

The cytosolic chaperonin T-complex protein 1-ring complex (TRiC) or chaperonin containing T-complex protein 1 (CCT) is essential in de novo folding of approximately 10% of the eukaryotic, newly translated polypeptides as well as misfolded proteins. There is a close link between the TRiC/CCT cytosolic chaperonin and neurodegenerative diseases (Lopez et al., 2015). A lot of research suggests that CCT plays neuroprotective roles in neurodegenerative diseases including Huntington’s disease (Lopez et al., 2015). Either overexpression of a single or all eight subunits (CCT1-8) or treatment of the substrate-binding apical domain of yeast CCT1 (ApiCCT1) prevented mutant Huntingtin aggregation and improved cellular and neuronal functions (Zhao et al., 2016). Importantly, our recent study has demonstrated that both CCT and ApiCCT could reduce mutant Huntingtin level and enhance both anterograde and retrograde axonal transport of brain-derived neurotrophic factor. These results led to restoration of the trophic status of striatal neurons from a bacterial artificial chromosome transgenic mouse model of Huntington’s disease (Zhao et al., 2016).
Axonal transport is regulated by many factors including microtubule-associated protein tau, which promotes tubulin polymerization and stabilizes microtubules. Impaired interaction between tau and microtubules plays a vital role in the pathogenesis of multiple neurodegenerative diseases (Wang and Mandelkow, 2016). Interestingly, tau phosphorylation is also observed in brains of several Huntington’s disease mouse models and Huntington’s disease patients (Gratuze et al., 2016). In a recent study, we explored if CCT subunit has any effect on axonal transport in a tau-dependent pathway (Chen et al., 2018b). We focused on the retrograde axonal transport of brain-derived neurotrophic factor, as neurotrophic factor-mediated signaling in the form of signaling endosome is essential in both the developing and the mature nervous system and dysregulation of trafficking of neurotrophic factors is tightly linked to disorders of the nervous system (Chen et al., 2018a). We found that the expression of a single CCT subunit (CCT5) significantly promoted retrograde axonal transport of brain-derived neurotrophic factor in primary cortical neurons. Mechanically, CCT regulated the level of cyclin-dependent kinase 5 (CDK5)/p35/p25 and, subsequently contributed to CCT-induced tau phosphorylation, which induced detachment of tau from microtubules (Chen et al., 2018b) (Figure 1).
Figure 1.
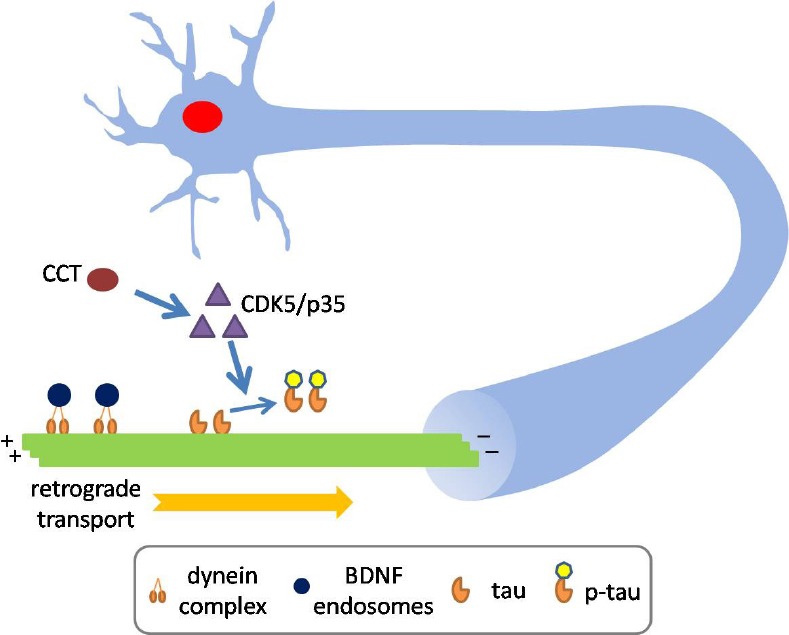
T-complex protein 1-ring complex (TRiC)/chaperonin containing T-complex protein 1 (CCT) subunit enhances retrograde axonal transport by modulating tau phosphorylation.
CCT enhances retrograde axonal transport of brain-derived neurotrophic factor (BDNF) in primary cortical neurons. The net effect on retrograde axonal transport of BDNF by CCT is the reduction in pauses with a concomitant increase in transport velocities. Importantly, the effect of CCT subunit on retrograde axonal transport of BDNF required tau expression. Mechanically, upregulation of CCT subunit increases the level of cyclin-dependent kinase 5 (CDK5)/p35/p25 in a tau-dependent manner, resulting in tau phosphorylation, which potentially induced tau detachment from microtubules.
CCT subunit modulates retrograde axonal transport in a tau-dependent manner: Lentivirus-mediated upregulation of CCT5 in primary cortical neurons resulted in an increase in instantaneous velocity (the absolute velocity calculated from moving time without pause time included) and a decrease in both the percentage of pause events and average pause duration of retrograde brain-derived neurotrophic factor axonal transport in microfluidic chamber. Consequently, the average retrograde velocity (the relative velocity calculated from moving time with pause time included) in CCT5-expressing neurons was significantly faster than observed in control neurons. Futhermore, CCT5 overexpression also increased the percentage of nonpause (motile) puncta. These results clearly indicate CCT5 overexpression promotes retrograde axonal brain-derived neurotrophic factor transport (Chen et al., 2018b). Importantly, by using the cortical neurons from tau–/– mice, in combination with lentivirus expressing wild-type tau (tauWT), we have proven that the effect on retrograde axonal transport by CCT subunit requires tau expression. Interestingly, reintroducing tauP301L, a pathogenic tau mutant, into tau–/– primary neurons could not rescue the effect of CCT5 on retrograde axonal transport, which highlights the differential regulation of tauWT and tauP301L by CCT subunit (Chen et al., 2018b).
Tau plays an important role in axonal transport of multiple cargoes including various vesicles and organelles. However, either overexpression or hyperphosphorylation of tau can also impair axonal transport. Mechanistically, tau as a microtubule-binding protein might compete with motor proteins for binding sites to microtubules and reduce the number of engaged motors per cargo for transport. As a cargo, tau also likely competes with other cargoes for availability of motor proteins; as a regulator, tau was also reported to regulate the release of cargo from kinesin involving protein phosphatase 1/glycogen synthase kinase 3β. Thus an overload of tau in axons may induce an adverse effect on axonal transport (Wang and Mandelkow, 2016). Consistently, hypophosphorylated as opposed to pseudophosphorylated tau displays an enhanced binding capacity to microtubules and induces pronounced axonal transport impairment in Drosophila (Talmat-Amar et al., 2011). Furthermore, hyperphosphorylation drives tau to detach from microtubules, possibly inducing destabilization of microtubules, which could also interrupt axonal transport (Wang and Mandelkow, 2016). Detached soluble and hyperphosphorylated tau tends to aggregate and potentially cause spatial clogging within axon(Wang and Mandelkow, 2016).
CCT subunit promotes CDK5 activity to enhance tau phosphorylation: Tau phosphorylation could modulate axonal transport (Wang and Mandelkow, 2016). In our study, CCT5 overexpression significantly increased the level of CDK5/p35/p25, contributing towards CCT-induced tau phosphorylation. Conversely, shRNA-mediated downregulation of CCT complex significantly lowered the level of CDK5/p35/p25 and ptau. It is noted that the CCT subunit exerted negligible effects on the phosphorylation of tauP301L. CCT5 overexpression had no apparent effect on either the activity or the level of glycogen synthase kinase 3β (Chen et al., 2018b), another important kinase involved in tau hyperphosphorylation in Alzheimer’s disease. Consistently, treatment with the CDK5 inhibitor roscovitine abolished the effect of CCT5 on retrograde axonal transport, pointing out that CDK5 is involved in CCT5-promoted retrograde axonal transport of brain-derived neurotrophic factor (Chen et al., 2018b).
Research has demonstrated that CDK5 could interact with dynein regulators including Ndel1/Lis1 to modulate retrograde axonal transport (Pandey and Smith, 2011). Although the CCT-induced increase of CDK5/p35 required tau expression (Chen et al., 2018b), the effect of CCT5 on the retrograde axonal transport of brain-derived neurotrophic factor may arise from either tau-dependent mechanism(s) or indirect mechanism(s) including CDK5-mediated Ndel1 phosphorylation or both. Our results argued against the involvement of glycogen synthase kinase 3β in CCT-induced phosphorylation of tau (Chen et al., 2018b). However, CCT complex modulates ~10% of the eukaryotic proteins, raising the possibility that other glycogen synthase kinase 3β effectors might be potentially affected and interact with the CDK5 pathway to enhance the CCT5-mediated beneficial effect on axonal transport. An important question is how the CCT subunit regulates the activity of CDK5 rather than glycogen synthase kinase 3β. CCT could potentially act at either transcription/translation or degradation step to impact protein level. Further study is needed to explore the underlying mechanism for the regulation of CDK5/p35 by CCT.
The roles of classical chaperone proteins such as heat shock proteins (heat shock protein 70, heat shock protein 90) in tau proteostasis have been extensively explored. Induction of heat shock protein 70 or induction of heat shock response through inhibition of heat shock protein 90 leads to ptau clearance through various mechanisms (Miyata et al., 2011). We found that CCT5 overexpression increased heat shock protein 70 levels in primary neurons (Chen et al., 2018b), however this could not explain the CCT5-induced increase in ptau, as heat shock protein 70 and its co-chaperone have been reported to facilitate the clearance of ptau (Miyata et al., 2011). However, heat shock protein proteins could cooperate with CCT complex in protein folding, thus whether heat shock protein is involved in CCT-mediated tau phosphorylation is yet to be elucidated.
Interestingly, other CCT subunits such as CCT3, CCT6 and CCT8 also increased the level of ptau, which points towards an effect on ptau by CCT that is not subunit specific. However, this effect only depends on the level of overexpressed individual subunits since upregulation of CCT5 did not affect the level of other CCT subunits (Chen et al., 2018b). Furthermore, almost all the transfected CCT5 subunits formed a complex in which tau was absent (Chen et al., 2018b). Thus, it is possible that the effect of CCT5 on tau phosphorylation requires an oligomeric complex that is either self-assembled by transfected CCT5 or assembled by CCT5 with other endogenous CCT subunits. Interestingly, our findings that CCT5 upregulation somehow prevents tau oligomerization despite an increase in ptau may explain why CCT5 upregulation promotes retrograde axonal transport.
CCT modulates tau binding to microtubules: Our study has shown that the increase in ptau by CCT attenuated binding of tau to microtubules and inhibited formation of tau-induced microtubule bundles (Chen et al., 2018b). It is conceivable that the reduction of tau binding to microtubules resulting from overexpression of CCT subunits makes microtubules more favorable for both motors and cargoes to move along. Indeed, this tau-dependent CCT modification of axonal transport was supported by tau–/– experiments; in which reintroducing tauWT rather than tauP301L, into tau–/– primary cortical neurons rescued the effect of CCT on axonal transport of brain-derived neurotrophic factor (Chen et al., 2018b). tauWT and tauP301L possess different biochemical, structural and conformational properties that potentially impact their aggregation and capacity to bind and stabilize microtubules. It is possible that these different properties of tauWT and tauP301L confer the differential effects of CCT on tauWT and tauP301L. On the one hand, CCT5 has negligible effect on tauP301L phosphorylation (Chen et al., 2018b). On the other hand, tauP301L displays weaker binding capacity to microtubules (Rodriguez-Martin et al., 2016). These factors together contribute towards the differential regulation of CCT on retrograde axonal transport of brain-derived neurotrophic factor in tauWT and tauP301L-expressing neurons.
Implication of CCT-promoted axonal transport in neurodegenerative diseases: In our previous study, both CCT3 and CCT5 rescued the axonal transport deficit in mutant Huntingtin-expressing neurons from an Huntington’s disease mouse model. These effects are primarily due to CCT-mediated clearance of mutant Huntingtin. In addition, ApiCCT delivery also rescued the axonal transport deficit in mutant Huntingtin-expressing neuron (Zhao et al., 2016). Since ApiCCT cannot regulate tau phosphorylation and the effect on retrograde axonal transport by CCT subunit in wild-type neurons relies on tau expression (Chen et al., 2018b), it is envisioned that ApiCCT might not affect axonal transport in wild-type neurons. Thus in comparison to Huntington’s disease neurons a different mechanism(s) is likely responsible for CCT5 to impact axonal transport via tau. Currently, we do not know if there is any link between the pathways involving CCT5/mutant Huntingtin and CCT5/tau in axonal transport in Huntington’s disease. Recent studies have demonstrated that mRNA levels of the TRiC/CCT complex were decreased in brain samples from Alzheimer’s disease patients in comparison with those from normal controls (Brehme et al., 2014). Interestingly, a previous study showed that in worms knockdown of the CCT subunit CCT1 attenuated the Aβ-induced toxicity (Khabirova et al., 2014). Thus, CCT might differentially act in Alzheimer’s disease and Huntington’s disease neurons. It is important to understand how decreased activity of CCT complex or individual CCT subunit impacts the proteostasis of tau, whose pathogenic species accumulates in Alzheimer’s disease. Although the physiological implications remain to be defined, our study has established that upregulation of CCT5 has an impact on tau-mediated functions such as axonal transport.
Conclusion and future perspective: Our present study has demonstrated that CCT5 expression affects tau phosphorylation and retrograde axonal transport of brain-derived neurotrophic factor (Figure 1) (Chen et al., 2018b). Although we show this effect on brain-derived neurotrophic factor, as tau is a universal regulator in axonal transport, it is likely that the microtubule-based transport of other cargoes is also affected. Further studies are needed to define this issue. A mouse model produced by pairing Huntington’s disease mouse and tau–/– mouse could help elucidate the contribution of tau in CCT5-mediated promotion of axonal transport in Huntington’s disease. Patients’ samples and Alzheimer’s disease mouse models might help figure out if any change of CCT subunit exists at protein level in Alzheimer’s disease and if so, whether these changes are linked to the pathology of Alzheimer’s disease including tau hyperphosphorylation.
I thank Prof. Chengbiao Wu (Department of Neurosciences, University of California San Diego, USA) and Mr. Christopher Carmona (Department of Bioengineering, University of California San Diego, USA) for helpful comments on the manuscript. Due to space limitations, the author regrets the omission of many important studies and their corresponding references.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by the author before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Yu J; T-Editor: Liu XL
References
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014;9:1135–1150. doi: 10.1016/j.celrep.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XQ, Sawa M, Mobley WC. Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic Biol Med. 2018a;114:52–61. doi: 10.1016/j.freeradbiomed.2017.10.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XQ, Fang F, Florio JB, Rockenstein E, Masliah E, Mobley WC, Rissman RA, Wu C. T-complex protein 1-ring complex enhances retrograde axonal transport by modulating tau phosphorylation. Traffic. 2018b;19:840–853. doi: 10.1111/tra.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratuze M, Cisbani G, Cicchetti F, Planel E. Is Huntington’s disease a tauopathy. Brain. 2016;139:1014–1025. doi: 10.1093/brain/aww021. [DOI] [PubMed] [Google Scholar]
- Khabirova E, Moloney A, Marciniak SJ, Williams J, Lomas DA, Oliver SG, Favrin G, Sattelle DB, Crowther DC. The TRiC/CCT chaperone is implicated in Alzheimer’s disease based on patient GWAS and an RNAi screen in Aβ-expressing Caenorhabditis elegans. PLoS One. 2014;9:e102985. doi: 10.1371/journal.pone.0102985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Dalton K, Frydman J. The mechanism and function of group II chaperonins. J Mol Biol. 2015;427:2919–2930. doi: 10.1016/j.jmb.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y, Koren J, Kiray J, Dickey CA, Gestwicki JE. Molecular chaperones and regulation of tau quality control: strategies for drug discovery in tauopathies. Future Med Chem. 2011;3:1523–1537. doi: 10.4155/fmc.11.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey JP, Smith DS. A Cdk5-dependent switch regulates Lis1/Ndel1/dynein-driven organelle transport in adult axons. J Neurosci. 2011;31:17207–17219. doi: 10.1523/JNEUROSCI.4108-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Martin T, Pooler AM, Lau DH, Morotz GM, De Vos KJ, Gilley J, Coleman MP, Hanger DP. Reduced number of axonal mitochondria and tau hypophosphorylation in mouse P301L tau knockin neurons. Neurobiol Dis. 2016;85:1–10. doi: 10.1016/j.nbd.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmat-Amar Y, Arribat Y, Redt-Clouet C, Feuillette S, Bouge AL, Lecourtois M, Parmentier ML. Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum Mol Genet. 2011;20:3738–3745. doi: 10.1093/hmg/ddr290. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- Zhao X, Chen XQ, Han E, Hu Y, Paik P, Ding Z, Overman J, Lau AL, Shahmoradian SH, Chiu W, Thompson LM, Wu C, Mobley WC. TRiC subunits enhance BDNF axonal transport and rescue striatal atrophy in Huntington’s disease. Proc Natl Acad Sci U S A. 2016;113:E5655–5664. doi: 10.1073/pnas.1603020113. [DOI] [PMC free article] [PubMed] [Google Scholar]