

Bacterial cell division proceeds through the coordinated assembly of the FtsZ-ring, or Z-ring, at the site of division. Assembly of the Z-ring requires polymerization of FtsZ, which is regulated by several proteins in the cell. In Escherichia coli, the Min system, which contains MinC, MinD, and MinE proteins, exhibits polar oscillation and inhibits the assembly of FtsZ at nonseptal locations. Here, we identify regions on the surface of MinC that are important for contacting FtsZ and destabilizing FtsZ polymers.
KEYWORDS: cell division, cytokinesis, divisome, patterning, tubulin
ABSTRACT
The Min system in Escherichia coli, consisting of MinC, MinD, and MinE proteins, regulates division site selection by preventing assembly of the FtsZ-ring (Z-ring) and exhibits polar oscillation in vivo. MinC antagonizes FtsZ polymerization, and in vivo, the cellular location of MinC is controlled by a direct association with MinD at the membrane. To further understand the interactions of MinC with FtsZ and MinD, we performed a mutagenesis screen to identify substitutions in minC that are associated with defects in cell division. We identified amino acids in both the N- and C-domains of MinC that are important for direct interactions with FtsZ and MinD in vitro, as well as mutations that modify the observed in vivo oscillation of green fluorescent protein (GFP)-MinC. Our results indicate that there are two distinct surface-exposed sites on MinC that are important for direct interactions with FtsZ, one at a cleft on the surface of the N-domain and a second on the C-domain that is adjacent to the MinD interaction site. Mutation of either of these sites leads to slower oscillation of GFP-MinC in vivo, although the MinC mutant proteins are still capable of a direct interaction with MinD in phospholipid recruitment assays. Furthermore, we demonstrate that interactions between FtsZ and both sites of MinC identified here are important for assembly of FtsZ-MinC-MinD complexes and that the conserved C-terminal end of FtsZ is not required for MinC-MinD complex formation with GTP-dependent FtsZ polymers.
IMPORTANCE Bacterial cell division proceeds through the coordinated assembly of the FtsZ-ring, or Z-ring, at the site of division. Assembly of the Z-ring requires polymerization of FtsZ, which is regulated by several proteins in the cell. In Escherichia coli, the Min system, which contains MinC, MinD, and MinE proteins, exhibits polar oscillation and inhibits the assembly of FtsZ at nonseptal locations. Here, we identify regions on the surface of MinC that are important for contacting FtsZ and destabilizing FtsZ polymers.
INTRODUCTION
The cell division pathway in prokaryotes a widely conserved and highly organized cellular process, whereby multiple cell division proteins cooperate to divide a single bacterial cell into two daughter cells (1). In Escherichia coli, cell division initiates with the assembly of the Z-ring, which is composed of the tubulin-like protein FtsZ and several FtsZ-interacting proteins, including FtsA and ZipA, at the site of septation (2, 3). After Z-ring assembly, additional proteins are sequentially recruited to the division site to insert and remodel the peptidoglycan leading to septation (4, 5). Normal cell growth and development in bacteria rely on protein-based mechanisms to regulate the location of the division machinery and restrict Z-ring position to the longitudinal center of the cell (6). Mislocalization of the division machinery promotes asymmetric cell division, generates daughter cells lacking a copy of the bacterial chromosome, and may lead to chromosomal severing and impaired chromosome segregation (7–10).
Division site selection in E. coli predominantly depends on two mechanisms: (i) nucleoid occlusion mediated by SlmA, which blocks Z-ring assembly over the bacterial chromosome by binding to DNA and FtsZ, and (ii) the Min system, which prevents Z-ring formation at the cell poles (3, 8, 11–18). E. coli lacking both the Min system and nucleoid occlusion is not viable in rich media, and min mutants of Neisseria gonorrhoeae and Escherichia coli O157:H7 are less virulent (8, 19, 20). E. coli min mutants display filamentation in liquid culture and produce anucleate minicells that arise from polar divisions (7). N. gonorrhoeae min mutants are enlarged compared to wild-type cells and undergo cell lysis (21, 22).
The Min system of E. coli is composed of three proteins, MinC, MinD, and MinE, that exhibit coordinated polar oscillation in vivo. MinC destabilizes GTP-dependent FtsZ polymers in vitro (23–25). In the current model, the MinC N-domain binds to the FtsZ-FtsZ subunit interface, preventing the head-to-tail assembly of FtsZ protomers (26, 27). Overexpression of the MinC C-domain, which is also the dimerization domain, inhibits cell division in vivo and has been suggested to inhibit lateral associations between FtsZ filaments, and the C-domain also contains the MinD binding site (25, 28–30). An amino acid on the MinC N-domain, G10, was previously reported to be important for FtsZ disassembly (23). In a recent study by the Lutkenhaus group, several additional N-domain residues (K9, F42, K35, and A39) on MinC were implicated in the FtsZ interaction (31).
MinD is a member of the Walker A cytoskeletal ATPase (WACA) protein family and contains a deviant Walker A motif (32, 33). MinD controls the cellular distribution of MinC through a direct interaction (34). MinD associates with the inner face of the cytoplasmic membrane in its ATP-bound dimer conformation (35). MinE promotes the dissociation of MinD from the membrane by stimulating ATP hydrolysis and release from the membrane surface (36, 37). Following MinD membrane displacement, MinE has been observed to remain at the membrane via an N-terminal membrane-targeting helix (38, 39), possibly serving to prevent rebinding of MinD at the same position. Indeed, reconstitution of MinCDE patterning in vitro reveals that MinD propagates at the leading edge of MinE waves (40).
It was previously demonstrated by our group and the Lowe group that MinC and MinD form copolymers in vitro in the presence of ATP (41, 42), likely containing alternating MinC and MinD dimers, as observed in a crystal structure of Aquifex aeolicus MinD in complex with the MinC dimerization domain (41). In addition to E. coli and A. aoelicus, MinCD copolymerization has been detected for Pseudomonas aeruginosa (43). After ATP-dependent assembly, MinCD copolymers remain stable but rapidly disassemble in the presence of MinE, suggesting that nucleotide hydrolysis and/or displacement of MinC with MinE mediates disassembly (41, 42). Although copolymers assemble robustly in vitro, oscillation of MinD in vivo is dependent on MinE and does not require MinC (34, 44). Moreover, cells expressing MinD heterodimers that bind to MinC but fail to form copolymers display wild-type morphology (45). Therefore, MinCD copolymer formation is not required for polar oscillation of MinD in vivo, and it is unclear if MinCD copolymers assemble in vivo.
To gain further mechanistic insight into the functional interactions of MinC, FtsZ, and MinD, we performed random mutagenesis on minC and constructed a library of strains expressing the randomly mutagenized genes from the chromosomal minC locus. By screening recombinants for morphological defects in vivo and testing purified MinC mutant proteins for protein-protein interactions with FtsZ and MinD in vitro, we mapped two distinct FtsZ binding sites on the surface of MinC, one at the cleft in the MinC N-domain and an additional region on the surface of the MinC C-domain. We show that FtsZ directly interacts with complexes of MinC and MinD in the absence and presence of GTP, the condition that stimulates FtsZ polymerization. Finally, we monitored the rate of MinC oscillation in vivo using green fluorescent protein (GFP)-MinC fusion proteins and observed impaired oscillation for both N- and C-domain mutant proteins. These results show that site-specific determinants on the MinC N- and C-domains contribute to FtsZ-MinC-MinD complex formation in the presence and absence of GTP, and that complex formation may modulate Min oscillation in vivo.
RESULTS
Identification and mapping of MinC regions important for cell division in vivo.
Cells deleted for minC fail to effectively regulate division site selection, leading to aberrant Z-ring placement and the production of minicells and short filamentous cells (7, 12, 46, 47). To identify residues on MinC important for regulating Z-ring placement in vivo, we generated a library of strains containing substitutions in the chromosomal copy of minC by PCR-based random mutagenesis. Mutagenized minC gene products were then inserted into a minC deletion strain (CL0030) containing a kanamycin resistance gene adjacent to parE, which encodes a DNA gyrase inhibitor and is under the control of a rhamnose-inducible promoter (Table 1); in this strain, the kan-Prha-parE functional cassette is located at the minC chromosomal locus. When expressed, ParE prevents cell growth, thereby providing selection for recombinants. Mutagenized minC from the library was inserted at the chromosomal minC locus. Successful recombinants were selected by growth on rhamnose and confirmed by sequencing.
TABLE 1.
E. coli strains and plasmids
| Strain or plasmid | Genotype | Source, reference, or construction |
|---|---|---|
| Strains | ||
| MG1655 | LAM-rph-1 | 76 |
| BL21 (λDE3) | F–
ompT gal dcm lon hsdSB(rB− mB−) λ(DE3[lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) |
EMD Millipore, USA |
| CL0030 | MG1655 ΔminC::kan-Prha-parE | pKD267; λred |
| CL0048 | MG1655 ΔminC::minC+ | CL0030; λred |
| CL0062 | MG1655 minC(G10D) | CL0030; λred |
| CL0064 | MG1655 minC(S12A) | CL0030; λred |
| CL0068 | MG1655 minC(S16D) | CL0030; λred |
| CL0070 | MG1655 minC(S12D) | CL0030; λred |
| CL0085 | MG1655 minC(S11A) | CL0030; λred |
| CL0093 | MG1655 minC(L194P) | CL0030; λred |
| CL0100 | MG1655 minC(S11D) | CL0030; λred |
| CL0156 | MG1655 minC(S134F) | CL0030; λred |
| CL0272 | MG1655 minC(L218Q) | CL0030; λred |
| CL0343 | MG1655 minC(I197T) | CL0030; λred |
| CL0369 | MG1655 minC(V147D) | CL0030; λred |
| CL0386 | MG1655 minC(A70P) | CL0030; λred |
| CL0389 | MG1655 minC(A155P) | CL0030; λred |
| CL0396 | MG1655 minC(V18D) | CL0030; λred |
| CL0397 | MG1655 minC(S69P) | CL0030; λred |
| CL0401 | MG1655 minC(E193V) | CL0030; λred |
| CL0403 | MG1655 minC(L203N) | CL0030; λred |
| CL0405 | MG1655 minC(A84T) | CL0030; λred |
| CL0406 | MG1655 minC(R172A) | CL0030; λred |
| CL0408 | MG1655 minC(Y201A) | CL0030; λred |
| CL0410 | MG1655 minC(M93K) | CL0030; λred |
| CL0413 | MG1655 minC(E193K) | CL0030; λred |
| CL0414 | MG1655 minC(E156A) | CL0030; λred |
| CL0416 | MG1655 minC(L174N) | CL0030; λred |
| CL0418 | MG1655 minC(G199D) | CL0030; λred |
| CL0428 | MG1655 pBAD-gfp-minC | CL0030; λred |
| CL0450 | MG1655 pBAD-gfp-minC(S16D) | CL0030; λred |
| CL0468 | MG1655 pBAD-gfp-minC(L194N) | CL0030; λred |
| Plasmids | ||
| pET-MinC | kan | 42 |
| pET-MinD | kan | 42 |
| pET-MinC(G10D) | kan | This study |
| pET-MinC(S11A) | kan | This study |
| pET-MinC(S11D) | kan | This study |
| pET-MinC(S12D) | kan | This study |
| pET-MinC(S16D) | kan | This study |
| pET-MinC(V18D) | kan | This study |
| pET-MinC(S69P) | kan | This study |
| pET-MinC(S134F) | kan | This study |
| pET-MinC(A155P) | kan | This study |
| pET-MinC(V165D) | kan | This study |
| pET-MinC(R172A) | kan | This study |
| pET-MinC(E193K) | kan | This study |
| pET-MinC(L194N) | kan | This study |
| pET-MinC(L194P) | kan | This study |
| pET-MinC(I197T) | kan | This study |
| pET-MinC(Y201A) | kan | This study |
| pKD46 | amp | 73 |
| pKD267 | kan | B. Wannera |
| pBAD33-gfp | cat | This study |
| pBAD33-gfp-minC | cat | This study |
| pBAD33-gfp-minC(S16D) | cat | This study |
| pBAD33-gfp-minC(L194N) | cat | This study |
J. Teramoto, K. A. Datsenko, and B. L. Wanner, unpublished.
During exponential growth in liquid culture, cells deleted for minC are filamentous, with a mean length of 5.28 ± 0.2 µm (n = 250), and produce minicells (23.7%) (Fig. 1a and Table 2). In contrast, wild-type cells containing functional minC have a mean length of 2.47 ± 0.05 µm (n = 250) and do not generate minicells (Fig. 1a and Table 2). We screened the library of minC mutants by microscopy for cell length defects and minicells and sequenced chromosomal minC from selected recombinants to identify amino acid substitutions that confer a functional defect. To confirm that this system can be used to successfully restore wild-type morphology to a minC deletion strain, we replaced the kan-Prha-parE cassette at the minC locus with a wild-type minC gene (ΔminC::minC+) and measured average cell length and expression of MinC by immunoblotting (Fig. 1). Overall cell morphology, cell length distribution, and MinC expression levels were similar between the wild-type MG1655 strain (minC+) and the restored minC recombinant strain (ΔminC::minC+), whereas the strain deleted for minC (ΔminC::kan-Prha-parE) contained many longer cells, minicells, and no detectable MinC (Fig. 1 and Table 2).
FIG 1.
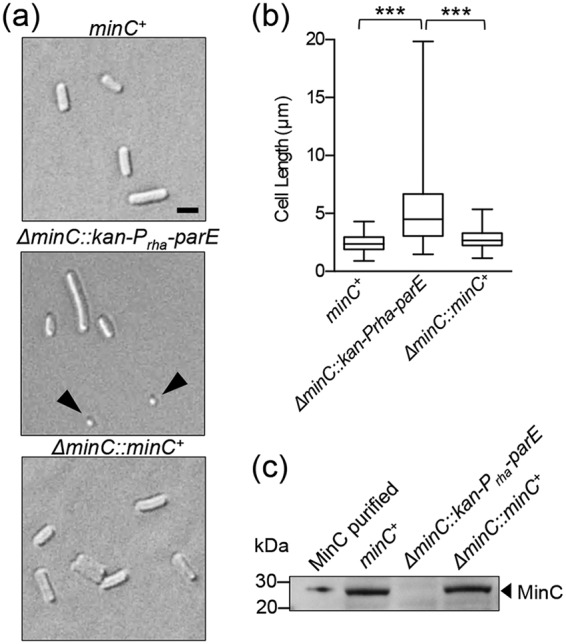
Cell morphology of strains deleted for minC and restored with minC PCR products. (a) Log-phase cultures of MG1655 (minC+), ΔminC::kan-Prha-parE, and ΔminC::minC+ were added to poly-l-lysine-coated coverslips and examined by DIC microscopy. Size bar, 2 µm. Arrowheads denote minicells. (b) Box-and-whisker plot of cell lengths for wild-type MG1655 (minC+), ΔminC::kan-Prha-parE, and ΔminC::minC+ strains (n = 250 cells for each). The box represents the interquartile range. The line in the middle of the box is plotted at the median (***, P < 0.0001). (c) Protein extracts (75 µg) from MG1655 (minC+), ΔminC::kan-Prha-parE, ΔminC::minC+, and purified MinC (10 ng) were immunoblotted with anti-MinC antisera as described in Materials and Methods.
TABLE 2.
Minicell percentages and cell lengths of E. coli strains
| Strain | Minicell percentagea | Mean cell lengtha (µm) | P valueb |
|---|---|---|---|
| MG1655 (minC+) | 0.0 | 2.47 ± 0.05 | |
| MG1655 ΔminC::minC+ | 0.0 | 2.78 ± 0.05 | |
| MG1655 ΔminC::gfp-minC | <0.5 | 2.41 ± 0.04 | |
| MG1655 ΔminC::kan-Prha-parE | 23.7 | 5.28 ± 0.20 | <0.01 |
| MG1655 minC(G10D) | 10.8 | 5.85 ± 0.18 | <0.01 |
| MG1655 minC(S12D) | 3.6 | 3.42 ± 0.11 | <0.01 |
| MG1655 minC(S16D) | 8.6 | 5.59 ± 0.20 | <0.01 |
| MG1655 minC(V18D) | 23.5 | 3.05 ± 0.12 | <0.01 |
| MG1655 minC(S134F) | 12.7 | 3.15 ± 0.10 | <0.01 |
| MG1655 minC(R172A) | 11.0 | 3.73 ± 0.16 | <0.01 |
| MG1655 minC(E193K) | 8.0 | 2.73 ± 0.12 | ≤0.02 |
| MG1655 minC(L194N) | 3.9 | 2.76 ± 0.17 | ≤0.05 |
| MG1655 minC(Y201A) | 4.8 | 2.51 ± 0.09 | NS |
Minicell percentage generated by analyzing images containing at least 215 cells for minicells. Mean cell length was calculated by measuring at least 215 cells; minicells were omitted from length measurements. Error values are reported as SEM.
P values for each data set are from comparisons to the wild-type strain and were calculated by Welch’s t test. NS, not significant.
Using the mutant library, we identified amino acid substitutions associated with defects in MinC function (see Table S1 in the supplemental material). Nearby surface-exposed amino acid residues were also targeted by site-directed mutagenesis. In addition, we constructed two mutations reported in the literature, G10D and R172A (23, 30, 48) (Table S1). Altogether, we characterized nine single-amino-acid substitutions in MinC associated with cell division defects, as indicated by length and/or minicell production in vivo (G10D, S12D, S16D, R172A, V18D, S134F, E193K, L194N, and Y201A) (Fig. 2a and b and Table 2); MinC mutant proteins characterized were reactive to MinC antisera (Fig. 2c). Several targeted mutations, including S11D, M93K, E156A, L203N, L174N, and G199D, did not produce a cell division defect and were not examined further (Fig. S1 and Table S1).
FIG 2.

Cell morphology and cell length distribution of strains expressing chromosomal minC mutants. (a) MG1655 (minC+) and ΔminC::kan-Prha-parE strains, and strains containing chromosomal minC mutant genes, were grown as described in Materials and Methods and examined by DIC microscopy. Size bar, 2 µm. (b) Box-and-whisker plots of cell lengths were plotted for wild-type and minC mutant strains. At least 215 cells were measured for each strain. The box represents the interquartile range. The line in the middle of the box is plotted at the median; all measured values were plotted. (c) Protein extracts (75 µg) from MG1655 (minC+) and ΔminC::kan-Prha-parE strains and strains containing chromosomal minC mutant genes were immunoblotted with anti-MinC antisera as described in Materials and Methods.
Residues on the MinC N- and C-domains mediate direct binding to FtsZ.
Residues identified in the screen map to both the MinC N- and C-terminal domains (Fig. 3a) and are depicted as Corey-Pauling-Koltun (CPK) spheres, based on the structural model of E. coli MinC generated by modeling the amino acid sequence onto the crystal structure available for Thermotoga maritima MinC (49, 50, 51). This is consistent with previous findings that the MinC N-domain prevents the sedimentation of FtsZ polymers, while the MinC C-domain abrogates lateral interactions between polymers (25, 29). Overexpression of either the MinC N-terminal domain (NTD) or the C-terminal dimerization domain (CTD) in vivo blocks cell division (24, 29).
FIG 3.

MinC mutant proteins are impaired for destabilizing FtsZ polymers, MinC-MinD copolymer formation, and recruitment to SUVs by MinD. (a) E. coli MinC modeled onto T. maritima MinC PDB entry 1HF2 (49); the dashed line represents the dimerization interface. MinC residues that were mutagenized are indicated as CPK spheres on one protomer. N- and C-terminal domains are labeled on the opposing protomer as NTD and CTD, respectively. (b and c) Inhibition of FtsZ polymerization by MinC and MinC mutant proteins was monitored by sedimentation of FtsZ polymers. FtsZ polymerization assays were performed as described in Materials and Methods in reaction mixtures containing FtsZ (6 µM) or the MinC or MinC mutant (0 to 4 µM), where indicated, and GTP (1 mM) with a GTP-regenerating system. Pellet-associated FtsZ was quantified by SDS-PAGE and densitometry. (d) MinC recruitment to SUVs by MinD was monitored by sedimentation assays as described in Materials and Methods in reaction mixtures containing MinD (4 µM), MinC (4 µM), or MinC mutant protein (4 µM), where indicated, SUVs (0.25 mg ml−1), and ATP (4 mM). SUV-associated MinC was quantified by SDS-PAGE and densitometry. Data from at least three replicates are shown as means ± standard errors of the means (*, P < 0.01; **, P < 0.001; ***, P < 0.0001). (e) MinC-MinD copolymer formation was visualized by transmission electron microscopy. Reaction mixtures containing MinD (8 µM), ATP (4 mM), and MinC (4 µM) or MinC mutant proteins (4 µM), where indicated, were applied to carbon-Formvar-coated grids, stained with uranyl acetate, and visualized by EM as described in Materials and Methods. Scale bar, 100 nm.
To determine if the minC mutations identified in the screen disrupt the interaction with FtsZ, we purified and assayed each MinC mutant protein for FtsZ destabilization in vitro. Purified FtsZ (6 µM) was polymerized with GTP (2 mM) under steady-state conditions in the presence of a GTP-regenerating system, and then increasing amounts of MinC (0 to 4 µM) were added. FtsZ polymers were collected by ultracentrifugation, and the amount of FtsZ in the pellet was quantified. MinC is a potent destabilizer of FtsZ polymers, and we detected that substoichiometric amounts of purified MinC (0.5 to 4 µM dimer) were sufficient to inhibit sedimentation of FtsZ (6 µM) polymers by 87% (Fig. 3b and c and Fig. S2). We next tested MinC mutant proteins for destabilization of FtsZ polymers in sedimentation assays. All mutant proteins purified similarly to wild-type MinC and eluted as a dimer by size exclusion chromatography, except for MinC(V18D), which was insoluble (data not shown). Our results show that all three MinC N-domain mutant proteins tested (G10D, S12D, and S16D) were defective for preventing sedimentation of dynamic FtsZ polymers and resulted in less than 5% inhibition at 1 µM (Fig. 3b).
We next tested if MinC proteins containing C-domain mutations prevented FtsZ polymer sedimentation. Compared to wild-type MinC, MinC(L194N) and MinC(R172A) were poor inhibitors of FtsZ sedimentation, resulting in 90% and 80% loss of function at 1 µM, respectively (Fig. 3c and Fig. S2), similar to MinC N-domain mutants (Fig. 3b and Fig. S2). MinC(Y201A) and MinC(E193K) were partially defective compared to wild-type MinC (55% and 35% loss of function, respectively) (Fig. 3c and Fig. S2). MinC(S134F), which contains a mutation in the known MinD binding site (S134) (30), was a potent inhibitor of FtsZ polymerization, similar to wild-type MinC (Fig. 3c and Fig. S2). Together, these results indicate that the two regions of MinC mapped in this study are largely responsible for destabilizing FtsZ dynamic polymers; one is located as a cleft on the MinC N-domain, and a second maps to the surface of the C-terminal dimerization domain outside the MinD interaction site (Fig. 3a and Fig. S3).
Mutations in the MinC CTD differentially impair recruitment to MinD-containing vesicles and copolymer formation with MinD.
MinD recruits MinC to small unilamellar vesicles (SUVs) in the presence of ATP in vitro (52, 53). To investigate if MinC mutant proteins are capable of binding to MinD, we tested them for recruitment to MinD-associated SUVs. MinC and MinC mutant proteins (4 µM) were added to reaction mixtures containing MinD (4 µM), SUVs (0.25 mg ml−1), and ATP (4 mM). The reaction mixtures were centrifuged at low speed to collect membrane-associated complexes, and the amount of MinC, wild-type or mutant protein, fractionating with the SUV pellet was quantified. Mutations in the MinC N-domain only modestly impaired binding of purified MinC to SUV-associated MinD (Fig. 3d). This is expected, since binding of MinC to MinD is mediated by the MinC C-terminal domain outside the dimerization interface and includes a conserved motif (133-RSGQ-136) (30). Accordingly, MinC(S134F) failed to fractionate with MinD (Fig. 3d), since S134 is present at the MinD interaction site (30, 41). MinC(E193K) and MinC(R172A) are located outside the MinD interaction site, based on the structural model (30, 41) (Fig. S3a and b); however, surprisingly they are defective for recruitment to SUVs via MinD by 97% and 63%, respectively (Fig. 3d). In a previous study, a MalE-MinC C-terminal domain fusion protein containing the R172A substitution also showed reduced phospholipid association with MinD, which is consistent with these observations (30). MinC(L194N) and MinC(Y201A) were only partially defective for recruitment to SUV-associated MinD (Fig. 3d). Together, our results confirm that mutations in the reported MinD interaction site, which includes S134, prevent recruitment of MinC to SUV-associated MinD and mutations in the MinC N-domain do not impair binding to MinD in the presence of SUVs. Furthermore, a large functional defect was observed for MinC(E193K), which failed to be recruited to MinD-SUV complexes but is outside the putative MinD interaction site (30, 41) (Fig. 3d and Fig. S3a and b).
Recently, our group and the Lowe group demonstrated that MinC and MinD form copolymers in the presence of ATP (41, 42). The crystal structure of an A. aeolicus copolymer containing alternating dimers of the MinC C-terminal domain and MinD has been reported, but the MinC N-domain is not present in the structure (Fig. S3a and b) (41). Therefore, we tested if MinC mutant proteins are capable of copolymer formation with MinD in the presence of ATP by negative staining and transmission electron microscopy (TEM), 90° angle light scatter, and sedimentation assays. MinC wild-type and mutant proteins (4 µM) were incubated with MinD (8 µM) and ATP (4 mM), and then reactions were applied to copper grids and analyzed by negative-staining TEM. We detected robust copolymer formation by wild-type MinC, MinD, and ATP, and we detected no copolymers when MinC was omitted (Fig. 3e). As expected, copolymers with MinD were observed for MinC(G10D), MinC(S12D), and MinC(S16D), which map to the MinC NTD (Fig. 3a and e). However, we did not detect copolymers by any of the MinC C-domain mutant proteins tested by TEM (Fig. S4a). MinC S134 is present at the MinC-MinD copolymer interface, based on the A. aeolicus crystal structure (41). Accordingly, we did not detect copolymers in reaction mixtures containing MinC(S134F) and MinD with ATP (Fig. S4a). Surprisingly, MinC C-terminal domain mutations, including R172A, E193K, L194N, and Y201A, which are located on the surface but are not present at the putative MinC-MinD copolymer interface or the dimerization interface, did not exhibit copolymers by TEM (Fig. 3a, Fig. S3a and b, and Fig. S4a) (41).
To confirm that MinC CTD mutant proteins do not copolymerize with MinD, we monitored 90° angle light scatter associated with reaction mixtures containing MinC mutant proteins, MinD, and ATP. The addition of ATP stimulated light scatter in reaction mixtures containing wild-type MinC and MinC NTD mutant proteins, but very little scatter was detected for MinC CTD mutant proteins (Fig. S4b). We next monitored copolymer formation using sedimentation assays. Copolymer assembly was stimulated by addition of ATP, and polymers were collected by ultracentrifugation. The amount of MinD present in copolymers with MinC wild-type or mutant protein was determined by SDS-PAGE and densitometry. As expected, we observed that the amount of MinD in pellet fractions was reduced by 68 to 86% in reaction mixtures containing MinC CTD mutant proteins (Fig. S4c). All MinC proteins tested here form stable dimers by size exclusion chromatography, indicating that the CTD is properly folded (data not shown). Finally, we also observed changes in the amounts of copolymers stimulated to form by addition of ATP in reaction mixtures containing MinD and MinC NTD mutant proteins. In light-scattering assays, MinC(G10D) and MinC(S12D) showed a larger amplitude increase by light scattering than wild-type MinC, suggesting that the mutant proteins are slightly more efficient at copolymer assembly with MinD than wild-type MinC (Fig. S4b). In contrast, MinC(S16D) showed a smaller amplitude increase with ATP and MinD by light scattering, suggesting that it is less efficient than wild-type MinC for copolymer formation; however, we observed copolymers of MinD and MinC(S16D) by TEM (Fig. 3e and Fig. S4b). All three MinC NTD mutant proteins were partially defective for assembly of large complexes collected by ultracentrifugation, although to a lesser extent than all of the MinC CTD mutant proteins. However, both light-scattering and ultracentrifugation assays may underreport small polymers. Together, these results suggest that there are additional residues in the MinC CTD, located outside of the putative MinD copolymer interface, that are important for assembly of MinC-MinD copolymers from E. coli.
The MinC-FtsZ interaction modulates MinC oscillation in vivo.
MinC fused to GFP (GFP-MinC) exhibits rapid pole-to-pole oscillation regulated by MinD and MinE (34, 54). To determine if MinC mutant proteins are capable of polar oscillation in vivo, we constructed gfp-minC and performed site-directed mutagenesis to incorporate two substitution mutations identified in the screen, gfp-minC(S16D) and gfp-minC(L194N). The gfp-minC wild-type and mutant genes then were reinserted into the ΔminC::kan-Prha-parE deletion strain at the native locus by lambda Red recombination. Cells were grown to early log phase, and GFP-MinC localization was examined by confocal fluorescence microscopy.
To detect GFP-MinC oscillation, we monitored fluorescence in live, dividing cells at 5-s intervals and measured the fluorescence amplitude across the long axis of the cell for each time series (Fig. 4a). We observed that GFP-MinC accumulates at one cell pole, redistributes to the opposing pole, and then returns, completing one oscillation cycle, in an average of 42.0 ± 8 s (n = 20) (Fig. 4b), with a minimum observed time of 35 s and a maximum observed time of 55 s, which is consistent with previous reports (34, 54). We plotted the amplitude of fluorescence as a function of cell length (in nanometers) at each interval and calculated the rate for directional movement of fluorescence foci along the longitudinal axis of the cell. In cells expressing GFP-MinC, this rate is 169.5 ± 5.4 nm s−1 (Fig. 4c).
FIG 4.

Oscillation of GFP-MinC and GFP-MinC mutant proteins. (a) Live cells expressing GFP-MinC from the chromosome were grown to log phase and imaged by confocal fluorescence microscopy as described in Materials and Methods. Frames were captured in 5-s increments, and the fluorescence intensity across the long axis of the cell for each frame was plotted for a representative cell. The cell image used for quantification is shown within each inset. Scale bar, 1 µm; A.U., arbitrary units. (b) Live cells containing chromosomal gfp-minC (top), gfp-minC(S16D) (middle), and gfp-minC(L194N) (bottom) were grown and imaged by confocal fluorescence microscopy as described in Materials and Methods. Panels showing one complete oscillation of GFP fluorescence are shown adjacent to a DIC image of each cell. Scale bar, 1 µm. (c) Box-and-whisker plot showing the rate of fluorescent focus tracking in live cells expressing chromosomal gfp-minC mutations. Live cells expressing chromosomal gfp-minC, gfp-minC(S16D), and gfp-minC(L194N) were grown and imaged by confocal fluorescence microscopy, and the rate of GFP movement (nm s−1) was determined as described in Materials and Methods. The box represents the interquartile range. The median rate of GFP movement is represented by a line within each box, and the mean is represented by a plus symbol. Twenty replicate cells that exhibited oscillation were analyzed for each strain (**, P < 0. 001; ***, P < 0.0001). (d) Live cells expressing chromosomal gfp-minC(S16D) were grown and imaged by confocal fluorescence microscopy as described in Materials and Methods. GFP-MinC(S16D) fluorescence localized at the septum is indicated by an arrow in the fluorescence panel. Scale bar, 1 µm.
It was previously reported that a chimera containing GFP and the MinC C-domain oscillated similarly to full-length MinC, suggesting that the MinD interaction only requires the C-domain of MinC (55); however, this fusion protein also transiently associated with the division septum, which was not observed by wild-type GFP-MinC. We next tested if GFP-MinC(S16D), which is competent for interacting with SUV-associated MinD but defective for destabilizing FtsZ polymers in vitro, oscillates in vivo. The rate of oscillation of GFP-MinC(S16D) is 26.6% lower (124.4 ± 9.6 nm min−1) than that of GFP-MinC (Fig. 4c). We also observed septal localization, or pausing, of GFP-MinC(S16D) in approximately 10% of cells (Fig. 4d) (n = 20 cells exhibiting oscillation).
We next tested if GFP-MinC(L194N) also exhibits impaired oscillation in vivo. In vitro, MinC(L194N) is also defective for destabilizing FtsZ polymers (Fig. 3c). Interestingly, we observed that 68% of cells expressing GFP-MinC(L194N) contained diffuse cytoplasmic fluorescence and failed to oscillate (data not shown), in contrast to GFP-MinC and GFP-MinC(S16D), where none or 12%, respectively, failed to oscillate. In the cells exhibiting GFP-MinC(L194N) oscillation, the rate was 37.7% lower (105.6 ± 7.4 nm min−1) than that of wild-type GFP-MinC (Fig. 4b and c). Interestingly, MinC(L194N) is recruited to SUVs by MinD, although slightly less so than wild-type MinC; however, it is defective for copolymer formation with MinD (Fig. 3d and Fig. S4). Together, these results show that mutations in the MinC N- and C-domains known to disrupt the FtsZ interaction described above also impair oscillation in vivo.
Recruitment of FtsZ to phospholipid vesicles by MinC and MinD.
Finally, we tested if FtsZ can be recruited to SUV-associated complexes of MinC and MinD using purified proteins in vitro. To detect complex formation between MinC, MinD, and FtsZ with SUVs, we used active, fluorescent FtsZ, labeled with Alexa Fluor 488 (FL-FtsZ). The current model suggests that FtsZ polymers first bind to the MinC C-domain via the FtsZ C-terminal end (CTE) (26, 27, 56). The Z-ring contains dynamic FtsZ polymers (57, 58); therefore, we monitored dynamic FtsZ polymers assembled with GTP, which we know are sensitive to disassembly by MinC (25, 26), for recruitment to SUV-associated complexes of MinC and MinD. Increasing amounts of FtsZ (0 to 250 pmol), polymerized with GTP and including a GTP-regenerating system, were added to reaction mixtures containing preassembled complexes of MinC (4 µM), MinD (4 µM), SUVs (0.25 mg ml−1), and ATP (4 mM). Complexes were collected by low-speed centrifugation, and the amount of FtsZ recruited was quantified by fluorescence intensity. Without MinC or MinD, few FtsZ polymers were detected in the pellet (Fig. 5a). We observed that MinC-MinD-SUV complexes recruit FtsZ in the presence of GTP, and that the recruitment was dependent on FtsZ concentration, with approximately 70 pmol, or 28% of the total FtsZ in the reaction mixture, recruited to SUVs (Fig. 5a). We next monitored SUV recruitment of stable FtsZ polymers in the presence of guanylyl 5’-α,β-methylenediphosphonate (GMPCPP). FtsZ-GMPCPP was not detected in pellets without MinC or MinD (Fig. 5b). As expected, we also observed concentration-dependent association of stable FtsZ polymers with Min complexes (Fig. 5b); however, the amount of FtsZ recruited to SUVs in the presence of GMPCPP was reduced by 70% compared to that in the presence of GTP (Fig. 5a and b). Previous reports suggested that the MinC N-domain interacts with an open FtsZ protofilament interface, and therefore this region should be also accessible in FtsZ monomers and dimers (27). To determine if MinC recruits FtsZ to SUV-associated complexes containing MinD via this site, we repeated the SUV recruitment assay in the presence of GDP to promote FtsZ dimer formation and also detected a concentration-dependent increase in the amount of FtsZ localizing to SUV-associated complexes containing MinD and wild-type MinC (Fig. 5c). Our results indicate that complexes of FtsZ-MinC-MinD can be assembled with both polymerized and nonpolymerized FtsZ, in contrast to previous reports (25, 26). A subsequent study also showed that FtsZ-GDP binds to MinC (59). To confirm that SUV recruitment is dependent on the MinC-MinD interaction and does not result from nonspecific trapping in SUVs, we repeated the SUV recruitment assay with GFP in the presence of MinC (4 µM), MinD (4 µM), ATP (4 mM), and, where indicated, GDP (2 mM), GMPCPP (0.2 mM), or GTP (1 mM) and a GTP-regenerating system. As expected, we detected no SUV-associated GFP in the pellet (Fig. S5). A recent report also indicated that MinD binds to FtsZ directly; however, we detected no recruitment of FtsZ to MinD-SUV complexes without MinC (data not shown), indicating that under these conditions, MinC is required for interaction with FtsZ (60).
FIG 5.

FtsZ complex formation with MinC, MinD, and SUVs. (a to c) FtsZ-MinC-MinD SUV sedimentation assays were assembled in reaction mixtures containing FtsZ (0 to 10 µM), SUVs (0.25 mg ml−1), MinC (4 µM), MinD (4 µM), GTP (1 mM) and a GTP-generating system, GMPCPP (0.2 mM), GDP (2 mM), and/or ATP (4 mM), where indicated, as described in Materials and Methods. SUV-associated FtsZ (pmol) in the presence or absence of MinC-MinD was collected by centrifugation as described in Materials and Methods and quantified by measuring the fluorescence associated with pellet fractions. (d to f) FtsZ-MinC-MinD SUV sedimentation assays were performed as described in Materials and Methods in reaction mixtures containing FtsZ (10 µM), SUVs (0.25 mg ml−1), MinD (4 µM), MinC (4 µM), MinC(S16D) (4 µM), MinC(L194N) (4 µM), GTP (1 mM) and a GTP-regenerating system, GMPCPP (0.2 mM), GDP (2 mM), and/or ATP (4 mM), where indicated. SUV-associated FtsZ (pmol) was collected by centrifugation as described in Materials and Methods and quantified by measuring the fluorescence associated with pellet fractions. The amount of FtsZ (pmol) shown is corrected for background. (g to i) FtsZ-MinC-MinD SUV sedimentation assays were performed as described in Materials and Methods in reaction mixtures containing FtsZ (10 µM) or FtsZ(ΔC4) (10 µM), FtsZ(ΔC9) (10 µM), FtsZ(ΔC18) (10 µM), FtsZ(I374V) (10 µM), SUVs (0.25 mg ml−1), MinD (4 µM), MinC (4 µM), ATP (4 mM), GTP (1 mM) and a GTP-regenerating system, GMPCPP (0.2 mM), and/or GDP (2 mM), where indicated. SUV-associated FtsZ was collected by centrifugation as described in Materials and Methods, quantified by measuring fluorescence, and corrected for background. Data from at least three replicates are shown as means ± SEM (*, P < 0.01; **, P < 0.001; ***, P < 0.0001).
To determine if the sites that we identified on the MinC N- and C-domains are important for recruitment of FtsZ under the conditions tested, we compared MinC(S16D) and MinC(L194N) to wild-type MinC in SUV sedimentation assays. In the presence of GTP and a regenerating system, we detected 40% less FtsZ associated with SUVs and MinD complexes with MinC(S16D) than with wild-type MinC and a modest but insignificant reduction with MinC(L194N) (Fig. 5d). In the presence of GMPCPP, we detected 82% less FtsZ in SUV-associated complexes containing MinD and MinC(L194N) (Fig. 5e) than with wild-type MinC but a small reduction associated with MinC(S16D)-containing complexes that was not significant (Fig. 5e). We next tested if MinC(S16D) and MinC(L194N) recruit nonpolymerized FtsZ to SUV-associated complexes with MinD. We detected 68% less FtsZ present in complexes with MinC(L194N) and 94% less FtsZ in complexes with MinC(S16D) than with wild-type MinC (Fig. 5f). Interestingly, these results show that in the presence of GDP, FtsZ interacts with both domains of MinC bound to MinD and SUVs. To confirm that MinC secondary structure was not perturbed by either of these substitutions, circular dichroism (CD) spectra of MinC(S16D) and MinC(L194N) were measured and shown to be similar to those of wild-type MinC (Fig. S6). Together, our results suggest that while stable FtsZ polymers require only the FtsZ-MinC CTD interaction to form a complex with MinC and MinD, nonpolymerized FtsZ requires contacts with each domain of MinC for complex assembly. Additionally, dynamic FtsZ polymers assembled with GTP are recruited to MinC much more efficiently than either FtsZ-GMPCPP or FtsZ-GDP, and mutations in either the MinC NTD or CTD only modestly impair recruitment of dynamic FtsZ polymers assembled with GTP. Notably, although MinC(S16D) and MinC(L194N) are able to efficiently recruit dynamic FtsZ polymers to MinD-associated SUVs, they are defective for disassembly of FtsZ polymers in vitro (Fig. 3b and c and 5d).
The FtsZ CTE is required for FtsZ-MinC-MinD complex formation with phospholipids.
The FtsZ CTE, consisting of residues 370 through 383, directly interacts with FtsA, ZipA, MinC, SlmA, ClpXP, and ZapD (18, 27, 58, 61–66). Several reports implicate the FtsZ CTE in mediating interactions with MinC (26, 64). In vivo, inhibition of cell division by the MinC CTD requires the FtsZ CTE (26), suggesting that FtsZ engages the MinC C-domain. Additionally, a GFP-MinC C-domain fusion protein is recruited to the Z-ring in vivo, and recruitment requires isoleucine 374 of the FtsZ CTE and R172 of MinC (26, 29, 30, 55, 67). Based on these observations, we investigated if recruitment of FtsZ to SUV-associated complexes containing MinC and MinD requires the FtsZ CTE in reaction mixtures containing fluorescent, truncated FtsZ [FL-FtsZ(ΔC18)], FtsZ [FL-FtsZ(ΔC9)], and FtsZ [FL-FtsZ(ΔC4)]. Surprisingly, all FtsZ C-terminal mutant proteins were efficiently recruited to SUV complexes containing MinC and MinD in the presence of GTP and a regenerating system, including FL-FtsZ(ΔC18) (Fig. 5g). However, FL-FtsZ(ΔC18) was not recruited to SUV complexes containing MinC and MinD in the presence of GMPCPP or GDP (Fig. 5h and i). Additionally, recruitment of both FtsZ(ΔC9) and FtsZ(ΔC4) mutant proteins in the presence of GTP, GMPCPP, or GDP was similar to that of FtsZ (Fig. 5g to i). Since residue I374 is present in both the FtsZ(ΔC9) and FtsZ(ΔC4) truncations but not in the FtsZ(ΔC18) mutant, we purified and tested FtsZ(I374V) for the interaction with MinCD. Interestingly, the I374V mutation alone did not impair recruitment of FtsZ to MinC-MinD-SUV complexes in the presence of GDP or GMPCPP in vitro, and we observed only a minor reduction with GTP (Fig. 5g to i). These results suggest that amino acids 366 through 374 of FtsZ are important for binding of MinC-MinD-SUV complexes to FtsZ dimers and GMPCPP-stabilized polymers but are not essential to FtsZ polymers assembled with GTP.
DISCUSSION
Mapping the FtsZ interaction sites on MinC.
We mapped two regions on the surface of MinC important for controlling septal localization using mutagenesis and site-specific recombination, followed by functional analyses in vivo and in vitro. Several purified MinC mutant proteins, with mutations in both the N-domain and the C-domain of MinC, failed to effectively prevent the GTP-dependent sedimentation of FtsZ in vitro but were recruited to SUVs by MinD (Fig. 3a to d), suggesting that cell division defects associated with chromosomal minC mutants are due to an impaired FtsZ interaction. Moreover, MinC mutant proteins with substitutions in the N-domain (S16D) or the C-domain (L194N) bind to dynamic FtsZ polymers but are defective for promoting polymer disassembly, in contrast to wild-type MinC. MinC is a potent inhibitor of FtsZ polymerization; disassembly of dynamic FtsZ polymers, assembled with GTP and a regenerating system, occurred with substoichiometric (1:12) amounts of MinC (0.5 µM MinC dimer and 6 µM FtsZ) (Fig. 3b and c; see also Fig. S2 in the supplemental material). This ratio is inconsistent with MinC promoting polymer disassembly via an FtsZ sequestration mechanism. Together, there are two distinct sites of FtsZ interaction on the surface of a single MinC protomer, with four total sites accessible on a dimer (Fig. 6a). Both interaction sites are important for destabilizing FtsZ polymers: one site is at an N-domain cleft (G10, S12, and S16) of a protomer, and another site is on the outer surface of the C-terminal dimerization domain (R172, E193, L194N, and Y201), as modeled here (Fig. 3a) and identified on a recent structure of the CTD of E. coli MinC (68). Importantly, MinC accommodates interactions simultaneously with MinD and FtsZ, and the regions do not appear to overlap based on FtsZ recruitment assays and structural modeling of the MinC-MinD interface from A. aeolicus (Fig. 5 and Fig. S3) (41). Our results are in agreement with a recent study of the MinC N-domain, which implicated nearby residues K9, F42, K35, and A39 in contacting FtsZ and are adjacent to the N-domain cleft described here (31). The phenotypic defects associated with MinC N- and C-domain mutations in this study do not result from overexpression, since MinC mutant proteins were expressed to nearly wild-type levels from the chromosome.
FIG 6.

Model for MinC engaging FtsZ assemblies. (a) Relative position of the FtsZ subunit interface and the MinC-FtsZ interaction sites. The distance between adjacent FtsZ protofilament interfaces (42 Å) and between MinC N-domain protomer clefts (76 Å) is shown. Although there is likely considerable flexibility in the position of the MinC NTD relative to the CTD, distances suggest that both MinC NTD clefts in a dimer would be unable to engage adjacent FtsZ protomers in a single polymer. E. coli FtsZ is modeled on the Methanococcusjannaschii FtsZ dimer (PDB entry 1W59) (75), and E. coli MinC is modeled on T. maritima MinC (PDB entry 1HF2) (49). GTP is indicated by light brown CPK spheres, and FtsZ(N280) and FtsZ(E276) are indicated by light blue and dark blue CPK spheres, respectively. MinC G10, S12, S16, R172, E193, L194, and Y201 are shown as yellow CPK spheres. (b) The MinC C-domain site recruits the FtsZ CTE of stable GMPCPP-FtsZ polymers. (c) Both the MinC N- and C-domain sites mediate binding to free GDP-FtsZ. (d) FtsZ polymers assembled with GTP are recruited by MinC. Recruitment does not require the CTE of FtsZ, although it may stabilize the interaction. Although neither the MinC NTD nor CTD site identified here is essential for recruitment, both are important for robust severing of FtsZ polymers.
Conservation of the MinC-MinD interface.
MinC and MinD assemble into copolymers in the presence of ATP in vitro (41, 42). The Lowe group reported a crystal structure for the A. aeolicus MinC CTD-MinD complex as alternating dimers arranged as a copolymer, revealing two MinC-MinD interaction sites (41). The first site contains a conserved RSGQ motif in MinC and helix 7 of MinD, while a secondary site is mediated by contacts between MinD helix 8 and MinC helix 3, although this helix does not appear to be present in E. coli MinC (41). To determine if the mutations identified here in MinC also impair copolymerization with MinD, we purified MinC mutant proteins and measured copolymer formation by EM, light scattering, and sedimentation assays. MinC(S134F) contains a mutation in the first predicted MinC-MinD interaction site (i.e., RSGQ) and is severely impaired for copolymer formation with MinD (Fig. S4). We mapped MinC R172, E193, L194, and Y201 onto a structural model of the E. coli MinC C-domain and observed that they are absent from both predicted MinC-MinD interfaces (Fig. S3a and b). Surprisingly, the purified C-domain mutant proteins were all impaired for copolymer formation; however, they form dimers (data not shown), suggesting that the C-domains are folded correctly, and they are recruited to SUVs by MinD, with the exception of MinC(E193K) (Fig. 3d and Fig. S4). This suggests that the interaction between MinC and MinD in the copolymer is different from the interaction with MinD at the membrane. The role of the copolymer during oscillation of the Min system or cell division is not yet understood, and a recent report suggests that copolymer formation is not required for division (45).
Slow oscillation by GFP-MinC mutant proteins.
Oscillation of GFP-MinD in vivo and, by extension, GFP-MinC requires MinE to promote ATP hydrolysis by MinD but does not require FtsZ (34, 44). Remarkably, we found that GFP-MinC mutant proteins impaired for disassembly of dynamic FtsZ polymers (S16D and L194N) oscillate 27% and 38% slower, respectively, in vivo than GFP-MinC (Fig. 3b and c and 4c). This is likely not due to a perturbed interaction with membrane-associated MinD, since purified MinC(S16D) and MinC(L194N) were recruited to MinD-bound SUVs (Fig. 3d). Both of these mutants are impaired for destabilizing dynamic FtsZ polymers, yet both recruit FtsZ polymers assembled with GTP to MinC-MinD-SUV complexes (Fig. 3b and c and 5d). In vivo, how could FtsZ binding modulate MinC oscillations? Pausing at the division septum could account for the slow oscillation of GFP-MinC(S16D). Consistent with this, we observed transient localization of GFP-MinC(S16D) at the Z-ring in 10% of cells (Fig. 4d), suggesting productive recruitment to dynamic FtsZ polymers in vivo. Failure to destabilize polymers at the Z-ring may lead to the prolonged localization observed. Accordingly, the MinC C-domain tagged with GFP also pauses at the septum, consistent with failure to disassemble the ring (55, 64). MinC(L194N) is also able to recruit FtsZ polymers assembled with GTP to MinD-associated SUVs (Fig. 5d), raising the possibility that MinC(L194N) also is recruited to FtsZ polymers in vivo, although we did not detect septal localization or pausing of GFP-MinC(L194N).
A recent report showed that the stoichiometry of MinE and MinD on the membrane may modulate MinE-stimulated stabilization and release of MinD (69). MinD oscillation in vivo and wave propagation in vitro occurs in the total absence of MinC (34, 38, 40, 44, 69). The Lutkenhaus group showed that expression of MinD heterodimers impaired for MinC copolymerization, but not MinC binding, restores wild-type morphology to a min deletion strain (45), suggesting that copolymer formation is not required to effectively regulate Z-ring formation in vivo under the conditions tested. However, engagement of MinD by MinC could serve to constrain the accessible population of MinD, through sequestration on the membrane surface or in the cytoplasmic pool, and therefore modulate the propagation of MinD across the phospholipid surface. If copolymers modulate the availability and membrane concentration of MinD, then the slow oscillation observed for MinC(L194N) could result from impaired copolymer formation with MinD (Fig. 4b and c and Fig. S4).
Destabilization of FtsZ polymers by MinC.
FtsZ assembles into dynamic polymers in the presence of GTP, with more than half of the FtsZ within filaments suggested to exist in a GDP-bound state (70, 71). MinC substoichiometrically destabilizes FtsZ polymers, without MinD, in a concentration-dependent manner, and destabilization requires active GTP hydrolysis (Fig. 3b and c). MinC does not lower the rate of GTP hydrolysis by FtsZ (23), which is consistent with MinC rapidly releasing FtsZ. In a model for how MinC destabilizes FtsZ polymers, the Lutkenhaus group proposed that the FtsZ CTEs in FtsZ filaments recruit the MinC CTD, thereby positioning the MinC NTD at the FtsZ helix 10 (H10) within the FtsZ subunit-subunit interface and promoting the breakage of polymers (27). More recently, it was suggested that potent disassembly activity by MinC in the presence of MinD occurs through a severing mechanism (31). Our results extend this model by revealing that nucleotide occupancy of FtsZ differentially affects engagement by MinC at each site mapped in this study. We further demonstrate that recruitment of FtsZ assembled with GTP to MinC does not require the CTE of FtsZ. Pausing of GFP-MinC(S16D) at the septum is consistent with a model in which a MinC NTD interaction with FtsZ in a filament promotes release of the FtsZ subunit from the filament by a severing mechanism. Recently, the Schwille group also demonstrated that MinC enhances the detachment rate of GDP-bound FtsZ from polymers (70).
FtsZ subunit turnover due to GTP hydrolysis occurs stochastically within filaments, resulting in exposed minus ends containing the H10 helix (70). The FtsZ H10 helix being exposed in GDP-bound FtsZ suggests that MinC should interact with GDP-FtsZ. A previous study from the Rivas group reported that MinC strongly binds GDP-FtsZ (59). Consistent with this, we detected complex formation between free GDP-FtsZ and SUV-associated MinC-MinD, which required both MinC NTD and CTD sites (Fig. 5c and f), but recruitment of GDP-FtsZ was much less efficient than recruitment of dynamic FtsZ polymers, consistent with a weaker interaction. Together, these findings suggest that a GDP-bound FtsZ subunit is conformationally distinct in the polymer compared to that in the dimer.
In the model of MinC engaging FtsZ, the MinC-FtsZ interaction is promoted by FtsZ GTP binding and hydrolysis, and although the interaction between the FtsZ CTE and MinC is not essential for recruitment of dynamic FtsZ polymers to MinC-MinD-SUV complexes, the CTE is important for binding of MinC to GMPCPP-stabilized FtsZ polymers and FtsZ-GDP dimers (Fig. 5g to i and 6). In the model, MinC residues R172, E193, L194, and Y201, which are clustered on the MinC C-domain, mediate an interaction with residues 366 to 374 of FtsZ. The cleft at the MinC N-domain, containing residues G10, S12, and S16, is important for binding to the FtsZ polymerization domain, likely at the protofilament interface (Fig. 6a). Both MinC NTD and CTD sites mapped in this study are important for promoting efficient FtsZ polymer disassembly or severing (Fig. 6d). Spatially, the distance between two MinC N-domain clefts in a dimer is approximately 80% larger than the distance between adjacent subunit interfaces in an FtsZ protofilament (76 Å and 42 Å, respectively) (Fig. 6a). Although the MinC N-domain is likely considerably more flexible in position than the C-domain due to a long tethering region between them, given this distance, it may be more likely that a MinC dimer would interact with, and subsequently destabilize, two separate FtsZ protofilaments, which is consistent with MinC acting as a potent inhibitor of FtsZ polymerization.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Lennox medium (LB) was inoculated with stationary-phase cultures of wild-type E. coli MG1655 or mutant strains, and cultures were grown at 30°C to an optical density at 600 nm (OD600) of approximately 0.5. Strains containing chromosomal gfp-minC were grown in the presence of l-arabinose (100 µM) to induce expression of gfp-minC (72).
Mutagenesis and screening.
Plasmids containing minC were mutagenized by amplification with the Mutazyme II error-prone polymerase (Agilent) and by site-directed mutagenesis (Agilent). Mutagenized minC gene products were introduced into the chromosomal copy of minC using λ-red recombination (73) (J. Teramoto, K. A. Datsenko, and B. L. Wanner, unpublished results). The MG1655 ΔminC::kan-Prha-parE strain containing pKD46 was grown as described in “Bacterial strains, plasmids, and growth conditions,” above, to an OD600 of approximately 0.4 in the presence of ampicillin (100 µg ml−1) and l-arabinose (20 mM). Cells were electroporated with mutagenized minC gene products, and recombinants containing chromosomal minC mutants were selected by growth on M9 minimal medium agar plates with 1% l-rhamnose. Subsequently, all recombinants were confirmed by sequencing. Recombinant strains containing mutagenized minC (1,020 strains) were isolated and screened for cell length by microscopy. Of the 240 isolates exhibiting a filamentous phenotype, 190 contained premature stop codons or missense mutations, 8 had single-nucleotide substitutions, and 42 contained two or three nucleotide substitutions, which were reconstructed as single substitutions.
Microscopy.
Wild-type and mutant cells were grown as described in “Bacterial strains, plasmids, and growth conditions,” above. Cells were collected by centrifugation at 5,000 × g, washed in Tris-buffered saline with EDTA (1 mM), and fixed to glass slides with poly-l-lysine-coated coverslips. Cells were imaged by differential interference contrast (DIC) microscopy with a Zeiss AxioCam HRc high-resolution camera. Images were prepared using Adobe Photoshop CS3, and cell lengths were measured using NIH ImageJ software. To monitor the oscillation of GFP-MinC mutant proteins, strains containing gfp-minC were grown as described above and applied to glass slides with agarose gel pads (5%, wt/vol) containing M9 minimal medium supplemented with 0.2% glucose. Live cells were visualized by confocal fluorescence microscopy with a Zeiss AxioImager M2 imaging system equipped with an LSM 700 confocal module with excitation and emission set to 488 and 525 nm, respectively. To determine the rate of fluorescence movement across the longitudinal cell axis (in nanometers per minute), images of live cells were captured in 5-s intervals for 20 frames. The peak fluorescence was determined for each image using NIH ImageJ software and plotted as a function of distance traveled from the originating pole.
Protein extraction and immunoblotting.
Wild-type and mutant strains were grown as described in “Bacterial strains, plasmids, and growth conditions,” above. Cells were collected by centrifugation at 5,000 × g and resuspended into 1 ml of LB (Lennox medium). Protein was extracted by the addition of 15% (vol/vol) trichloroacetic acid, and protein concentration was determined by the bicinchoninic acid assay (BCA). Protein extracts (75 µg) were analyzed by SDS-PAGE and immunoblotted with antibodies directed against MinC. Anti-MinC antiserum was generated in rabbits using purified wild-type MinC (Noble Life Sciences, Inc.).
Expression and purification of proteins.
Native MinC, MinD, and FtsZ wild-type and mutant proteins were overexpressed in E. coli BL21 (λDE3) and purified as described previously (42, 61, 74). Size exclusion chromatography performed as part of the purification confirmed dimerization of MinC and MinC mutant proteins. Protein concentrations are reported as FtsZ monomers, MinC dimers, and MinD dimers. Where indicated, circular dichroism spectra were obtained for reaction mixtures containing MinC (1 µM) or MinC mutant proteins (1 µM) in assembly buffer on a Jasco J-1100 CD spectrophotometer at room temperature using a 0.1-mm quartz cuvette. Data were collected from 200 to 250 nm and averaged over 5 scans.
FtsZ polymerization assays with MinC.
Purified FtsZ was incubated with GTP (2 mM) and a GTP-regenerating system containing acetyl phosphate (15 mM) and acetate kinase (25 µg ml−1) in the presence of increasing MinC or MinC mutant protein concentrations (0 to 4 µM). Reactions were performed in assembly buffer containing 2-(N-morpholino)ethanesulfonic acid (MES) (50 mM, pH 6.5), KCl (100 mM), MgCl2 (10 mM), and polymers were collected by centrifugation at 129,000 × g for 30 min at 23°C in a Beckman TLA 120.1 rotor. Pellets containing polymers were resuspended in boiling lithium dodecyl sulfate (LDS) loading buffer and analyzed by SDS-PAGE and densitometry using NIH ImageJ.
Phospholipid recruitment assays with MinD and MinC.
MinD (4 µM) was added to reaction mixtures containing SUVs (0.25 mg ml−1), MinC (4 µM), or MinC mutants (4 µM), where indicated, and ATP (4 mM) in assembly buffer. Small unilamellar vesicles from E. coli extracts (Avanti Polar lipids) were prepared as described by Conti et al. (42). Reaction mixtures were incubated at 30°C for 10 min, and phospholipids were collected by centrifugation at 14,000 × g for 15 min at 23°C. SUV-containing pellets were resuspended in LDS loading buffer and analyzed by SDS-PAGE and densitometry.
Where indicated, purified FtsZ or FtsZ mutant proteins labeled with Alexa Fluor 488 or GFP were premixed with GMPCPP (0.2 mM), GDP (2 mM), or GTP (1 mM) and a GTP-regenerating system containing acetyl phosphate (15 mM) and acetate kinase (25 µg ml−1), where indicated, and increasing amounts of FtsZ or GFP with nucleotide were added to reaction mixtures containing MinCD complexes (4 µM each) preassembled onto SUVs as described above. Active, fluorescent FtsZ wild-type and mutant proteins were generated by labeling with Alexa Fluor 488 succinimidyl ester and cycling through GTP-dependent polymerization as described previously (61). Reaction mixtures containing GMPCPP or GTP were incubated for 5 min at 23°C, and SUVs were collected by centrifugation at 10,000 × g for 2 min 23°C. Reaction mixtures containing GDP were incubated for 10 min at 30°C, and SUVs were collected by centrifugation at 14,000 × g for 15 min at 23°C. SUV-containing pellets were resuspended in assembly buffer, and pellet-associated fluorescence was measured with excitation and emission wavelengths set for 490 nm and 525 nm, respectively (Alexa Fluor 488), or 395 nm and 510 nm, respectively (GFP).
Copolymer assays with MinC and MinD.
MinD (8 µM) was added to reaction mixtures containing MinC (4 µM) or MinC mutant proteins (4 µM), where indicated, in assembly buffer. ATP (4 mM) was added to initiate copolymer formation, and reaction mixtures were incubated for 5 min at 23°C. Polymers were collected by centrifugation at 129,000 × g for 30 min at 23°C in a Beckman TLA 120.1 rotor. Pellets were resuspended in LDS loading buffer (1×), and the amount of pellet-associated MinD was determined by SDS-PAGE and densitometry.
To monitor MinCD copolymer formation by light scatter, MinC (4 µM) or MinC mutant proteins (4 µM), where indicated, were added to reaction mixtures containing MinD (8 µM) in assembly buffer. Angle light scatter at 90° was monitored with excitation and emission wavelengths set to 450 nm. After 5 min, ATP (4 mM) was added and light scatter was monitored for an additional 30 min.
Electron microscopy.
Reaction mixtures containing MinD (8 µM), MinC (4 µM), and MinC mutant proteins (4 µM), where indicated, and ATP (4 mM) in assembly buffer were applied to 300-mesh carbon-Formvar-coated grids, fixed with glutaraldehyde (1%), and stained with uranyl acetate (2%). Samples were imaged by transmission electron microscopy using an FEI Tecnai G2 Spirit BioTWIN 80-kV instrument equipped with an SIS Morada 11-megapixel camera.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shannon May, Eric DiBiasio, Cathy Trebino, and Evelyn Siler for helpful discussions and critical reading of the manuscript, Barry Wanner for pKD267, Janet Atoyan for microscopy and sequencing assistance, and Neil Greene for providing helpful comments and considerations. We also thank Marc Llaguno and Xinran Liu at the Center for Cellular and Molecular Imaging at Yale School of Medicine for TEM.
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM118927 to J. Camberg. Fluorescence microscopy and sequencing were performed at the Rhode Island Genomics and Sequencing Center, which is supported in part by the National Science Foundation (MRI grant no. DBI-0215393 and EPSCoR grant nos. 0554548 and EPS-1004057), the U.S. Department of Agriculture (grant nos. 2002-34438-12688, 2003-34438-13111, and 2008-34438-19246), and the University of Rhode Island. CD was performed at the RI-INBRE core research facility, which is supported in part by an Institutional Development Award (IDeA), Network for Biomedical Research Excellence, from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103430.
The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00374-18.
REFERENCES
- 1.Egan AJ, Vollmer W. 2013. The physiology of bacterial cell division. Ann N Y Acad Sci 1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- 2.Lutkenhaus J, Pichoff S, Du S. 2012. Bacterial cytokinesis: from Z ring to divisome. Cytoskeleton (Hoboken) 69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lutkenhaus J. 2007. Assembly dynamics of the bacterial MinCDE system and spatial regulation of the Z ring. Annu Rev Biochem 76:539–562. doi: 10.1146/annurev.biochem.75.103004.142652. [DOI] [PubMed] [Google Scholar]
- 4.Adams DW, Errington J. 2009. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat Rev Microbiol 7:642–653. doi: 10.1038/nrmicro2198. [DOI] [PubMed] [Google Scholar]
- 5.Erickson HP, Anderson DE, Osawa M. 2010. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev 74:504–528. doi: 10.1128/MMBR.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapiro L, Losick R. 1997. Protein localization and cell fate in bacteria. Science 276:712–718. doi: 10.1126/science.276.5313.712. [DOI] [PubMed] [Google Scholar]
- 7.de Boer PA, Crossley RE, Rothfield LI. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- 8.Bernhardt TG, de Boer PA. 2005. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol Cell 18:555–564. doi: 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsang MJ, Bernhardt TG. 2015. Guiding divisome assembly and controlling its activity. Curr Opin Microbiol 24:60–65. doi: 10.1016/j.mib.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akerlund T, Gullbrand B, Nordstrom K. 2002. Effects of the Min system on nucleoid segregation in Escherichia coli. Microbiology 148:3213–3222. doi: 10.1099/00221287-148-10-3213. [DOI] [PubMed] [Google Scholar]
- 11.Mulder E, Woldringh CL. 1989. Actively replicating nucleoids influence positioning of division sites in Escherichia coli filaments forming cells lacking DNA. J Bacteriol 171:4303–4314. doi: 10.1128/jb.171.8.4303-4314.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu XC, Margolin W. 1999. FtsZ ring clusters in min and partition mutants: role of both the Min system and the nucleoid in regulating FtsZ ring localization. Mol Microbiol 32:315–326. doi: 10.1046/j.1365-2958.1999.01351.x. [DOI] [PubMed] [Google Scholar]
- 13.Cho H, McManus HR, Dove SL, Bernhardt TG. 2011. Nucleoid occlusion factor SlmA is a DNA-activated FtsZ polymerization antagonist. Proc Natl Acad Sci U S A 108:3773–3778. doi: 10.1073/pnas.1018674108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tonthat NK, Arold ST, Pickering BF, Van Dyke MW, Liang S, Lu Y, Beuria TK, Margolin W, Schumacher MA. 2011. Molecular mechanism by which the nucleoid occlusion factor, SlmA, keeps cytokinesis in check. EMBO J 30:154–164. doi: 10.1038/emboj.2010.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rowlett VW, Margolin W. 2013. The bacterial Min system. Curr Biol 23:R553–R556. doi: 10.1016/j.cub.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 16.Monahan LG, Liew AT, Bottomley AL, Harry EJ. 2014. Division site positioning in bacteria: one size does not fit all. Front Microbiol 5:19. doi: 10.3389/fmicb.2014.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shih YL, Zheng M. 2013. Spatial control of the cell division site by the Min system in Escherichia coli. Environ Microbiol 15:3229–3239. doi: 10.1111/1462-2920.12119. [DOI] [PubMed] [Google Scholar]
- 18.Du S, Lutkenhaus J. 2014. SlmA antagonism of FtsZ assembly employs a two-pronged mechanism like MinCD. PLoS Genet 10:e1004460. doi: 10.1371/journal.pgen.1004460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parti RP, Biswas D, Wang M, Liao M, Dillon JA. 2011. A minD mutant of enterohemorrhagic E. coli O157:H7 has reduced adherence to human epithelial cells. Microb Pathog 51:378–383. doi: 10.1016/j.micpath.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Parti RP, Biswas D, Helgeson S, Michael FS, Cox A, Dillon JA. 2011. Attenuated virulence of min operon mutants of Neisseria gonorrhoeae and their interactions with human urethral epithelial cells. Microbes Infect 13:545–554. doi: 10.1016/j.micinf.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Ramirez-Arcos S, Szeto J, Beveridge T, Victor C, Francis F, Dillon J. 2001. Deletion of the cell-division inhibitor MinC results in lysis of Neisseria gonorrhoeae. Microbiology 147:225–237. doi: 10.1099/00221287-147-1-225. [DOI] [PubMed] [Google Scholar]
- 22.Szeto J, Ramirez-Arcos S, Raymond C, Hicks LD, Kay CM, Dillon JA. 2001. Gonococcal MinD affects cell division in Neisseria gonorrhoeae and Escherichia coli and exhibits a novel self-interaction. J Bacteriol 183:6253–6264. doi: 10.1128/JB.183.21.6253-6264.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Z, Mukherjee A, Pichoff S, Lutkenhaus J. 1999. The MinC component of the division site selection system in Escherichia coli interacts with FtsZ to prevent polymerization. Proc Natl Acad Sci U S A 96:14819–14824. doi: 10.1073/pnas.96.26.14819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu Z, Lutkenhaus J. 2000. Analysis of MinC reveals two independent domains involved in interaction with MinD and FtsZ. J Bacteriol 182:3965–3971. doi: 10.1128/JB.182.14.3965-3971.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dajkovic A, Lan G, Sun SX, Wirtz D, Lutkenhaus J. 2008. MinC spatially controls bacterial cytokinesis by antagonizing the scaffolding function of FtsZ. Curr Biol 18:235–244. doi: 10.1016/j.cub.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 26.Shen B, Lutkenhaus J. 2009. The conserved C-terminal tail of FtsZ is required for the septal localization and division inhibitory activity of MinC(C)/MinD. Mol Microbiol 72:410–424. doi: 10.1111/j.1365-2958.2009.06651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen B, Lutkenhaus J. 2010. Examination of the interaction between FtsZ and MinCN in E. coli suggests how MinC disrupts Z rings. Mol Microbiol 75:1285–1298. doi: 10.1111/j.1365-2958.2010.07055.x. [DOI] [PubMed] [Google Scholar]
- 28.Szeto TH, Rowland SL, King GF. 2001. The dimerization function of MinC resides in a structurally autonomous C-terminal domain. J Bacteriol 183:6684–6687. doi: 10.1128/JB.183.22.6684-6687.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiomi D, Margolin W. 2007. The C-terminal domain of MinC inhibits assembly of the Z ring in Escherichia coli. J Bacteriol 189:236–243. doi: 10.1128/JB.00666-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou H, Lutkenhaus J. 2005. MinC mutants deficient in MinD- and DicB-mediated cell division inhibition due to loss of interaction with MinD, DicB, or a septal component. J Bacteriol 187:2846–2857. doi: 10.1128/JB.187.8.2846-2857.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park KT, Dajkovic A, Wissel M, Du S, Lutkenhaus J. 2018. MinC and FtsZ mutant analysis provides insight into MinC/MinD-mediated Z Ring disassembly. J Biol Chem 16:5834–5846. doi: 10.1074/jbc.M117.815894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowe J, Amos LA. 2009. Evolution of cytomotive filaments: the cytoskeleton from prokaryotes to eukaryotes. Int J Biochem Cell Biol 41:323–329. doi: 10.1016/j.biocel.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 33.Michie KA, Lowe J. 2006. Dynamic filaments of the bacterial cytoskeleton. Annu Rev Biochem 75:467–492. doi: 10.1146/annurev.biochem.75.103004.142452. [DOI] [PubMed] [Google Scholar]
- 34.Raskin DM, de Boer PA. 1999. MinDE-dependent pole-to-pole oscillation of division inhibitor MinC in Escherichia coli. J Bacteriol 181:6419–6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutkenhaus J, Sundaramoorthy M. 2003. MinD and role of the deviant Walker A motif, dimerization and membrane binding in oscillation. Mol Microbiol 48:295–303. doi: 10.1046/j.1365-2958.2003.03427.x. [DOI] [PubMed] [Google Scholar]
- 36.Hu Z, Lutkenhaus J. 2001. Topological regulation of cell division in E. coli. spatiotemporal oscillation of MinD requires stimulation of its ATPase by MinE and phospholipid. Mol Cell 7:1337–1343. doi: 10.1016/S1097-2765(01)00273-8. [DOI] [PubMed] [Google Scholar]
- 37.Park KT, Wu W, Battaile KP, Lovell S, Holyoak T, Lutkenhaus J. 2011. The Min oscillator uses MinD-dependent conformational changes in MinE to spatially regulate cytokinesis. Cell 146:396–407. doi: 10.1016/j.cell.2011.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vecchiarelli AG, Li M, Mizuuchi M, Mizuuchi K. 2014. Differential affinities of MinD and MinE to anionic phospholipid influence Min patterning dynamics in vitro. Mol Microbiol 93:453–463. doi: 10.1111/mmi.12669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsieh CW, Lin TY, Lai HM, Lin CC, Hsieh TS, Shih YL. 2010. Direct MinE-membrane interaction contributes to the proper localization of MinDE in E. coli. Mol Microbiol 75:499–512. doi: 10.1111/j.1365-2958.2009.07006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loose M, Fischer-Friedrich E, Ries J, Kruse K, Schwille P. 2008. Spatial regulators for bacterial cell division self-organize into surface waves in vitro. Science 320:789–792. doi: 10.1126/science.1154413. [DOI] [PubMed] [Google Scholar]
- 41.Ghosal D, Trambaiolo D, Amos LA, Lowe J. 2014. MinCD cell division proteins form alternating copolymeric cytomotive filaments. Nat Commun 5:5341. doi: 10.1038/ncomms6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conti J, Viola MG, Camberg JL. 2015. The bacterial cell division regulators MinD and MinC form polymers in the presence of nucleotide. FEBS Lett 589:201–206. doi: 10.1016/j.febslet.2014.11.047. [DOI] [PubMed] [Google Scholar]
- 43.Huang H, Wang P, Bian L, Osawa M, Erickson HP, Chen Y. 2018. The cell division protein MinD from Pseudomonas aeruginosa dominates the assembly of the MinC-MinD copolymers. J Biol Chem 293:7786–7795. doi: 10.1074/jbc.RA117.001513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raskin DM, de Boer PA. 1999. Rapid pole-to-pole oscillation of a protein required for directing division to the middle of Escherichia coli. Proc Natl Acad Sci U S A 96:4971–4976. doi: 10.1073/pnas.96.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park KT, Du S, Lutkenhaus J. 2015. MinC/MinD copolymers are not required for Min function. Mol Microbiol 98:895–909. doi: 10.1111/mmi.13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teather RM, Collins JF, Donachie WD. 1974. Quantal behavior of a diffusible factor which initiates septum formation at potential division sites in Escherichia coli. J Bacteriol 118:407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camberg JL, Hoskins JR, Wickner S. 2011. The interplay of ClpXP with the cell division machinery in Escherichia coli. J Bacteriol 193:1911–1918. doi: 10.1128/JB.01317-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Labie C, Bouche F, Bouche JP. 1990. Minicell-forming mutants of Escherichia coli: suppression of both DicB- and MinD-dependent division inhibition by inactivation of the minC gene product. J Bacteriol 172:5852–5855. doi: 10.1128/jb.172.10.5852-5855.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cordell SC, Anderson RE, Lowe J. 2001. Crystal structure of the bacterial cell division inhibitor MinC. EMBO J 20:2454–2461. doi: 10.1093/emboj/20.10.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46:W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guex N, Peitsch MC, Schwede T. 2009. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30:S162–S173. doi: 10.1002/elps.200900140. [DOI] [PubMed] [Google Scholar]
- 52.Lackner LL, Raskin DM, de Boer PA. 2003. ATP-dependent interactions between Escherichia coli Min proteins and the phospholipid membrane in vitro. J Bacteriol 185:735–749. doi: 10.1128/JB.185.3.735-749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu Z, Saez C, Lutkenhaus J. 2003. Recruitment of MinC, an inhibitor of Z-ring formation, to the membrane in Escherichia coli: role of MinD and MinE. J Bacteriol 185:196–203. doi: 10.1128/JB.185.1.196-203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu Z, Lutkenhaus J. 1999. Topological regulation of cell division in Escherichia coli involves rapid pole to pole oscillation of the division inhibitor MinC under the control of MinD and MinE. Mol Microbiol 34:82–90. doi: 10.1046/j.1365-2958.1999.01575.x. [DOI] [PubMed] [Google Scholar]
- 55.Johnson JE, Lackner LL, de Boer PA. 2002. Targeting of (D)MinC/MinD and (D)MinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ rings. J Bacteriol 184:2951–2962. doi: 10.1128/JB.184.11.2951-2962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dajkovic A, Lutkenhaus J. 2006. Z ring as executor of bacterial cell division. J Mol Microbiol Biotechnol 11:140–151. doi: 10.1159/000094050. [DOI] [PubMed] [Google Scholar]
- 57.Stricker J, Maddox P, Salmon ED, Erickson HP. 2002. Rapid assembly dynamics of the Escherichia coli FtsZ-ring demonstrated by fluorescence recovery after photobleaching. Proc Natl Acad Sci U S A 99:3171–3175. doi: 10.1073/pnas.052595099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Viola MG, LaBreck CJ, Conti J, Camberg JL. 2017. Proteolysis-dependent remodeling of the tubulin homolog FtsZ at the division septum in Escherichia coli. PLoS One 12:e0170505. doi: 10.1371/journal.pone.0170505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hernandez-Rocamora VM, Garcia-Montanes C, Reija B, Monterroso B, Margolin W, Alfonso C, Zorrilla S, Rivas G. 2013. MinC shortens FtsZ protofilaments by preferentially interacting with GDP-bound subunits. J Biol Chem 288:24625–24635. doi: 10.1074/jbc.M113.483222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taviti AC, Beuria TK. 2017. MinD directly interacting with FtsZ at the H10 helix suggests a model for robust activation of MinC to destabilize FtsZ polymers. Biochem J 474:3189–3205. doi: 10.1042/BCJ20170357. [DOI] [PubMed] [Google Scholar]
- 61.Camberg JL, Viola MG, Rea L, Hoskins JR, Wickner S. 2014. Location of dual sites in E. coli FtsZ important for degradation by ClpXP; one at the C-terminus and one in the disordered linker. PLoS One 9:e94964. doi: 10.1371/journal.pone.0094964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mosyak L, Zhang Y, Glasfeld E, Haney S, Stahl M, Seehra J, Somers WS. 2000. The bacterial cell-division protein ZipA and its interaction with an FtsZ fragment revealed by X-ray crystallography. EMBO J 19:3179–3191. doi: 10.1093/emboj/19.13.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conti J, Viola MG, Camberg JL. 2018. FtsA reshapes membrane architecture and remodels the Z-ring in Escherichia coli. Mol Microbiol 107:558–576. doi: 10.1111/mmi.13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma X, Margolin W. 1999. Genetic and functional analyses of the conserved C-terminal core domain of Escherichia coli FtsZ. J Bacteriol 181:7531–7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Durand-Heredia J, Rivkin E, Fan G, Morales J, Janakiraman A. 2012. Identification of ZapD as a cell division factor that promotes the assembly of FtsZ in Escherichia coli. J Bacteriol 194:3189–3198. doi: 10.1128/JB.00176-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schumacher MA, Huang KH, Zeng W, Janakiraman A. 2017. Structure of the Z ring-associated protein, ZapD, bound to the C-terminal domain of the tubulin-like protein, FtsZ, suggests mechanism of Z ring stabilization through FtsZ cross-linking. J Biol Chem 292:3740–3750. doi: 10.1074/jbc.M116.773192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson JE, Lackner LL, Hale CA, de Boer PA. 2004. ZipA is required for targeting of DMinC/DicB, but not DMinC/MinD, complexes to septal ring assemblies in Escherichia coli. J Bacteriol 186:2418–2429. doi: 10.1128/JB.186.8.2418-2429.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang S, Shen Q, Wang S, Song C, Lei Z, Han S, Zhang X, Zheng J, Jia Z. 2017. Characterization of C-terminal structure of MinC and its implication in evolution of bacterial cell division. Sci Rep 7:7627. doi: 10.1038/s41598-017-08213-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vecchiarelli AG, Li M, Mizuuchi M, Hwang LC, Seol Y, Neuman KC, Mizuuchi K. 2016. Membrane-bound MinDE complex acts as a toggle switch that drives Min oscillation coupled to cytoplasmic depletion of MinD. Proc Natl Acad Sci U S A 113:E1479–E1488. doi: 10.1073/pnas.1600644113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arumugam S, Petrasek Z, Schwille P. 2014. MinCDE exploits the dynamic nature of FtsZ filaments for its spatial regulation. Proc Natl Acad Sci U S A 111:E1192–E1200. doi: 10.1073/pnas.1317764111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen Y, Erickson HP. 2009. FtsZ filament dynamics at steady state: subunit exchange with and without nucleotide hydrolysis. Biochemistry 48:6664–6673. doi: 10.1021/bi8022653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Camberg JL, Hoskins JR, Wickner S. 2009. ClpXP protease degrades the cytoskeletal protein, FtsZ, and modulates FtsZ polymer dynamics. Proc Natl Acad Sci U S A 106:10614–10619. doi: 10.1073/pnas.0904886106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oliva MA, Cordell SC, Lowe J. 2004. Structural insights into FtsZ protofilament formation. Nat Struct Mol Biol 11:1243–1250. doi: 10.1038/nsmb855. [DOI] [PubMed] [Google Scholar]
- 76.Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.