

To divide, most bacteria first construct a protein machine at the plane of division and then recruit the machinery that will synthesize the division septum. In Escherichia coli, this first stage involves the assembly of FtsZ polymers at midcell, which directly bind to membrane-associated proteins FtsA and ZipA to form a discontinuous ring structure. Although FtsZ directly binds both FtsA and ZipA, it is unclear why FtsZ requires two separate membrane tethers. Here, we uncover a new direct interaction between the tethers, which involves a helix within FtsA that is adjacent to its ATP binding pocket. Our findings imply that in addition to their known roles as FtsZ membrane anchors, FtsA and ZipA may regulate each other’s structure and function.
KEYWORDS: Escherichia coli, cell division, cross-linking, ftsA, ftsZ, zipA
ABSTRACT
The initiation of Escherichia coli cell division requires three proteins, FtsZ, FtsA, and ZipA, which assemble in a dynamic ring-like structure at midcell. Along with the transmembrane protein ZipA, the actin-like FtsA helps to tether treadmilling polymers of tubulin-like FtsZ to the membrane. In addition to forming homo-oligomers, FtsA and ZipA interact directly with the C-terminal conserved domain of FtsZ. Gain-of-function mutants of FtsA are deficient in forming oligomers and can bypass the need for ZipA, suggesting that ZipA may normally function to disrupt FtsA oligomers, although no direct interaction between FtsA and ZipA has been reported. Here, we use in vivo cross-linking to show that FtsA and ZipA indeed interact directly. We identify the exposed surface of FtsA helix 7, which also participates in binding to ATP through its internal surface, as a key interface needed for the interaction with ZipA. This interaction suggests that FtsZ’s membrane tethers may regulate each other’s activities.
IMPORTANCE To divide, most bacteria first construct a protein machine at the plane of division and then recruit the machinery that will synthesize the division septum. In Escherichia coli, this first stage involves the assembly of FtsZ polymers at midcell, which directly bind to membrane-associated proteins FtsA and ZipA to form a discontinuous ring structure. Although FtsZ directly binds both FtsA and ZipA, it is unclear why FtsZ requires two separate membrane tethers. Here, we uncover a new direct interaction between the tethers, which involves a helix within FtsA that is adjacent to its ATP binding pocket. Our findings imply that in addition to their known roles as FtsZ membrane anchors, FtsA and ZipA may regulate each other’s structure and function.
INTRODUCTION
To divide, Escherichia coli cells first coassemble FtsA, ZipA, and FtsZ in a circumferential but discontinuous ring structure at midcell (1). FtsA and ZipA anchor FtsZ filaments to the inner membrane to form the proto-ring, which then recruits another set of conserved proteins in a hierarchical and roughly temporal order (FtsEX-FtsK-FtsBLQ-FtsW-FtsI-FtsN) to form the divisome (2, 3). Dynamic treadmilling by FtsZ polymers around the ring structure guides the formation of the division septum (4, 5). The divisome also orchestrates the invagination of the outer membrane and inner membrane in concert with synthesis of the division septum to complete cytokinesis (6, 7).
FtsA is a widely conserved bacterial homolog of actin. In E. coli, the essential FtsA protein binds directly to FtsZ and tethers FtsZ polymers to the inner face of the cytoplasmic membrane through a C-terminal amphipathic helix (8). FtsA assembles into dimeric and higher oligomeric forms in vivo (4, 9–13) and assembles into curved filaments on lipid membranes in vitro (14–16) that regulate the assembly and dynamics of FtsZ polymers (13, 16–18).
The other proto-ring protein, ZipA, harbors an N-terminal transmembrane domain and a cytoplasmic FtsZ binding interface in its C-terminal domain and consequently also tethers FtsZ polymers to the cytoplasmic membrane (19). In vitro, ZipA, like FtsA, tethers FtsZ polymers to lipid membranes and permits dynamic behavior that likely mimics the treadmilling observed in vivo (17, 20, 21). The loss of either FtsA or ZipA in E. coli cells allows FtsZ rings to still form but blocks cell division from progressing further (22–24). The loss of both FtsA and ZipA blocks most FtsZ rings from forming (25), presumably because these are the only two essential membrane tethers for FtsZ in E. coli. ZipA is not conserved outside the gammaproteobacteria, but other likely membrane anchors for FtsZ function in addition to FtsA, such as FzlC in Caulobacter (26) and EzrA and SepF in Gram-positive species (27, 28). Indeed, the ability of the widely conserved SepF protein to substitute for FtsA in Bacillus subtilis and still allow cell division (29) suggests that SepF may take the place of FtsA in species that lack it (30).
Although ZipA is normally essential for E. coli cell division, products of hypermorphic alleles of the FtsA gene, called FtsA*, can permit cell division in the absence of ZipA (11, 31). Genetic, cytological, and biochemical studies suggest that FtsA*-like proteins are deficient in oligomerizing and that this deficiency results in the gain of function (11, 16, 18). Although FtsA* and FtsA*-like mutants can also bypass the requirement for other cell division proteins such as FtsK and can suppress other divisome defects (18, 32–34), zipA is the only essential cell division gene that can be completely bypassed by FtsA*, with virtually no cell division phenotype (31, 35). One hypothesis to explain the ZipA bypass proposed that FtsA*-like proteins mimic the action of ZipA (11). If true, then ZipA should inhibit FtsA oligomerization. However, there is no evidence to date for a direct interaction between ZipA and FtsA. If such an interaction existed, it might provide support for the idea that one normal function of ZipA is to convert FtsA into an FtsA*-like state during the cell division process.
To investigate whether FtsA can bind directly to ZipA in vivo and to maximize chances of detecting a potentially transient interaction, we employed in vivo site-specific cross-linking. Because E. coli FtsA contains nine cysteines, ruling out the use of disulfide cross-linkers, we turned to the genetically encoded photoactivatable amino acid p-benzoyl-l-phenylalanine (BpA) (36). BpA has the advantage that it cross-links target aliphatic residues within a very short linker length (3 to 4 Å), can be inserted at any engineered amber stop codon, and has been successful in pinpointing specific residues that engage in protein-protein interactions, including some E. coli cell division proteins (37, 38). Here, we show that an exposed helix of FtsA near the ATP binding pocket and FtsA-FtsA interaction site can form cross-links with ZipA in vivo.
RESULTS
Engineering cross-linkable derivatives of FtsA.
To investigate potentially weak in vivo interactions between FtsA and ZipA and identify interacting residues, we chose to use BpA as a genetically encoded cross-linker. To generate sites for BpA in FtsA, we initially engineered a series of single TAG (amber) codons to replace charged residues in FtsA that are predicted to lie at the surface and be involved in protein-protein interactions. Five of these, R122, E124, R126, R153, and R177, lie in domain 1C and are candidates for coordinating FtsA-FtsA interactions, whereas R202, R260, R286, and R300 are in regions of the protein implicated in FtsA-FtsA or FtsA-FtsZ interactions. In each of these constructs, these codon replacements represent the sole amber codon in ftsA, as the stop codon at the end of native ftsA is TAA. Cells expressing FLAG-FtsA with these engineered TAG codons in a plasmid require pEVOL, a compatible plasmid that expresses the orthogonal tRNA and tRNA synthetase, inducible with l-arabinose, which can incorporate BpA at the residue changed to TAG (39). By growing cells in the presence of BpA, a proportion of FtsA protein incorporates BpA at the targeted residue. The exposure of these cells to long-wave UV light triggers the formation of a single covalent linkage between the BpA residue on FtsA and a target amino acid, if it is within ∼3 to 4 Å.
To confirm that the mutant FtsA proteins properly incorporated BpA at their respective sites, we cotransformed cells carrying a chromosomal thermosensitive ftsA allele (ftsA12, in strain WM1115) with pEVOL and pDSW210F expressing FLAG-tagged FtsA amber mutants to determine the capacity of FtsA-BpA derivatives to suppress the thermosensitivity (Ts) of WM1115. While optimizing the experimental conditions, we found that high concentrations of l-arabinose or BpA caused cell filamentation, indicating that cell division was inhibited. As we wanted division to proceed normally in cells being probed for interactions between cell division proteins, we ended up using lower inducer concentrations to minimize division defects.
To test the ability of the BpA-substituted amber mutants to complement, we spotted various dilutions of cells onto LB agar plates at permissive (30°C) or nonpermissive (42°C) temperatures supplemented with ampicillin (Ap), chloramphenicol (Cm), 100 µM l-arabinose, 100 µM BpA, and different isopropyl-β-d-thiogalactopyranoside (IPTG) concentrations (see Fig. S1A in the supplemental material). As expected, wild-type FtsA produced from pDSW210F was toxic when induced at >100 µM IPTG and completely inviable at 1 mM IPTG at 30°C (40). In contrast, none of the mutant proteins were toxic even at 1 mM IPTG (Fig. S1A). At the nonpermissive temperature of 42°C, three mutants (R122X, R153X, and R300X) failed to suppress ftsA12, indicating that they were inactive. However, all the other mutants complemented ftsA12 at some level of IPTG, indicating that they were functional but were not toxic like wild-type FtsA (Fig. S1A). Interestingly, the substitution at R286, which is the same residue altered in FtsA*, fully complemented with no added IPTG, whereas the other substitutions all required at least 10 µM IPTG for optimal complementation.
To determine how efficiently the various mutants incorporated BpA, we grew the same WM1115 (ftsA12) strains containing the various derivatives used for the viability assays in the presence or absence of BpA, followed by UV cross-linking. We then probed cell extracts for the expressed FLAG-FtsA on Western blots by using anti-FLAG antibody. All mutants except R122 displayed a band corresponding to full-length FLAG-FtsA-BpA (∼45 kDa) only when grown in medium containing BpA (Fig. S1B, “+” lanes), indicating that BpA was incorporated into the FtsA amber derivatives. The band intensities for R153X and R300X were lower than the others, suggesting that their inability to complement (Fig. S1A) might be caused by protein instability. In addition to the full-length BpA-containing proteins, we also detected protein bands from the various derivatives that migrated at smaller sizes than expected if their translation were terminated at the engineered premature TAG codon. In the samples grown in the presence of BpA, the bands of truncated protein that were large enough to be retained by SDS-PAGE (R153X, R177X, R202X, R260X, R286X, and R300X) were less abundant than the respective full-length protein (Fig. S1B), with R202X, R260X, and R286X incorporating 64%, 78%, and 87%, respectively, of the total detectable FtsA into the full-length protein, suggesting that BpA was incorporated efficiently into these proteins under these conditions.
BpA at residue 260 of FtsA can cross-link with ZipA or additional FtsA subunits.
In the initial screening, we used anti-FLAG to detect whether the FLAG-tagged BpA derivatives of FtsA shifted into higher-molecular-weight protein bands that might form cross-linked species (CLS) under these conditions, but we observed only very faint potential CLS in the “+ UV” lanes. However, when we probed a blot of the same samples with anti-ZipA, we observed a strikingly strong UV-dependent CLS for the R260X protein, suggesting that the BpA at residue 260 might be able to cross-link with native ZipA (Fig. S1C, asterisk). No UV-dependent CLS were observed for any of the BpA derivatives in this initial screen by using antibodies against FtsZ or FtsN (Fig. S1D and E). Using anti-ZipA, we further confirmed that FtsAR260-BpA generated CLS containing ZipA only in the presence of BpA and only when irradiated by UV (Fig. 1B, lane 8, compare with lanes 5 to 7). When we used anti-FLAG on a blot with the same samples to detect FLAG-FtsA, this time we detected a faint higher-molecular-weight CLS in the sample with BpA and UV (Fig. 1A, lane 8), probably because there was less nonspecific staining. In addition, the R260 amber derivatives of FtsA that failed to incorporate BpA were detectable as prematurely truncated proteins (Fig. 1A, lanes 5 to 8); the amount of truncated protein was reduced, as expected, in the presence of BpA (Fig. 1A, lanes 7 and 8, compare with lanes 5 and 6).
FIG 1.
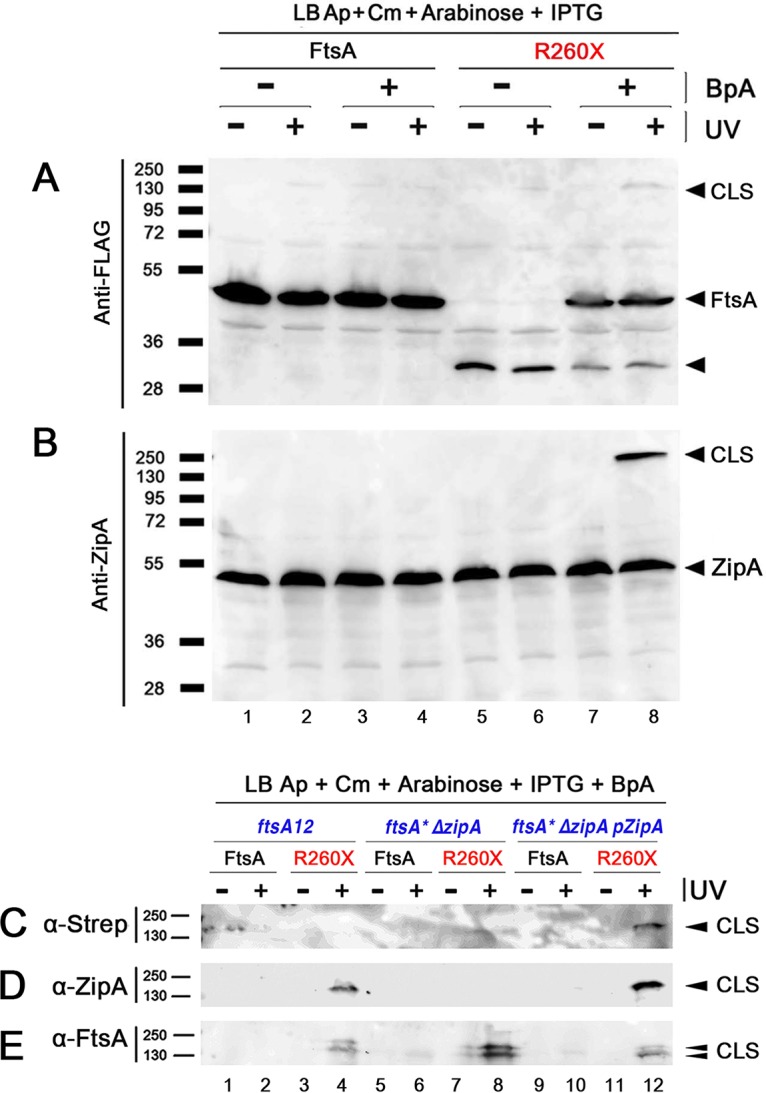
FtsA-R260-BpA specifically cross-links with ZipA. (A and B) Strains expressing either FLAG-FtsA or FLAG-FtsA with an amber codon replacing R260 (R260X) were grown with the antibiotics specified, with or without BpA, and either subjected to UV cross-linking or not. Protein extracts were separated by SDS-PAGE, followed by Western blotting using anti-FLAG to detect the FLAG-FtsA (A) or anti-ZipA (B). Full-length FtsA or ZipA bands are indicated by arrowheads, as are the most prominent cross-linked species (CLS). The band representing FtsA protein terminated at codon 260 is indicated by an unlabeled arrowhead. (C to E) Strains expressing FtsA or FtsAR260X, carrying ftsA12 (WM1115), ΔzipA ftsA* (WM1657) or WM1657 with ZipA-Strep-Tag produced from plasmid pPK9 (selection with spectinomycin), were grown with BpA and either UV cross-linked or not, and their proteins were separated by SDS-PAGE. Western blots were probed with anti-Strep-Tag to detect ZipA-Strep-Tag (C), anti-ZipA (D), or anti-FtsA (E). Bands representing CLS are indicated by arrowheads. Molecular weight markers in kilodaltons are shown to the left of all blots.
Although the predicted molecular weights for FtsA and ZipA are 45.3 and 36.5 kDa, respectively, ZipA migrates aberrantly at ∼50 kDa in SDS-PAGE (19), probably because of its internally disordered P/Q-rich domain. Even accounting for the slow migration of ZipA, the size of the CLS (∼135 kDa) we observed after SDS-PAGE was not the expected ∼95-kDa size of one FtsA molecule cross-linked to one ZipA. To resolve this issue, we first wanted to confirm that the CLS corresponded to FtsA cross-linked to ZipA and not a larger ∼90-kDa protein that potentially cross-reacted with anti-ZipA.
We addressed this in several ways. To determine whether the CLS detected with anti-ZipA actually represented ZipA, we repeated the cross-linking by expressing FtsAR260-BpA not only in WM1115 but also in WM1657, a mutant lacking zipA that harbors the ftsA* allele (which bypasses zipA), and WM1657 transformed with a zipA-expressing plasmid (pPK9-ZipA-StrepTag, compatible with pEVOL and pDSW210F-FtsA) as an isogenic zipA+ control. As expected, in WM1115 (ftsA12 zipA+), CLS were observed by probing Western blots with anti-ZipA (Fig. 1D, lane 4) or anti-FtsA (Fig. 1E, lane 4). The CLS, as mentioned above, were only detectable after incubating with BpA and after UV irradiation. Importantly, in the absence of ZipA (WM1657 ftsA* ΔzipA), CLS were detected only by anti-FtsA (Fig. 1E, lane 8) and not by anti-ZipA or anti-Strep-Tag (Fig. 1C and D, lane 8). When ZipA was restored back to the ΔzipA strain by expressing it from a plasmid (WM1657/pPK9-ZipA-Strep-Tag), CLS were observed under all conditions (Fig. 1C to E, lane 12). These results strongly suggested that the CLS correspond to FtsA-ZipA, but we also noticed that ∼135-kDa CLS were generated even in the absence of ZipA (Fig. 1E, lane 8). This raised the possibility that three FtsAR260-BpA monomers could cross-link as a trimer (45 × 3 = 135 kDa) or that FtsA could, in the absence of ZipA, cross-link to another protein of ∼90 kDa.
A search for ∼90-kDa proteins involved in E. coli cell division revealed that PBP1A and/or PBP1B, both 89-kDa proteins that could theoretically interact with FtsA, might be targets of FtsA cross-linking. To test this idea genetically, we constructed mutants lacking either the mrcA or mrcB genes encoding PBP1A or PBP1B, respectively (deleting both is lethal) and transformed them with pEVOL and the plasmid carrying FLAG-FtsAR260X. Both ΔmrcA and ΔmrcB strains still generated the same CLS at intensities at least as high as the mrc+ parent strain (see Fig. S2), suggesting that PBP1A or PBP1B is not crucial for forming the CLS under these conditions.
Finally, to confirm this and to explore whether other potential targets of FtsA-R260-BpA cross-linking existed, we purified CLS from the WM1657 ΔzipA strain expressing ZipA-Strep-Tag from pPK9-ZipA-Strep-Tag (WM5952), only after exposure to BpA and UV light, with anti-Strep-Tag affinity chromatography. We identified three bands in the 135-kDa area after SDS-PAGE and silver staining and analyzed the contents of the excised bands by mass spectrometry. The contents of the three bands were similar, with band 2 being the most prominent. The analysis identified FtsA and ZipA as top hits, but no other known cell division-related protein was identified, including PBP1A or PBP1B (see Fig. S3). These results suggest that the CLS contains only FtsA-R260-BpA cross-linked to FtsA and/or ZipA. It remains possible that other protein partners interact with FtsA in the absence of ZipA.
Residues in FtsA helix 7 are part of an FtsA-ZipA-interacting domain.
According to the atomic structure of Thermotoga maritima FtsA, R260 lies within helix 7 (41) (see Fig. S4). Although R260 is not well conserved in FtsA proteins from diverse species, several nearby residues in helix 7, such as E255 and K258, are highly conserved (Fig. 2D; see also Fig. S5) and interact with the adenosine moiety of ATP (41). To determine whether residues immediately adjacent to R260 might be involved in the FtsA-ZipA interaction, we constructed a new set of FtsA amber codons to replace K258, V259, H261, and G262. As for previous mutants, we tested the ability of these amber mutant FtsA proteins to complement ftsA12 in WM1115 at 30°C and 42°C in the presence of BpA (Fig. 2A). The results at 30°C were similar to those shown in Fig. S1A and B: wild-type (WT) FtsA was toxic at high induction levels, but none of the BpA derivatives were toxic (Fig. 2A, mutants row and data not shown). As shown at the bottom of Fig. 2A, FtsAR260X and H261X complemented but needed higher IPTG levels than WT FtsA. On the other hand, substitutions at V259 and G262 complemented poorly, with only a slight increase in viability at higher IPTG levels. The K258 amber mutant complemented slightly better but still was unable to complement without the addition of IPTG.
FIG 2.

Residues on the outer face of helix 7 of FtsA preferentially cross-link with ZipA. (A) WM1115 strains expressing FtsA amber substitutions at all the residues within helix 7 were grown in the presence of BpA and tested for their ability to complement ftsA12 at 42°C. After growth at 30°C, all strains shown were serially diluted 10-fold, spotted on plates containing different concentrations of IPTG to induce expression of the ftsA derivatives at different levels plus arabinose to induce pEVOL and BpA, and incubated either at 30°C or 42°C for 24 h. To save space, the “Mutants” row shows one of the mutants that grew at all IPTG levels at 30°C, because all of the other mutants behaved similarly. (B and C) The same strains in panel A were UV cross-linked, and protein extracts were separated by SDS-PAGE. Western blots were probed with either anti-FtsA (B) or anti-ZipA (C), and arrowheads indicate the general position of CLS. Molecular weight markers in kilodaltons are shown to the left. (D) Shown at the left is the position of each residue in FtsA helix 7 (including P250, which is outside the helix) and the relative ability of BpA at each position to cross-link with ZipA. The protein sequence of ZipA from E. coli was used to generate a model PDB file with the Phyre2 server and subsequently analyzed in UCSF Chimera. Summarized in the table at the right is the ability of each amber substitution under the conditions of the experiments in panels A and C to cross-link with ZipA and to complement at 42°C, along with the relative conservation of each substituted residue. The scoring system for CLS reflects the intensity of CLS bands, from ++++ (strongest) to − (undetectable). Scoring for complementation reflects the ability to grow at no IPTG like WT FtsA (++++), weak at no IPTG but strong growth with 10 µM IPTG (+++), weak with 10 µM IPTG but stronger growth at higher IPTG levels (++), or little growth at any induction level (+). Scoring for conservation depends on the degree of conservation across the species aligned in Fig. S5 in the supplemental material: ++++, identity across all species; +++, identity across most species; ++, identity in at least 4 other species in the list; +, some identity or similar characteristics in other species; −, no obvious conservation.
We then tested cells expressing these amber substitution derivatives under permissive growth conditions in the presence of BpA for the ability to cross-link ZipA. As before, CLS specific for FtsA expression were observed only in the presence of BpA and UV cross-linking. Consistent with our previous data, FtsAR260-BpA showed a strong ∼135-kDa CLS via anti-ZipA (Fig. 2C, lane 12) and a prominent CLS in the same part of the gel via anti-FtsA (Fig. 2B, lane 12), visible here as a doublet. As a doublet, CLS was also visible with anti-FtsA in other cases (e.g., Fig. 1E); we suspect that a portion of the protein sometimes undergoes cleavage near the FLAG-FtsA junction and these two populations then form cross-links, resulting in variations in CLS molecular weights. An additional weak higher-molecular-weight CLS at ∼200 kDa was also detectable by anti-FLAG or anti-ZipA and was reproducible under different conditions (see below). However, although a weak CLS was visible for H261X probed with anti-ZipA (Fig. 2C, lane 13), analogous BpA-specific CLS bands above background were not detectable with anti-ZipA for K258X, V259X, or G262X despite detectable CLS bands with anti-FtsA (Fig. 2A and B, lanes 10, 11, and 14).
One possible reason for the sharp drop in cross-linking efficiency on either side of R260 as detected by anti-ZipA is that residue 260 is an exclusive FtsA-ZipA contact point. However, this is unlikely given that strong cross-linking is achieved when BpA replaces the arginine at this position. A more likely scenario is that the interaction with ZipA only occurs on the exposed surface of the helix, oriented away from the FtsZ binding face of FtsA (Fig. 2D). If true, we considered that other exposed residues in helix 7 distal from R260 might exhibit a similar capacity to generate ZipA-containing CLS as R260.
To test this hypothesis, we selected two putative exposed residues, D253 and A256, on the same side of the helix as R260 (Fig. 2D). When we tested amber codon replacements of these two residues in the cross-linking assay, we reproducibly detected prominent CLS with anti-ZipA, and they were specific for the presence of BpA and UV irradiation, although the CLS bands were weaker than those for R260X (Fig. 2C, lanes 5 and 8). These data support the idea that multiple residues in FtsA helix 7 can cross-link with ZipA, provided they are on the exposed face of the helix pointing away from the FtsZ binding face of FtsA. Notably, BpA substitutions at both D253 and A256 complemented the ftsA12 mutant nearly as well as WT FtsA (Fig. 2A and D).
To further test our hypothesis and more fully characterize the role of helix 7 in protein-protein interactions, we generated single amber mutants for the remaining residues in the helix (Fig. 2D). BpA substitutions at P251, A254, and E255, all predicted to face inward, showed no detectable CLS with anti-ZipA (Fig. 2B and C, lanes 3, 6, and 7), although all three substitutions showed CLS of various intensities with anti-FtsA (Fig. 2B and C, lanes 3, 6, and 7), suggesting that they can interact with another molecule, probably FtsA, more readily than with ZipA. The BpA substitution at S252, which is at the top of the helix and is predicted to face outward, behaved similarly to substitutions at D253, A256, and R260 in that it showed robust CLS bands at ∼135 kDa with both anti-FtsA and anti-ZipA and complemented ftsA12 efficiently (Fig. 2A to C, lane 4). Altogether, our data indicate that only residues on the outward-facing surface of FtsA helix 7 are capable of generating strong FtsA-ZipA cross-links (Fig. 2D).
Disruptions in helix 7 differentially inhibit FtsA-FtsA and FtsA-ZipA interactions.
Given the key involvement of helix 7 in generating CLS, we wanted to determine the role of some of the residues that cross-link with ZipA (D253, A256, and R260) in FtsA function in vivo. Using pDSW210F-FtsA as the template for site-directed mutagenesis, we generated D253R, D253W, A256R, A256W, R260E, and R260W substitutions to test the effects of charge changes and the introduction of bulky hydrophobic residues at these positions. These mutants in pDSW210F were transformed into wild-type (MG1655) and ftsA12 (WM1115) strains to assess their function under permissive (30°C) and nonpermissive (42°C) conditions.
Notably, the D253R and R260E opposite-charge mutants mainly behaved like wild-type FtsA, complementing ftsA12 at 42°C and exhibiting toxic effects with IPTG concentrations higher than 100 µM (Fig. 3A and B). D253R consistently conferred slightly more toxicity than WT FtsA. In contrast, A256R was unable to complement and was not toxic (Fig. 3A and B), indicating that it is nonfunctional. As A256 is not well conserved in other FtsA proteins, it is possible that replacing a small neutral residue with a bulkier charged residue at the center of the helix (A256R) causes more dramatic effects on structure and function. Indeed, the A256R protein was undetectable by immunoblotting even with high levels of IPTG induction (see Fig. S6A). Interestingly, replacing D253 or A256 with W showed no toxicity and was able to suppress ftsA12, similar to the phenotype of ftsA*-like alleles (Fig. 3A and B). In contrast, R260W complemented ftsA12 weakly at low levels but was extremely toxic under all conditions. Given their FtsA*-like nontoxic phenotype, we asked whether the D253W and A256W replacements could bypass ZipA, but they did not (see Fig. S7).
FIG 3.

Disruption of FtsA helix 7 reduces FtsA-ZipA interaction. (A and B) Various FtsA constructs with residue changes in helix 7 were expressed in ftsA12(Ts) cells (WM1115) and tested for viability at different concentrations of inducer under either permissive (A) or nonpermissive (B) conditions. (C) A subset of these residue changes were converted into BpA-containing derivatives and tested for their ability to cross-link with ZipA. Protein extracts from cultures supplemented with BpA and cross-linked with UV were separated by SDS-PAGE and probed on Western blots with anti-FtsA or anti-ZipA. FtsA, ZipA, and cross-linked species (CLS) are highlighted. Blots contain spliced lanes from the same gel. Molecular weight markers in kilodaltons are shown at the left.
As all BpA substitutions are by definition already missense changes like those mentioned above, we chose to generate a set of small deletions (ΔD253, ΔA256, ΔR260, ΔA256-R260, and ΔD253-R260) to investigate further the role of these residues in cross-linking with ZipA. We first tested their capacity to suppress ftsA12. At 42°C, the single residue deletions at the ends of helix 7, ΔD253 and ΔR260, suppressed ftsA12 without toxicity (Fig. 3A and B). In contrast, FtsA derivatives harboring ΔA256, ΔA256-R260, or ΔD253-R260 complemented ftsA12 poorly, either not at all (Δ256-260) or required 500 µM IPTG to achieve weak growth (ΔA256 and ΔA253-260) (Fig. 3B). All three of these deletion proteins were expressed and inducible by IPTG but at approximately half the levels of the intact FtsA, which correlate with the phenotypes observed (Fig. S7). Combined with the amino acid substitution data described above, these data suggest that A256 itself is not required for proper FtsA function, but even a single residue deletion in the middle of helix H7 can potentially disrupt the helical structure and inactivate FtsA function in vivo.
We next determined the effects of the deletions in helix 7 on the ability of FtsA to form cross-links with ZipA. To use R260-BpA as the donor residue for cross-linking, we combined the D253R, D253W, or ΔD253 mutant with R260X. Conversely, we combined ΔR260 with D253X so that D253-BpA would serve as the cross-linking residue. When we probed Western blots with anti-FtsA, CLS were detected for R260-BpA as expected (Fig. 3C, lane 2), as well as for R260-BpA plus ΔD253 (Fig. 3C, lane 5), but band intensities were significantly reduced for R260-BpA plus D253R (Fig. 3C, lane 3) and further reduced for R260-BpA plus D253W (Fig. 3C, lane 4). By analogy with R260-BpA plus ΔD253, significant CLS were observed for D253-BpA plus ΔR260 (Fig. 3C, lane 6). In contrast, by using the anti-ZipA antibody, strong CLS were detected for R260-BpA plus D253R as well as the control R260-BpA (Fig. 3C, lanes 8 and 9), but only weak CLS were detected for R260-BpA plus D253W (Fig. 3C, lane 10) and almost undetectable for R260-BpA plus ΔD253 and D253-BpA plus ΔR260 (Fig. 3C, lanes 11 and 12). Altogether, the data suggest that alterations in helix 7 can differentially affect FtsA-FtsA and FtsA-ZipA interactions, especially as the levels of endogenous ZipA and plasmid-expressed FtsA are relatively unchanged in the different strains used for cross-linking (Fig. 3C). In particular, deletion of D253 when BpA replaces R260, or deletion of R260 when BpA replaces D253, can drastically inhibit the ability of FtsA to interact with ZipA, possibly because two residues at the interface are affected simultaneously.
FtsA*-like mutants cross-link similarly to wild-type FtsA.
The overproduction of ZipA inhibits cell division, but FtsA* confers resistance to these toxic effects (31). Although the mechanism of resistance is not known, it is possible that the lower oligomerization capacity or higher membrane binding capacity of FtsA* (11, 42) plays a role. We reasoned that if this were the case, FtsA*-R260-BpA might exhibit reduced cross-linking to ZipA compared with that of FtsA-R260-BpA. We engineered a double replacement carrying R260X and R286W (FtsA* allele) to test this idea. However, when this double replacement was produced from pDSW210F, we saw no significant differences in the generation of CLS by FtsA-R260-BpA versus that by FtsA*-R260-BpA (Fig. 4B), either in the ftsA12 strain background (WM1115, at the permissive temperature) or a strain with ftsA* replacing ftsA in the chromosome (WM1657). As shown in Fig. 1, WM1657 also lacks ZipA, as seen by the lack of CLS detectable by anti-ZipA (Fig. 4B, lane 6), but the ZipA-containing CLS is restored upon ectopic expression of ZipA from a plasmid (Fig. 4B, lane 8).
FIG 4.

FtsA* forms CLS in the absence of ZipA and cross-links as well as FtsA. Cross-linking experiments with FtsAR260-Bpa or FtsA containing both R286W (FtsA* allele) and R260-BpA tested whether the presence of the R286W allele (ftsA*), which decreases FtsA oligomerization, influences the relative levels of cross-linking to ZipA or another FtsA. The zipA+ strain WM1115 or the ftsA* ΔzipA strain WM1657, with or without pPK9-ZipA to overproduce ZipA, was used to express either FtsAR260-BpA (lanes 1 and 2) or FtsA*R260-BpA (lanes 3 to 8) in the presence of BpA, UV cross-linked or not, followed by separation of proteins by SDS-PAGE and probing Western blots with anti-FtsA (A) or anti-ZipA (B). Un-cross-linked FtsA and ZipA, as well as major and minor CLS bands, are denoted with arrowheads. Molecular weight markers are shown at the left. Lanes 1 and 2 and lanes 3 to 8 were from separate gels.
These results suggest that the ZipA-resistant phenotype of FtsA* is not a consequence of poor interactions with ZipA. Moreover, the strong CLS observed from the ΔzipA strain detected by anti-FtsA (Fig. 4A) indicates that our cross-linking conditions may not be able to distinguish between the strong oligomerization activity of FtsA and the weaker oligomerization predicted for FtsA*. This may be because FtsA’s oligomerization activity is regulated over the course of cell division, and our cross-linking experiments, which use relatively low levels of protein that do not significantly perturb cell division, may capture its time-averaged oligomerization activity instead of one specific oligomerization state. Finally, these results clearly demonstrate that FtsA*-R260-BpA (Fig. 4A) or FtsA-R260-BpA (Fig. 1D and E, lane 8) is capable of cross-linking to other proteins, probably other FtsA subunits, independently of ZipA.
FtsA interacts with ZipA in bacterial two-hybrid assays.
To provide independent support for the FtsA-ZipA interaction observed by cross-linking, we investigated whether FtsA and ZipA could give a positive signal in a bacterial two-hybrid assay. This assay relies on the ability of the T18 and T25 domains of adenylate cyclase to generate beta-galactosidase in cells and thus a Lac+ phenotype that can be scored colorimetrically on agar plates, only when proteins fused to these two domains interact sufficiently closely to bring the two domains together. When we combined pUT18c-FtsA with pKNT25-ZipA, the colonies were strongly positive, consistent with the ability of FtsA and ZipA to interact in cells (Fig. 5, A5).
FIG 5.
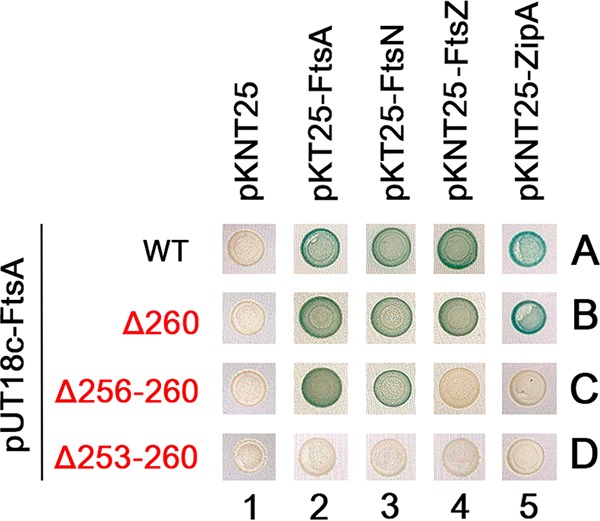
Bacterial two-hybrid assay for ZipA-FtsA interactions. BTH101 cells containing pUT18c derivatives carrying full-length FtsA (WT) or small deletions within helix 7 plus pKNT25 or pKT25 derivatives fused to FtsA, FtsN, FtsZ, or ZipA (or empty vector) were spotted onto LB X-Gal plates with antibiotics and 50 µM IPTG, incubated for 24 h at 30°C, and photographed. All six replicates for each pair had identical phenotypes, and so only one example of each pair is shown.
We then asked whether disruptions of residues in helix 7 of FtsA might disrupt the two-hybrid signal in the same way they inhibited FtsA-ZipA cross-linking. When pUT18c-FtsAΔ260 was combined with pKNT25-ZipA, the Lac+ phenotype was at least as strong as that with WT FtsA, indicating that deleting only R260 had no effect on the interaction (Fig. 5, B5). This is different from the result for FtsA A253-BpA R260 in cross-linking experiments, which showed a significant diminution of CLS; however, this could easily be explained by the simultaneous loss of both A253 and R260 in the cross-linking experiment (A253 is changed to BpA), as opposed to only the loss of R260 in the bacterial two-hybrid assay.
When we tested a deletion of 5 residues (Δ256-260) in helix 7, which we showed earlier cannot complement ftsA12, it no longer interacted with ZipA in the two-hybrid assay (Fig. 5, C5). To rule out the possibility that this pUT18c-FtsA Δ256-260 construct is inactive, we combined it with T25 versions of FtsN, FtsA, and FtsZ and found that it gave positive two-hybrid interactions with FtsA and FtsN but not with FtsZ (Fig. 5, C2 to 4). The ability of the deleted FtsA to interact with FtsN or another FtsA indicated that the loss of interaction with ZipA was fairly specific, although the loss of interaction with FtsZ also suggested that the deletion resulted in allosteric conformational changes. A larger deletion of residues 253 to 260, essentially removing the entire helix 7, abolished all interactions, including those with FtsA, FtsN, and FtsZ (Fig. 5, D2 to 5), perhaps because the removal of helix 7 destabilizes the entire protein structure when expressed from pUT18c. Yet, FtsA Δ253-260 expressed from pWM2785 retains some ability to complement ftsA12, suggesting in that context, it is not completely inactive.
Role of the Z ring in FtsA-ZipA interaction.
Although the FtsA-ZipA cross-linking data indicate that the in vivo interaction is direct, we asked whether this interaction is dependent on an intact Z ring. We took two approaches. In the first approach, we tested FtsAR260-BpA for the ability to cross-link to ZipA after a relatively brief treatment with nalidixic acid (NA), an inhibitor of DNA gyrase that rapidly induces the SOS response and disassembly of Z rings (43). By introducing the FtsAR260X and pEVOL plasmids into a functional ftsZ-mCerulean chromosomal reporter strain (44) and adding BpA to the medium, we confirmed that many midcell FtsZ-mCerulean rings remained after growth in BpA, resuspension of the culture in buffer, and UV cross-linking (see Fig. S8C). ZipA-green fluorescent protein (GFP) rings also persisted at midcell after UV cross-linking in experiments expressing that fusion protein (Fig. S8D). In contrast, nearly all cells grown in parallel but in the presence of NA for 1 h lacked Z rings (Fig. S8C). When we compared anti-FtsA or anti-ZipA immunoblots after UV irradiation, we observed that the ∼135-kDa CLS containing ZipA and FtsA from the NA-treated cells were present at levels equivalent to the CLS from untreated cells (Fig. S8A and B, compare lanes 6 and 8). We also repeated the cross-linking experiment in our WT strain background containing native FtsZ and again observed equivalent CLS bands with or without NA treatment (Fig. S8A and B, compare lanes 2 and 4).
In a more targeted approach, we tested FtsAR260-BpA for the ability to cross-link to ZipA in a ftsZ84(Ts) strain (WM1125), which forms Z rings and divides fairly well at 30°C but mostly lacks Z rings when shifted to the nonpermissive temperature of 42°C. Comparing the formation of CLS in a WT strain with that in the ftsZ84 strain, we found that CLS formed readily at both 30°C and 42°C, with more intense bands at 42°C for both the WT and ftsZ84 backgrounds (Fig. S8E), with either anti-FtsA (lanes 1 to 4) or anti-ZipA (lanes 5 to 8). Even with the expression of FtsAR260-BpA, cells of this mutant divided fairly well at 30°C, although not at 42°C, as expected (Fig. S8F). Taken together, these results suggest that the FtsA-ZipA interaction through FtsA’s helix 7 can occur independently of the Z ring.
DISCUSSION
Previous work has established that FtsA and ZipA both interact directly with FtsZ. Here, we used photo-cross-linking in growing and dividing cells, supported by bacterial two-hybrid analysis, to show that FtsA and ZipA closely interact with each other. The exposed face of helix 7 of FtsA is important for the interaction with ZipA, as the residues on the exposed face that are replaced with BpA are able to form cross-links with ZipA. Residues substituted for BpA on the opposite face of helix 7 are still capable of forming cross-links with another protein, probably another FtsA subunit, but not ZipA. Based on the T. maritima FtsA structure, several residues on the inside face of helix 7 are also important for coordinating the adenosine moiety of ATP. These predicted inside face residues in helix 7 (A254, E255, K258, and G262) are the most conserved (Fig. 2D; see also Fig. S5 in the supplemental material), have the lowest ability of any of the helix 7 substitutions to complement ftsA12 when replaced with BpA, and are unable to cross-link with ZipA. In contrast, individual substitution or deletion of several of the exposed residues in helix 7 still permits FtsA to cross-link with ZipA. Unlike the inside face residues, the exposed residues are not as conserved, and FtsA proteins with BpA substitutions at these residues are still able to complement ftsA12. Deletion of A256, an exposed residue at the center of the helix, causes more defects in function than deletion of R253 or R260, which are at the ends of the helix. However, simultaneously perturbing both of the latter residues, such as deleting R253 and replacing R260 with BpA, or vice versa, abolished detectable cross-linking with ZipA. Together, these data suggest that FtsA-ZipA interactions may involve multiple residues within helix 7 and that the integrity of the helix as a whole is required for binding to ZipA. Larger deletions, not surprisingly, have more significant defects that are harder to interpret. For example, the Δ256-260 deletion of FtsA cannot cross-link to ZipA, complement an ftsA12 mutant efficiently, or interact with FtsZ in a bacterial two-hybrid assay. Yet, FtsA Δ256-260 likely retains much of its tertiary structure, because it is expressed at approximately half of WT levels from a plasmid and shows a positive interaction with FtsA and FtsN in the two-hybrid assay.
Typically, cross-linking between two proteins results in a CLS after SDS-PAGE that is the predicted size of the heterodimer. However, despite the fact that FtsA is 45 kDa and ZipA is 36 kDa (but migrates aberrantly at 50 kDa), we consistently observed CLS in the ∼135-kDa range instead of the predicted ∼95 kDa. In fact, although we often observed homodimer-sized FtsA (90 kDa) bands after SDS-PAGE, we never detected a homodimer-sized band that was specific to BpA photo-cross-linking. So how can we explain the ∼135-kDa CLS, along with the secondary weak bands at ∼190 kDa that we sometimes observed? One possible model is that two FtsA subunits separately cross-link to one molecule of ZipA. At 45 + 45 + 50, this would add up to 140 kDa, the approximate size of the CLS. But the problem with that idea is the persistence of the ∼135-kDa CLS detected by anti-FtsA either in strains where FtsA-ZipA cross-linking is undetectable (e.g., with BpA substitutions at interior-facing residues such as P251 or E255) or in FtsA* strains that completely lack ZipA. In that case, the ∼135-kDa CLS is either a homotrimer of FtsA or FtsA cross-linked to another protein that coincidentally happens to be ∼90 kDa. Although PBP1A or PBP1B is probably not involved, we cannot be sure at this point of the arrangement of FtsA and/or ZipA in the cross-linked complexes we detect by SDS-PAGE. As both FtsA and ZipA are strongly bound to the cytoplasmic membrane in the cell, it is possible that the cross-linking traps them in a structure that is either two FtsA molecules plus one ZipA molecule or three FtsA molecules. The presence of FtsZ polymers in the cell may contribute to these complexes, particularly as a cross-linking-dependent homodimer for FtsA was never detected. It should be noted that one potential alternative, 1 FtsA cross-linked to 2 ZipA, should not be possible, as each FtsA-BpA should be able to cross-link with only one target.
We consider two possible explanations of the trimer size that involve lateral interactions and/or twisting of oligomeric subunits at the membrane surface. In the lateral interaction example, an FtsA trimer structure might feature a typical head-to-tail homodimer (15, 45) (Fig. S4), whose bottom subunit cannot bind to the top subunit through helix 7 but only to a third subunit that is bound to the side. Such a lateral subunit, in addition to accepting a cross-link from helix 7 from the “bottom” BpA-containing subunit of the homodimer, might itself be able to form a BpA cross-link to the “top” subunit of the same homodimer, which may be twisted relative to the bottom subunit (45) and/or blocked by the membrane by oligomeric constraints (16) that prevent further cross-linking to a fourth subunit. This hypothetical FtsA trimer would be reminiscent of the trimer nucleus of actin that guides assembly of the double-stranded actin filament (46), although there is no evidence to date that FtsA filaments share this property. The other hypothetical case might solely involve a significant twist of subunits within the membrane-bound miniring, such that the top and bottom subunits within a 3-subunit FtsA oligomer are unable to donate or accept a cross-link. In this case, relative twisting of the FtsA subunits with respect to the membrane could sterically block cross-linking sites, effectively capping the oligomer. In this scenario, ZipA could compete with FtsA for binding to the oligomer and might similarly cap the oligomer, terminating it at 2 FtsA plus 1 ZipA. Such subunit twisting by FtsA might also explain the previously observed spatial constraints imposed on FtsZ protofilament binding to membrane-bound FtsA minirings (16). Clearly, more investigation is needed to clarify these potential structures in vivo.
The initial rationale for this work was the idea, proposed initially by Pichoff et al. that ZipA binding might disrupt FtsA oligomerization, helping to drive FtsA into a more open FtsA*-like state and thus explaining why FtsA* can bypass ZipA (11). Although such disruption by ZipA has not yet been confirmed, the involvement of helix 7, which seems to bind ZipA on one face and ATP on the other, does suggest a potential mechanism. In this model, ZipA binding to helix 7 would deform the ATP binding pocket, altering ATP hydrolysis or binding affinity, and consequently influencing FtsA’s oligomerization state and possibly other activities. Although future refinement of the relationship between FtsA’s ATP binding, ATPase activity, and oligomerization is clearly necessary, one possible scenario is that the interaction between ZipA and membrane-bound FtsA minirings disrupts the minirings, converting them into FtsA*-like short oligomers that can then recruit downstream division proteins. However, the ability of BpA at each residue in helix 7 to form a homotrimer-sized CLS, whether or not ZipA can be cross-linked, suggests that many FtsA oligomers exist despite the presence of ZipA, at least when averaged over the cell population, and that ZipA binding to FtsA needs to compete with FtsA self-interactions. Our data suggest that these interactions may occur independently of the Z ring.
A major challenge in understanding bacterial cell division is that the flexibility and modularity of the divisome likely depend on multiple weak protein-protein interactions (47). In addition, cells in an unsynchronized population are all in different stages, making it difficult to capture specific stages using biochemical methods. Cross-linking at least provides a way to capture transient interactions, and future use of cross-linking methods in synchronized cells may allow a finer temporal view of these protein-protein interactions that have so far been evasive. In addition, future cross-linking studies will benefit from FtsA mutants that cannot oligomerize at all or are resistant to depolymerization as well as a better idea of the residues on ZipA that are targeted.
MATERIALS AND METHODS
Plasmids, strains, growth conditions, and microscopy.
The plasmids and strains used in this study are listed in Table 1. For most genetic experiments, cells were grown at either the permissive (30°C) or nonpermissive (42°C) temperature in Luria-Bertani (LB) agar or broth. Transformants were selected and maintained by adding ampicillin (Ap; 50 µg · ml−1), chloramphenicol (Cm; 20 µg · ml−1), spectinomycin (Spc; 50 µg · ml−1), glucose (0.2%), and variable concentrations of l-arabinose or IPTG when necessary. Nalidixic acid (NA) was used at a final concentration of 40 µg · ml−1.
TABLE 1.
E. coli strains and plasmids
| Plasmid or strain | Description | Source or reference |
|---|---|---|
| Plasmids | ||
| pDSW210 | Derivative of pBR322 with weakened Ptrc promoter | 49 |
| pWM2785 | pDSW210 expressing FLAG-FtsA | 10 |
| pEVOL | Cmr P15a plasmid expressing tRNA synthetase and tRNA for pBpA | 39 |
| pPK9 | pSC101 derivative, Spcr carrying araC and BAD promoter | P. Christie |
| pPK9-ZipA | pPK9 with zipA-Strep-Tag inserted between NotI and HindIII | This study |
| pUT18c | High-copy-no. bacterial two-hybrid vector, Apr | 50 |
| pKT25 | Medium-copy-no. bacterial two-hybrid vector, Kanr | 50 |
| pKNT25 | Medium-copy-no. bacterial two-hybrid vector, Kanr | 50 |
| pKNT25-ZipA | Bacterial two-hybrid plasmid expressing ZipA | 51 |
| pKNT25-FtsZ | Bacterial two-hybrid plasmid expressing FtsZ | 50 |
| pWM3014 | pKT25-FtsA | 10 |
| pWM3183 | pKT25-FtsN | 10 |
| pWM3021 | pUT18c-FtsA | 10 |
| pWM6134 | pUT18c-FtsA ΔR260 | This study |
| pWM6135 | pUT18c-FtsA ΔA256-R260 | This study |
| pWM6136 | pUT18c-FtsA ΔD253-R260 | This study |
| pWM5265 | pDSW210-ZipA-GFP | 21 |
| Strains | ||
| WM5163 | MG1655 WT E. coli K-12 strain | Lab collection |
| WM1074 | lac-deficient derivative of MG1655 (ilvG rpb-50 rph-1 ΔlacU169) | Lab collection |
| WM1115 | WM1074 ftsA12(Ts) | Lab collection |
| WM1125 | WM1074 ftsZ84(Ts) | Lab collection |
| WM1657 | ftsA-R286W ΔzipA::aph Kanr | This study |
| WM5855 | WM1115/pEVOL/pDSW210 | This study |
| WM5856 | WM1115/pEVOL/pWM2785 | This study |
| WM5886 | WM1115/pEVOL/pWM2785-R122X | This study |
| WM5857 | WM1115/pEVOL/pWM2785-E124X | This study |
| WM5858 | WM1115/pEVOL/pWM2785-R126X | This study |
| WM5887 | WM1115/pEVOL/pWM2785-R153X | This study |
| WM5888 | WM1115/pEVOL/pWM2785-R177X | This study |
| WM5889 | WM1115/pEVOL/pWM2785-R202X | This study |
| WM5890 | WM1115/pEVOL/pWM2785-R260X | This study |
| WM5891 | WM1115/pEVOL/pWM2785-R286X | This study |
| WM5892 | WM1115/pEVOL/pWM2785-R300X | This study |
| WM5998 | WM1115/pEVOL/pWM2785-P251X | This study |
| WM5999 | WM1115/pEVOL/pWM2785-S252X | This study |
| WM5964 | WM1115/pEVOL/pWM2785-D253X | This study |
| WM6000 | WM1115/pEVOL/pWM2785-A254X | This study |
| WM6001 | WM1115/pEVOL/pWM2785-E255X | This study |
| WM5965 | WM1115/pEVOL/pWM2785-A256X | This study |
| WM6002 | WM1115/pEVOL/pWM2785-I257X | This study |
| WM5908 | WM1115/pEVOL/pWM2785-K258X | This study |
| WM5909 | WM1115/pEVOL/pWM2785-V259X | This study |
| WM5910 | WM1115/pEVOL/pWM2785-H261X | This study |
| WM5911 | WM1115/pEVOL/pWM2785-G262X | This study |
| WM5949 | WM1657/pEVOL/pWM2785 | This study |
| WM5950 | WM1657/pEVOL/pWM2785-R260X | This study |
| WM5951 | WM1657/pEVOL/pWM2785/pPK9-ZipA | This study |
| WM5952 | WM1657/pEVOL/pWM2785-R260X/pPK9-ZipA | This study |
| WM5970 | WM1115/pWM2785(R286W)-R260X | This study |
| WM5971 | WM1657/pWM2785(R286W)-R260X | This study |
| WM5972 | WM1657/pWM2785(R286W)-R260X/pPK9-ZipA | This study |
| WM5991 | WM1115 ΔmrcA::kan/pWM2785-R260X (P1 JW3359 × WM5890) | This study |
| WM5992 | WM1115 ΔmrcB::kan/pWM2785-R260X (P1 JW0145 × WM5890) | This study |
| WM6101 | WM1115/pWM2785(D253R) | This study |
| WM6102 | WM1115/pWM2785(D253W) | This study |
| WM6103 | WM1115/pWM2785(A256R) | This study |
| WM6104 | WM1115/pWM2785(A256W) | This study |
| WM5935 | WM1115/pWM2785(R260A) | This study |
| WM5936 | WM1115/pWM2785(R260E) | This study |
| WM5937 | WM1115/pWM2785(R260W) | This study |
| WM6105 | WM1115/pWM2785(Δ253) | This study |
| WM6106 | WM1115/pWM2785(Δ256) | This study |
| WM6107 | WM1115/pWM2785(Δ260) | This study |
| WM6108 | WM1115/pWM2785(Δ256-260) | This study |
| WM6109 | WM1115/pWM2785(Δ253-260) | This study |
| BTH101 | F− cya-99 araD139 galE15 galK16 rpsL1 hsdR2 mcrA1 mcrB1 | 52 |
| JW3359 | ΔmrcA::kan strain | Keio collection |
| JW0145 | ΔmrcB::kan strain | Keio collection |
| WM6234 | ftsZ-55-mCer-56 sandwich fusion replacing chromosomal ftsZ | 44 |
| WM6015 | WM1125/pEVOL/pWM2785-R260X | This study |
| WM6019 | WM5163/pEVOL/pWM2785-R260X | This study |
| WM6253 | WM6234/pEVOL/pWM2785-R260X | This study |
| WM5446 | MG1655/pDSW210-ZipA-GFP | Lab collection |
For microscopic analysis, 10 µl of cells after UV irradiation were spread onto an agarose-covered glass microscope slide and topped with a glass coverslip. Cells were imaged with a 100× oil objective on an Olympus BX60 microscope outfitted with a Hamamatsu ORCA charge-coupled-device (CCD) camera, either by differential interference contrast (DIC) or by fluorescence.
Construction of ftsA amber mutants in pWM2785 (pDSW210F-FtsA).
A single amber codon (TAG) was added at different positions of FtsA by using site-directed mutagenesis. Primers were designed by using the Agilent QuikChange primer design tool with pWM2785 as the template for PCR. Mutagenized PCR products were digested with DpnI for 1 h at 37°C, transformed into Top10 competent cells, and selected on LB Ap plates supplemented with 0.2% glucose at 37°C. Each mutant was confirmed by DNA sequencing. Sequences of the mutagenic primers will be provided upon request. Deletions within helix 7 in pUT18c-FtsA were constructed by cloning the deleted ftsA genes from pDSW210F constructs as PstI-EcoRI fragments into pUT18c.
Other plasmid and strain constructions.
For the expression of E. coli zipA from a third plasmid compatible with both colE1 (pDSW210F) and p15a (pEVOL) plasmids, we used pPK9, a spectinomycin-resistant pSC101 derivative containing an arabinose-inducible promoter. The zipA gene was C-terminally fused to Strep-Tag and cloned into pPK9 as a NotI plus HindIII fragment that carries its own ribosome binding site and is oriented downstream of the araBAD promoter.
To generate BpA cross-linking strains that lacked PBP1A or PBP1B, P1 bacteriophage lysates were first made from Keio collection strains JW3359 and JW0145, which carry ΔmrcA::kan and ΔmrcB::kan, respectively. Each phage was then used to transduce WM5589 to Kanr, and the presence of the Kanr cassette replacing mrcA or mrcB was confirmed.
Bacterial two-hybrid assay.
The cya-deficient strain BTH101 was cotransformed with pUT18c and pKT25 (or pKNT25) derivatives, and transformants were selected with Ap and kanamycin (Kan). Six individual colonies carrying plasmid pairs were grown at 30°C with selection, and cultures were spotted on LB plates supplemented with Ap, Kan, 40 µg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside), and 50 µM IPTG. Plates were incubated for 24 h at 30°C and photographed.
BpA incorporation and cell viability measurements.
To test the proper incorporation of BpA into the FtsA amber mutant proteins and to determine the functionality of the resulting FtsA-BpA derivatives, WM1115 (ftsA12) was cotransformed with pEVOL and pWM2785 (with or without single amber mutations), and purified cotransformants were grown for 16 h at 30°C in LB broth supplemented with Ap and Cm. Cells were reinoculated in fresh medium (1:250 dilution) and incubated for 2 h at 30°C. Undiluted and serially diluted cells (10−1 to 10−5) were spotted onto LB plates supplemented with Ap, Cm, 100 µM l-arabinose, and 100 µM BpA with a range of IPTG concentrations (10 to 1,000 µM) and incubated for 20 h at 30°C or 42°C. Proper incorporation of BpA was evaluated by the capacity of the FtsA-BpA derivatives to suppress thermosensitivity of the ftsA12 allele in WM1115 on plates incubated at 42°C.
In vivo cross-linking and analysis by Western blotting.
Cells cotransformed with pEVOL and plasmids expressing the target protein (with or without amber mutations) were grown at 30°C for 16 h in 3 ml of LB supplemented with Ap and Cm. These cells were then diluted 1:100 into 7 ml of fresh medium and incubated for 2.5 h at 30°C, at which point 100 µM (each) IPTG, l-arabinose, and BpA were added and incubated for 2.5 h at 30°C. Cells were harvested by centrifugation at 5,000 rpm for 10 min at 4°C, and the pellets were resuspended in 1 ml of ice-cold PBS and transferred to multiwell plates. For UV cross-linking, plates containing the resuspended cells were exposed to long-wave UV (365 nm) irradiation for 15 min at 4°C. Cells were subsequently transferred to 2-ml microcentrifuge tubes and harvested by centrifugation at 15,000 rpm for 2 min at room temperature (RT). Pellets were resuspended in 300 µl of resuspension buffer (750 mM 6-aminocaproic acid, 50 mM bis-Tris, pH 7) (48) supplemented with 2% lysozyme plus protease inhibitor cocktail and incubated at RT while rotating for 30 min. After incubation, SDS and n-dodecyl-beta-d-maltopyranoside were added to a final concentration of 1% and incubated at RT for 30 min with rotation. Next, cells were subjected to three cycles of 5 min of boiling and 5 min of freezing and centrifuged for 30 min at RT. The supernatant (solubilized protein extract [SPE]) was transferred to a 1.5-ml microcentrifuge tube and stored at 4°C. For immunoblot analysis, samples were added to 5× protein loading buffer containing 5% beta-mercaptoethanol and then boiled for 5 min and centrifuged for 3 min. Proteins were separated by SDS-PAGE (8%) and transferred to nitrocellulose membranes for Western blotting. Primary antibodies (anti-FtsZ, anti-FtsN, anti-FtsA, anti-ZipA, anti-FLAG, and anti-Strep-Tag) were all used at 1:5,000 dilution in Tris-buffered saline with Tween 20 (TBS-T) plus 5% BSA, as was the secondary goat anti-rabbit antibody. Western transfer was carried out at 200 mA for 70 min to optimize detection of the large CLS.
Purification and identification of CLS.
For purification of CLS for mass spectrometry, WM5952 (WM1657/pEVOL/pWM2785-R260X/pPK9-ZipA) was grown at 30°C in 1.5 liters of LB supplemented with Ap, Kan, and Spc. Inducers and BpA were added, the cells were cross-linked by UV light, and the SPE was isolated as described above. For the pulldown experiment, SPE was incubated with magnetic Strep-Tag beads (MagStrep type 3 XT beads; IBA Lifesciences) for 16 h at 4°C. The flowthrough fractions were collected, and the magnetic beads were washed five times with W buffer (100 mM Tris HCl [pH 8], 150 mM NaCl, 1 mM EDTA) according to the manufacturer’s instructions. Washed beads were resuspended in 50 µl of phosphate-buffered saline (PBS) plus protein loading buffer, boiled for 5 min, and centrifuged for 3 min, and the entire sample was used for 8% SDS-PAGE. The gel was silver stained using the Silver Stain kit from Pierce (Thermo Fisher) according to the manufacturer’s instructions. Bands corresponding to CLS were excised and analyzed by mass spectrometry at the M.D. Anderson Cancer Center Proteomics and Metabolomics Facility.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marcin Krupka, Steven Distelhorst, and Kara Schoenemann for contributing plasmids and valuable ideas, Harold Erickson for the kind gift of the strain expressing ftsZ-mCerulean from the native ftsZ locus, Peter Christie and Rasika Harshey for plasmids, and Anna Konovalova for valuable advice on the cross-linking system.
This work was supported by grant GM61074 from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00579-18.
REFERENCES
- 1.Haeusser DP, Margolin W. 2016. Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nat Rev Microbiol 14:305–319. doi: 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lutkenhaus J, Pichoff S, Du S. 2012. Bacterial cytokinesis: from Z ring to divisome. Cytoskeleton (Hoboken) 69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.den Blaauwen T, Hamoen LW, Levin PA. 2017. The divisome at 25: the road ahead. Curr Opin Microbiol 36:85–94. doi: 10.1016/j.mib.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bisson-Filho AW, Hsu Y-P, Squyres GR, Kuru E, Wu F, Jukes C, Sun Y, Dekker C, Holden S, VanNieuwenhze MS, Brun YV, Garner EC. 2017. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355:739–743. doi: 10.1126/science.aak9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang X, Lyu Z, Miguel A, McQuillen R, Huang KC, Xiao J. 2017. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355:744–747. doi: 10.1126/science.aak9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray AN, Egan AJF, Van’t Veer IL, Verheul J, Colavin A, Koumoutsi A, Biboy J, Altelaar AFM, Damen MJ, Huang KC, Simorre J-P, Breukink E, den Blaauwen T, Typas A, Gross CA, Vollmer W. 2015. Coordination of peptidoglycan synthesis and outer membrane constriction during Escherichia coli cell division. Elife 4:e07118. doi: 10.7554/eLife.07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du S, Lutkenhaus J. 2017. Assembly and activation of the Escherichia coli divisome. Mol Microbiol 105:177–187. doi: 10.1111/mmi.13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pichoff S, Lutkenhaus J. 2005. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol 55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- 9.Feucht A, Lucet I, Yudkin MD, Errington J. 2001. Cytological and biochemical characterization of the FtsA cell division protein of Bacillus subtilis. Mol Microbiol 40:115–125. doi: 10.1046/j.1365-2958.2001.02356.x. [DOI] [PubMed] [Google Scholar]
- 10.Shiomi D, Margolin W. 2007. Dimerization or oligomerization of the actin-like FtsA protein enhances the integrity of the cytokinetic Z ring. Mol Microbiol 66:1396–1415. doi: 10.1111/j.1365-2958.2007.05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pichoff S, Shen B, Sullivan B, Lutkenhaus J. 2012. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA’s self-interaction competes with its ability to recruit downstream division proteins. Mol Microbiol 83:151–167. doi: 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szwedziak P, Wang Q, Bharat TAM, Tsim M, Löwe J. 2014. Architecture of the ring formed by the tubulin homologue FtsZ in bacterial cell division. Elife 3:e04601. doi: 10.7554/eLife.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conti J, Viola MG, Camberg JL. 2018. FtsA reshapes membrane architecture and remodels the Z-ring in Escherichia coli. Mol Microbiol 107:558–576. doi: 10.1111/mmi.13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lara B, Rico AI, Petruzzelli S, Santona A, Dumas J, Biton J, Vicente M, Mingorance J, Massidda O. 2005. Cell division in cocci: localization and properties of the Streptococcus pneumoniae FtsA protein. Mol Microbiol 55:699–711. doi: 10.1111/j.1365-2958.2004.04432.x. [DOI] [PubMed] [Google Scholar]
- 15.Szwedziak P, Wang Q, Freund SM, Löwe J. 2012. FtsA forms actin-like protofilaments. EMBO J 31:2249–2260. doi: 10.1038/emboj.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krupka M, Rowlett VW, Morado DR, Vitrac H, Schoenemann KM, Liu J, Margolin W. 2017. Escherichia coli FtsA forms lipid-bound minirings that antagonize lateral interactions between FtsZ protofilaments. Nat Commun 8:15957. doi: 10.1038/ncomms15957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loose M, Mitchison TJ. 2014. The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat Cell Biol 16:38–46. doi: 10.1038/ncb2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoenemann KM, Krupka M, Rowlett VW, Distelhorst SL, Hu B, Margolin W. 2018. Gain-of-function variants of FtsA form diverse oligomeric structures on lipids and enhance FtsZ protofilament bundling. Mol Microbiol 109:676–693. doi: 10.1111/mmi.14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hale CA, de Boer PA. 1997. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88:175–185. doi: 10.1016/S0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez-Diaz DA, García-Soriano DA, Raso A, Mücksch J, Feingold M, Rivas G, Schwille P. 2018. Treadmilling analysis reveals new insights into dynamic FtsZ ring architecture. PLoS Biol 16:e2004845. doi: 10.1371/journal.pbio.2004845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krupka M, Sobrinos-Sanguino M, Jimenez M, Rivas G, Margolin W. 2018. Escherichia coli ZipA organizes FtsZ polymers into dynamic ring-like protofilament structures. mBio 9:e1008-18. doi: 10.1128/mBio.01008-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Addinall SG, Bi E, Lutkenhaus J. 1996. FtsZ ring formation in fts mutants. J Bacteriol 178:3877–3884. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Mukherjee A, Lutkenhaus J. 1999. Recruitment of ZipA to the division site by interaction with FtsZ. Mol Microbiol 31:1853–1861. doi: 10.1046/j.1365-2958.1999.01322.x. [DOI] [PubMed] [Google Scholar]
- 24.Hale CA, de Boer PA. 2002. ZipA is required for recruitment of FtsK, FtsQ, FtsL, and FtsN to the septal ring in Escherichia coli. J Bacteriol 184:2552–2556. doi: 10.1128/JB.184.9.2552-2556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pichoff S, Lutkenhaus J. 2002. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J 21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meier EL, Razavi S, Inoue T, Goley ED. 2016. A novel membrane anchor for FtsZ is linked to cell wall hydrolysis in Caulobacter crescentus. Mol Microbiol 101:265–280. doi: 10.1111/mmi.13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haeusser DP, Garza AC, Buscher AZ, Levin PA. 2007. The division inhibitor EzrA contains a seven-residue patch required for maintaining the dynamic nature of the medial FtsZ ring. J Bacteriol 189:9001–9010. doi: 10.1128/JB.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duman R, Ishikawa S, Celik I, Strahl H, Ogasawara N, Troc P, Lowe J, Hamoen LW. 2013. Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proc Natl Acad Sci U S A 110:E4601–E4610. doi: 10.1073/pnas.1313978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishikawa S, Kawai Y, Hiramatsu K, Kuwano M, Ogasawara N. 2006. A new FtsZ-interacting protein, YlmF, complements the activity of FtsA during progression of cell division in Bacillus subtilis. Mol Microbiol 60:1364–1380. doi: 10.1111/j.1365-2958.2006.05184.x. [DOI] [PubMed] [Google Scholar]
- 30.Gupta S, Banerjee SK, Chatterjee A, Sharma AK, Kundu M, Basu J. 2015. The essential protein SepF of mycobacteria interacts with FtsZ and MurG to regulate cell growth and division. Microbiology 161:1627–1638. doi: 10.1099/mic.0.000108. [DOI] [PubMed] [Google Scholar]
- 31.Geissler B, Elraheb D, Margolin W. 2003. A gain of function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci U S A 100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geissler B, Margolin W. 2005. Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Mol Microbiol 58:596–612. doi: 10.1111/j.1365-2958.2005.04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samaluru H, SaiSree L, Reddy M. 2007. Role of SufI (FtsP) in cell division of Escherichia coli: evidence for its involvement in stabilizing the assembly of the divisome. J Bacteriol 189:8044–8052. doi: 10.1128/JB.00773-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ortiz C, Casanova M, Palacios P, Vicente M. 2017. The hypermorph FtsA* protein has an in vivo role in relieving the Escherichia coli proto-ring block caused by excess ZapC. PLoS One 12:e0184184. doi: 10.1371/journal.pone.0184184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geissler B, Shiomi D, Margolin W. 2007. The ftsA* gain-of-function allele of Escherichia coli and its effects on the stability and dynamics of the Z ring. Microbiology 153:814–825. doi: 10.1099/mic.0.2006/001834-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin JW, Martin AB, King DS, Wang L, Schultz PG. 2002. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc Natl Acad Sci U S A 99:11020–11024. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van den Berg van Saparoea HB, Glas M, Vernooij IGWH, Bitter W, den Blaauwen T, Luirink J. 2013. Fine-mapping the contact sites of the Escherichia coli cell division proteins FtsB and FtsL on the FtsQ protein. J Biol Chem 288:24340–24350. doi: 10.1074/jbc.M113.485888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan F, Yu J, Yu J, Liu Y, Li Y, Feng X-H, Huang KC, Chang Z, Ye S. 2018. Lateral interactions between protofilaments of the bacterial tubulin homolog FtsZ are essential for cell division. Elife 7:e35578. doi: 10.7554/eLife.35578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young TS, Ahmad I, Yin JA, Schultz PG. 2010. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol 395:361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 40.Herricks JR, Nguyen D, Margolin W. 2014. A thermosensitive defect in the ATP binding pocket of FtsA can be suppressed by allosteric changes in the dimer interface. Mol Microbiol 94:713–727. doi: 10.1111/mmi.12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Den Ent F, Löwe J. 2000. Crystal structure of the cell division protein FtsA from Thermotoga maritima. EMBO J 19:5300–5307. doi: 10.1093/emboj/19.20.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shiomi D, Margolin W. 2008. Compensation for the loss of the conserved membrane targeting sequence of FtsA provides new insights into its function. Mol Microbiol 67:558–569. doi: 10.1111/j.1365-2958.2007.06085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cambridge J, Blinkova A, Magnan D, Bates D, Walker JR. 2013. A replication-inhibited unsegregated nucleoid at mid-cell blocks Z-ring formation and cell division independently of SOS and the SlmA nucleoid occlusion protein in Escherichia coli. J Bacteriol 196:36–49. doi: 10.1128/JB.01230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore DA, Whatley ZN, Joshi CP, Osawa M, Erickson HP. 2017. Probing for binding regions of the FtsZ protein surface through site-directed insertions: discovery of fully functional FtsZ-fluorescent proteins. J Bacteriol 199:e00553-16. doi: 10.1128/JB.00553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujita J, Maeda Y, Nagao C, Tsuchiya Y, Miyazaki Y, Hirose M, Mizohata E, Matsumoto Y, Inoue T, Mizuguchi K, Matsumura H. 2014. Crystal structure of FtsA from Staphylococcus aureus. FEBS Lett 588:1879–1885. doi: 10.1016/j.febslet.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 46.Pfaendtner J, Branduardi D, Parrinello M, Pollard TD, Voth GA. 2009. Nucleotide-dependent conformational states of actin. Proc Natl Acad Sci U S A 106:12723–12728. doi: 10.1073/pnas.0902092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.den Blaauwen T. 2013. Prokaryotic cell division: flexible and diverse. Curr Opin Microbiol 16:738–744. doi: 10.1016/j.mib.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Trip EN, Scheffers D-J. 2015. A 1 MDa protein complex containing critical components of the Escherichia coli divisome. Sci Rep 5:18190. doi: 10.1038/srep18190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol 181:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karimova G, Dautin N, Ladant D. 2005. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol 187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HK, Harshey RM. 2016. A diguanylate cyclase acts as a cell division inhibitor in a two-step response to reductive and envelope stresses. mBio 7:e00822-16. doi: 10.1128/mBio.00822-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.