulates the expression of cell surface molecules involved in cell adhesion, migration, and invasiveness, particularly HGFR (hepatocyte growth factor receptor), also called Met, ephrins (EFNs) and Eph receptors (EPHs), and NCAM in various cell types.168 Studies of the role of RAGE in RMS have shown that high PAX7 levels in ERMS cells are partly dependent on reduced and/or absent RAGE signalling and are major cause of their uncontrolled proliferation and the acquisition of a metastatic behaviour.8, 169, 170 As mentioned earlier, HMGB1–RAGE interaction on myoblasts results in enhanced myogenic differentiation with simultaneous cell proliferation arrest, repression of PAX7 transcription, and enhanced PAX7 degradation by a proteasomal‐dependent mechanism69, 71, 78, 171 and apoptosis induction.8 Indeed, an inverse relationship exists between RAGE and myogenin protein levels and those of PAX7 in muscle precursor cells170 (Figures 3 and 5 ). Interestingly, inoculation of myoblasts stably overexpressing RAGEΔcyto into immunocompromised mice results in much larger and more aggressive tumour formation compared with inoculation of myoblasts stably overexpressing fl‐RAGE.8 Tumour masses derived from RAGE‐defective myoblasts appeared earlier and were characterized by large central areas of necrosis and peripheral neovascularization compared to masses formed by myoblasts harbouring fl‐RAGE. Injected cells invaded the neighbouring skeletal muscle tissue in the case of RAGEΔcyto myoblasts only, and the masses obtained in this case showed hallmarks of ERMSs.8
Figure 5.
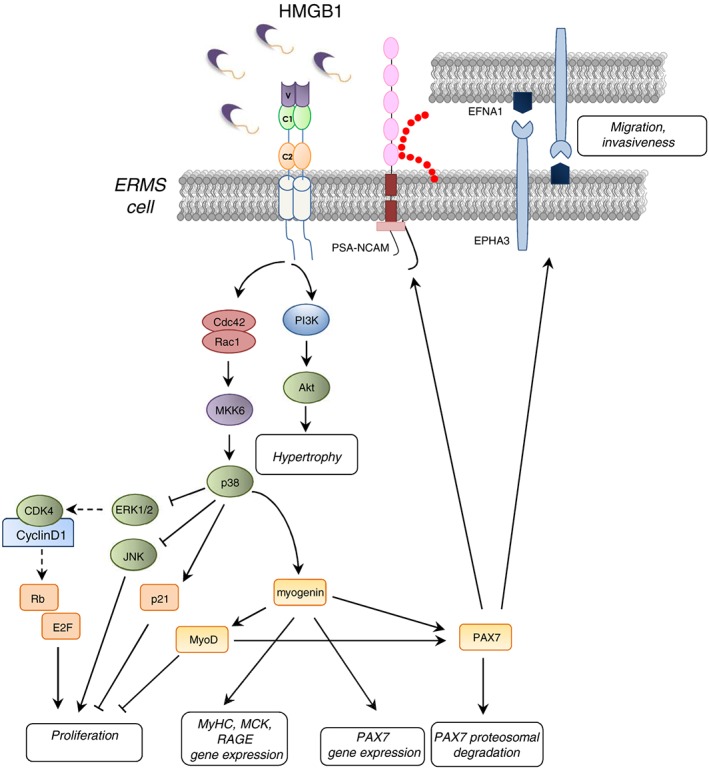
Schematic representation of the effects of enforced expression of RAGE in TE671 ERMS cells. RAGE engaged by HMGB1 stimulates myogenic differentiation through activation of MKK6/p38 MAPK and myotube hypertrophy through PI3‐K/Akt. HMGB1/RAGE‐dependent activation of p38 MAPK also causes inactivation of ERK1/2 and JNK with consequent inhibition of proliferation and decrease in cell survival. Induction of RAGE in ERMS cells also results in the expression of myogenin and MyoD so as to cause repression of PAX7 expression and PAX7 proteasomal degradation, ultimately leading to reduction of proliferation, enhancement of apoptosis, and myogenic differentiation. Finally, the reduction of Pax7 expression by the RAGE/myogenin axis causes a reduction of cell migration and invasiveness in ERMS.
Studies of several RMS cell lines showed that the ability of RMS cells to undergo myogenic differentiation appears to be dependent on the amount of functionally active RAGE.171 Indeed, TE671 cells stably transfected with RAGE showed HMGB1‐dependent activation of Cdc42–Rac1–MKK6–p38 MAPK and PI3‐K/Akt pathways, leading to myogenin and MyHC expression and MCK induction and partially reduction of JNK's anti‐myogenic activity (Figure 5 ). Thus, RAGE expression in TE671 cells is sufficient to restore the myogenic programme through sustained activation of p38 MAPK, which is deregulated in ERMS cells.171
Notably, ectopic expression of CDO or BOC in ERMS cells also restores the ability of rhabdomyosarcoma cells to undergo terminal myogenic differentiation, leading to enhanced expression of sarcomeric proteins and formation of MHC‐positive myotubes.172 These observations indicate that the Ig superfamily members RAGE, CDO, and BOC may have a central role in the inverse relationship between cell differentiation and transformation occurring in myogenic precursors during muscle development.
Restoration of RAGE activity in TE671 cells also resulted in dramatic morphological changes (e.g. cell hypertrophy and stress fibre formation) consistent with their reduced proliferation and invasiveness and increased adhesiveness. Further, the RAGE‐dependent decrease in TE671 cell aggressive potential caused a reduction of in vivo tumour growth and incidence.171 Thus, functional inactivation of RAGE or repression of RAGE expression in myoblasts might confer an aggressive potential on myoblasts and contribute to RMS genesis via PAX7‐dependent upregulation of the ephrin receptor, EPHA3, and ephrin A1 (EFNA1) and a predominance of polysialilated (PSA)‐NCAM, all of which have a role in sustaining cell migration and invasiveness169 (Figure 5 ). In this latter context, measurements of RAGE levels have been proposed for ERMS grade diagnosis. However, inoculation of Ager −/− myoblasts into NOD–SCID mice did not result in tumour development, and no RMS formation could be observed in Ager −/−mice irrespective of their age, suggesting that RAGE absence or reduced activity should be accompanied by other factors/conditions for RMS formation to occur.169 Interestingly, RAGE was found absent or expressed at very low levels in a panel of 31 human ERMSs.170 Further investigation should establish the clinical correlation between RAGE expression in ERMS primary tumours and patient outcome, with particular attention to metastasis occurrence.
RAGE is generally considered as a factor promoting proliferation, survival, migration, and invasion of tumour cells, as is the cases with glioma, melanoma, and pancreas, prostate, and colon cancers173 (see Box 3 ). Nevertheless, RAGE expression in non‐small cell lung carcinoma and oral or esophageal squamous cell carcinomas negatively correlates with tumour growth and depth of invasion174, 175 (see Box 3), similar to ERMS cells. The reason why RAGE has opposite roles depending on the tumour cell type should be further investigated, and this ambiguous behaviour suggests that caution should be exerted in setting RAGE‐targeted anticancer protocols.
RAGE inhibitors
The involvement of RAGE in the pathophysiology of several diseased conditions has prompted studies aimed to identify endogenous inhibitors and synthesize chemical inhibitors. Indeed, a variety of RAGE antagonists are now available for preclinical and clinical studies.176
To date, several small‐molecule inhibitors have been developed to contrast RAGE activity in several diseases that bind either the extracellular or intracellular domain. Among the extracellular inhibitors of RAGE, TTP488 ([3‐(4‐{2‐butyl‐1‐[4‐(4‐chlorophenoxy)‐phenyl]‐1H‐imidazole‐4‐yl}‐phenoxy)‐propyl]‐diethylamine 1), also known as azeliragon or PF‐04494700, is an orally bioavailable small molecule able to cross the blood–brain barrier (BBB).177 TTP488 is able to inhibit the interaction between RAGE and Aβ, S100B, HMGB1, and AGEs and preclinical studies have demonstrated that TTP488 efficiently contrasts complications due to RAGE activity in diabetes, cardiovascular disease, cancer, and AD.42, 178 On the basis of the results obtained in a mouse model of AD, in which TTP488 administration inhibited inflammation, neuronal Aβ accumulation, and neurocognitive decline, a phase III clinical trial of TTP488 is currently in progress in an AD cohort of 800 human patients.179 Several derivatives of TTP488, set up to reduce the toxic proprieties of this inhibitor when used at elevated doses, have been developed and demonstrated able to inhibit the disease in mouse model of AD.180, 181 However, none of these derivatives have progressed to human clinical trial yet.
Similarly, the compound FPS‐ZM1 (N‐benzyl‐N‐cyclohexyl‐4‐chlorobenzamide 12) was selected through the screening of 5000 compounds in the search for inhibitors of RAGE/Aβ interaction. FPS‐ZM1 is able to cross the BBB and blocks inflammatory signalling in the mouse brain, reduces Aβ accumulation, and improves cognitive performance without causing toxic side effects in mice, even at high doses.182 The efficacy of FPS‐ZM1 was explored in other animal models of neuropathology (such as hippocampal inflammation and brain stroke), heart failure, asthma, and cancer progression/metastasis of lung and liver tumours.183, 184 The results demonstrated that the administration of FPS‐ZM1 reduces or abrogates the pathogenic effects caused by RAGE activity by reducing AGE–RAGE interactions. Further, small‐molecule inhibitors have been identified, which interfere with the RAGE–mDia‐1 interaction. In vitro and in vivo RAGE−Dia‐1 competitive inhibitors displayed inhibitory effects on AGE–RAGE signal transduction by reducing inflammation, vascular cell migration, and pro‐inflammatory cytokines.185 Although these results are promising, further studies are needed to establish chemical refinement, tolerance, selectivity, and pharmacokinetics of these compounds and to test whether intracellular and extracellular inhibitors are effective therapeutics in several diseases.
Conclusions
Increasing evidence suggests that RAGE signalling plays an important role in skeletal muscle during the development, dictating the fate of myogenic precursor cells. Once activated by S100B or HMGB1, RAGE regulates the fine balance between apoptosis, proliferation, and differentiation in myoblasts so that absent or reduced RAGE signalling in activated muscle SCs might concur to RMS genesis. Accordingly, restoring RAGE signalling can be a potential therapeutic tool in ERMSs by promoting terminal differentiation, reducing proliferation, and repressing the expression of molecules responsible for cell migration and invasiveness. Also, the timely expression of RAGE during skeletal muscle development and regeneration, together with its absence in healthy adult muscle tissue, highlights the role of this receptor in building muscle mass. The fact that Ager −/− mice do not show overt muscle defects and do not spontaneously develop RMS suggests that other receptors belonging to the same superfamily (e.g. NCAM, CDO, and BOC) might compensate for the lack of RAGE activity.
Several studies demonstrated that RAGE is re‐expressed in muscle under acute or chronic inflammatory conditions, and the local environment appears to determine the outcome of RAGE signalling. In acutely injured myofibres, re‐expressed RAGE supports the restoration of tissue homeostasis and concurs to tissue repair by promoting myoblast proliferation and differentiation and the resolution of inflammation. On the contrary, in the course of muscle diseases characterized by chronic inflammation, RAGE signalling appears to concur to the pathogenesis by amplifying and perpetuating the inflammatory response, inducing apoptosis of myocytes, and causing lethal accumulation of oxidative stress leading to myofibre damage. Thus, RAGE is likely to represent one main factor intervening in the fine regulation of the balance between injury and repair: in acute muscle injury, short‐term RAGE signalling triggered by relatively low concentrations of RAGE ligands results in beneficial effects, whereas in chronic muscle injury conditions, long‐term RAGE signalling triggered by high concentrations of RAGE ligands is deleterious. Although additional studies are required to better elucidate the role of RAGE and its ligands in myopathies, blockade of ligand–RAGE interaction results in reduction of inflammation and ROS‐induced myofibre damage, thus representing a potential therapeutic strategy.
Although several studies suggest that AGE–RAGE is able to activate well‐known signalling pathways and transcriptional factors inducing muscle atrophy, membrane damage, and insulin resistance in diabetes, these findings suffer from some limitations. The techniques used in in vivo studies are not able to differentiate the type of AGEs capable of recruiting RAGE, and supraphysiological doses of albumin were used in in vitro studies to provide a heterogeneous level of AGEs. Additional studies are needed to discern the true physiological relevance of such findings in the context of RAGE activation in diabetes.
Interestingly, CML–RAGE interactions have been reported to stimulate ROS formation determining systemic oxidative stress and inflammation and loss of muscle mass in obesity and during ageing. However, the causal relationship between high levels of specific AGEs and sarcopenia/obesity is still elusive. The data obtained so far suggest that RAGE is involved in the pathogenesis of these two important medical problems through common mechanisms that await elucidation, though. Moreover, the opposite behaviour of RAGE in skeletal muscle might be explained by the presence and expression levels of different types of RAGE‐binding proteins in physiological and pathological conditions. We can hypothesize that the different effects of RAGE in muscle precursor cells in acute injury versus chronic inflammation/elevated ROS conditions might be explained by a differential availability of RAGE adaptor proteins and/or physical/functional interactions between RAGE and receptors shown to share with RAGE certain ligands. The discovery of these binding partners in skeletal muscle might hopefully elucidate the role of RAGE in maintaining or altering muscle homeostasis. The behaviour of RAGE in skeletal muscle may be also affected by the state of RAGE phosphorylation, which has been demonstrated to dictate both the downstream signalling of RAGE and the translocation of the receptor to the nucleus or mitochondria, where RAGE might exert yet unidentified function(s) in skeletal muscle.
A few studies have shown that RAGE signalling contributes to the remodelling of skeletal muscle metabolism by reducing protein reserve in chronic stress conditions, such as cancer. Considering that the majority of metabolic disorders induce muscle degenerative diseases and that high serum levels of RAGE ligands correlate positively with cancer severity in a variety of tumour types, it will be important to explore RAGE's activity in tumour‐induced metabolic disturbances and RAGE's role(s) in the crosstalk between metabolic diseases and cancer.
In this respect, several reports suggest that modulation of RAGE activity might be of therapeutic importance for skeletal muscle in various disease states that have chronic inflammation and oxidative stress as common stigmata. Interestingly, a number of RAGE inhibitors (such as TTP488 and FPS‐ZM1) have been developed, and some of them resulted well tolerated in human clinical trials. Potentially, these molecules might be useful for skeletal muscle diseases as well. However, caution should be exerted with the use of pharmacological blockers of RAGE signalling, since the complete absence of RAGE activity might severely compromise SC function and skeletal muscle regeneration. Modulation rather than inhibition of RAGE activity might represent a successful approach to restore muscle homeostasis in different pathological conditions affecting skeletal muscle tissue.
Conflict of interest
The authors declare no conflict of interest.
Funding
The authors were supported by the Association Française contre les Myopathies (projects 12992 and 16812), the Associazione Italiana per la Ricerca sul Cancro (project 17581), the Ministero dell'Istruzione, dell'Università e della Ricerca, Italy (PRIN 2009WBFZYM_002, PRIN 2010R8JK2X_004, and PRIN 2012N8YJC3), and the Fondazione Cassa di Risparmio di Perugia (projects 2012.0241.021, 2015.0325.021, and 2016‐0136.021).
Acknowledgement
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017.202
Riuzzi, F. , Sorci, G. , Sagheddu, R. , Chiappalupi, S. , Salvadori, L. , and Donato, R. (2018) RAGE in the pathophysiology of skeletal muscle. Journal of Cachexia, Sarcopenia and Muscle, 9: 1213–1234. 10.1002/jcsm.12350.
Contributor Information
Francesca Riuzzi, Email: francesca.riuzzi@unipg.it.
Rosario Donato, Email: rosario.donato@unigp.it.
References
- 1. Frontera WR, Ochala J. Skeletal muscle: a brief review of structure and function. Calcif Tissue Int 2015;96:183–195. [DOI] [PubMed] [Google Scholar]
- 2. Schnyder S, Handschin C. Skeletal muscle as an endocrine organ: PGC‐1α, myokines and exercise. Bone 2015;80:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013;6:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev 2011;91:1447–1531. [DOI] [PubMed] [Google Scholar]
- 5. Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 1992;267:14998–15004. [PubMed] [Google Scholar]
- 6. Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 2005;83:876–886. [DOI] [PubMed] [Google Scholar]
- 7. Xie J, Méndez JD, Méndez‐valenzuela V. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal 2013;25:2185–2197. [DOI] [PubMed] [Google Scholar]
- 8. Riuzzi F, Sorci G, Donato R. The amphoterin (HMGB1)/Receptor for Advanced Glycation End Products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiveness: functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J Biol Chem 2006;281:8242–8253. [DOI] [PubMed] [Google Scholar]
- 9. Riuzzi F, Beccafico S, Sagheddu R, Chiappalupi S, Giambanco I, Bereshchenko O, et al. Levels of S100B protein drive the reparative process in acute muscle injury and muscular dystrophy. Sci Rep 2017;7:12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Halaby DM, Mornon JPE. The immunoglobulin superfamily: an insight on its tissular, species, and functional diversity. J Mol Evol 1998;46:389–400. [DOI] [PubMed] [Google Scholar]
- 11. Hudson BI, Carter AM, Harja E, Harja E, Kalea AZ, Arriero M, et al. Identification, classification, and expression of RAGE gene splice variants. FASEB J 2008;22:1572–1580. [DOI] [PubMed] [Google Scholar]
- 12. Sugaya K, Fukagawa T, Matsumoto K, Mita K, Takahashi E, Ando A, et al. Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int‐3. Genomics 1994;23:408–419. [DOI] [PubMed] [Google Scholar]
- 13. Sessa L, Gatti E, Zeni F, Antonelli A, Catucci A, Koch M, et al. The receptor for advanced glycation end‐products (RAGE) is only present in mammals, and belongs to a family of cell adhesion molecules (CAMs). PLoS One 2014;9:e86903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 2001;108:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yatime L, Betzer C, Jensen RK, Mortensen S, Jensen PH, Andersen GR. The structure of the RAGE:S100A6 complex reveals a unique mode of homodimerization for S100 proteins. Structure 2016;24:2043–2052. [DOI] [PubMed] [Google Scholar]
- 16. Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem 2007;282:31317–31331. [DOI] [PubMed] [Google Scholar]
- 17. Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis‐associated carcinogenesis. Carcinogenesis 2008;29:2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, et al. N ‐Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J Neurochem 2002;80:998–1008. [DOI] [PubMed] [Google Scholar]
- 19. Lee EJ, Park JH. Receptor for Advanced Glycation Endproducts (RAGE), its ligands, and soluble RAGE: potential biomarkers for diagnosis and therapeutic targets for human renal diseases. Genomics Inform 2013;11:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999. 25;97:889–901 [DOI] [PubMed] [Google Scholar]
- 21. Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane‐bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 2008;22:3716–3727. [DOI] [PubMed] [Google Scholar]
- 22. Schmidt AM. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem 1992;267:14987–14997. [PubMed] [Google Scholar]
- 23. Schmidt AM, Hori O, Cao R, Yan SD, Brett J, Wautier JL, et al. RAGE: a novel cellular receptor for advanced glycation end products. Diabetes 1996;45:S77–S80. [DOI] [PubMed] [Google Scholar]
- 24. Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei‐Jadidi R, et al. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J 2003;375:581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xue J, Rai V, Singer D, Chabierski S, Xie J, Reverdatto S, et al. Advanced glycation end product recognition by the receptor for AGEs. Structure 2011;19:722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xue J, Ray R, Singer D, Böhme D, Burz DS, Rai V, et al. The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal‐derived AGEs. Biochemistry 2014;53:3327–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fatchiyah F, Hardiyanti F, Widodo N. Selective inhibition on RAGE‐binding AGEs required by bioactive peptide alpha‐S2 case in protein from goat Ethawah breed milk: study of biological modeling. Acta Inform Med 2015;23:90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid‐beta peptide neurotoxicity in Alzheimer's disease. Nature 1996;382:685–691. [DOI] [PubMed] [Google Scholar]
- 29. Rouhiainen A, Kuja‐Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, et al. Regulation of monocyte migration by amphoterin (HMGB1). Blood 2004;104:1174–1182. [DOI] [PubMed] [Google Scholar]
- 30. Yamamoto Y, Yamamoto H. Controlling the receptor for advanced glycation end‐products to conquer diabetic vascular complications. J Diabetes Investig 2012;3:107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co‐expression of rage and amphoterin in the developing nervous system. J Biol Chem 1995;270:25752–25761. [DOI] [PubMed] [Google Scholar]
- 32. Chavakis T, Bierhaus A, Al‐Fakhri N, Schneider D, Witte S, Linn T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med 2003;198:1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, et al. Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol 2011;186:3248–3257. [DOI] [PubMed] [Google Scholar]
- 34. Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med 2013;210:2447–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sasaki N, Takeuchi M, Chowei H, Kikuchi S, Hayashi Y, Nakano N, et al. Advanced glycation end products (AGE) and their receptor (RAGE) in the brain of patients with Creutzfeldt–Jakob disease with prion plaques. Neurosci Lett 2002;326:117–120. [DOI] [PubMed] [Google Scholar]
- 36. Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta ‐ Mol Cell Res 2013;1833:101–109. [DOI] [PubMed] [Google Scholar]
- 37. Chavakis T, Bierhaus A, Nawroth PP. RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect 2004;6:1219–1225 [DOI] [PubMed] [Google Scholar]
- 38. Kim S, Kim SY, Pribis JP, Lotze M, Mollen KP, Shapiro R, et al. Signaling of high mobility group box 1 (HMGB1) through toll‐like receptor 4 in macrophages requires CD14. Mol Med 2013;19:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 40. Sorci G, Giovannini G, Riuzzi F, Bonifazi P, Zelante T, Zagarella S, et al. The danger signal S100B integrates pathogen‐ and danger‐sensing pathways to restrain inflammation. PLoS Pathog 2011;7:e1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fritz G. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci 2011;36:625–632. [DOI] [PubMed] [Google Scholar]
- 42. Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med 2018;69:349–364. [DOI] [PubMed] [Google Scholar]
- 43. Sakatani S, Yamada K, Homma C, Munesue S, Yamamoto Y, Yamamoto H, et al. Deletion of RAGE causes hyperactivity and increased sensitivity to auditory stimuli in mice. PLoS One 2009;4:e8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol 2013;94:55–68. [DOI] [PubMed] [Google Scholar]
- 45. Kang R, Tang D, Schapiro NE, Loux T, Livesey KM, Billiar TR, et al. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014;33:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morales ME, Rojas RA, Monasterio AV, González BI, Figueroa CI, Manques MB, et al. Expression of RAGE in Helicobacter pylori infested gastric biopsies. Rev Med Chil 2013;141:1240–1248. [DOI] [PubMed] [Google Scholar]
- 47. Karimi J, Goodarzi MT, Tavilani H, Khodadadi I, Amiri I. Increased receptor for advanced glycation end products in spermatozoa of diabetic men and its association with sperm nuclear DNA fragmentation. Andrologia 2012;44:280–286. [DOI] [PubMed] [Google Scholar]
- 48. Stopper H, Schinzel R, Sebekova K, Heidland A. Genotoxicity of advanced glycation end products in mammalian cells. Cancer Lett 2003;190:151–156. [DOI] [PubMed] [Google Scholar]
- 49. Mallidis C, Agbaje IM, Rogers DA, Glenn JV, Pringle R, Atkinson AB, et al. Advanced glycation end products accumulate in the reproductive tract of men with diabetes. Int J Androl 2009;32:295–305. [DOI] [PubMed] [Google Scholar]
- 50. Kumar V, Fleming T, Terjung S, Gorzelanny C, Gebhardt C, Agrawal R, et al. Homeostatic nuclear RAGE–ATM interaction is essential for efficient DNA repair. Nucleic Acids Res 2017;45:10595–10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, et al. Structural basis for ligand recognition and activation of RAGE. Structure 2010;18:1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yatime L, Andersen GR. Structural insights into the oligomerization mode of the human receptor for advanced glycation end‐products. FEBS J 2013;280:6556–6568. [DOI] [PubMed] [Google Scholar]
- 53. Zong H, Madden A, Ward M, Mooney MH, Elliott CT, Stitt AW. Homodimerization is essential for the receptor for advanced glycation end products (RAGE)‐mediated signal transduction. J Biol Chem 2010;285:23137–23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, et al. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One 2011;6:e23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sakaguchi M, Kinoshita R, Putranto EW, Ruma IMW, Sumardika IW, Youyi C, et al. Signal diversity of receptor for advanced glycation end products. Acta Med Okayama 2017;71:459–465. [DOI] [PubMed] [Google Scholar]
- 56. Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D'Agati V, et al. Interaction of the RAGE cytoplasmic domain with diaphanous‐1 is required for ligand‐stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem 2008;283:34457–34468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bianchi R, Kastrisianaki E, Giambanco I, Donato R. S100B protein stimulates microglia migration via RAGE‐dependent up‐regulation of chemokine expression and release. J Biol Chem 2011;286:7214–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yamamoto K, Murata H, Putranto EW, Kataoka K, Motoyama A, Hibino T, et al. DOCK7 is a critical regulator of the RAGE‐Cdc42 signaling axis that induces formation of dendritic pseudopodia in human cancer cells. Oncol Rep 2013;29:1073–1079. [DOI] [PubMed] [Google Scholar]
- 59. Sakaguchi M, Murata H, Aoyama Y, Hibino T, Putranto EW, Ruma IM, et al. DNAX‐activating protein 10 (DAP10) membrane adaptor associates with receptor for advanced glycation end products (RAGE) and modulates the RAGE‐triggered signaling pathway in human keratinocytes. J Biol Chem 2014;289:23389–23402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. lowik A, Merres J, Elfgen A, Jansen S, Mohr F, Wruck CJ, et al. Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)‐and amyloid beta 1‐42‐induced signal transduction in glial cells. Mol Neurodegener 2012;7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ichiki T, Koga T, Okuno T, Saeki K, Yamamoto Y, Yamamoto H, et al. Modulation of leukotriene B4 receptor 1 signaling by receptor for advanced glycation end products (RAGE). FASEB J 2016;30:1811–1822. [DOI] [PubMed] [Google Scholar]
- 62. Li JH, Wang W, Huang XR, Oldfield M, Schmidt AM, Cooper ME, et al. Advanced glycation end products induce tubular epithelial‐myofibroblast transition through the RAGE‐ERK1/2 MAP kinase signaling pathway. Am J Pathol 2004;164:1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sorci G, Riuzzi F, Arcuri C, Giambanco I, Donato R. Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol Cell Biol 2004;24:4880–4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Buckingham M, Bajard L, Chang T, Buckingham M, Bajard L, Chang T, et al. The formation of skeletal muscle: from somite to limb. J Anat 2003;202:59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chargé SBP, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev 2004;84:209–238. [DOI] [PubMed] [Google Scholar]
- 66. Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr Physiol 2015;5:1027–1059. [DOI] [PubMed] [Google Scholar]
- 67. Hernández‐Hernández JM, García‐González EG, Brun CE, Rudnicki MA. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin Cell Dev Biol 2017;72:10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zammit PS. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol 2017;72:19–32. [DOI] [PubMed] [Google Scholar]
- 69. Zammit PS. Pax7 and myogenic progression in skeletal muscle satellite cells. J Cell Sci 2006;119:1824–1832. [DOI] [PubMed] [Google Scholar]
- 70. Seale P, Sabourin LA, Girgis‐Gabardo A, Mansouri A, Gruss P, Rudnicki MA. Pax7 is required for the specification of myogenic satellite cells. Cell 2000;102:777–786. [DOI] [PubMed] [Google Scholar]
- 71. Olguin HC, Olwin BB. Pax‐7 up‐regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self‐renewal. Dev Biol 2004;275:375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR. Muscle satellite cells adopt divergent fates: a mechanism for self‐renewal? J Cell Biol 2004;166:347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X, et al. microRNA‐1 and microRNA‐206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J Cell Biol 2010;190:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dey BK, Gagan J, Dutta A. miR‐206 and ‐486 induce myoblast differentiation by downregulating Pax7. Mol Cell Biol 2011;31:203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Olguin HC, Yang Z, Tapscott SJ, Olwin BB. Reciprocal inhibition between Pax7 and muscle regulatory factors modulates myogenic cell fate determination. J Cell Biol 2007;177:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kuang S, Chargé SB, Seale P, Huh M, Rudnicki MA. Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J Cell Biol 2006;172:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fidziańska A, Goebel HH. Human ontogenesis. 3. Cell death in fetal muscle. Acta Neuropathol 1991;81:572–577. [DOI] [PubMed] [Google Scholar]
- 78. Riuzzi F, Sorci G, Sagheddu R, Donato R. HMGB1‐RAGE regulates muscle satellite cell homeostasis through p38‐MAPK‐ and myogenin‐dependent repression of Pax7 transcription. J Cell Sci 2012;125:1440–1454. [DOI] [PubMed] [Google Scholar]
- 79. Riuzzi F, Sorci G, Beccafico S, Donato R. S100B engages RAGE or bFGF/FGFR1 in myoblasts depending on its own concentration and myoblast density. Implications for muscle regeneration. PLoS One 2012;7:e28700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee J, Hong F, Kwon S, Kim SS, Kim DO, Kang HS, et al. Activation of p38 MAPK induces cell cycle arrest via inhibition of Raf/ERK pathway during muscle differentiation. Biochem Biophys Res Commun 2002;298:765–771. [DOI] [PubMed] [Google Scholar]
- 81. Chen X, Li Y. Role of matrix metalloproteinases in skeletal muscle: migration, differentiation, regeneration and fibrosis. Cell Adh Migr 2009;3:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, Tubaro C, et al. The many faces of S100B protein: when an extracellular factor inactivates its own receptor and activates another one. Ital J Anat Embryol 2010;115:147–151. [PubMed] [Google Scholar]
- 83. Dattilo BM, Fritz G, Leclerc E, Vander KCW, Heizmann CW, Chazin WJ. The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry 2007;46:6957–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Charlton CA, Mohler WA, Blau HM. Neural cell adhesion molecule (NCAM) and myoblast fusion. Dev Biol 2000;221:112–119. [DOI] [PubMed] [Google Scholar]
- 85. Lyons GE, Moore R, Yahara O, Buckingham ME, Walsh FS. Expression of NCAM isoforms during skeletal myogenesis in the mouse embryo. Dev Dyn 1992;194:94–104. [DOI] [PubMed] [Google Scholar]
- 86. Dormoy‐Raclet V, Cammas A, Celona B, Lian XJ, van der Giessen K, Zivojnovic M, et al. HuR and miR‐1192 regulate myogenesis by modulating the translation of HMGB1 mRNA. Nat Commun 2013;4:2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rigamonti E, Zordan P, Sciorati C, Rovere‐Querini P, Brunelli S. Macrophage plasticity in skeletal muscle repair. Biomed Res Int 2014;2014:560629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol 2013;183:1352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhang C, Wang C, Li Y, Miwa T, Liu C, Cui W, Song WC, Du J. Complement C3a signaling facilitates skeletal muscle regeneration by regulating monocyte function and trafficking. Nat Commun 2017;8:2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. De Mori R, Straino S, Di Carlo A, Mangoni A, Pompilio G, Palumbo R, et al. Multiple effects of high mobility group box protein 1 in skeletal muscle regeneration. Arterioscler Thromb Vasc Biol 2007;27:2377–2383. [DOI] [PubMed] [Google Scholar]
- 91. Sachdev U, Cui X, Tzeng E. HMGB1 and TLR4 mediate skeletal muscle recovery in a murine model of hindlimb ischemia. J Vasc Surg 2013;58:460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med 2012;209:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tirone M, Tran NL, Ceriotti C, Gorzanelli A, Canepari M, Bottinelli R, et al. High mobility group box 1 orchestrates tissue regeneration via CXCR4. J Exp Med 2018;215:303–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dutt V, Gupta S, Dabur R, Injeti E, Mittal A. Skeletal muscle atrophy: potential therapeutic agents and their mechanisms of action. Pharmacol Res 2015;99:86–100. [DOI] [PubMed] [Google Scholar]
- 95. Sandri M. Protein breakdown in muscle wasting: role of autophagy‐lysosome and ubiquitin‐proteasome. Int J Biochem Cell Biol 2013;45:2121–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 2005;37:1974–1984. [DOI] [PubMed] [Google Scholar]
- 97. Fukada SI. The roles of muscle stem cells in muscle injury, atrophy and hypertrophy. J Biochem 2018;163:353–358. [DOI] [PubMed] [Google Scholar]
- 98. McKenna CF, Fry CS. Altered satellite cell dynamics accompany skeletal muscle atrophy during chronic illness, disuse, and aging. Curr Opin Clin Nutr Metab Care 2017;20:447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas‐Ahner J, et al. NF‐κB‐mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013;123:4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, et al. Insulin‐like growth factor‐1 (IGF‐1) inversely regulates atrophy‐induced genes via the phosphatidylinositol 3‐kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 2005;280:2737–2744. [DOI] [PubMed] [Google Scholar]
- 101. Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, et al. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1‐dependent ubiquitylation. J Cell Biol 2009;185:1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Li YP, Schwartz RJ, Waddell ID, Holloway BR, Reid MB. Skeletal muscle myocytes undergo protein loss and reactive oxygen‐mediated NF‐kappaB activation in response to tumor necrosis factor alpha. FASEB J 1998;12:871–880. [DOI] [PubMed] [Google Scholar]
- 104. Ding H, Zhang G, Sin KW, Liu Z, Lin RK, Li M, et al. Activin A induces skeletal muscle catabolism via p38β mitogen‐activated protein kinase. J Cachexia Sarcopenia Muscle 2017;8:202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Clavel S, Siffroi‐Fernandez S, Coldefy AS, Boulukos K, Pisani DF, Dérijard B. Regulation of the intracellular localization of Foxo3a by stress‐activated protein kinase signaling pathways in skeletal muscle cells. Mol Cell Biol 2010;30:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Andersen H, Schmitz O, Nielsen S. Decreased isometric muscle strength after acute hyperglycaemia in type 1 diabetic patients. Diabet Med 2005;22:1401–1407. [DOI] [PubMed] [Google Scholar]
- 107. Umegaki H. Sarcopenia and frailty in older patients with diabetes mellitus. Geriatr Gerontol Int 2016;16:293–299. [DOI] [PubMed] [Google Scholar]
- 108. Aguiari P, Leo S, Zavan B, Vindigni V, Rimessi A, Bianchi K, et al. High glucose induces adipogenic differentiation of muscle‐derived stem cells. Proc Natl Acad Sci U S A 2008;105:1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nguyen MH, Cheng M, Koh TJ. Impaired muscle regeneration in ob/ob and db/db mice. ScientificWorldJournal 2011;11:1525–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Snow LM, Fugere NA, Thompson LV. Advanced glycation end‐product accumulation and associated protein modification in type II skeletal muscle with aging. J Gerontol A Biol Sci Med Sci 2007;62:1204–1210. [DOI] [PubMed] [Google Scholar]
- 111. Snow LM, Thompson LV. Influence of insulin and muscle fiber type in nepsilon‐(carboxymethyl)‐lysine accumulation in soleus muscle of rats with streptozotocin‐induced diabetes mellitus. Pathobiology 2009;76:227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chiu CY, Yang RS, Sheu ML, Chan DC, Yang TH, Tsai KS, et al. Advanced glycation end‐products induce skeletal muscle atrophy and dysfunction in diabetic mice via a RAGE‐mediated, AMPK‐down‐regulated, Akt pathway. J Pathol 2016;238:470–482. [DOI] [PubMed] [Google Scholar]
- 113. Howard AC, McNeil AK, Xiong F, Xiong WC, McNeil PL. A novel cellular defect in diabetes: membrane repair failure. Diabetes 2011;60:3034–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Nagai R, Murray DB, Metz TO, Baynes JW. Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes 2012;61:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Boden G. Obesity, insulin resistance and free fatty acids. Curr Opin Endocrinol Diabetes Obes 2011;18:139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell 2012;148:852–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Guo Q, Mori T, Jiang Y, Hu C, Osaki Y, Yoneki Y, et al. Methylglyoxal contributes to the development of insulin resistance and salt sensitivity in Sprague‐Dawley rats. J Hypertens 2009;27:1664–1671. [DOI] [PubMed] [Google Scholar]
- 118. Wang X, Jia X, Chang T, Desai K, Wu L. Attenuation of hypertension development by scavenging methylglyoxal in fructose‐treated rats. J Hypertens 2008;26:765–772. [DOI] [PubMed] [Google Scholar]
- 119. Li W, Xu H, Hu Y, He P, Ni Z, Xu H, et al. Edaravone protected human brain microvascular endothelial cells from methylglyoxal‐induced injury by inhibiting AGEs/RAGE/oxidative stress. PLoS One 2013;8:e76025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Berlanga J, Cibrian D, Guillén I, Freyre F, Alba JS, Lopez‐Saura P, et al. Methylglyoxal administration induces diabetes‐like microvascular changes and perturbs the healing process of cutaneous wounds. Clin Sci (Lond) 2005;109:83–95. [DOI] [PubMed] [Google Scholar]
- 121. Yao D, Brownlee M. Hyperglycemia‐induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010;59:249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ma H, Li SY, Xu P, Babcock SA, Dolence EK, Brownlee M, et al. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up‐regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med 2009;13:1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 123. Bourajjaj M, Stehouwer CD, van Hinsbergh VW, Schalkwijk CG. Role of methylglyoxal adducts in the development of vascular complications in diabetes mellitus. Biochem Soc Trans 2003;31:1400–1402. [DOI] [PubMed] [Google Scholar]
- 124. Uribarri J, Cai W, Ramdas M, Goodman S, Pyzik R, Chen X, et al. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: potential role of AGER1 and SIRT1. Diabetes Care 2011;34:1610–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Aftab MF, Afridi SK, Ghaffar S, Murtaza M, Khan M, Karim A, Khan KM, Waraich RS. A bis‐Schiff base of isatin improves methylglyoxal mediated insulin resistance in skeletal muscle cells. Arch Pharm Res. 2015;Oct 30. [DOI] [PubMed] [Google Scholar]
- 126. Cassese A, Esposito I, Fiory F, Barbagallo AP, Paturzo F, Mirra P, et al. In skeletal muscle advanced glycation end products (AGEs) inhibit insulin action and induce the formation of multimolecular complexes including the receptor for AGEs. J Biol Chem 2008;283:36088–36099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. He C, Sabol J, Mitsuhashi T, Vlassara H. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 1999;48:1308–1315. [DOI] [PubMed] [Google Scholar]
- 128. Hofmann SM, Dong HJ, Li Z, Cai W, Altomonte J, Thung SN, et al. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes 2002;51:2082–2089. [DOI] [PubMed] [Google Scholar]
- 129. Vlassara H. The AGE‐receptor in the pathogenesis of diabetic complications. Diabetes Metab Res Rev 2001;17:436–443. [DOI] [PubMed] [Google Scholar]
- 130. Miele C, Riboulet A, Maitan MA, Oriente F, Romano C, Formisano P, et al. Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin‐induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha‐mediated mechanism. J Biol Chem 2003;278:47376–47387. [DOI] [PubMed] [Google Scholar]
- 131. de la Maza MP, Uribarri J, Olivares D, Hirsch S, Leiva L, Barrera G, et al. Weight increase is associated with skeletal muscle immunostaining for advanced glycation end products, receptor for advanced glycation end products, and oxidation injury. Rejuvenation Res 2008;11:1041–1048. [DOI] [PubMed] [Google Scholar]
- 132. Melton LJ 3rd, Khosla S, Riggs BL. Epidemiology of sarcopenia. Mayo Clin Proc 2000; Suppl:S10‐2; discussion S12–3. [PubMed] [Google Scholar]
- 133. Sakuma K, Aoi W, Yamaguchi A. Molecular mechanism of sarcopenia and cachexia: recent research advances. Pflugers Arch 2017;469:573–591. [DOI] [PubMed] [Google Scholar]
- 134. Walston JD. Sarcopenia in older adults. Curr Opin Rheumatol 2012;24:623–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell‐autonomous loss of stem cell self‐renewal in skeletal muscle of aged mice. Nat Med 2014;20:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Brack AS, Rando TA. Intrinsic changes and extrinsic influences of myogenic stem cell function during aging. Stem Cell Rev 2007;3:226–237. [DOI] [PubMed] [Google Scholar]
- 137. Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 2014;20:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sousa‐Victor P, Gutarra S, García‐Prat L, Rodriguez‐Ubreva J, Ortet L, Ruiz‐Bonilla V, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014;506:316–321. [DOI] [PubMed] [Google Scholar]
- 139. Beccafico S, Riuzzi F, Puglielli C, Mancinelli R, Fulle S, Sorci G, et al. Human muscle satellite cells show age‐related differential expression of S100B protein and RAGE. Age (Dordr) 2011;33:523–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tubaro C, Arcuri C, Giambanco I, Donato R. S100B protein in myoblasts modulates myogenic differentiation via NF‐kappaB‐dependent inhibition of MyoD expression. J Cell Physiol 2010;223:270–282. [DOI] [PubMed] [Google Scholar]
- 141. Morozzi G, Beccafico S, Bianchi R, Riuzzi F, Bellezza I, Giambanco I, et al. Oxidative stress‐induced S100B accumulation converts myoblasts into brown adipocytes via an NF‐κB/YY1/miR‐133 axis and NF‐κB/YY1/BMP‐7 axis. Cell Death Differ 2017;24:2077–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Grillari J, Katinger H, Voglauer R. Aging and the ubiquitinome: traditional and non‐traditional functions of ubiquitin in aging cells and tissues. Exp Gerontol 2006;41:1067–1079. [DOI] [PubMed] [Google Scholar]
- 143. Dalal M, Ferrucci L, Sun K, Beck J, Fried LP, Semba RD. Elevated serum advanced glycation end products and poor grip strength in older community‐dwelling women. J Gerontol A Biol Sci Med Sci 2009;64:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Haus JM, Carrithers JA, Trappe SW, Trappe TA. Collagen, cross‐linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol 2007;103:2068–2076. [DOI] [PubMed] [Google Scholar]
- 145. Semba RD, Nicklett EJ, Ferrucci L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol A Biol Sci Med Sci 2010;65:963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Finsterer J, Löscher WN, Wanschitz J, Quasthoff S, Grisold W. Secondary myopathy due to systemic diseases. Acta Neurol Scand 2016;134:388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med 2013;45:4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Mercuri E, Muntoni F. Muscular dystrophies. Lancet 2013;381:845–860. [DOI] [PubMed] [Google Scholar]
- 149. Haslbeck KM, Friess U, Schleicher ED, Bierhaus A, Nawroth PP, Kirchner A, et al. The RAGE pathway in inflammatory myopathies and limb girdle muscular dystrophy. Acta Neuropathol 2005;110:247–254. [DOI] [PubMed] [Google Scholar]
- 150. Macaione V, Aguennouz M, Rodolico C, Mazzeo A, Patti A, Cannistraci E, et al. RAGE‐NF‐kappaB pathway activation in response to oxidative stress in facioscapulohumeral muscular dystrophy. Acta Neurol Scand 2007;115:115–121. [DOI] [PubMed] [Google Scholar]
- 151. Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001;280:E685–E694. [DOI] [PubMed] [Google Scholar]
- 152. Winokur ST, Barrett K, Martin JH, Forrester JR, Simon M, Tawil R, et al. Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul Disord 2003;13:322–333. [DOI] [PubMed] [Google Scholar]
- 153. Mandel DE, Malemud CJ, Askari AD. Idiopathic inflammatory myopathies: a review of the classification and impact of pathogenesis. Int J Mol Sci 2017;18:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ulfgren AK, Grundtman C, Borg K, Alexanderson H, Andersson U, Harris HE, et al. Down‐regulation of the aberrant expression of the inflammation mediator high mobility group box chromosomal protein 1 in muscle tissue of patients with polymyositis and dermatomyositis treated with corticosteroids. Arthritis Rheum 2004;50:1586–1594. [DOI] [PubMed] [Google Scholar]
- 155. Askanas V, Engel WK, Nogalska A. Pathogenic considerations in sporadic inclusion‐body myositis, a degenerative muscle disease associated with aging and abnormalities of myoproteostasis. J Neuropathol Exp Neurol 2012;71:680–693. [DOI] [PubMed] [Google Scholar]
- 156. Muth IE, Zschüntzsch J, Kleinschnitz K, Wrede A, Gerhardt E, Balcarek P, et al. HMGB1 and RAGE in skeletal muscle inflammation: implications for protein accumulation in inclusion body myositis. Exp Neurol 2015;271:189–197. [DOI] [PubMed] [Google Scholar]
- 157. Yan SS, Chen D, Yan S, Guo L, Du H, Chen JX. RAGE is a key cellular target for Abeta‐induced perturbation in Alzheimer's disease. Front Biosci (Schol Ed) 2012;4:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Rüster M, Franke S, Späth M, Pongratz DE, Stein G, Hein GE. Detection of elevated N epsilon‐carboxymethyllysine levels in muscular tissue and in serum of patients with fibromyalgia. Scand J Rheumatol 2005;34:460–463. [DOI] [PubMed] [Google Scholar]
- 159. Phillips WD, Vincent A. Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Res 2016;5:1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Uzawa A, Kanai T, Kawaguchi N, Oda F, Himuro K, Kuwabara S. Changes in inflammatory cytokine networks in myasthenia gravis. Sci Rep 2016;6:25886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Molin CJ, Westerberg E, Punga AR. Profile of upregulated inflammatory proteins in sera of Myasthenia gravis patients. Sci Rep 2017;7:39716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med 2013;13:24–57. [PMC free article] [PubMed] [Google Scholar]
- 163. Luo Y, Yoneda J, Ohmori H, Sasaki T, Shimbo K, Eto S, et al. Cancer usurps skeletal muscle as an energy repository. Cancer Res 2014;74:330–340. [DOI] [PubMed] [Google Scholar]
- 164. Pappo AS, Dirksen U. Rhabdomyosarcoma, Ewing sarcoma, and other round cell sarcomas. J Clin Oncol 2018;36:168–179. [DOI] [PubMed] [Google Scholar]
- 165. Saab R, Spunt SL, Skapek SX. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr Top Dev Biol 2011;94:197–234. [DOI] [PubMed] [Google Scholar]
- 166. Hettmer S, Wagers AJ. Muscling in: uncovering the origins of rhabdomyosarcoma. Nat Med 2010;16:171–173. [DOI] [PubMed] [Google Scholar]
- 167. Calhabeu F, Hayashi S, Morgan JE, Relaix F, Zammit PS. Alveolar rhabdomyosarcoma‐associated proteins PAX3/FOXO1A and PAX7/FOXO1A suppress the transcriptional activity of MyoD‐target genes in muscle stem cells. Oncogene 2013;32:651–662. [DOI] [PubMed] [Google Scholar]
- 168. Blake J, Ziman MR. Aberrant PAX3 and PAX7 expression. A link to the metastatic potential of embryonal rhabdomyosarcoma and cutaneous malignant melanoma? Histol Histopathol 2003;18:529–539. [DOI] [PubMed] [Google Scholar]
- 169. Chiappalupi S, Riuzzi F, Fulle S, Donato R, Sorci G. Defective RAGE activity in embryonal rhabdomyosarcoma cells results in high PAX7 levels that sustain migration and invasiveness. Carcinogenesis 2014;35:2382–2392. [DOI] [PubMed] [Google Scholar]
- 170. Riuzzi F, Sorci G, Sagheddu R, Sidoni A, Alaggio R, Ninfo V, et al. RAGE signaling deficiency in rhabdomyosarcoma cells causes upregulation of PAX7 and uncontrolled proliferation. J Cell Sci 2014;127:1699–1711. [DOI] [PubMed] [Google Scholar]
- 171. Riuzzi F, Sorci G, Donato R. RAGE expression in rhabdomyosarcoma cells results in myogenic differentiation and reduced proliferation, migration, invasiveness, and tumor growth. Am J Pathol 2007;171:947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Wegorzewska M, Krauss RS, Kang JS. Overexpression of the immunoglobulin superfamily members CDO and BOC enhances differentiation of the human rhabdomyosarcoma cell line RD. Mol Carcinog 2003;37:1–4. [DOI] [PubMed] [Google Scholar]
- 173. Sparvero LJ, Asafu‐Adjei D, Kang R, Tang D, Amin N, Im J, et al. RAGE (receptor for advanced glycation endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 2009;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Bartling B, Hofmann HS, Weigle B, Silber RE, Simm A. Down‐regulation of the receptor for advanced glycation end‐products (RAGE) supports non‐small cell lung carcinoma. Carcinogenesis 2005;26:293–301. [DOI] [PubMed] [Google Scholar]
- 175. Tateno T, Ueno S, Hiwatashi K, Matsumoto M, Okumura H, Setoyama T, et al. Expression of receptor for advanced glycation end products (RAGE) is related to prognosis in patients with esophageal squamous cell carcinoma. Ann Surg Oncol 2009;16:440–446. [DOI] [PubMed] [Google Scholar]
- 176. Bongarzone S, Savickas V, Luzi F, Gee AD. Targeting the receptor for advanced glycation endproducts (RAGE): a medicinal chemistry perspective. J Med Chem 2017;60:7213–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Sabbagh MN, Agro A, Bell J, Aisen PS, Schweizer E, Galasko D. PF‐04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis Assoc Disord 2011;25:206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Galasko D, Bell J, Mancuso JY, Kupiec JW, Sabbagh MN, van Dyck C, Thomas RG, Aisen PS; Alzheimer's Disease Cooperative Study. Clinical trial of an inhibitor of RAGE–Aβ interactions in Alzheimer disease. Neurology 2014;82:1536–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Burstein AH, Grimes I, Galasko DR, Aisen PS, Sabbagh M, Mjalli AM. Effect of TTP488 in patients with mild to moderate Alzheimer's disease. BMC Neurol 2014;14:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Han YT, Choi GI, Son D, Kim NJ, Yun H, Lee S, et al. Ligand‐based design, synthesis, and biological evaluation of 2‐aminopyrimidines, a novel series of receptor for advanced glycation end products (RAGE) inhibitors. J Med Chem 2012;55:9120–9135. [DOI] [PubMed] [Google Scholar]
- 181. Lee YS, Kim H, Kim YH, Roh EJ, Han H, Shin KJ. Synthesis and structure‐activity relationships of tri‐substituted thiazoles as RAGE antagonists for the treatment of Alzheimer's disease. Bioorg Med Chem Lett 2012;22:7555–7561. [DOI] [PubMed] [Google Scholar]
- 182. Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B, et al. A multimodal RAGE‐specific inhibitor reduces amyloid β‐mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 2012;122:1377–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Hong Y, Shen C, Yin Q, Sun M, Ma Y, Liu X. Effects of RAGE‐specific inhibitor FPS‐ZM1 on Amyloid‐β metabolism and AGEs‐induced inflammation and oxidative stress in rat hippocampus. Neurochem Res 2016;41:1192–1199. [DOI] [PubMed] [Google Scholar]
- 184. Wang D, Liu K, Wake H, Teshigawara K, Mori S, Nishibori M. Anti‐high mobility group box‐1 (HMGB1) antibody inhibits hemorrhage‐induced brain injury and improved neurological deficits in rats. Sci Rep 2017;7:46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, et al. Small molecule inhibition of ligand‐stimulated RAGE‐DIAPH1 signal transduction. Sci Rep 2016;6:22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. López‐Díez R, Rastrojo A, Villate O, Aguado B. Complex tissue‐specific patterns and distribution of multiple RAGE splice variants in different mammals. Genome Biol Evol 2013;5:2420–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Ding Q, Keller JN. Splice variants of the receptor for advanced glycosylation end products (RAGE) in human brain. Neurosci Lett 2005;373:67–72. [DOI] [PubMed] [Google Scholar]
- 188. Chuah YK, Basir R, Talib H, Tie TH, Nordin N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int J Inflam 2013;2013:403460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Riehl A, Németh J, Angel P, Hess J. The receptor RAGE: bridging inflammation and cancer. Cell Commun Signal 2009;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Bierhaus A, Stern DM, Nawroth PP. RAGE in inflammation: a new therapeutic target? Curr Opin Investig Drugs 2006;7:985–991. [PubMed] [Google Scholar]
- 191. Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol 2005;61:1–9. [DOI] [PubMed] [Google Scholar]
- 192. Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, et al. S100B's double life: intracellular regulator and extracellular signal. Biochim Biophys Acta 2009;1793:1008–1022. [DOI] [PubMed] [Google Scholar]
- 193. Huttunen HJ, Kuja‐Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem 2000;275:40096–40105. [DOI] [PubMed] [Google Scholar]
- 194. Businaro R, Leone S, Fabrizi C, Sorci G, Donato R, Lauro GM, et al. S100B protects LAN‐5 neuroblastoma cells against Abeta amyloid‐induced neurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. J Neurosci Res 2006;83:897–906. [DOI] [PubMed] [Google Scholar]
- 195. Jin X, Yao T, Zhou Z, Zhu J, Zhang S, Hu W, et al. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF‐κB pathway. Biomed Res Int 2015;732450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Malik P, Chaudhry N, Mittal R, Mukherjee TK. Role of receptor for advanced glycation end products in the complication and progression of various types of cancers. Biochim Biophys Acta 2015;1850:1898–1904. [DOI] [PubMed] [Google Scholar]
- 197. Ahmad S, Khan H, Siddiqui Z, Khan MY, Rehman S, Shahab U, et al. AGEs, RAGEs and s‐RAGE; friend or foe for cancer. Semin Cancer Biol 2018;49:44–55. [DOI] [PubMed] [Google Scholar]
- 198. Logsdon CD, Fuentes MK, Huang EH, Arumugam T. RAGE and RAGE ligands in cancer. Curr Mol Med 2007;7:777–789. [DOI] [PubMed] [Google Scholar]
- 199. Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 2010;28:367–388. [DOI] [PubMed] [Google Scholar]
- 200. Yu YX, Pan WC, Cheng YF. Silencing of advanced glycosylation and glycosylation and product‐specific receptor (RAGE) inhibits the metastasis and growth of non‐small cell lung cancer. Am J Transl Res 2017;9:2760–2774. [PMC free article] [PubMed] [Google Scholar]
- 201. Landesberg R, Woo V, Huang L, Cozin M, Lu Y, Bailey C, et al. The expression of the receptor for glycation endproducts (RAGE) in oral squamous cell carcinomas. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2008;105:617–624. [DOI] [PubMed] [Google Scholar]
- 202. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]