

See Noebels (doi:10.1093/brain/awy356) for a scientific commentary on this article.
The mechanisms underlying ‘sudden unexpected death in epilepsy’ (SUDEP) are unclear. Using behavioural, neurophysiological and neuroimaging data, Loonen et al. reveal characteristics specific to fatal seizures in homozygous Cacna1a S218L mice and show that brainstem spreading depolarization may play a critical role in SUDEP.
Keywords: sudden unexpected death in epilepsy, semiology, Cav2.1 channels, mouse model, spreading depolarization
Abstract
Sudden unexpected death in epilepsy (SUDEP) is a fatal complication of epilepsy in which brainstem spreading depolarization may play a pivotal role, as suggested by animal studies. However, patiotemporal details of spreading depolarization occurring in relation to fatal seizures have not been investigated. In addition, little is known about behavioural and neurophysiological features that may discriminate spontaneous fatal from non-fatal seizures. Transgenic mice carrying the missense mutation S218L in the α1A subunit of Cav2.1 (P/Q-type) Ca2+ channels exhibit enhanced excitatory neurotransmission and increased susceptibility to spreading depolarization. Homozygous Cacna1aS218L mice show spontaneous non-fatal and fatal seizures, occurring throughout life, resulting in reduced life expectancy. To identify characteristics of fatal and non-fatal spontaneous seizures, we compared behavioural and electrophysiological seizure dynamics in freely-behaving homozygous Cacna1aS218L mice. To gain insight on the role of brainstem spreading depolarization in SUDEP, we studied the spatiotemporal distribution of spreading depolarization in the context of seizure-related death. Spontaneous and electrically-induced seizures were investigated by video monitoring and electrophysiological recordings in freely-behaving Cacna1aS218L and wild-type mice. Homozygous Cacna1aS218L mice showed multiple spontaneous tonic-clonic seizures and died from SUDEP in adulthood. Death was preceded by a tonic-clonic seizure terminating with hindlimb clonus, with suppression of cortical neuronal activity during and after the seizure. Induced seizures in freely-behaving homozygous Cacna1aS218L mice were followed by multiple spreading depolarizations and death. In wild-type or heterozygous Cacna1aS218L mice, induced seizures and spreading depolarization were never followed by death. To identify temporal and regional features of seizure-induced spreading depolarization related to fatal outcome, diffusion-weighted MRI was performed in anaesthetized homozygous Cacna1aS218L and wild-type mice. In homozygous Cacna1aS218L mice, appearance of seizure-related spreading depolarization in the brainstem correlated with respiratory arrest that was followed by cardiac arrest and death. Recordings in freely-behaving homozygous Cacna1aS218L mice confirmed brainstem spreading depolarization during spontaneous fatal seizures. These data underscore the value of the homozygous Cacna1aS218L mouse model for identifying discriminative features of fatal compared to non-fatal seizures, and support a key role for cortical neuronal suppression and brainstem spreading depolarization in SUDEP pathophysiology.
See Noebels (doi:10.1093/brain/awy356) for a scientific commentary on this article.
Introduction
Sudden unexpected death in epilepsy (SUDEP) is a tragic and poorly understood complication of epilepsy (Nashef et al., 2012), affecting primarily young adults (Massey et al., 2014; Harden et al., 2017). Rare recordings in SUDEP cases demonstrated post-ictal generalized EEG suppression and respiratory depression prior to death (Lhatoo et al., 2010; Ryvlin et al., 2013). Risk of SUDEP appears higher in those with poorly controlled tonic-clonic seizures (Surges et al., 2009; Hesdorffer et al., 2011), especially during sleep (Lamberts et al., 2012; Ryvlin et al., 2013). In addition, mutations in a subset of ion channel genes have been linked to SUDEP risk (Goldman et al., 2009; Bagnall et al., 2016; Shmuely et al., 2016).
Mechanisms of SUDEP are largely unknown. Homozygous Kv1.1 knockout, and heterozygous Nav1.1 knockout mice, as well as mice with a R176Q missense mutation in RyR2 have been proposed as SUDEP models as the mutant mice exhibit seizures followed by cardiorespiratory arrest (Glasscock et al., 2010; Moore et al., 2014; Aiba and Noebels, 2015; Aiba et al., 2016). In these models, evoked cortical seizures were associated with respiratory arrest, followed by spreading depolarization in the dorsal brainstem and death. Seizure-related hypoxia was proposed as a critical trigger of brainstem spreading depolarization, resulting in loss of brainstem control over respiration inducing further hypoxia and eventually cardiac arrest (Aiba and Noebels, 2015).
Transgenic mice carrying the familial hemiplegic migraine type 1 S218L gain-of-function missense mutation in the Cacna1a gene, which encodes the α1A subunit of voltage-gated Cav2.1 (P/Q-type) Ca2+ channels (van den Maagdenberg et al., 2010), exhibit enhanced excitatory neurotransmitter release and increased susceptibility to induced cortical spreading depolarization (van den Maagdenberg et al., 2004, 2010; Eikermann-Haerter et al., 2009, 2011; Tottene et al., 2009; Vecchia et al., 2015; Cain et al., 2017). Spreading depolarization can propagate to distinct subcortical regions in homozygous Cacna1aS218L mice, but is typically constrained to the cortex in wild-type mice (Eikermann-Haerter et al., 2011; Cain et al., 2017). Patients with the CACNA1A gain-of-function S218L mutation suffer from hemiplegic migraine that can be associated with seizures and lethality following minor head trauma (Ophoff et al., 1996; Kors et al., 2001; Stam et al., 2009). Homozygous Cacna1aS218L mice display spontaneous seizures and fatal events that can occur throughout life and have been associated with epileptic attacks (van den Maagdenberg et al., 2010).
We here compared behavioural and electrophysiological non-fatal and fatal seizure dynamics in Cacna1aS218L mice under freely-behaving conditions to assess their value as a SUDEP model and investigate SUDEP mechanisms. We used diffusion-weighted MRI (DW-MRI) to investigate seizure-related propagation of spreading depolarization to distinct brain regions in anaesthetized mice and studied the incidence of brainstem spreading depolarization during spontaneous seizures in freely-behaving homozygous Cacna1aS218L mice.
Materials and methods
Animals
Knock-in Cacna1aS218L mice harbouring the human missense mutation S218L in the mouse Cacna1a gene were generated using a gene-targeting approach (van den Maagdenberg et al., 2010). Mice (mutant and wild-type littermates), backcrossed to C57BL/6J for at least 10 generations, of 2.5 to 6.5 months of age were used. Mutant homozygous Cacna1aS218L mice were investigated, except for evoked seizure experiments in freely-behaving mice, for which both heterozygous and homozygous Cacna1aS218L mice were used. Male mice were used, except for behavioural recordings in naïve (non-implanted) mice and DW-MRI experiments under anaesthesia, for which male and female mice were used.
Mice were kept under standard housing conditions with a 12-h light/dark cycle and food and water ad libitum. Mice freely moving during experiments were housed individually. Experimental procedures were carried out during the light period between 10 am and 5 pm. Experiments were approved by local ethical committees in accordance with recommendations of the European Communities Council Directive (2010/63/EU) (Leiden) or the Canadian Council for Animal Care guidelines (Vancouver). Experiments were carried out in accordance with ARRIVE guidelines. All efforts were made to minimize animal suffering.
Surgery for electrode placement
Cortical electrodes [single or paired 75 µm platinum (Pt)/iridium (Ir), PT6718; Advent Research Materials] were implanted under isoflurane anaesthesia (induction 4%; maintenance 1.5% in oxygen-enriched air) at the following coordinates (mm to bregma): 0.5 anterior/2.0 lateral/0.6 depth [left and right sensorimotor cortex (M1/S1); paired Pt/Ir]; 3.5 posterior/2.0 lateral/0.5 depth [right visual cortex (V1); single Pt/Ir]; two ball-tip electrodes were placed above the cerebellum and served as reference (Ag) and ground (Ag/AgCl). For cortical stimulation experiments, the left M1/S1 electrodes were implanted at 0.2 mm depth. For ECG recordings, 75 or 150 µm Pt/Ir electrodes were placed subcutaneously over the left latissimus dorsi muscle and the right trapezius and/or latissimus dorsi and tunnelled to the skull. For brainstem direct current (DC) recordings, electrodes were implanted at the following coordinates (mm to bregma): 4.8 posterior/0.8 lateral/3.7 depth [oral pontine reticular nucleus (PnO); single Pt/Ir] and 6.7 posterior/1.3 lateral/3.8 depth [medullary reticular formation (MRF); single Pt/Ir]. Electrodes were connected to a 7-channel pedestal (E363/0 socket contacts and MS373 pedestal, Plastics One) and secured to the skull using dental cement (DiaDent Europe). Carprofen (5 mg/kg, s.c.) and bupremorphine (Temgesic® 0.1 mg/kg, s.c.; only in homozygous Cacna1aS218L mice) were administered for postoperative pain relief.
Electrophysiological recordings in freely-behaving mice
Wild-type and heterozygous Cacna1aS218L mice were allowed to recover for 1 week following surgery (except for a subset of heterozygous Cacna1aS218L mice that were studied on the day of surgery). Homozygous Cacna1aS218L mice were given a shorter recovery period of 1 h to minimize chances of missing fatal seizures, which mostly occurred within 5 days after surgery (Fig. 1A). Recordings in freely-behaving mice were performed as described previously (Houben et al., 2017). Electrophysiological signals were 3× pre-amplified and fed into separate DC potential (500 Hz low-pass; 10× gain) and alternating current (AC) potential [0.05–500 Hz; 800× gain for electrocorticogram (ECoG); 400× gain for ECG; 200× gain for stimulation experiments] amplifiers. Signals were digitized (Power 1401 and Spike2 software, CED) at 1000 Hz (DC-potential) or 5000 Hz (ECoG and ECG). Differential signals from paired cortical electrodes were used to detect multi-unit activity (MUA; 500–5000 Hz; 12 000–36 000× amplification; 25 000 Hz sampling rate). Video recordings were made using a CCD camera (acA1300–60gmNIR, 30 frames/s, Basler). Locomotor activity was recorded using a custom-built passive infrared motion detection sensor.
Figure 1.

Survival of implanted homozygous Cacna1aS218L mice and fatal seizure duration in implanted and naïve mice. (A) Survival plot of 26 implanted homozygous Cacna1aS218L mice showing that the majority of mice died within 1 week after surgery. Note that during recovery from surgery (shaded grey), approximately half of the mice (n = 12) died. In the remaining mice (n = 14), which were monitored in the ECoG set-up for 2 weeks, fatal seizures occurred in all, except one that survived the recording period. Note, one additional mouse died following status epilepticus and was excluded from further analysis and not included in the plot. (B) Duration of spontaneous fatal seizure behaviour was not different for naïve and implanted mice (P = 0.50; data shown as mean ± SD).
Analysis of electrophysiological data
Electrophysiological data were analysed offline using Spike2 software and custom-written MATLAB scripts (The Mathworks). AC signals were inspected for electrographic seizures, defined as rhythmic discharges (spikes, sharp waves and/or slow waves) with a duration of >5 s. For peri-ictal power analyses, AC signals were artefact-rejected, digitally low-pass filtered (Chebyshev IIR eighth order filter) and down-sampled to 500 Hz. Next, a Fast Fourier Transform was performed using a Hamming window of 4 s. Total power was calculated for different spectral bands: delta (1–5 Hz), theta (5–10 Hz), alpha (10–15 Hz), beta (15–30 Hz), gamma (30–45 Hz) and total (1–100 Hz). Spreading depolarizations (in cortex or brainstem) were detected by the presence of a transient negative DC shift with amplitudes >5 mV on at least two recording locations within 60 s from one another. Terminal depolarization was identified by a prolonged DC-shift without recovery in all channels.
Wave marks were identified from MUA recordings in Spike2 using a detection window of 1 ms to extract the population firing rate. Thresholds were set offline above noise level, defined as overall activity during a 1-min period after the terminal depolarization. Consistency of the MUA signal over the recording period was verified for all recordings by comparison of average firing rates across vigilance states. For comparison across animals, ECoG and MUA data were normalized by the respective activity in a 5-min baseline period of continuous non-rapid eye movement (non-REM) sleep. For ECG analyses, R-peak detection was performed in MATLAB.
Electrically induced seizures in freely-behaving mice
In the majority of homozygous Cacna1aS218L mice (8/11), seizures were induced on the day of surgery, except for a small separate group of mutant mice (n = 3), in which seizures were induced 2 weeks after surgery (by comparing both groups of homozygous mutants it was assessed whether immediate effects of local tissue damage due to the surgical procedure confounded seizure outcome). In the majority of heterozygous Cacna1aS218L mice (7/9) and all wild-type mice (n = 7), seizures were induced 2 weeks after surgery, except for a small separate group of heterozygous Cacna1aS218L mice (n = 2) that were stimulated on the day of surgery (for comparison of seizure outcome between heterozygous and homozygous Cacna1aS218L mice stimulated on the day of surgery).
Electrical stimulation [15-s train consisting of 1-ms bipolar pulses at a frequency of 8 Hz at incrementally increasing current steps (0.25–4 mA) from a constant-current stimulus isolator (A395, WPI)] was administered every 5 min to determine the threshold for afterdischarges (>5 s) and/or cortical spreading depolarizations recorded in contralateral M1/S1. For experiments performed 2 weeks after surgery, threshold stimulation was repeated the next day for five times with 20-min intervals. If afterdischarges and/or cortical spreading depolarizations lasted more than 10 min, no subsequent stimuli were administered. For experiments performed on the day of surgery, stimulation sessions were combined due to high mortality.
Behavioural analysis
Video recordings of the 24 h preceding a fatal seizure were inspected for seizure-related behaviour. Single seizures or seizure clusters lasting more than 30 min were regarded as status epilepticus, which qualifies as an exclusion criterion for SUDEP (Nashef et al., 2012) and were therefore excluded from further analysis. Seizure severity was determined using the Racine scale (Racine, 1972) with slight adaptation whereby the most severe stage 5 also included jumping, circling and wild-running behaviour. Subconvulsive stage 1/2 seizures were sporadically observed during video analysis but were not considered for further analysis due to low detection sensitivity and absence of ECoG changes. Convulsive stage 3/4 seizures could reliably be detected by video analysis, and showed similar but milder changes of ECoG as observed for stage 5 seizures. Specific behavioural components during the fatal and last non-fatal seizure occurring at least 1 h before the fatal seizure, were analysed using software for quantitative behavioural assessment (Observer XT, Noldus Information Technology). Vigilance state was determined by inspection of V1 power spectra and behavioural activity, assessed from video and movement sensor, and categorized as (i) awake (high theta and low delta ECoG activity, during behavioural activity); (ii) non-REM sleep (high delta activity, no movements); and (iii) REM sleep (high theta and low delta activity following a period of non-REM sleep, no movements).
Naïve mice were continuously videotaped. Recordings were assessed for spontaneous seizures in the 24 h preceding a fatal seizure and analysed as described above.
Diffusion-weighted MRI during induced seizures in anaesthetized mice
Spreading depolarization is accompanied by an influx of water to the intracellular compartment of brain cells that causes a decrease in MRI apparent diffusion coefficient (de Crespigny et al., 1998). Here, a recently developed DW-MRI protocol that allows spreading depolarization visualization with high time-resolution (Cain et al., 2017) was combined with ECoG recordings and cortical electrical stimulation. Animals were anaesthetized using isoflurane (2–5% in 100% oxygen) during surgical preparation and once implanted transferred to dexmedetomidine/fentanyl/midazolam and spontaneous breathing of normal air for at least 25 min prior to DW-MRI. Carbon fibre electrodes (WPI) were used, given their compatibility with MRI: two in left M1/S1 for stimulation, one in right M1/S1 for ECoG, and one as reference electrode in the cerebellum (coordinates as described above). MRI experiments were performed on a 7 T animal scanner (Bruker). A quadrature radiofrequency coil with 70-mm inner diameter volume was used for pulse transmission and the MRI signal was received with a 14-mm diameter actively decoupled surface coil. The mouse was laid supine in the MRI cradle with electrodes fed through the centre of the radiofrequency coil. Stimulation electrodes were connected to a constant-current unit and stimulator (Grass Technologies) outside the scanner. ECoG electrodes were connected to a W2100 amplifier/receiver outside the scanner (Multichannel Systems) and data were acquired at 0.1–1000 Hz. Respiratory rate, heart rate and body temperature were monitored during scanning with a Model 1025 Control/Gating system (SA Instruments). DW-MRI was acquired using diffusion-weighted spin-echo planar imaging (EPI) with a b-value of 1800 s/mm2 (echo time/repetition time = 29/2000 ms, four shots, field of view = 2 × 2 cm, matrix size = 64 × 64, slice thickness = 1.25 mm, eight interleaved slices) resulting in an 8-s time resolution.
Electrical stimulation was performed as described above until afterdischarges and/or cortical spreading depolarizations were observed during 15-min DW-MRI scans. Stimulation was applied at 1, 6 and 11 min with increasing stimulation intensity. If afterdischarges were not observed, a second scan with higher intensity stimulations was performed, and similarly for a third scan, if required [scan set 1 (0.25, 0.5, 1), scan set 2 (2, 4, 8), scan set 3 (10, 15, 20) mA]. If the animal survived the threshold voltage, stimulation was reapplied at 20-min intervals for 1 h. Seizures were identified by motion artefacts resulting from clonus during DW-MRI acquisition and epileptiform spiking (2–10 Hz) in the ECoG. DW-MRI data were post-processed in MATLAB and ImageJ software (NIH) to mask non-brain structures, apply colour maps and measure voxel intensity in defined brain regions (Cain et al., 2017). Identification of DW-MRI spreading depolarization events was defined by an increase in signal intensity exceeding 2× the standard deviation (SD) of baseline DW-MRI intensity, sustained for a minimum of 24 s in a single region of interest and visually demonstrating a propagating wave pattern into adjacent structures.
Histology
Following experiments, surviving animals were euthanized by CO2 and transcardially perfused [0.1 M phosphate-buffered saline followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer]. Brains were removed, post-fixed in 4% PFA for 2.5 h at room temperature and sucrose-processed. For mice that died during experiments, brains were post-fixed in 4% PFA for 24 h at 4°C. Brains were sagittally sectioned on a sliding microtome (Leica) and sections (40 µm) were Nissl stained to verify electrode locations.
Statistical analysis
Data visualization and statistical testing were performed using MATLAB and Graphpad Prism (GraphPad). Unless indicated otherwise, data were presented as mean ± standard error of the mean (SEM) and analysed using two-tailed paired or unpaired t-tests, Mann-Whitney U-test, or repeated measures one-way or two-way ANOVA, where appropriate. A P-value < 0.05 was considered significant.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
Homozygous Cacna1aS218L mice exhibit hallmark features of SUDEP
First, we analysed the occurrence of spontaneous death in homozygous Cacna1aS218L mice (n = 27; all males, age 2.5–5.5 months) with implanted electrodes. Twelve mice died within the 1-h recovery period following surgery, i.e. before the start of electrophysiological recordings. Video analysis (when available) confirmed death following seizure activity. Of the mice that survived the immediate post-surgical period (n = 15), fatal seizures were observed immediately preceding definitive behavioural arrest and death (n = 13; Fig. 1A). One animal survived the 2-week recording period, and one died following status epilepticus (and was excluded from further analysis).
Chronic electrode implantation may influence seizure characteristics by causing brain damage, thus fatal seizure behaviour was also studied in non-implanted animals. Among naïve (non-implanted) homozygous Cacna1aS218L mice with fatal events (n = 11 in six males and five females), in eight mice seizure behaviour was observed immediately preceding definitive behavioural arrest, similar to the fatal seizure phenotype in implanted mice. In the remaining mice (n = 3), seizure-related behaviour was followed by a prolonged period (3–22 h) of immobility before death. In both implanted and naïve mice, the duration of fatal seizure behaviour immediately prior to cessation of motor activity was comparable (Fig. 1B). Most seizures (12/13 in implanted and 7/11 in naïve mice) were graded stage 5 (Racine, 1972).
Video inspection of fatal seizures in implanted mice revealed respiratory movements indicative of gasps (between 2 and 5) following behavioural stage 5 seizure activity and preceding terminal depolarization (in 6/12 mice with sufficient quality of video recordings). Concomitant recording of cortical neuronal MUA, ECoG, and behaviour revealed suppression of cortical neuronal activity starting during seizure behaviour and continuing up to the terminal depolarization (Fig. 2A). Ictal asystole could be excluded as cause of death in a separate group of mice (n = 5) with cortical and thoracic leads. Electrical cardiac activity was evident minutes after the terminal depolarization (Fig. 2B), with electrocardiographic arrest 315–810 s after terminal depolarization. Periods of ictal bradycardia and atrioventricular block were observed during seizure behaviour for all fatal cases, but ventricular fibrillation did not occur until minutes after terminal depolarization.
Figure 2.

Electrophysiological recordings during spontaneous fatal seizures in homozygous Cacna1aS218L mice. (A) Example of cortical MUA and ECoG (DC and AC) recordings during a fatal seizure (behavioural onset indicated by the vertical dashed line). MUA (histogram) decreased during seizure behaviour (clonic behaviour in grey, tonic behaviour in black). MUA and AC ECoG were attenuated in the minute preceding the terminal depolarization (TD; indicated by an arrow). Note that the pre-ictal transition of AC ECoG amplitude parallels vigilance state (non-REM sleep followed by wakefulness). (B) ECG recordings during a fatal seizure in another mouse showing cardiac electrical activity following TD. Electrocardiographic arrest (not shown) occurred 7 min after the terminal depolarization. The onset of seizure-related behaviour preceded the plotted time series. AC = alternating current; bps = beats per second; DC = direct current; IHR = instantaneous heart rate.
Non-fatal and fatal seizures in homozygous Cacna1aS218L mice show subtle behavioural differences
To identify discriminative features of fatal seizures, behavioural characteristics were compared between non-fatal and fatal seizures in the 24-h period preceding death in implanted homozygous Cacna1aS218L mice. Fatal seizures were typically preceded by multiple behavioural non-fatal seizures (range 1–22/mouse; median 12), of which 41% were classified as stage 3/4 (n = 49 in nine mice), and 59% as stage 5 (n = 71 in 11 mice). To allow for comparison of non-fatal and fatal seizures independent of seizure severity, only stage 5 seizures were included for further analysis. Non-fatal seizure incidence rates were variable in different animals but seizure frequency did not differ between 0–12 h versus 12–14 h preceding the fatal seizure (P = 0.36; Fig. 3A). Non-fatal seizure duration (averaged per animal) was comparable to fatal seizure duration (191 ± 30 and 131 ± 14 s, respectively; P = 0.37).
Figure 3.
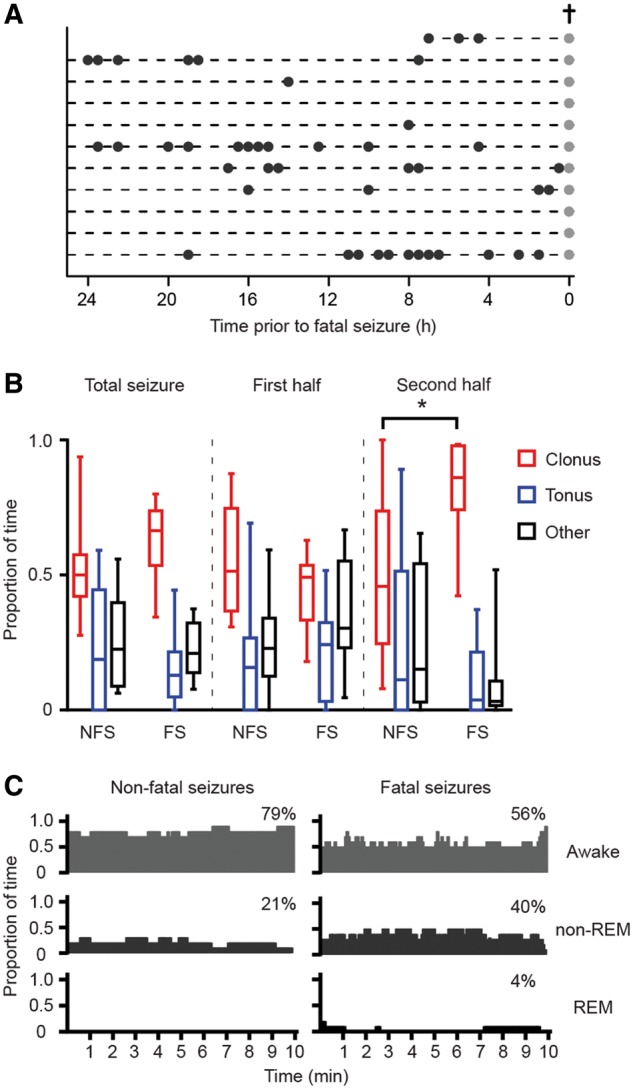
Frequency of spontaneous non-fatal and fatal seizures (stage 5) and (pre-)ictal behaviour in implanted homozygous Cacna1aS218L mice. (A) The distribution of non-fatal seizures (black dots) in implanted mice over the 24 h preceding the fatal seizure (grey dots; cross). (B) Fatal seizures were associated with a proportional increase of clonic behaviour during the second half of the seizure at the cost of tonic and other behaviours, when compared to non-fatal seizures (n = 11 per group; *P = 0.001; data are shown as median, interquartile intervals, minimum and maximum). (C) Vigilance state preceding non-fatal and fatal seizures (percentages indicate fraction of total time) was not significantly different (P > 0.99). FS = fatal seizures; NFS = non-fatal seizures.
Both fatal and non-fatal stage 5 seizures were associated with generalized tonic and clonic behaviour and loss of balance, and a subset with circling, jumping and/or wild-running. To detect subtle behavioural differences between the fatal and the last non-fatal seizure (occurring at least 1 h before the fatal seizure), the timing and duration of tonic, clonic, and other seizure behaviours was analysed in implanted mice (n = 11; Fig. 3B). An increase in clonic behaviour was observed in the second half of the fatal seizure. Indeed, closer inspection revealed that all fatal seizures ended with myoclonic jerks of fore- and hindlimbs, gradually decreasing in frequency over a period of 12–36 s. Forelimb clonus always ceased 5–30 s before termination of hindlimb clonus. Non-fatal seizures (n = 11) ended with various seizure-related behaviours [alternating tonic-clonic activity of all limbs (n = 4); exclusive forelimb clonus (n = 4); forelimb and hindlimb clonus (n = 3)]. This characteristic pattern of fatal seizure termination, ending with a hindlimb clonus, was also present in naïve (non-implanted) Cacna1aS218L mice.
The majority of reported human SUDEP events occur at night (Lamberts et al., 2012; Ryvlin et al., 2013) thus nocturnal patterns in seizure incidence and vigilance state were determined in implanted mice in the 10 min preceding seizures. Stage 5 non-fatal and fatal seizures were equally distributed over the light, dark, and dusk/dawn (transition) phases (Fig. 3C) (non-fatal: 49%, 44% and 7%, respectively; fatal: 50%, 42% and 8%, respectively; P = 0.91). Animals were more often asleep prior to fatal seizures, compared to the last non-fatal seizure, although this difference did not reach statistical significance.
Fatal seizures are associated with specific ECoG dynamics and (post-)ictal neuronal suppression
In implanted homozygous Cacna1aS218L mice, rhythmic high-amplitude cortical epileptiform events were never observed during spontaneous non-fatal or fatal seizures (stage 5) but only occurred during status epilepticus (one non-fatal and one fatal). Rather, seizures were associated with reduced ECoG amplitude followed by a period of rhythmic theta band activity (Fig. 4B) and medium-amplitude spikes during pronounced clonic behaviour, which often coincided with cortical neuronal suppression (Fig. 4C). Group analysis of (pre)ictal ECoG dynamics confirmed attenuation of total ECoG power, which, for fatal seizures, resulted in complete ECoG suppression during and following the behavioural seizure and never recovered (Fig. 4D). For non-fatal seizures, such suppression of ECoG power was rarely observed (3/54 seizures; example in Supplementary Fig. 1). A decrease in normalized ECoG delta band activity was observed during the first minute of seizure-related behaviour for all seizures (Fig. 4E), whereas normalized theta band power was significantly higher during the first minute for fatal compared to non-fatal seizures (Fig. 4F).
Figure 4.

Different ECoG and cortical multi-unit activity during spontaneous non-fatal and fatal seizures in homozygous Cacna1aS218L mice. (A) Example cortical MUA and AC ECoG recordings of a fatal seizure [behavioural onset indicated by a red dashed line; clonic behaviour is shown in grey, tonic behaviour in black; terminal depolarization (TD) indicated by an arrow]. Details of ictal AC ECoG and MUA indicated by dashed boxes are shown in B and C. (B) Detailed inspection of the AC ECoG reveals oscillatory activity corresponding to theta band frequencies and (C) spike-wave complexes (indicated by arrowheads) not exceeding normal AC ECoG amplitude, that coincide with loss of cortical MUA. (D–F) Spectral analyses of primary sensorimotor (M1/S1) AC ECoG during non-fatal (red) and fatal (black) stage 5 seizures (n = 9) for total power (D), delta/total ratio (E) and theta/total ratio (F). Power during the first 60 s of all seizures (indicated by black bars) was compared with baseline power (1 h pre-ictal; baseline power levels for non-fatal/fatal seizures are indicated by red/black dashed lines). In addition, non-fatal and fatal seizures were compared over the same ictal period (insets; data are shown as median, interquartile intervals, minimum and maximum). Normalized total power and delta/total power ratio were attenuated (D and E; P = 0.001 and P = 0.010 for non-fatal and fatal seizures, respectively). Theta/total ratio was increased only for fatal seizures, compared to both baseline and non-fatal seizures (F; P < 0.001 and *P = 0.003, respectively). For all fatal seizures, cessation of seizure-related behaviour is indicated by vertical blue bars. (G and H) Normalized cortical MUA dynamics in relation to onset [G(i)] and end [G(ii)] of seizure-related behaviour. Black bars indicate 120-s time windows used for comparison of non-fatal and fatal seizures in (H). MUA decreased during fatal seizures (G) and was significantly reduced when compared to non-fatal seizures during the last 120 s of seizure-related behaviour (H; *P = 0.018; data are shown as median, interquartile intervals, minimum and maximum). AC = alternating current.
Cortical neuronal activity was increased in the seconds to minutes preceding the behavioural onset of the fatal seizure, although not significantly different from non-fatal seizures [Fig. 4G(i) and H]. Notably, only during fatal seizures, neuronal activity was significantly attenuated during the last 120 s of seizure-related behaviour [Fig 4G(ii) and H]. This pattern was followed by low to absent neuronal activity during the post-ictal phase.
Induced seizures in homozygous Cacna1aS218L mice are associated with multiple cortical spreading depolarizations and death
Seizures, induced by stimulation of the sensorimotor cortex, were compared between freely-behaving homozygous Cacna1aS218L mice [stimulated on the day of surgery (n = 8) or 2 weeks after surgery (n = 3)], heterozygous Cacna1aS218L mice [stimulated on the day of surgery (n = 2) or 2 weeks after surgery (n = 7)], and wild-type mice [stimulated 2 weeks after surgery (n = 7)].
Cortical stimulation resulted in severe seizures in all homozygous Cacna1aS218L mice (starting immediately to 15.5 min after stimulation) that were fatal in seven mice. Six mice died following single or multiple stage 5 seizures 2–73 min after stimulation (median 5 min); one mouse died following status epilepticus. Cortical stimulation never resulted in fatal seizures in heterozygous Cacna1aS218L mice or wild-type mice. Epileptiform afterdischarges were observed in all heterozygous Cacna1aS218L mice and wild-type mice (Fig. 5A and B), but only in 2 of 11 homozygous Cacna1aS218L mice.
Figure 5.
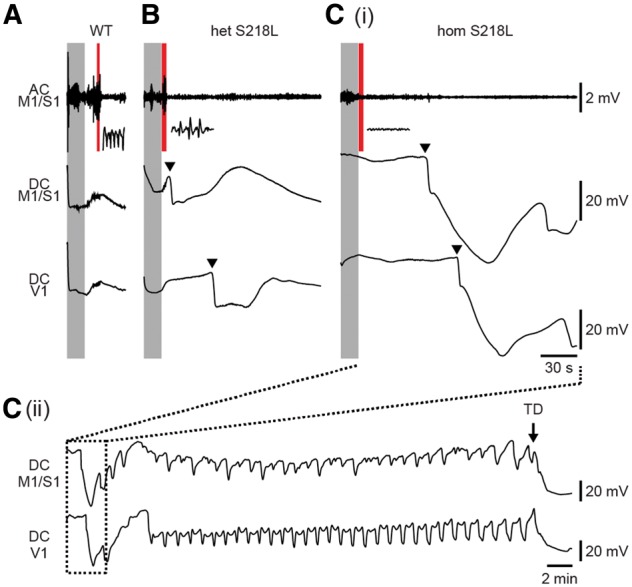
Multiple cortical spreading depolarizations associated with induced seizures followed by death in homozygous Cacna1aS218L mice. Representative ECoG (AC and DC) recordings showing that stimulation (shaded grey) was followed by afterdischarges in all wild-type (WT) mice (7/7; example in A) and heterozygous Cacna1aS218L (het S218L) mice (9/9; example in B), while only in a minority of homozygous Cacna1aS218L (hom S218L) mice [2/11; example in C(i)], as indicated by insets (corresponding to the shaded red time windows). Cortical spreading depolarization (indicated by arrowheads in example traces) was observed directly following afterdischarges in all but one heterozygous Cacna1aS218L mice. In homozygous Cacna1aS218L mice, multiple cortical spreading depolarizations occurred, followed by a terminal depolarization [C(ii); TD; indicated by an arrow]. Fatal outcome was observed in 7 of 11 homozygous Cacna1aS218L mice but none of the wild-type and heterozygous Cacna1aS218L mice. AC = alternating current; DC = direct current.
Afterdischarges were followed by a single cortical spreading depolarization, recorded contralateral to the stimulation electrode, in all but one heterozygous Cacna1aS218L mice whereas no cortical spreading depolarizations were observed in wild-type mice (Fig. 5A and B). Severe seizure behaviour in homozygous Cacna1aS218L mice was, however, associated with multiple cortical spreading depolarizations (n = 2–38 waves, median 21, in 9/11 mice; example in Fig. 5C). These findings indicate that fatal seizure outcome in homozygous Cacna1aS218L mice may relate to an enhanced propensity to seizure-associated spreading depolarization.
Induced fatal seizures in homozygous Cacna1aS218L mice are associated with brainstem spreading depolarization
DW-MRI was used to assess the spatiotemporal propagation of spreading depolarizations in response to cortically induced seizures in anaesthetized homozygous Cacna1aS218L (n = 7) and wild-type (n = 3) mice. Spreading depolarization occurrence, visualized by MRI-diffusional changes associated with spreading depolarization-related cell swelling, was confirmed by simultaneous (ipsilateral) cortical DC recording (Supplementary Fig. 2). Stimulation resulted in epileptiform afterdischarges (2–10 Hz), concurrent with DW-MRI movement artefacts directly following stimulation. In wild-type mice stimulation resulted only in non-fatal seizures (n = 10), while in homozygous Cacna1aS218L mice both non-fatal seizures (n = 15) and fatal seizures (n = 4) were observed.
DW-MRI data (from the hemisphere ipsilateral to the stimulation site) showed cortical spreading depolarization with all fatal (4/4) and most non-fatal seizures (13/15) in homozygous Cacna1aS218L mice and with most non-fatal seizures (7/10) in wild-type mice. Cortical spreading depolarizations were first observed in the dorsal sensorimotor cortex and propagated to the visual cortex with different timing delays between genotypes: wild-type: 43 ± 6 s; Cacna1aS218L non-fatal: 26 ± 3 s; Cacna1aS218L fatal: 20 ± 2 s (wild-type versus Cacna1aS218L non-fatal: P = 0.016; wild-type versus Cacna1aS218L fatal: P = 0.018; Cacna1aS218L non-fatal versus Cacna1aS218L fatal: P = 0.72). For fatal seizures, cortical spreading depolarizations were followed by spreading depolarizations in subcortical structures including striatum (14 ± 13 s, indicating the time delay following spreading depolarization in the sensorimotor cortex), hippocampus (16 ± 1 s), superior and inferior colliculus (24 ± 5 s) and brainstem (31 ± 4 s) (Fig. 6A, B and E). Within the brainstem, spreading depolarizations in the medulla was detected after spreading depolarization appearance in the pons (3/4 fatal seizures; delay of 1–3 scan repetitions; 8–24 s). In one animal medullary and pontine spreading depolarization were detected at the same time point. For non-fatal seizures in homozygous Cacna1aS218L mice, cortical spreading depolarizations were followed by spreading depolarizations in striatum (n = 8; 59 ± 11 s), hippocampus (n = 6; 46 ± 10 s), and colliculi (n = 5; 45 ± 13 s), but never in the brainstem (Fig. 6C and E). In all but one wild-type mice, spreading depolarizations were not observed in subcortical structures (Fig. 6D). Cortical spreading depolarization occurrence in the contralateral cortex was only rarely observed in the DW-MRI experiments following non-fatal seizures (2/10) in wild-type mice, in line with ECoG from freely-behaving wild-type mice in which contralateral cortical spreading depolarizations were not observed following stimulation. In summary, subcortical spreading depolarization propagation is more extensive in homozygous Cacna1aS218L mice with brainstem spreading depolarization only observed during fatal seizures.
Figure 6.

DW-MRI visualization of spreading depolarization following induced seizures in anaesthetized homozygous Cacna1aS218L mice. (A) Coronal brain maps corresponding to DW-MRI images in B–D. (B) Representative DW-MRI data following an induced fatal and (C) non-fatal seizure in a homozygous Cacna1aS218L (hom S218L) mouse, and (D) a non-fatal seizure in a wild-type (WT) mouse. Data are shown for a single representative time point in each slice, capturing a snapshot of spreading depolarization (SD). Note that in B, all coronal maps are shown at three time points to demonstrate spatiotemporal propagation of spreading depolarization, whereas in C and D different sections at three time points are depicted to show the limited subcortical spread of spreading depolarization in non-fatal seizures. (E) Time course data of wild-type mice (n = 3; seven spreading depolarizations) and Cacna1aS218L mice (n = 7; 13 non-fatal spreading depolarizations; four fatal spreading depolarizations) in relation to seizure onset (dashed line) are shown for distinct brain regions obtained from DW-MRI data. In Cacna1aS218L mice, spreading depolarization appeared in the brainstem only during fatal seizures and was constrained to striatum, amygdala, hippocampus and colliculi during non-fatal seizures. Cortical spreading depolarization occurred in all wild-type and Cacna1aS218L mice. A.u. = arbitrary units; Cb = cerebellum; Cx = cortex; DC = direct current; Hc = hippocampus; Md = medulla oblongata; Po = pons; SC = superior colliculus; St = striatum; Th = thalamus.
Respiratory rate and heart rate, monitored during evoked seizure experiments, were increased after non-fatal seizures in homozygous Cacna1aS218L and wild-type mice (Fig. 7). Both parameters returned to baseline within 1 min in wild-type mice, whereas in homozygous Cacna1aS218L mice respiratory rate fluctuated for an extended period of time (2–3 min) before returning to baseline. In fatal seizures, respiratory slowing was followed by respiratory arrest that preceded cardiac arrest. Respiratory arrest occurred near-concurrently with the observation of spreading depolarization in the brainstem (Fig. 7A), either shortly before (12 and 5 s) or after (11 and 33 s) brainstem spreading depolarization (recognizing that the time resolution of DW-MRI was limited to 8 s).
Figure 7.
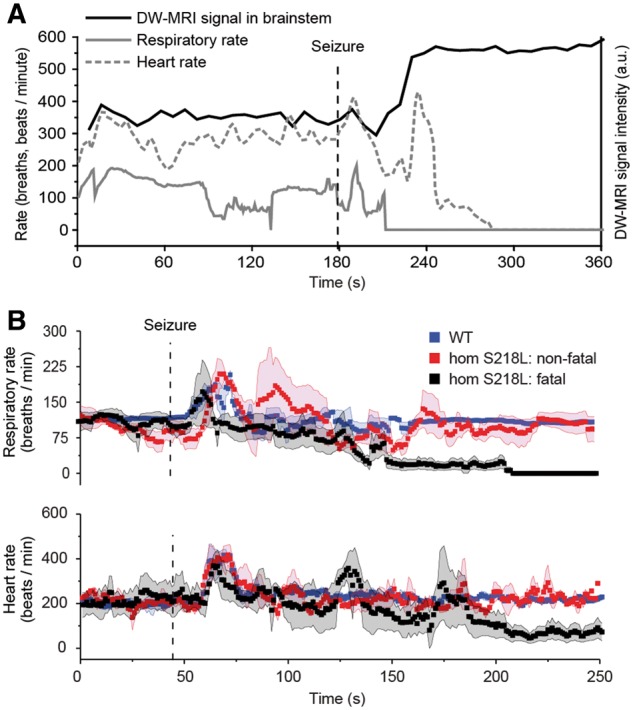
Physiological function following induced seizures during DW-MRI in anaesthetized homozygous Cacna1aS218L mice. (A) Representative example from a single animal of respiratory rate, heart rate and spreading depolarization aligned temporally in relation to seizure onset (dashed line). (B) Physiological parameters (aligned with spreading depolarization dynamics shown in Fig. 6E) from homozygous Cacna1aS218L (hom S218L) mice, following non-fatal (n = 10) and fatal seizures (n = 4), and wild-type (WT) mice, following non-fatal seizures (n = 15). Simultaneous respiratory (upper trace) and heart rate (lower trace) data acquired during seizures from the same animals showing that, for fatal seizures, respiratory rate decreased until complete arrest and preceded cardiac arrest. A.u. = arbitrary units. Data shown as mean ± SD.
As the amygdala has been implicated in seizure-related respiratory arrest (Dlouhy et al., 2015), one could argue that respiratory arrest might be caused by spreading depolarization occurring in the amygdala. Although spreading depolarization in the amygdala was observed following cortical spreading depolarization in Cacna1aS218L mice (n = 13) and in wild-type mice (n = 2), this did not coincide with profound respiratory changes (Supplementary Fig. 3).
Brainstem spreading depolarization occurs during spontaneous fatal seizures in homozygous Cacna1aS218L mice
To study the incidence of brainstem spreading depolarization during spontaneous seizures in homozygous Cacna1aS218L mice, brainstem DC ECoG recordings from the pons (PnO) and medulla (MRF) were combined with V1 ECoG and ECG in freely-behaving mice. Three mice had a fatal seizure (duration 185–270 s)—after a recording period of 1–3.5 days—that was always associated with brainstem spreading depolarization. Brainstem spreading depolarization had an onset during seizure behaviour (24–60 s preceding seizure termination) and was followed by ECoG suppression, bradycardia and a cortical terminal depolarization (Fig. 8). Gasping was absent during fatal seizures. A delay in MRF spreading depolarization was observed in two mice (9 and 23 s after PnO spreading depolarization), whereas in one mouse spreading depolarization occurred simultaneously in PnO and MRF. Brainstem spreading depolarization was not observed during non-fatal seizures (n = 8) or during interictal recordings preceding the fatal seizure.
Figure 8.

Brainstem DC recordings during a spontaneous fatal seizure in a homozygous Cacna1aS218L mouse. Example of a spreading depolarization (indicated by arrowheads) that occurred during seizure behaviour in the oral pontine reticular nucleus (PnO) and subsequently in the medullary reticular formation (MRF), followed by visual cortex (V1) AC ECoG suppression, bradycardia and cortical terminal depolarization (TD; indicated by an arrow). Note that ictal V1 AC ECoG amplitude was greatly reduced following cortical spreading depolarization. AC = alternating current; bps = beats per second; DC = direct current; IHR = instantaneous heart rate.
Discussion
Here we describe features that discriminate spontaneous and induced fatal seizures from non-fatal events in homozygous Cacna1aS218L mice. Characteristics specific to fatal seizures included: (i) an increase in clonic activity during later phases of the seizure; (ii) early (ictal) and post-ictal suppression of cortical neuronal activity; (iii) subcortical spreading depolarization spread following induced seizures culminating in brainstem spreading depolarization with respiratory arrest, and eventually cardiac arrest; and (iv) brainstem spreading depolarization during spontaneous seizures preceding complete ECoG suppression and cardiac arrest.
DW-MRI revealed that cortically-induced non-fatal seizures were associated with spreading depolarization that remained predominantly restricted to the ipsilateral cortex in wild-type mice, but invaded subcortical structures in Cacna1aS218L mice. This is in agreement with previous findings in homozygous Cacna1aS218L mice wherein induced cortical spreading depolarization was followed by striatal and hippocampal (Cain et al., 2017) and, occasionally, thalamic invasion (Eikermann-Haerter et al., 2011). Simultaneous cortical DC recordings in a subset of DW-MRI experiments confirm that measured changes in signal intensity indeed result from cortical spreading depolarization events.
The results from both induced and spontaneous seizures in Cacna1aS218L mice indicate that only fatal seizures were associated with brainstem spreading depolarization, observed in the pons and medulla. This coincided with a decrease in respiratory activity during fatal seizures, suggesting that brainstem spreading depolarization induces apnoea. Although we were unable to measure ictal respiratory activity systematically in freely-behaving mice, few post-ictal respiratory gasps were observed in half of the mice whereas in the other half no respiratory activity was observed, indicating limited auto-resuscitation. This supports the hypothesis that seizure-induced spreading depolarization causes death by suppression of brainstem respiratory control. Indeed, in animals with confirmed brainstem spreading depolarization during the fatal seizure no gasps were observed. Our data contrast with previous studies, where apnoea clearly preceded brainstem spreading depolarization, and hypoxia was proposed as a trigger for brainstem spreading depolarization (Aiba and Noebels, 2015; Aiba et al., 2016). Differences in timing between respiratory changes and spreading depolarization might be explained by methodology, since previous studies relied on electrodes inserted in the dorsocaudal medulla only (Aiba and Noebels, 2015; Aiba et al., 2016). Future studies should indicate whether brainstem spreading depolarization indeed precedes and causes respiratory arrest.
Head trauma due to electrode implantation is likely to have influenced the features of spontaneous fatal seizures in recorded homozygous Cacna1aS218L mice. Whereas surgery contributed to seizure frequency and caused a reduction of lifespan in homozygous Cacna1aS218L mice, spontaneous seizure behaviour was similar in naïve and implanted animals. In addition, cortical stimulation experiments on the day of surgery and after a 2-week recovery period both resulted in fatal seizures in the majority of homozygous Cacna1aS218L mice, indicating that acute post-surgery complications are not key contributors to fatal seizure outcome in our model.
All fatal seizures and only a minority of non-fatal seizures in freely-behaving Cacna1aS218L mice were associated with a late ictal and post-ictal cortical suppression pattern, that resembles post-ictal generalized EEG suppression (PGES), a consistent observation in clinical SUDEP recordings (Lhatoo et al., 2010; Ryvlin et al., 2013). Global (Alexandre et al., 2015) and/or local (Farrell et al., 2016) hypoxia could underlie the suppression of cortical neuronal activity. Indeed, in fatal seizures in a mouse model of Dravet syndrome, an epileptic encephalopathy associated with high SUDEP risk (Shmuely et al., 2016), apnoea preceded PGES (Kim et al., 2018). Periods of apnoea and bradycardia followed by a terminal apnoea and asystole have been reported in human SUDEP cases (Ryvlin et al., 2013). Our ECG recordings during spontaneous and induced fatal seizures showed intermittent bradycardia. This may indicate autonomic disturbances due to brainstem hyperexcitability, spreading depolarization, and/or direct effects of intermittent hypoxia on cardiac function and warrants further investigation.
In anaesthetized mice in the DW-MRI study fatal outcome occurred within minutes following stimulation, whereas in freely-behaving mice the delay showed high variation with particularly long delays in some animals. This discrepancy may be explained by consequences of anaesthesia, resulting in a reduced respiratory drive in anaesthetized animals (Tremoleda et al., 2012) and possible transient hypoxia that can affect seizure and spreading depolarization threshold (Wei et al., 2014). In freely-behaving mice, occurrence of cortical spreading depolarization following induced seizures will limit spread of seizure activity. With an expected lower cortical spreading depolarization susceptibility under anaesthesia (Kudo et al., 2013), electrical seizure induction during the DW-MRI experiments may have involved a more extensive seizure network including brainstem areas. Since brainstem spreading depolarization precedes cortical spreading depolarization during severe hypoxia (Richter et al., 2010), mild hypoxic conditions in anaesthetized mice may have increased spreading depolarization risk in brainstem areas. The short delay between spreading depolarizations in cortical and subcortical structures in the DW-MRI study would be in line with a contribution of local hyperexcitability, as recent evidence has shown that brainstem epileptiform activity can occur concurrently with fatal seizures (Salam et al., 2017). Future studies of fatal seizures in this and other SUDEP mouse models are required to confirm the path of spreading depolarization propagation from subcortical structures to the brainstem, as suggested by our DW-MRI studies in anaesthetized Cacna1aS218L mice.
We present Cacna1aS218L mice as a relevant and validated model for SUDEP since: (i) homozygous Cacna1aS218L mice displayed both non-fatal and fatal seizures, allowing for identification of SUDEP-related seizure dynamics of potential clinical value for anticipating and preventing SUDEP; and (ii) mice displayed fatal seizures at various ages reflecting clinical reports of SUDEP incidence rates (Massey et al., 2014; Harden et al., 2017). A fatal seizure outcome did not occur in heterozygous Cacna1aS218L or wild-type mice. Electrographically, cortical epileptiform activity was limited to incidental low-amplitude spikes during myoclonic behaviour. This contrasts the pronounced epileptiform activity seen in e.g. Kv1.1 and Nav1.1 SUDEP mouse models (Glasscock et al., 2010; Kalume et al., 2013). However, human semiological, EEG and ECoG data indicate heterogeneity of SUDEP phenomena that involve absence of epileptiform activity and include observations suggesting brainstem involvement (Surges et al., 2009; Massey et al., 2014; Devinsky et al., 2016). Multiple mechanisms may therefore contribute to SUDEP, and it is conceivable that mechanisms may differ between patients. Clinical reports of monitored SUDEP cases indicated that most fatal events are preceded by tonic-clonic seizures, while in a minority there is no preceding seizure (Ryvlin et al., 2013; Lhatoo et al., 2016; Devinsky et al., 2018). For those cases in which death was not preceded by epileptiform activity, a brainstem mechanism seems plausible (Lhatoo et al., 2016; Devinsky et al., 2018). Practical constraints limit electrography of brainstem areas in human epilepsy. Nevertheless, increased blood flow in the cerebellum, thalamus, and midbrain observed during and following seizure spread in patients with epilepsy supports brainstem involvement in tonic-clonic seizures (Blumenfeld et al., 2009). Clinical observations also support a link between SUDEP risk and brainstem involvement. Convulsive seizures with tonic arm extension, thought to coincide with spread of activity to the brainstem, were found to be strongly associated with PGES (Alexandre et al., 2015), which is associated with increased SUDEP risk.
Stage 5 seizures generally showed attenuation of the ECoG, with a relative dominance of theta band activity during fatal seizures. ECoG attenuation during seizures has also been reported for idiopathic generalized epilepsy during the tonic phase (Seneviratne et al., 2012). The seizure-related behaviour in Cacna1aS218L mice appears similar to that described for audiogenic seizures in genetic epilepsy-prone rats (GEPRs), in which ECoG attenuation was observed during tonic hindlimb extension (Naritoku et al., 1992; Jobe et al., 1995). Behavioural similarities include wild-running, jumping, tonic, and clonic behaviours, which for GEPR models have been linked to involvement of the brainstem reticular formation (Faingold, 2012). Brainstem networks are sufficient for the expression of running and tonic seizure components, whereas generalized clonic seizures require forebrain circuitry (Kreindler et al., 1958; Browning and Nelson, 1986). The observed behavioural and ECoG characteristics in Cacna1aS218L mice thereby support an important role for the brainstem in the seizure mechanism. We hypothesize that an intricate balance exists between epileptiform activity and spreading depolarization in Cacna1aS218L mice, as indicated by the frequent absence of afterdischarges following cortical stimulation, that may differ for different brain regions and induce seizure activity in brainstem networks while suppressing forebrain seizure activity. Fatal seizures in Cacna1aS218L mice specifically terminated with clonic behaviour. As brainstem seizure activity may suppress clonic seizure behaviour related to the forebrain (Merrill et al., 2005), the clonic behaviour during the second part of fatal seizures in Cacna1aS218L mice may result from suppression of brainstem networks by spreading depolarization. To the best of our knowledge, extensive semiological descriptions of fatal seizures in SUDEP cases that include detailed EEG analyses and timings of tonic and clonic activity are lacking. It therefore remains to be determined whether early cortical neuronal suppression, theta band activity and clonic activity are specific for spontaneous fatal seizures in the Cacna1aS218L model or represent a hallmark of SUDEP.
In conclusion, Cacna1aS218L mice appear a valid model to study SUDEP mechanisms. Behavioural, neurophysiological and imaging data revealed characteristics specific to fatal seizures, implicating a role for cortical neuronal suppression and brainstem spreading depolarization in SUDEP pathophysiology. This adds to the fundamental understanding of the processes that underlie seizure termination and risk for SUDEP, and may aid identification of predictive SUDEP biomarkers.
Supplementary Material
Acknowledgements
The authors thank Mr. Ludo Broos and Dr. Thijs Houben for assistance with histology and electrophysiology.
Glossary
Abbreviations
- DW-MRI
diffusion-weighted MRI
- ECoG
electrocorticography
- M1/S1
primary sensorimotor cortex
- MUA
multi-unit activity
- REM
rapid eye movement
- SUDEP
sudden unexpected death in epilepsy
- V1
primary visual cortex
Funding
Financial support was provided by the Dutch National Epilepsy Foundation (2017–10, R.D.T., A.M.J.M.v.d.M., E.A.T.), EU IAPP Program “BRAINPATH” (612360, A.M.J.M.v.d.M., E.A.T.); EU Marie Curie Career Integration Grant (294233, E.A.T.) and a CURE SUDEP research award (280560, R.D.T., A.M.J.M.v.d.M., E.A.T.). S.M.C. received support from a BC Epilepsy Society research award and the CURE - Taking Flight Award. T.P.S. is supported by an operating grant from the Canadian Institutes of Health Research (#10677), the Canada Research Chair in Biotechnology and Genomics-Neurobiology, and a Brain Canada Multi-Investigator Research Initiative Grant with matching support from Genome British Columbia, the Michael Smith Foundation for Health Research, and the Koerner Foundation.
Competing interests
The authors report no competing interests.
References
- Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med 2015; 7(282): 282ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiba I, Wehrens XH, Noebels JL. Leaky RyR2 channels unleash a brainstem spreading depolarization mechanism of sudden cardiac death. Proc Natl Acad Sci U S A 2016; 113(33): E4895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre V, Mercedes B, Valton L, Maillard L, Bartolomei F, Szurhaj W et al. Risk factors of postictal generalized EEG suppression in generalized convulsive seizures. Neurology 2015; 85(18): 1598–603. [DOI] [PubMed] [Google Scholar]
- Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI et al. Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol 2016; 79(4): 522–34. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, Varghese GI, Purcaro MJ, Motelow JE, Enev M, McNally KA et al. Cortical and subcortical networks in human secondarily generalized tonic-clonic seizures. Brain 2009; 132(Pt 4): 999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning RA, Nelson DK. Modification of electroshock and pentylenetetrazol seizure patterns in rats after precollicular transections. Exp Neurol 1986; 93(3): 546–56. [DOI] [PubMed] [Google Scholar]
- Cain SM, Bohnet B, LeDue J, Yung AC, Garcia E, Tyson JR et al. In vivo imaging reveals that pregabalin inhibits cortical spreading depression and propagation to subcortical brain structures. Proc Natl Acad Sci U S A 2017; 114(9): 2401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crespigny A, Rother J, van Bruggen N, Beaulieu C, Moseley ME. Magnetic resonance imaging assessment of cerebral hemodynamics during spreading depression in rats. J Cereb Blood Flow Metab 1998; 18(9): 1008–17. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Friedman D, Duckrow RB, Fountain NB, Gwinn RP, Leiphart JW et al. Sudden unexpected death in epilepsy in patients treated with brain-responsive neurostimulation. Epilepsia 2018; 59(3): 555–61. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 2016; 15(10): 1075–88. [DOI] [PubMed] [Google Scholar]
- Dlouhy BJ, Gehlbach BK, Kreple CJ, Kawasaki H, Oya H, Buzza C et al. Breathing Inhibited When Seizures Spread to the Amygdala and upon Amygdala Stimulation. J Neurosci 2015; 35(28): 10281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Dilekoz E, Kudo C, Savitz SI, Waeber C, Baum MJ et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest 2009; 119(1): 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Yuzawa I, Qin T, Wang Y, Baek K, Kim YR et al. Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J Neurosci 2011; 31(15): 5755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold CL. Brainstem Networks: Reticulo-Cortical Synchronization in Generalized Convulsive Seizures. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th edn. Bethesda (MD); 2012. [PubMed] [Google Scholar]
- Farrell JS, Gaxiola-Valdez I, Wolff MD, David LS, Dika HI, Geeraert BL et al. Postictal behavioural impairments are due to a severe prolonged hypoperfusion/hypoxia event that is COX-2 dependent. Elife 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci 2010; 30(15): 5167–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med 2009; 1(2): 2ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harden C, Tomson T, Gloss D, Buchhalter J, Cross JH, Donner E et al. Practice guideline summary: Sudden unexpected death in epilepsy incidence rates and risk factors: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2017; 88(17): 1674–80. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Tomson T, Benn E, Sander JW, Nilsson L, Langan Y et al. Combined analysis of risk factors for SUDEP. Epilepsia 2011; 52(6): 1150–9. [DOI] [PubMed] [Google Scholar]
- Houben T, Loonen IC, Baca SM, Schenke M, Meijer JH, Ferrari MD et al. Optogenetic induction of cortical spreading depression in anesthetized and freely behaving mice. J Cereb Blood Flow Metab 2017; 37(5): 1641–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobe PC, Mishra PK, Adams-Curtis LE, Deoskar VU, Ko KH, Browning RA et al. The genetically epilepsy-prone rat (GEPR). Ital J Neurol Sci 1995; 16(1–2): 91–9. [DOI] [PubMed] [Google Scholar]
- Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 2013; 123(4): 1798–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Bravo E, Thirnbeck CK, Smith-Mellecker LA, Kim SH, Gehlbach BK et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol 2001; 49(6): 753–60. [DOI] [PubMed] [Google Scholar]
- Kreindler A, Zuckermann E, Steriade M, Chimion D. Electro-clinical features of convulsions induced by stimulation of brain stem. J Neurophysiol 1958; 21(5): 430–6. [DOI] [PubMed] [Google Scholar]
- Kudo C, Toyama M, Boku A, Hanamoto H, Morimoto Y, Sugimura M et al. Anesthetic effects on susceptibility to cortical spreading depression. Neuropharmacology 2013; 67: 32–6. [DOI] [PubMed] [Google Scholar]
- Lamberts RJ, Thijs RD, Laffan A, Langan Y, Sander JW. Sudden unexpected death in epilepsy: people with nocturnal seizures may be at highest risk. Epilepsia 2012; 53(2): 253–7. [DOI] [PubMed] [Google Scholar]
- Lhatoo SD, Faulkner HJ, Dembny K, Trippick K, Johnson C, Bird JM. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol 2010; 68(6): 787–96. [DOI] [PubMed] [Google Scholar]
- Lhatoo SD, Nei M, Raghavan M, Sperling M, Zonjy B, Lacuey N et al. Nonseizure SUDEP: Sudden unexpected death in epilepsy without preceding epileptic seizures. Epilepsia 2016; 57(7): 1161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol 2014; 10(5): 271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill MA, Clough RW, Jobe PC, Browning RA. Brainstem seizure severity regulates forebrain seizure expression in the audiogenic kindling model. Epilepsia 2005; 46(9): 1380–8. [DOI] [PubMed] [Google Scholar]
- Moore BM, Jerry Jou C, Tatalovic M, Kaufman ES, Kline DD, Kunze DL. The Kv1.1 null mouse, a model of sudden unexpected death in epilepsy (SUDEP). Epilepsia 2014; 55(11): 1808–16. [DOI] [PubMed] [Google Scholar]
- Naritoku DK, Mecozzi LB, Aiello MT, Faingold CL. Repetition of audiogenic seizures in genetically epilepsy-prone rats induces cortical epileptiform activity and additional seizure behaviors. Exp Neurol 1992; 115(3): 317–24. [DOI] [PubMed] [Google Scholar]
- Nashef L, So EL, Ryvlin P, Tomson T. Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia 2012; 53(2): 227–33. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996; 87(3): 543–52. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 1972; 32(3): 281–94. [DOI] [PubMed] [Google Scholar]
- Richter F, Bauer R, Lehmenkuhler A, Schaible HG. The relationship between sudden severe hypoxia and ischemia-associated spreading depolarization in adult rat brainstem in vivo. Exp Neurol 2010; 224(1): 146–54. [DOI] [PubMed] [Google Scholar]
- Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A et al. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol 2013; 12(10): 966–77. [DOI] [PubMed] [Google Scholar]
- Salam MT, Montandon G, Genov R, Devinsky O, Del Campo M, Carlen PL. Mortality with brainstem seizures from focal 4-aminopyridine-induced recurrent hippocampal seizures. Epilepsia 2017; 58(9): 1637–44. [DOI] [PubMed] [Google Scholar]
- Seneviratne U, Cook M, D’Souza W. The electroencephalogram of idiopathic generalized epilepsy. Epilepsia 2012; 53(2): 234–48. [DOI] [PubMed] [Google Scholar]
- Shmuely S, Sisodiya SM, Gunning WB, Sander JW, Thijs RD. Mortality in Dravet syndrome: A review. Epilepsy Behav 2016; 64(Pt A): 69–74. [DOI] [PubMed] [Google Scholar]
- Stam AH, Luijckx GJ, Poll-The BT, Ginjaar IB, Frants RR, Haan J et al. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J Neurol Neurosurg Psychiatry 2009; 80(10): 1125–9. [DOI] [PubMed] [Google Scholar]
- Surges R, Thijs RD, Tan HL, Sander JW. Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms. Nat Rev Neurol 2009; 5(9): 492–504. [DOI] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M et al. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron 2009; 61(5): 762–73. [DOI] [PubMed] [Google Scholar]
- Tremoleda JL, Kerton A, Gsell W. Anaesthesia and physiological monitoring during in vivo imaging of laboratory rodents: considerations on experimental outcomes and animal welfare. EJNMMI Res 2012; 2(1): 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004; 41(5): 701–10. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE et al. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol 2010; 67(1): 85–98. [DOI] [PubMed] [Google Scholar]
- Vecchia D, Tottene A, van den Maagdenberg AM, Pietrobon D. Abnormal cortical synaptic transmission in CaV2.1 knockin mice with the S218L missense mutation which causes a severe familial hemiplegic migraine syndrome in humans. Front Cell Neurosci 2015; 9: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Ullah G, Schiff SJ. Unification of neuronal spikes, seizures, and spreading depression. J Neurosci 2014; 34(35): 11733–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.