

Abstract
Objective
To uncover the microRNA (miRNA) interactome of the osteoarthritis (OA) pathophysiological process in the cartilage.
Methods
We performed RNA sequencing in 130 samples (n=35 and n=30 pairs for messenger RNA (mRNA) and miRNA, respectively) on macroscopically preserved and lesioned OA cartilage from the same patient and performed differential expression (DE) analysis of miRNA and mRNAs. To build an OA-specific miRNA interactome, a prioritisation scheme was applied based on inverse Pearson’s correlations and inverse DE of miRNAs and mRNAs. Subsequently, these were filtered by those present in predicted (TargetScan/microT-CDS) and/or experimentally validated (miRTarBase/TarBase) public databases. Pathway enrichment analysis was applied to elucidate OA-related pathways likely mediated by miRNA regulatory mechanisms.
Results
We found 142 miRNAs and 2387 mRNAs to be differentially expressed between lesioned and preserved OA articular cartilage. After applying prioritisation towards likely miRNA-mRNA targets, a regulatory network of 62 miRNAs targeting 238 mRNAs was created. Subsequent pathway enrichment analysis of these mRNAs (or genes) elucidated that genes within the ‘nervous system development’ are likely mediated by miRNA regulatory mechanisms (familywise error=8.4×10−5). Herein NTF3 encodes neurotrophin-3, which controls survival and differentiation of neurons and which is closely related to the nerve growth factor.
Conclusions
By an integrated approach of miRNA and mRNA sequencing data of OA cartilage, an OA miRNA interactome and related pathways were elucidated. Our functional data demonstrated interacting levels at which miRNA affects expression of genes in the cartilage and exemplified the complexity of functionally validating a network of genes that may be targeted by multiple miRNAs.
Keywords: osteoarthritis, microRNAs, data integration, regulatory networks, mRNA
Key messages.
What is already known about this subject?
Dysfunctional microRNA–messenger RNA (miRNA–mRNA) interactions have been demonstrated to mark osteoarthritis (OA) pathophysiology.
Targeting dysfunctional miRNA–mRNA interactions by miR mimics or antagomirs fulfil an important therapeutic promise.
What does this study add?
A data integration framework to systematically identify miRNAs involved in OA pathophysiology.
A first comprehensive miRNA interactome of OA pathophysiology.
How might this impact on clinical practice or future developments?
The OA miRNA interactome provides a roadmap to pinpoint candidates for future miRNA-based therapies.
Introduction
Osteoarthritis (OA) is an age-related, disabling joint disease characterised by progressive heterogeneous changes in articular cartilage and subchondral bone. OA is the most common arthritic disease causing serious restrictions in major daily life activities, yet without an effective treatment.1 At the tissue level, it has been demonstrated that OA pathophysiology is marked by altered gene expression regulation.2–5 This may be triggered by dysfunctional adaptation processes of the bone and/or cartilage on inevitable challenges occurring during life, due to ageing,6 genetic make-up7 or mechanical (over)loading.8
A substantial number of mechanisms, commonly referred to as epigenetics, are known to dynamically regulate changes in gene expression, particularly relevant in postmitotic cells such as chondrocytes. Epigenetic variation includes DNA methylation at CpG sites, histone modifications, and expression of non-coding RNAs such as microRNAs (<22 base pairs; miRNA) and long non-coding RNAs (<200 base pairs).9–11 miRNAs are known to play an important role in post-translational regulation of gene expression via antisense binding to messenger RNA (mRNA), whereas their dysfunction has been demonstrated to mark many complex diseases including OA.12 Notably, targeting dysfunctional miRNA–mRNA interactions has emerged as an important therapeutic promise for preclinical development as exemplified by successfully applied miRNA mimics or anti-miRs in cancer.13
With respect to OA, an increasing number of studies report on differential expression (DE) of miRNAs with ongoing OA pathophysiology.12 14 Nevertheless, the majority of these studies report on candidate miRNA and mRNA interactions, such as miR-140 with ADAMTS5, MMP13 and IGFBP5.15 16 To explore the full miRNA interactome of OA pathophysiology and explore its full potential to dynamically regulate gene expression in articular cartilage, we performed genome-wide miRNA and mRNA sequencing in preserved and OA-affected articular cartilage samples. To prioritise the most likely genes sensitive to the OA process that are targeted by miRNAs, we applied a stepwise integrative approach to our partly overlapping miRNA and mRNA sequencing data sets of preserved and lesioned OA cartilage, integrated with data from publicly available databases such as those with target predictions as well as those with experimentally validated data.
Methods
Sample description
Preserved and lesioned cartilage samples from the same donor were obtained from the Research in Articular osteoArthritis Cartilage (RAAK) study consisting of patients with OA who underwent joint replacement surgery due to an end-stage disease.4 In the current study, cartilage samples of 63 patients were included (online supplementary table-S1).
annrheumdis-2018-213882supp001.xlsx (9.2KB, xlsx)
Small RNA and mRNA sequencing
Sequencing of high-quality miRNA and mRNA was performed on, respectively, the Illumina HiSeq 2500 and the HiSeq 2000/4000. A detailed description on alignment, mapping and normalisation is available in online supplementary materials.
annrheumdis-2018-213882supp002.docx (44.4KB, docx)
DE analysis
DE analysis was performed on paired samples of both data sets, that is, 30 paired samples (14 knees and 16 hips) for miRNA and 35 paired samples for mRNA (28 knees and 7 hips; figure 1). miRNA DE analysis was also performed while stratifying for joint (14 paired knees and 16 paired hips) (online supplementary materials). Benjamini-Hochberg multiple testing corrected-p values with a significance cut-off of 0.05 are reported as false discovery rate (FDR).
Figure 1.
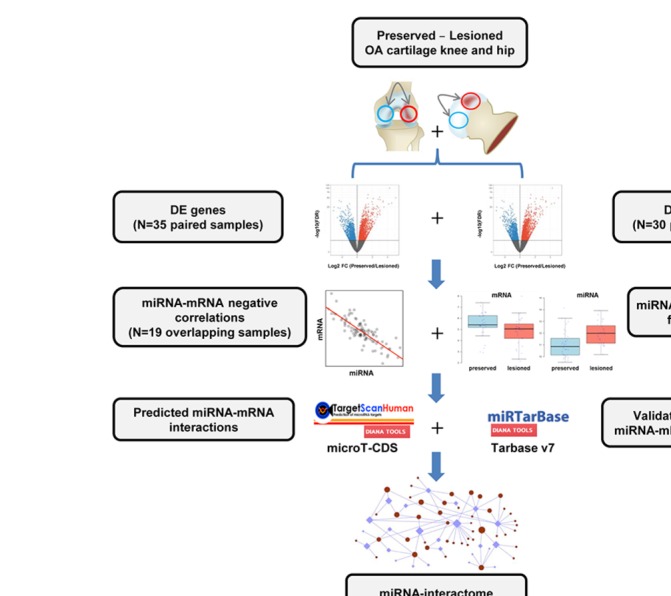
Data integration approach. Relative log normalised miRNA and mRNA expression matrices were concatenated. Next, differentially expressed miRNAs and genes were correlated and integrated according to the opposite direction of its FC. Further, miRNA–mRNA interactions from prediction tools and experimentally validated databases were integrated. Finally, target genes that followed these criteria were considered to build an OA miRNA–mRNA network. DE, differential expression; FC, fold change; miRNA, microRNA; mRNA, messenger RNA; OA, osteoarthritis.
Prioritisation of miRNA–mRNA targeting pairs
To generate an OA-specific miRNA interactome, we applied an integrated stepwise prioritisation approach (figure 1) to the identified set of DE miRNAs and mRNAs based on (1) significant negative Pearson’s correlation (|r|>0.5 and p<0.05) between miRNA and mRNA levels (19 overlapping samples, 15 patients); (2) opposite direction of fold change (FC) of mRNA and miRNA between the paired samples; (3) miRNA–mRNA target prediction from TargetScan17 and microT-CDS18; and (4) experimentally validated miRNA–mRNA target pairs downloaded from miRTarBase V.7.019 and TarBase V.7.20 Prioritised miRNA–mRNA target pairs were integrated into an miRNA–mRNA network based on their correlation and FCs (online supplementary materials).
Pathway enrichment
Pathway enrichment analysis was performed using the online tool DAVID21 while selecting Gene Ontology terms for biological processes (GOTERM_BP_DIRECT). Bonferroni multiple testing-corrected p values with a significance cut-off of 0.05 are reported as familywise error rate (FWER). Enrichment analyses of the DE genes with FC ≥2 were performed separately for further comparison. To specifically identify the miRNA regulated pathways in OA cartilage, enrichments of miRNA-target genes were performed using all significant DE genes (FDR<0.05) as background.
Functional validation
Primary chondrocytes isolated from three independent donors were transfected with antagomirs and miR mimics for miR-143–3 p, miR-329–3 p and miR-99a-3p using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s protocol. Reverse transcriptase-quantitative PCR (RT-qPCR) was performed adjusting for the housekeeping genes GAPDH and ARP (for further details see online supplementary materials).
Data availability
FASTQ files are available on ArrayExpress E-MTAB-7313. The code to reproduce the analysis is available on https://git.lumc.nl/rcoutinhodealmeida/miRNAmRNA.
Results
To identify the miRNA–mRNA regulatory landscape in OA cartilage, we performed a stepwise approach to integrate the miRNA (n=30 pairs) and mRNA (n=35 pairs) sequencing data sets of preserved and lesioned OA cartilage, samples containing n=19 overlapping samples (figure 1).
Differentially expressed miRNAs between lesioned and preserved OA cartilage
We found 142 DE miRNAs (FDR<0.05) between lesioned and preserved OA cartilage with absolute FCs ranging from 1.2 to 4.9 (figure 2A, online supplementary table-S2). The most significant DE miRNAs were miR-127–3 p (FC=0.5, FDR=1.1×10−6) and miR-451a (FC=2.3, FDR=1.2×10−6). As shown in figure 2A, the majority of DE miRNAs were upregulated in lesioned as compared with preserved OA cartilage (91 out of 142 miRNAs) with miR-206, recently reported in relation to OA,22 displaying the largest FC (FC=4.9, FDR=3.5×10−6). Conversely miR-504–5 p (FC=0.4, FDR=2×10−5) showed the largest fold decrease in lesioned OA cartilage (figure 2A, online supplementary table-S2). Next to miR-206, we found 40 other DE miRNAs that have consistently been associated with OA in functional follow-up studies, for example miR-140–5 p (FC=1.4, FDR=0.04), miR-143–3 p (FC=2.1, FDR=0.0001), miR-146a (FC=0.5, FDR=0.01) and miR-155–5 p (FC=1.8, FDR=0.005).16 23–25 Additionally, we identified 102 DE miRNAs not previously reported in OA, such as miR-95–3 p (FC=4.3, FDR=2.8×10−8), miR-3934–5 p (FC=1.9, FDR=1.8×10−6) and miR-99a-3p (FC=0.6, FDR=0.004). Moreover, several members of particular miRNA families are found to be differentially expressed, such as the miR-320 and let-7 family (online supplementary table-S2). DE expression was confirmed for four out of four miRNAs by applying RT-qPCR in independent paired preserved and lesioned OA cartilage samples (n=21): miR-127–3 p (FC=0.8, p=0.012), miR-451a (FC=2.3, p=0.024), miR-99a-3p (FC=0.8, p=0.0007) and miR143-3p (FC=1.8, p=0.07) (online supplementary figure-S1).
Figure 2.
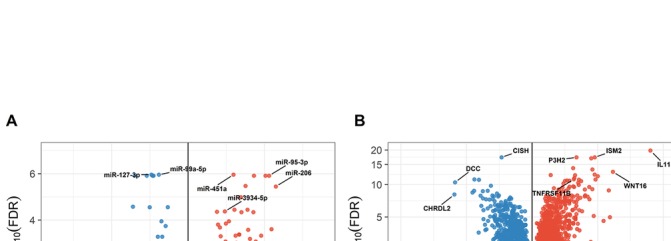
Paired differential expression analyses between preserved and lesioned OA cartilage. (A) Volcano plot with the differentially expressed miRNAs. (B) Volcano plot with the differentially expressed genes. Blue circles represent downregulated miRNAs (A) or genes (B); circles are red when they are upregulated. Labelled are the top differentially expressed genes and miRNAs, as well as the known and novel discovered ones. FC, fold change; FDR, false discovery rate; miRNA, microRNA; OA, osteoarthritis.
annrheumdis-2018-213882supp003.xlsx (20.4KB, xlsx)
annrheumdis-2018-213882supp004.tif (2.8MB, tif)
To explore whether we could detect joint site-specific miRNAs, stratified analyses for hip (n=16 pairs) and knee joints (n=15 pairs) were performed. Despite the relative equal number of samples, we found in the hip 117 (online supplementary table-S3) and in the knee 22 (online supplementary table-S4) significant DE miRNAs (FDR<0.05) between lesioned and preserved OA cartilage. Of these, 14 DE miRNAs were specific for hip cartilage (eg, miR-122–5 p: FC=5.14, FDR=6.6×10−5) and five for knee cartilage (online supplementary figure-S2). Notably miR-451a was the most significantly differentially expressed in the hip (FC=3.3, FDR=5.0×10−8) and total (FC=2.3, FDR=1.2×10−6) data set, yet not differentially expressed in the knee data set.
annrheumdis-2018-213882supp005.xlsx (17.2KB, xlsx)
annrheumdis-2018-213882supp006.xlsx (9.4KB, xlsx)
annrheumdis-2018-213882supp007.tif (111.6KB, tif)
Genes differentially expressed between lesioned and preserved OA cartilage
We identified 2387 DE mRNAs or genes (FDR<0.05) between lesioned and preserved OA cartilage (figure 2B, online supplementary table-S5). Of these, 1188 genes were downregulated in lesioned compared with preserved OA cartilage, and 1199 upregulated (online supplementary table-S5). As shown in figure 2B, the most significantly downregulated gene was CISH (FC=0.5, FDR=4.7×10−18), encoding the cytokine inducible SH2 containing protein which is known as an important suppressor of cytokine signalling through the JAK-STAT5 pathway. The most significantly upregulated gene was IL11 encoding interleukin-11, which also showed the largest FC in lesioned as compared with preserved OA cartilage (FC=22.7, FDR=1.5×10−20). The genes DCC (FC=0.1, FDR=3.3×10−11) and CHRDL2 (FC=0.1, FDR=7×10−9) showed the largest fold decrease in lesioned as compared with preserved OA cartilage (figure 2B, online supplementary table-S5). We found several previously reported OA-related genes, such as WNT16 (FC=8.4, FDR=1.1×10−13) and TNFRSF11B (FC=3.0, FDR=7.1×10−12),26 but also revealed new DE genes with OA, such as P3H2 (FC=3.2, FDR=4.7×10−18) and ISM2 (FC=5.2, FDR=4.7×10−18). As shown in table 1, enrichment analyses of the DE genes with FC≥2 (n=372) revealed significant enrichment (FWER <0.05) for pathways earlier reported in relation to OA pathophysiology (eg, ‘extracellular matrix organization’, ‘skeletal system development’, ‘cell adhesion’ and ‘positive regulation of gene expression’).
Table 1.
Pathways enrichment analysis of differentially expressed genes
| Term | P values | FWER | Fold enrichment | Genes |
| Extracellular matrix organisation | 1.82E-07 | 5.99E-05 | 5.20 | MATN4, POSTN, VIT, COL9A1, LAMB3, TNFRSF11B, FBLN1, COL19A1, COL7A1, FOXF1, TNR, SERPINE1, TGFBI, LAMC2, VCAN, SPP1, FN1 |
| Skeletal system development | 3.07E-06 | 1.01E-03 | 5.69 | BMP3, NOG, SOX11, POSTN, NPR3, FRZB, PAX1, TNFRSF11B, COL19A1, GDF10, VCAN, BMPR1B, BMP6 |
| Cell adhesion | 8.94E-05 | 2.93E-02 | 2.74 | AMTN, POSTN, AJAP1, CDH6, TNFAIP6, LAMB3, WISP2, COL19A1, COL7A1, LSAMP, TNR, MSLN, TGFBI, DSC3, RELN, LAMC2, VCAN, CHL1, SPP1, FN1, AOC3 |
| Positive regulation of gene expression | 1.30E-04 | 4.25E-02 | 3.43 | ODAM, WNT16, NOG, TESC, SOX11, IQGAP3, KIT, HMGA2, RIMS1, NTRK3, INHBA, FBLN1, ANK3, NGF, FN1 |
FWER, familywise error rate.
annrheumdis-2018-213882supp008.xlsx (212KB, xlsx)
OA-specific miRNA interactome
To generate an OA-specific miRNA interactome of the most likely miRNA–mRNA target pairs, the integrated stepwise prioritisation approach outlined in figure 1 was applied using 19 samples for which we had both miRNA and mRNA sequencing data. In online supplementary table-S6 we provided the 331 prioritised miRNA–mRNA target pairs, including their target predictions and/or experimental validations from respective databases. Prioritised miRNA–mRNA target pairs were integrated into an miRNA–mRNA network based on their correlation and FCs as such generating the OA-specific miRNA interactome (figure 3). The fact that 62 DE miRNAs were interacting with 238 DE target genes reflects that miRNAs are bound to target many genes in a complex structure. As shown in figure 3, the network consists of two large clusters of connected miRNA–mRNA pairs, one in which the miRNAs are downregulated (blue miRNA nodes) and another in which the miRNAs are upregulated (red miRNA nodes). Notably within the ‘downregulated miRNA cluster’ is the previously unknown OA-related miR-99a-3p that targets 36 DE genes, with 3 of them showing a strong correlation: FZD1 (r=−0.73, p=0.0001), ITGB5 (r=−0.70, p=0.00031) and GDF6 (r=−0.70, p=0.00039) (online supplementary table-S6). Furthermore, these three genes each correlates with at least one other targeting miRNA (figure 3). A notable example in the ‘upregulated miRNA cluster’ is the previously identified miR-143–3 p that targets 16 DE genes. Among these, at least three genes, DCAKD (r=−0.71, p=0.0002), AMIGO1 (r=−0.70, p=0.0003) and SMAD3 (r=−0.68, p=0.0008), show a strong correlation to miR-143–3 p, which additionally shares target genes with miR-10a-5p and miR-21–5 p, being TNS3, THRA and GID8. Of note in the ‘upregulated miRNA cluster’ is also the miRNA family miR-320, targeting the mRNA of 30 genes including MANF and CISD2, which are correlated with all miRNAs from this respective family. Besides these two large clusters, there are 15 small clusters, mostly with one miRNA targeting few mRNAs (figure 3). For example, miR-493–3 p forms a separate cluster with its 10 target DE genes. By applying RT-qPCR in n=21 independent preserved OA cartilage samples, we confirmed correlation between the miRNA–mRNA expression for miR-99a-3p with FZD1 (r=−0.54, p=0.02) and with GDF6 (r=−0.58, p=0.01) (online supplementary figure-S3).
Figure 3.

OA miRNA–mRNA interactome. Network of differentially expressed miRNAs targeting differentially expressed genes between unaffected (preserved) and lesioned OA cartilage. Diamonds are miRNAs and circles genes; edges denote that an miRNA targets the connected gene. The size of the nodes is proportional to the number of edges (interactions). Node colour characterises the strength and the direction of the expression change between unaffected (preserved) and lesioned OA cartilage. Edge thickness corresponds to Pearson’s correlation between the miRNA and gene across all samples. miRNA, microRNA; mRNA, messenger RNA; OA, osteoarthritis.
annrheumdis-2018-213882supp009.xlsx (51.7KB, xlsx)
annrheumdis-2018-213882supp010.tif (2.8MB, tif)
Functional validation of miRNA–mRNA target pairs
To study the downstream effects of highlighted miRNAs, we studied the effects of miR-143–3 p antagomir and mimic. This miRNA shows singular connections to the GHR, SMAD3, AMIGO1 and DCAKD genes (figure 4A). As shown in figure 4A, transfection with the miR-143–3 p mimic resulted in consistent downregulation of its singular target genes AMIGO1 (p=0.071), SMAD3 (p=0.032), GHR (p=0.115) and DCAKD (p=0.089). The antagomir of miR-143–3 p did not result in a clear response to the respective target genes. Furthermore, the effects of antagomirs and miR-mimics of miR-99a-3p and miR-329–3 p, both correlating to the WNT9A, FZD1 and GDF6 genes (figure 4B), were analysed. As shown in figure 4B, the miR-99a and miR-329–3 p mimics and antagomirs resulted in consistent changes in the WNT9A, FZD1 and GDF6 gene expression; however, the direction of effects was variable. Most notable is the inverse effect of the miR mimics on GDF6 expression (FC=1.8, p=0.036 and FC=0.6, p=0.059 for miR-329–3 p and miR-99a-3p, respectively). The strongest effect was observed for miR-329–3 p mimic on WNT9A expression (FC=0.25, p=0.036). To further explore these interactions, we correlated the expression levels of WNT9A, GDF6 and FZD1 on transfections with mock controls, antagomirs and mimics of miR-329–3 p and miR-99a-3p (online supplementary figure-S4). Notable is the reduced correlation between WNT9A and FZD1 particularly on transfection with, respectively, the miR-329–3 p mimic and the miR-99a-3p antagomir illustrating the different interacting levels at which expression of these genes is controlled by miR-99a-3p and miR-329–3 p.
Figure 4.
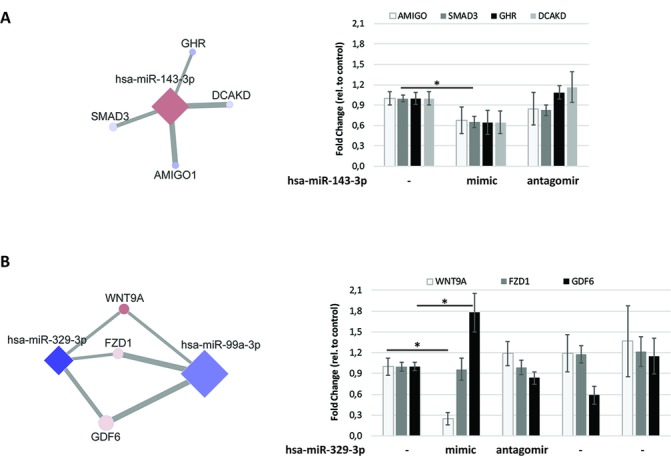
Functional validation miRNA–mRNA interactions. (A) Expression of AMIGO1, SMAD3, GHR and DCAKD in primary chondrocytes transfected with miR-143–3 p mimic or antagomir compared with control as determined by RT-qPCR. (B) Expression of WNT9A, FZD1 and GDF6 in primary chondrocytes transfected with miR-329–3 p or miR-99a-3p mimic or antagomir compared with control as determined by RT-qPCR. Data shown are the average±SE of the mean for three independent donors (*p<0.05). miRNA, microRNA; mRNA, messenger RNA; RT-qPCR, reverse transcriptase-quantitative PCR.
annrheumdis-2018-213882supp011.tif (59.4KB, tif)
miRNA-regulated gene pathways
To find specific miRNA-regulated gene pathways involved in OA pathophysiology, we analysed the 238 prioritised mRNAs likely targeted by DE miRNAs in OA cartilage for enrichment in biological processes using the 2387 DE genes as background. As shown in table 2, 10 pathways were significantly enriched, including ‘positive regulation of GTPase activity’ (FWER=9.8×10−6) and ‘nervous system development’ (FWER=8.4×10−5). Notable genes involved in the latter were NLGN1 (FC=0.61, FDR=0.014), which plays a role in synapse function, and NTF3 (FC=2.7, FDR=6.6×10−6), which controls survival and differentiation of neurons.
Table 2.
Pathway analysis of differentially expressed target genes
| Term | P values | FWER | Fold enrichment | Genes |
| Positive regulation of GTPase activity | 4.60E-08 | 9.80E-06 | 7.68 | SNX18, PDGFA, ARHGEF17, S100A10, IRS1, RAB3IP, ELMO1, THY1, FZD10, RGS4, RASA3, ST5, TBC1D20, SRGAP1 |
| Nervous system development | 3.93E-07 | 8.37E-05 | 7.07 | GLRB, NTF3, PCDHB4, NLGN1, NINJ1, EVL, SLC7A5, CSGALNACT1, BZW2, TPP1, DLG3, CRIM1, DCLK1 |
| Protein phosphorylation | 5.66E-06 | 1.20E-03 | 5.46 | RPS6KA5, CCNE1, CCND1, PKN3, WNK4, PHKG2, MKNK2, TESK2, PRKACA, PRKACB, BMPR1B, DCLK1, TRIB2 |
| IRE1-mediated unfolded protein response | 6.84E-06 | 1.46E-03 | 102.27 | TPP1, SRPRB, SEC61A1, ADD1 |
| Stimulatory C-type lectin receptor signalling pathway | 6.84E-06 | 1.46E-03 | 102.27 | RPS6KA5, PSMD1, PRKACA, PRKACB |
| Extracellular matrix organisation | 9.93E-06 | 2.11E-03 | 5.16 | PDGFA, LUM, ADAMTSL4, TNC, ITGB4, SPINT1, ITGB5, ITGA3, CSGALNACT1, COL9A1, ITGA5, COL27A1, TGFBI |
| Signal transduction | 1.96E-05 | 4.17E-03 | 3.06 | PTPRK, OSTF1, NTF3, CYTL1, MAPKAPK3, PDE3B, TNFSF13, CDS1, IRS1, SUFU, PLAUR, TMED4, MYD88, EPS8, PKN3, SMOC1, PDE8B, IGFBP1, PRKACB, RASA3, SRGAP1 |
| Tumour necrosis factor-mediated signalling pathway | 1.11E-04 | 2.36E-02 | 42.61 | TNFRSF12A, PSMD1, TNFRSF19, TNFSF13 |
| Activation of protein kinase A activity | 1.50E-04 | 3.19E-02 | 153.41 | PRKAR2B, PRKACA, PRKACB |
| Cellular response to BMP stimulus | 2.18E-04 | 4.65E-02 | 34.09 | SPINT1, TMEM100, BMPR1B, BMP6 |
*p<0.05.
BMP, bone morphogenic protein; FWER, familywise error rate.
Discussion
By integrating overlapping RNA sequencing of mRNA and miRNA in paired preserved and lesioned OA cartilage samples, we presented the first comprehensive, OA-specific, miRNA interactome. Hereto, we identified 142 miRNAs and 2387 genes with significant DE between lesioned and preserved cartilage. By a strict prioritisation scheme, we created a novel OA-associated miRNA interactome of 62 miRNAs and their 238 likely target mRNAs. Subsequent pathway analyses of the miRNA targeted genes showed significant enrichment for genes that act, among others, within ‘positive regulation of GTPase-activity’ and ‘nervous system development’. To allow biological interpretation of some of the highlighted clusters, functional validation experiments were performed with antagomirs and mimics. We observed that mimics of miR-143–3 p, with singular correlation to the GHR, SMAD3, AMIGO1 and DCAKD genes, had consistent inverse effects on gene expression. On the other hand, antagomirs and mimics of miR-99a-3p and miR-329–3 p, both paired with WNT9A, FZD1 and GDF6 genes (figure 4B), had consistent effects on gene expression but with variable directions. Together, our data suggest that interacting levels of miRNAs collectively affect gene expression in the cartilage, yet exemplifies the complexity of functional validation of miRNA–mRNA networks.
Among the enriched miRNA targeted genes in the nervous system development pathway were NLGN1 and NTF3. NLGN1 encodes neuroligin 1, which is a postsynaptic adhesion molecule involved in the regulation of glutamatergic transmission. More recently it was shown that NLGN1 is expressed during chondrogenesis and marks cellular identity of articular chondrocytes.27 NTF3 encodes neurotrophin-3, a member of the neurotrophin family that controls survival and differentiation of mammalian neurons. The protein is closely related to both nerve growth factor and brain-derived neurotrophic factor. In our data set we prioritised the NTF3 gene as a likely target of miR-502–3 p and involved in OA pathophysiology. This since NTF3 is highly significantly upregulated (FC=2.7, FDR=6.6×10−6) and miR-502–3 p is significantly downregulated (FC=0.8, FDR=0.04) in lesioned OA cartilage, the expression of NTF3 and miR-502–3 p was inversely correlated (r=−0.57, p=0.007), and they are a predicted mRNA–miRNA target pair (miTG score=0.473) (online supplementary table-S6). Targeting such a dysfunctional miRNA–mRNA interaction may represent a therapeutic promise for preclinical development, for example, by applying miRNA mimics of miR-502–3 p. As exemplified by our functional validation, the direct miRNA–mRNA target interaction should, however, be carefully assessed, for example, by luciferase assays. Moreover, we advocate that a systems medicines approach of antagomirs or miR mimics transfections followed by RNA sequencing is preferably taken to obtain a thorough understanding of all biological mechanisms involved. Finally, bioinformatics tools need to be developed that take into account, or take advantage of, the fact that the miRNA ‘seed’ sequence (nucleotides 2 and 7) can target the 3’UTR region of multiple mRNAs28 or may bind to other parts of the gene.29 Together, our miRNA interactome represents a comprehensive legacy to directly probe miRNAs of interest with their likely downstream signalling pathways, target predictions and/or experimental validations from respective databases. The fact that some of these tools use publication criteria as experimental validation of miRNA–mRNA target pairs should raise awareness that this could confound complex miRA–mRNA target interactions rather than illuminating them.
Within the interactome, miR-99a-3p, not previously associated with OA, targets the highest number of genes (n=36), with 20 of those genes targeted only by miR-99a-3p, while 16 genes were also targeted by other DE miRNAs. Of the latter, GDF6 is targeted by three other miRNAs (miR-329–3 p, miR-339–5 p, miR-532–3 p), with miR329-3p having the strongest inverse correlation (r=−0.67). GDF6 is a member of the transforming growth factor-beta super family whose members are essential for normal formation of the bones and joints in the limbs, skull and axial skeleton.30 Also notable is that GDF6 is an important paralogue of GDF5, the most consistently OA susceptibility gene found to date.31 Moreover, GDF5 has recently been identified as one of the key genes able to stratify two OA subgroups of knee articular cartilage based on expression levels.32
In our DE miRNA data set, we identified many of the previously reported, OA-associated, miRNAs such as miR-20622 and miR-140.15 16 However, in our miRNA interactome, these miRNAs did not necessarily correlate to their previously reported target genes. For example, miR-140–5 p in our miRNA interactome is only connected to RGS4 (figure 3) and not to ADAMTS5, MMP13 and IGFBP5, as reported previously by Tardif et al.15 16 To explore this further, we report in online supplementary table-S7 our miRNA and mRNA expression data of the most consistently reported miRNA–mRNA target pairs, for example, as reported in a recent review.33 As shown in online supplementary table-S7, miR-140–5 p has only very modest correlations to these previous reported target genes, likely reflecting their indirect effects. Moreover, some of the previously reported miRNAs are not among the ones presented in the miRNA interactome due to our strict prioritisation approach (figure 3). For example, miR-27a-3p is highly upregulated in lesioned OA cartilage (FC=1.8, FDR=5.0×10−4), but is not present in our miRNA interactome because it has significant positive correlation to MMP13 (r=0.5, p=3.6×10−2), and this gene does not show significant DE in preserved versus lesioned OA cartilage (FC=0.9, FDR=8.34×10−1).
annrheumdis-2018-213882supp012.xlsx (2.4MB, xlsx)
Another example of earlier reported miRNA associated to OA pathophysiology is miR-206. In a recent study, it was shown that increased expression of miR-206 significantly inhibited proliferation of chondrocytes while promoting expression of catabolic enzymes and apoptosis-inducing factors, suggesting that inhibition of miR-206 may control cartilage degradation in OA.22 In our data set miR-206 indeed exhibits a high and significant upregulation in lesioned compared with preserved OA cartilage (FC=4.9, FDR=2.6×10−6) and, based on the here identified interactions with CFH, IFIH1, NINJ1, GPM6A, L3MBTL4, COLEC12, PLCD3, ACSF2, CYTL1, RHNO1, FIBIN and C22orf39, we advocate that these genes may be involved in this process (figure 3). Taken together, the field of miRNA biology has demonstrated that miRNAs are bound to target multiple mRNAs in a network and, via dysregulation, causal to complex diseases,34 including OA.35 Moreover, targeting dysfunctional miRNA–mRNA interactions has emerged as an important therapeutic promise for preclinical development. As such, the here identified miRNA interactome of OA articular cartilage may represent a first important step to fulfil this promise. Nevertheless, our functional validation experiments highlighted that additional high-throughput (ie, high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP)) functional analyses towards systems medicines approaches are necessary to demonstrate direct binding of miRNAs to specific target genes and concurrent downstream changes in all mRNA expression levels.
Acknowledgments
We thank all study participants of the RAAK study. The Leiden University Medical Center has and is supporting the RAAK study. We thank Elwin Verheijen for mRNA and miRNA isolations.
Footnotes
RCdA and YFMR contributed equally.
Handling editor: Professor Josef S Smolen
Contributors: All authors have made substantial contributions to the completion of this study. Study concept and design: RCdA, YFMR, AM, MR, IM. Data analyses: RCdA, YFMR, AM, HM, SMK. Acquisition of material and data: RCdA, WdH, NL, MvH, EH, HEDS, ARR, PES, RGHHN, IM, YFMR. Preparation of the manuscript: RCdA, YFMR, AM, MR, IM. Critical reviewing and approval of the manuscript: all authors.
Funding: The study was funded by the Foundation for Research in Rheumatology (FOREUM), Dutch Arthritis Society (DAA_10_1-402), BBMRI-NL Complementation project (CP2013-83), Ana Fonds (O2015-27), and Dutch Scientific Research Council NWO /ZonMW VICI scheme (nr 91816631/528).
Competing interests: None declared.
Patient consent for publication: Obtained.
Ethics approval: Cohorts ethical approval for the RAAK study (Ramos et al, PlosOne 2014) was obtained from the medical ethics committee of the LUMC (P08.239).
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: All data from the study are available by public access within the text or online.
References
- 1. Woolf AD, Erwin J, March L. The need to address the burden of musculoskeletal conditions. Best Pract Res Clin Rheumatol 2012;26:183–224. 10.1016/j.berh.2012.03.005 [DOI] [PubMed] [Google Scholar]
- 2. Xu Y, Barter MJ, Swan DC, et al. . Identification of the pathogenic pathways in osteoarthritic hip cartilage: commonality and discord between hip and knee OA. Osteoarthritis Cartilage 2012;20:1029–38. 10.1016/j.joca.2012.05.006 [DOI] [PubMed] [Google Scholar]
- 3. den Hollander W, Ramos YF, Bomer N, et al. . Transcriptional associations of osteoarthritis-mediated loss of epigenetic control in articular cartilage. Arthritis Rheumatol 2015;67:2108–16. 10.1002/art.39162 [DOI] [PubMed] [Google Scholar]
- 4. Ramos YF, den Hollander W, Bovée JV, et al. . Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PLoS One 2014;9:e103056 10.1371/journal.pone.0103056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang L, Yang M, Marks P, et al. . Serum non-coding RNAs as biomarkers for osteoarthritis progression after ACL injury. Osteoarthritis Cartilage 2012;20:1631–7. 10.1016/j.joca.2012.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lotz MK, Caramés B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol 2011;7:579–87. 10.1038/nrrheum.2011.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meulenbelt I. Osteoarthritis year 2011 in review: genetics. Osteoarthritis Cartilage 2012;20:218–22. 10.1016/j.joca.2012.01.007 [DOI] [PubMed] [Google Scholar]
- 8. O'Conor CJ, Case N, Guilak F. Mechanical regulation of chondrogenesis. Stem Cell Res Ther 2013;4:61 10.1186/scrt211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003;33 Suppl:245–54. 10.1038/ng1089 [DOI] [PubMed] [Google Scholar]
- 10. Slieker RC, Bos SD, Goeman JJ, et al. . Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 2013;6:26 10.1186/1756-8935-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Laurent L, Wong E, Li G, et al. . Dynamic changes in the human methylome during differentiation. Genome Res 2010;20:320–31. 10.1101/gr.101907.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramos YF, Meulenbelt I. The role of epigenetics in osteoarthritis: current perspective. Curr Opin Rheumatol 2017;29:119–29. 10.1097/BOR.0000000000000355 [DOI] [PubMed] [Google Scholar]
- 13. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 2017;16:203–22. 10.1038/nrd.2016.246 [DOI] [PubMed] [Google Scholar]
- 14. Vicente R, Noël D, Pers YM, et al. . Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat Rev Rheumatol 2016;12:496 10.1038/nrrheum.2016.119 [DOI] [PubMed] [Google Scholar]
- 15. Tardif G, Pelletier JP, Fahmi H, et al. . NFAT3 and TGF-β/SMAD3 regulate the expression of miR-140 in osteoarthritis. Arthritis Res Ther 2013;15:R197 10.1186/ar4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tardif G, Hum D, Pelletier J-P, et al. . Regulation of the IGFBP-5 and MMP-13 genes by the microRNAs miR-140 and miR-27a in human osteoarthritic chondrocytes. BMC Musculoskelet Disord 2009;10:1–11. 10.1186/1471-2474-10-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agarwal V, Bell GW, Nam JW, et al. . Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015;4 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paraskevopoulou MD, Georgakilas G, Kostoulas N, et al. . DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res 2013;41:W169–W173. 10.1093/nar/gkt393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chou CH, Chang NW, Shrestha S, et al. . miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res 2016;44(D1):D239–D247. 10.1093/nar/gkv1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vlachos IS, Paraskevopoulou MD, Karagkouni D, et al. . DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res 2015;43:D153–D159. 10.1093/nar/gku1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiao X, Sherman BT, Huang daW, et al. . DAVID-WS: a stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012;28:1805–6. 10.1093/bioinformatics/bts251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ni Z, Shang X, Tang G, et al. . Expression of miR-206 in Human Knee Articular Chondrocytes and Effects of miR-206 on Proliferation and Apoptosis of Articular Chondrocytes. Am J Med Sci 2018;355:240–6. 10.1016/j.amjms.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 23. Matsukawa T, Sakai T, Yonezawa T, et al. . MicroRNA-125b regulates the expression of aggrecanase-1 (ADAMTS-4) in human osteoarthritic chondrocytes. Arthritis Res Ther 2013;15:R28–11. 10.1186/ar4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liang ZJ, Zhuang H, Wang GX, et al. . MiRNA-140 is a negative feedback regulator of MMP-13 in IL-1β-stimulated human articular chondrocyte C28/I2 cells. Inflamm Res 2012;61:503–9. 10.1007/s00011-012-0438-6 [DOI] [PubMed] [Google Scholar]
- 25. Haseeb A, Makki MS, Khan NM, et al. . Deep sequencing and analyses of miRNAs, isomiRs and miRNA induced silencing complex (miRISC)-associated miRNome in primary human chondrocytes. Sci Rep 2017;7:15178 10.1038/s41598-017-15388-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramos YF, Bos SD, van der Breggen R, et al. . A gain of function mutation in TNFRSF11B encoding osteoprotegerin causes osteoarthritis with chondrocalcinosis. Ann Rheum Dis 2015;74:1756–62. 10.1136/annrheumdis-2013-205149 [DOI] [PubMed] [Google Scholar]
- 27. Ferguson GB, Van Handel B, Bay M, et al. . Mapping molecular landmarks of human skeletal ontogeny and pluripotent stem cell-derived articular chondrocytes. Nat Commun 2018;9:3634 10.1038/s41467-018-05573-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brennecke J, Stark A, Russell RB, et al. . Principles of microRNA-target recognition. PLoS Biol 2005;3:e85 10.1371/journal.pbio.0030085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hausser J, Zavolan M. Identification and consequences of miRNA-target interactions--beyond repression of gene expression. Nat Rev Genet 2014;15:599–612. 10.1038/nrg3765 [DOI] [PubMed] [Google Scholar]
- 30. Settle SH, Rountree RB, Sinha A, et al. . Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol 2003;254:116–30. 10.1016/S0012-1606(02)00022-2 [DOI] [PubMed] [Google Scholar]
- 31. Reynard LN, Loughlin J. The genetics and functional analysis of primary osteoarthritis susceptibility. Expert Rev Mol Med 2013;15:e2 10.1017/erm.2013.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Soul J, Dunn SL, Anand S, et al. . Stratification of knee osteoarthritis: two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann Rheum Dis 2018;77:423 10.1136/annrheumdis-2017-212603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sondag GR, Haqqi TM. The Role of MicroRNAs and Their Targets in Osteoarthritis. Curr Rheumatol Rep 2016;18:56 10.1007/s11926-016-0604-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nat Rev Genet 2016;17:719–32. 10.1038/nrg.2016.134 [DOI] [PubMed] [Google Scholar]
- 35. Ntoumou E, Tzetis M, Braoudaki M, et al. . Serum microRNA array analysis identifies miR-140-3p, miR-33b-3p and miR-671-3p as potential osteoarthritis biomarkers involved in metabolic processes. Clin Epigenetics 2017;9:1–15. 10.1186/s13148-017-0428-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2018-213882supp001.xlsx (9.2KB, xlsx)
annrheumdis-2018-213882supp002.docx (44.4KB, docx)
annrheumdis-2018-213882supp003.xlsx (20.4KB, xlsx)
annrheumdis-2018-213882supp004.tif (2.8MB, tif)
annrheumdis-2018-213882supp005.xlsx (17.2KB, xlsx)
annrheumdis-2018-213882supp006.xlsx (9.4KB, xlsx)
annrheumdis-2018-213882supp007.tif (111.6KB, tif)
annrheumdis-2018-213882supp008.xlsx (212KB, xlsx)
annrheumdis-2018-213882supp009.xlsx (51.7KB, xlsx)
annrheumdis-2018-213882supp010.tif (2.8MB, tif)
annrheumdis-2018-213882supp011.tif (59.4KB, tif)
annrheumdis-2018-213882supp012.xlsx (2.4MB, xlsx)
Data Availability Statement
FASTQ files are available on ArrayExpress E-MTAB-7313. The code to reproduce the analysis is available on https://git.lumc.nl/rcoutinhodealmeida/miRNAmRNA.