

Summary
Cas9 nucleases can be programmed with single guide RNAs (sgRNAs) to mediate gene editing. High CRISPR/Cas9-mediated gene knockout efficiencies are essential for genetic screens and critically depend on the properties of the sgRNAs used. The specificity of an sgRNA is defined by its targeting sequence. Here, we discovered that two short sequence motifs at the 3′ end of the targeting sequence are almost exclusively present in inefficient sgRNAs of published sgRNA-activity datasets. By specific knock-in of sgRNA target sequences with or without these motifs and quantitative measurement of knockout efficiency, we show that the presence of these motifs in sgRNAs per se results in a 10-fold reduction of gene knockout frequencies. Mechanistically, the cause of the low efficiency differs between the two motifs. These sequence motifs are relevant for future sgRNA design approaches and studies of Cas9-DNA interactions.
Keywords: CRISPR/Cas9, gene targeting, knockout efficiency, sgRNA design, sgRNA efficiency, sgRNA motif, scaffold RNA, CrispRGold, CRISPR screening
Graphical Abstract
Highlights
-
•
TT- and GCC-motifs are enriched in inefficient sgRNAs in published sgRNA-activity datasets
-
•
These sequence motifs are located in the 4 PAM-proximal bases of the targeting sequence
-
•
Their presence is sufficient to block CRISPR/Cas9-mediated gene editing
-
•
A modified scaffold RNA neutralizes the negative impact of the TT-motif
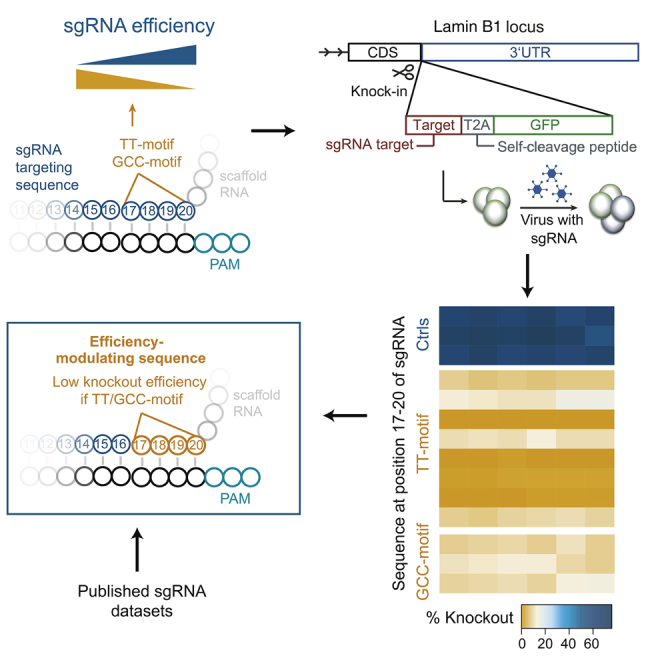
CRISPR/Cas9-mediated gene editing efficiency critically depends on the properties of the sgRNAs used. Graf et al. show that the presence of two position-specific sequence motifs in the targeting sequence of sgRNAs per se results in a severe reduction of gene knockout frequencies.
Introduction
The CRISPR/Cas9 technology is a powerful tool for genome editing (Cong et al., 2013, Jinek et al., 2012). Cas9 nucleases can be programmed with single guide RNAs (sgRNAs) to generate gene knockouts by inducing frameshift mutations in protein-coding genes. High CRISPR/Cas9-mediated knockout efficiencies are essential for genetic screens because of their impact on the signal-to-noise ratio, and they critically depend on the properties of the sgRNAs used. sgRNAs consist of a targeting sequence, defining their specificity, followed by the scaffold RNA at the 3′ end. Previous studies based on large-scale measurements of sgRNA activity have identified targeting sequence features that affect the on-target efficiency. Targeting sequences with a very high (≥80%) or low (≤35%) guanine-cytosine (GC) content overall were less effective. Furthermore, Cas9 has been shown to preferentially bind sgRNAs with purines in the four protospacer adjacent motif (PAM)-proximal bases of the targeting sequence, whereas pyrimidines and, especially, thymines (Ts) were disfavored (Wang et al., 2014). Along these lines, guanine (G) and cytosine (C) were found to be preferred and unfavorable, respectively, as the last base before the PAM (Doench et al., 2014). Overall, a bias against thymines at the 3′ end of the targeting sequence has been observed (Doench et al., 2014, Moreno-Mateos et al., 2015, Xu et al., 2015). While these features based on enrichments with respect to efficiency helped to improve sgRNA design criteria, they, by definition, do not allow researchrs to predict with certainty whether an sgRNA is inefficient.
Using sgRNAs designed with the sgRNA design tool CrispRGold and Cas9-transgenic mice, we achieved high homozygous knockout frequencies in primary B cells and other hematopoietic cells (Chu et al., 2016). Unexpectedly, one sgRNA reproducibly led to very low knockout frequencies. This sgRNA had a short T-rich sequence at its 3′ end that was not present in functional sgRNAs. Following up on this observation, we show here that this sequence belongs to one of two sequence motifs that are almost exclusively present in low-efficiency sgRNAs in published sgRNA-activity datasets. Using CRISPR-mediated specific knock-in of sgRNA target sequences followed by gene targeting, we demonstrate that the presence of these motifs results in drastically decreased knockout frequencies and that the mechanisms leading to these low efficiencies differ between the two motifs.
Results
In the course of developing efficient CRISPR/Cas9-mediated genetic screens in primary cells, we previously transduced wild-type and Cas9-transgenic activated B cells with retroviruses encoding sgRNAs targeting 12 surface markers, leading to an average knockout frequency of ∼80% (Chu et al., 2016). One sgRNA against CD22 (CD22-2) led to an extremely low knockout frequency of ∼10%, despite matching the stringent design criteria that otherwise defined efficient sgRNAs. This effect could not be explained by the positional nucleotide composition of the targeting sequence, as we had observed high knockout frequencies with all nucleotide types at every position of the targeting sequence (Figure S1A). Moreover, a second sgRNA targeting CD22 that was only 9 bps upstream of CD22-2 worked with high efficiency, indicating that the low knockout efficiency of CD22-2 was not a peculiarity of the genomic locus. We thus looked at more complex features of the CD22-2 sgRNA and identified a T-rich sequence at the 3′ end of the targeting sequence as unique in our sgRNA pool. By testing additional sgRNAs with such T-rich sequences at this position (targeting CD22 and B220), we discovered that all of these sgRNAs led to very low knockout frequencies (Figure 1A). These data suggested that specific T-rich sequences within the last four bases of the targeting sequence are sufficient to severely impede knockout efficiency. To confirm this observation with an independent dataset, we re-analyzed the surface marker knockout-based sgRNA-activity dataset reported by Doench et al. (2014). Consistent with our results, T-rich sequences were significantly overrepresented in the least active sgRNAs (percentage rank ≤25%) compared to the most active ones (percentage rank ≥75%) (Figure 1B). Based on these observations, we could refine the T-rich efficiency-modulating sequence at the 3′ end of the targeting sequence to consist of a TT-dinucleotide and at least one pyrimidine (TT + Y) or of four pyrimidines with at least two Ts (2T + 2Y), termed TT-motif hereafter. In addition, this motif analysis led to the discovery of four additional efficiency-modulating sequences at the same position that share a GCC core (DGCC and GCCT) (termed GCC-motif hereafter). In the dataset of Doench et al. (2014), these sequences were almost exclusively present in the least active sgRNAs (Figure 1B). sgRNAs containing the TT- or GCC-motif had a significantly lower overall knockout activity rank than the entire set of sgRNAs (Figure 1C).
Figure 1.

sgRNA Sequence Motifs Blocking Efficient CRISPR/Cas9-Mediated Gene Editing
(A) Flow cytometry plots showing the knockout frequencies (lower right gate) of CD22 (top) and B220 (bottom) in primary B cells isolated from Cas9-transgenic (GFP+) animals 4 days post-transduction with retroviruses encoding the indicated sgRNAs. The sequences in the four PAM-proximal bases of the targeting sequence (hereafter called efficiency-modulating sequence [EMS]) are shown in orange (T-rich) or blue (control); the PAM is shown in gray. Data are representative for two independent experiments.
(B) Number of sgRNAs with the indicated sequences in the EMS in the inefficient (last 25%) or efficient (top 25%) sgRNAs reported by Doench et al. (2014).
(C) Boxplots of the sgRNA activities reported by Doench et al. (2014), considering all of the sgRNAs (black) or the sgRNAs with the indicated motifs in the EMS (orange). The top, middle, and bottom lines of the boxplot represent the 25th, 50th, and 75th percentiles, respectively; the whiskers represent the max and min values. Subgroups were compared to the control set using ordinary one-way ANOVA (∗∗∗∗p < 0.0001).
(D) Scheme of validation experiment. sgRNA target sequences followed by T2A and GFP were targeted in-frame into the Lamin B1 locus of a mouse B cell tumor cell line using CRISPR/Cas9. GFP+ cells were subcloned, transduced using retroviruses encoding the respective sgRNAs, and cultured for 8 days before analysis.
(E) Example of GFP knockout measurement (GFP KO gate) by flow cytometry 8 days post-transduction with the indicated sgRNA, as in the experimental system shown in (D).
(F) Heatmap of the GFP knockout frequencies in the cell lines with the indicated sequences in the EMS 8 days post-transduction with the respective sgRNAs. Sequences matching the TT- and GCC-motifs are shown in orange.
(G) Knockout frequencies in Cas9-transgenic primary B cells 4 days post-transduction with sgRNAs, normalized to the higher knockout frequency of the two sgRNAs used. The sgRNAs matching the TT- or GCC-motifs are encircled, and the respective sequences indicated. Data are based on two independent experiments and adapted from Chu et al. (2016).
(H) Schematic diagram of the targeting sequence of the sgRNA (blue and orange) bound to the DNA (black). The four PAM-proximal nucleotides of the targeting sequence (orange) were called EMS due to their potential for modulating knockout frequencies. The predicted CRISPR/Cas9-mediated DNA cut is indicated.
To prove that these motifs are solely responsible for the low knockout efficiency, we removed all of the other variables (e.g., locus context) by targeting sgRNA target sequences into the Lamin B1 (Lmnb1) locus of a Cas9-expressing mouse tumor cell line and expressing the respective sgRNAs using retroviruses (Sander et al., 2012). To do so, we designed a unique sgRNA target sequence that is not present in the mouse genome and targeted 22 variants of this sequence (each variant was followed by a self-cleavage peptide and GFP) in front of the 3′ UTR of the Lmnb1 gene using CRISPR/Cas9 (Figure 1D). This system was functional and led to reproducible knockout frequencies (Figures 1E and S1B). Consistent with previous reports, purines in front of the PAM led to slightly increased knockout frequencies. In contrast, all of the sgRNAs with the GCC- and TT-motifs led to drastically (∼10-fold) decreased knockout frequencies (Figures 1F and S1C). We verified that the target sequences in GFP+ cells with a TT-motif (TTCA) were unmodified, showing that the respective sgRNA did not lead to gene editing (Figure S1D). Moreover, these efficiency-modulating motifs accounted for all of the low-efficiency sgRNAs in our previously published surface marker knockout experiment (Figure 1G) (Chu et al., 2016). These data show that the TT- and GCC-motif in the four PAM-proximal bases of the targeting sequence per se can render sgRNAs inefficient in CRISPR-mediated gene editing (Figure 1H). These sgRNA design restrictions were thus implemented in the CrispRGold sgRNA design tool (https://crisprgold.mdc-berlin.de).
Addressing the underlying mechanism of the low knockout efficiencies, we performed follow-up experiments using two control sgRNAs (Ctrl1/Ctrl2) and two sgRNAs per TT- and GCC-motif (TT1/TT2 and GC1/GC2, respectively) (Figure 2A). We first folded all of the sgRNAs in silico and did not find a structural feature that was peculiar to the motif sgRNAs (Figure S2A) (Lorenz et al., 2011). To find out whether the motifs directly impede Cas9-mediated DNA cleavage, we performed in vitro cleavage assays using ribonucleoprotein particles (RNPs) consisting of Cas9 and synthetic sgRNAs. All of the sgRNAs efficiently cleaved the target DNA in vitro, indicating that the motifs do not directly block Cas9-mediated DNA cleavage (Figures 2B and S2B). In addition, transfection of the synthetic sgRNAs into the respective cell lines rescued the knockout efficiency in the case of the TT-motif, but not the GCC-motif (Figure 2C). In these experiments, we also observed that the TT-motif sgRNAs were less efficient than the control sgRNAs at low doses (Figure S2C). These results suggested that the TT-motif sgRNAs were inefficient due to low doses when expressed from viral vectors. Indeed, the TT1/TT2 sgRNAs were transcribed less efficiently in vitro, leading to levels that were similar to the levels obtained with an sgRNA having an RNA polymerase III termination signal (five Ts) at the 3′ end of the targeting sequence (Figure 2D). Thus, T-rich sequences at the 3′ end of the targeting sequence led to reduced transcription, as it had been suggested before, likely due to the proximal tetra-T sequence at the 5′ end of the scaffold RNA (Doench et al., 2014). Inspired by previous reports on scaffold optimization, we designed variants of the scaffold RNA by mutating position five (T5A) or three and four (TT3AA) and the corresponding complementary nucleotides in the stem structure to interrupt the T-stretch (Figure 2E) (Chen et al., 2013, Dang et al., 2015, Hsu et al., 2013). The T5A scaffold fully restored the knockout activity of the TT-motif sgRNAs (Figure 2F). In contrast, the TT3AA scaffold only partially restored the knockout activity of these sgRNAs. Both mutated scaffold variants did not restore the knockout activity of GC1/GC2, further indicating that the low efficiency of these sgRNAs has other causes (Figure 2G).
Figure 2.

Mechanism of Motifs Blocking Efficient CRISPR/Cas9-Mediated Gene Editing
(A) Targeting sequences of the sgRNAs used to study mechanism of motifs. The four PAM-proximal bases are highlighted in blue and orange.
(B) In vitro cleavage assay using ribonucleoprotein particles (RNPs) with the indicated sgRNAs and amplified target sequences.
(C) Knockout frequencies in vivo 2 days post-electroporation with the indicated synthetic sgRNAs.
(D) sgRNAs produced by in vitro transcription of the indicated sgRNAs and a negative control sgRNA having five Ts at the 3′ end of the targeting sequence (5-T).
(E) Scheme of the 3′ end of the targeting sequence and the 5′ end of the scaffold RNA. The four Ts in the scaffold were mutated to the indicated variants (T5A and TT3AA).
(F) Knockout frequencies 8 days post-transduction, with sgRNAs consisting of the indicated targeting sequences and variants of scaffold RNAs.
(G) Heatmap of the knockout frequencies obtained with the mutated scaffolds as in (F) in three clones (Cl) per condition.
(H) In vitro cleavage assay using Ctrl1 and the Ctrl1 target site in the presence of increasing levels (0×, 0.5×, 1×, 2×, 4×, 8×, and 8×) of the indicated competing sgRNAs.
(I) Heatmap of the knockout frequencies 8 days post-electroporation, with increasing doses of the indicated synthetic sgRNAs.
(J) Quantification of target sites bound to Cas9. Cas9 was immunoprecipitated 16 h post-transfection with sgRNA-encoding plasmids. Data are representative for two independent experiments.
To test whether the GCC-motif sgRNAs are efficiently loaded into Cas9 or potentially have a higher off-rate, we performed Cas9-loading and in vitro cleavage competition assays. All sgRNAs were efficiently loaded into Cas9 in vitro (Figure S2D). In addition, in the sgRNA competition assay, increasing doses of GC1 and GC2 prevented the Ctrl1 sgRNA from cleaving its target in a dose-dependent manner, similar to TT2 (which served as control here), which indirectly indicated that GC1 and GC2 were efficiently loaded into Cas9 (Figure 2H). Of note, even when the GCC-motif sgRNAs were preloaded into Cas9 in vitro, they showed poor knockout activity in vivo when delivered as RNPs to the cell lines by electroporation (Figure S2E). These data suggested that the GCC-motif sgRNAs inefficiently recruit Cas9 to the target site in vivo. Increased doses of synthetic sgRNAs to the respective cell lines did not lead to increased knockout frequencies for the GCC-motif sgRNAs, as was the case for the control and TT-motif sgRNAs (Figure 2I). This was consistent with a decreased binding of the sgRNA/Cas9 complex to the respective target sites in the case of the GCC-motif, as measured by the immunoprecipitation of Cas9 and qPCR (Figure 2J). These data show that the TT-motif sgRNAs are ineffective because of the low expression when virally expressed, which can be overcome by alternative expression methods or mutated scaffold RNAs, whereas the GCC-motif sgRNAs show a general deficiency in accessing their target sites in vivo. As a consequence, the TT-motif sgRNAs should be avoided in polymerase III (Pol III)-based gene editing experiments requiring high sgRNA expression levels, especially in the context of genetic screens, where they can lead to false-negatives. In contrast, the GCC-motif sgRNAs appear to be inefficient irrespective of the delivery method and should thus be generally avoided.
Discussion
Given the strong impact of the two sequence motifs reported here on gene editing efficiency, these motifs are relevant for future sgRNA design approaches and studies of the Cas9-mediated gene editing mechanism. Of note, both motifs are located in the sgRNA “seed” region (the PAM-proximal 10–12 bases of the targeting sequence) that is important for pairing with the target DNA (Jinek et al., 2012). The bias against T-rich sequences at the end of the targeting sequence has previously been observed and explained as a possible consequence of RNA polymerase III termination at such T-rich sequence stretches, which would include the four Ts following the initial G of the scaffold RNA (Doench et al., 2014). An alternative explanation of the TT-motif is the reported negative impact of Ts among the last four bases at the 3′ end of the targeting sequence on sgRNA loading into Cas9 (Wang et al., 2014). We show here that the low efficiency of the TT-motif sgRNAs is indeed related to their reduced expression level, amplifying the decreased efficiency at low dosage potentially caused by a reduced loading into Cas9. The strong reduction in transcription caused by the TT-motif in vitro was unexpected, as efficient halting and release of RNA by RNA polymerase III is thought to require at least five Ts in a row (Arimbasseri and Maraia, 2015). However, the fact that bacteria did not change this scaffold feature through evolution is not surprising, given the differences in RNA polymerases between bacteria and eukaryotes and the fact that the targeting sequence and the scaffold RNA are transcribed from different loci in bacteria (Jinek et al., 2012). Halting and release of RNA polymerase III is context dependent. In fact, even differences in promoters have been shown to affect RNA polymerase III termination efficiencies on T-stretches (Gao et al., 2018). We show that a mutated version of the scaffold (T5A) may restore the knockout activity of the TT-motif sgRNAs. This scaffold has previously been shown to improve knockout efficiency in a motif-unrelated context (Chen et al., 2013, Dang et al., 2015, Hsu et al., 2013). Thus, the T5A scaffold RNA is an interesting candidate to replace the standard scaffold RNA in virus-based CRISPR screens.
The case of the low efficiency of the GCC-motif sgRNAs is more complex. The potential underlying mechanisms range from inefficient loading over non-specific binding to off-targets to co-factor-dependent mechanistic problems. Our results suggest that these sgRNAs are either inactivated in vivo (e.g., by RNA-binding proteins) or incompetent to correctly scan and bind the target site (e.g., due to enhanced unspecific binding to off-targets or interference of the GC-rich sequence with proper PAM recognition). High-throughput experiments will be needed to investigate these possibilities.
While the present study shows a consistent negative impact of the motifs on knockout efficiency in human and mouse cell lines as well as primary mouse B cells, cell type-specific variations may exist. Furthermore, while our study did not include experiments involving dCas9, the in vitro data suggest that the motifs may also reduce target gene regulation in CRISPRi (interference) and CRISPRa (activation) experiments, as they do not interfere at the level of DNA cutting (Gilbert et al., 2013, Qi et al., 2013).
Although significant progress has been made in sgRNA design during the last few years, our data uncover definable sgRNA features that on their own block knockout efficiency (Doench et al., 2016). Additional such features may exist that per se modulate knockout efficiency. A further de-convolution of overall sgRNA efficiency may thus lead to an improved understanding of sgRNA activity.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC-B220 (Clone RA3-6B2) | BioLegend | Cat#103212; RRID:AB_312997 |
| BV786-CD19 (Clone 6D5) | BioLegend | Cat#115543; RRID:AB_11218994 |
| PE-CD22 (Clone OX-97) | BioLegend | Cat#126112; RRID:AB_2561632 |
| anti-flag M2 antibody | Sigma | Cat#F1804; RRID:AB_262044 |
| Bacterial and Virus Strains | ||
| DH5α | Thermofisher | Cat#18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| aCD40 | BioLegend | Cat#102802 |
| IL-4 | Peprotech | Cat#214-14 |
| IL-21 | Peprotech | Cat#210-21 |
| Cas9 | Homemade | N/A |
| Puromycin | Sigma | Cat#P8833 |
| Critical Commercial Assays | ||
| CD43 (Ly-48) MicroBeads, mouse | Miltenyi Biotec | Cat#130-049-801 |
| FugeneHD transfection reagents | Promega | Cat#E2312 |
| MEGAscript T7 transcription Kit | Thermo Fisher Scientific | Cat#AM1354 |
| Alt-R® CRISPR-Cas9 sgRNA | IDT | N/A |
| Alt-R® CRISPR-Cas9 tracrRNA | IDT | N/A |
| SYBR Green PCR Master mix | Applied Biosystems | Cat#4309155 |
| Zero Blunt TOPO PCR Cloning Kit | Invitrogen | Cat#450245 |
| Experimental Models: Cell Lines | ||
| Plat-E packaging cells | Cell Biolabs | RV-101 |
| 40LB feeder cells | Nojima et al., 2011 | N/A |
| Murine Burkitt lymphoma cell line | Sander et al., 2012 | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 | Taconic | B6NTac |
| R26-Cas9iGFP/+ | Chu et al., 2016 | N/A |
| Oligonucleotides | ||
| T5A-gRNA scaffold sequence: GTTTAAGAGCTAGAAATAGCAAGTTTAAATAAG GCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC |
This paper | N/A |
| TT3AA-gRNA scaffold sequence: GTAATAGAGCTAGAAATAGCAAGTTATTATA AGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGC |
This paper | N/A |
| ChiP-qPCR forward primer: AATCTTAACTGTTTACAGGCCTAGGTCAGCT | This paper | N/A |
| ChiP-qPCR reverse primer: CTCCACGTCACCGCATGTT | This paper | N/A |
| Knock-in genotyping forward primer: AATCTTAACTGTTTACAGGCCTAGGTC AGCT |
This paper | N/A |
| Knock-in genotyping reverse primer: TTACTTGTACAGCTCGTCCATGCC | This paper | N/A |
| In vitro T7 transcription template forward primer: TTAATACGACTCACTATAGG (+sgRNA) | This paper | N/A |
| In vitro T7 transcription template reverse primer: AAAAGCACCGACTCGGTGCC | This paper | N/A |
| Recombinant DNA | ||
| MSCV_hU6_CcdB_PGK_Puro_T2A_BFP | Chu et al., 2016 | N/A |
| pTV_Lmnb1_BsmBI_T2A_GFP | This paper | N/A |
| Software and Algorithms | ||
| Prism 7.0a | GraphPad | https://www.graphpad.com/ |
| FlowJo v10 | LLC | https://www.flowjo.com/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Klaus Rajewsky (Klaus.Rajewsky@mdc-berlin.de).
Experimental Model and Subject Details
Mice
C57BL/6 mice were purchased from Taconic. R26-Cas9iGFP mice were generated by CRISPR/Cas9 and zygote injection (Chu et al., 2016). R26-Cas9iGFP/+ were generated by crossing R26-Cas9iGFP to C57BL/6 mice. All mice were kept in specific pathogen-free facilities. All mice used were male and ∼24 weeks old. All animal care and procedures were approved by the Institutional Animal Care and the LaGeSo Berlin.
Cell lines
The Burkitt lymphoma cell line (male) was generated before (Sander et al., 2012). Stable Cas9 expression was achieved by retroviral transduction and subcloning to obtain homogeneous Cas9 expression. Cells were cultured in B cell medium (DMEM high glucose, 10% FCS (GIBCO), L-Glutamine (2 mM, GIBCO), HEPES (2 mM, GIBCO), Non-essential amino acids (1x, GIBCO), sodium pyruvate (2 mM, GIBCO), Penicillin/Streptomycin (100 U/ml, GIBCO), β-mercaptoethanol (52 μM, Sigma)).
Primary cells
Primary B cells were isolated from spleens of adult C57BL/6 and R26-Cas9iGFP/+ mice by CD43-depletion (Miltenyi) according to the manufacturer’s protocol. Cells were cultured in B cell medium.
Method Details
Knockout in primary B cells
Knockout experiments in primary B cells were performed as described before (Chu et al., 2016). Briefly, primary B cells from C57BL/6 and R26-Cas9iGFP/+ mice were mixed at a 1:4 ratio. These cells were then activated with anti-CD40 (2 μg/ml, BioLegend) and IL-4 (20 ng/ml, Peprotech) for two days, spin-transduced with retroviruses encoding the sgRNAs and cultured for four days on 40LB feeder cells with IL-21 (20 ng/ml, Peprotech) until analysis.
Motif analysis
sgRNA sequences were extracted from Doench et al. (Doench et al., 2014) and Chu et al. (Chu et al., 2016) and the sequence variants in the last four bases of the targeting sequence were counted.
Validation of low-efficiency motifs in B cell tumor cell line
The constructs containing the sgRNA target sites, T2A and GFP were targeted in-frame into the endogenous Lmnb1 locus (in front of the 3′ UTR) of a mouse Burkitt lymphoma cell line (Sander et al., 2012) by homologous directed repair and CRISPR/Cas9 (using GTCTTGACAAGTTCACATAA as sgRNA target in Lmnb1 and 1.4kb up-stream and 2kb downstream targeting arms). The GFP positive cells were sub-cloned by FACS single cell sorting (BD AriaII/III). Correct targeting of each clone was assessed by PCR and Sanger sequencing. sgRNA targeting sequences were cloned into the BbsI sites of the MSCV_U6_CcdB_PGK_Puro_T2A_BFP vector as described before (Chu et al., 2016). Retroviruses were produced in Plat-E cells. Cell line sub-clones were transduced using spin-transduction in 96-well plates, selected with 1.25 μg/mL puromycin (Sigma) and acquired using BD Fortessa with 96-well HTS unit eight days later.
Sequence analysis
GFP+ and GFP- cells were sorted from heterozygous clones (in parallel to the analysis by FACS) using BD AriaII/III and directly lysed. PCR amplicons were sub-cloned using TOPO cloning and sequenced using Sanger sequencing.
Sequence folding
The secondary structures of the sgRNAs were predicted with the RNAfold tool (Vienna Package, Lorenz et al.). The secondary structures shown in the figures were adapted from these predictions.
In vitro cleavage assay
The substrate DNA (target site) was amplified by PCR and incubated for 1h at 37°C with the synthetic sgRNA (IDT) and Cas9 (homemade). The reaction was stopped using 1 μl of stop solution (30% glycerol, 1.2% SDS, 250 mM EDTA) at 37°C for 15 min. The cleaved products were separated by gel electrophoresis.
In vivo knockout using synthetic sgRNAs
The indicated amounts of sgRNAs (1x corresponding to 50 pmol) were electroporated into the cell lines using a 4D-Nucleofector (Lonza). To generate RNPs, 600 ng Cas9 proteins (homemade) and 100 pmol of synthetic sgRNAs were incubated at room temperature for 15 min prior to electroporation.
In vitro transcription
In vitro transcription was performed using the MEGAscript T7 Transcription Kit (Applied Biosystems) according to the manufacturer’s protocol.
ChIP and qPCR on target site
The plasmids encoding the sgRNAs were transfected into the cell lines. 14h post transfection, the cells were fixed and used for IP using an anti-flag M2 antibody (Sigma). The target site was amplified using the SYBR Green PCR Master mix (Applied Biosystems) according to the manufacturer’s protocol. The target site Cts were normalized to genomic Actb measurements.
Quantification and Statistical Analysis
P values were computed with Prism (version 7.0a) using ANOVA (with Bonferroni correction for multiple testing and assuming Gaussian distributions). Four asterisks indicate a p value below 0.0001. P values were confirmed using Mann-Whitney and Kolmogorov-Smirnov tests. In the case of primary cells, one mouse per genotype was used per biological replicate. Knockout frequencies were quantified using flow cytometry and FlowJo (version 10, LLC) by gating on GFP- cells. The number of biological replicates for each type of experiment is outlined in the figure legends or shown as data points in figures.
Acknowledgments
This work was supported by the European Research Council (ERC Advanced Grant 268921 to K.R.). We thank H.P. Rahn for excellent fluorescence-activated cell sorting (FACS)-related support.
Author Contributions
R.G. designed and analyzed the experiments and, with the help of K.R., wrote the manuscript. X.L. performed the experiments with the help of V.T.C. and developed the scaffold-rescue experiments. R.G. supervised the project.
Declaration of Interests
The authors declare no competing interests.
Published: January 29, 2019
Footnotes
Supplemental Information includes two figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.024.
Supplemental Information
References
- Arimbasseri A.G., Maraia R.J. Mechanism of Transcription Termination by RNA Polymerase III Utilizes a Non-template Strand Sequence-Specific Signal Element. Mol. Cell. 2015;58:1124–1132. doi: 10.1016/j.molcel.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.-W., Park J., Blackburn E.H., Weissman J.S., Qi L.S., Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V.T., Graf R., Wirtz T., Weber T., Favret J., Li X., Petsch K., Tran N.T., Sieweke M.H., Berek C. Efficient CRISPR-mediated mutagenesis in primary immune cells using CrispRGold and a C57BL/6 Cas9 transgenic mouse line. Proc. Natl. Acad. Sci. USA. 2016;113:12514–12519. doi: 10.1073/pnas.1613884113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y., Jia G., Choi J., Ma H., Anaya E., Ye C., Shankar P., Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015;16:280. doi: 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Hartenian E., Graham D.B., Tothova Z., Hegde M., Smith I., Sullender M., Ebert B.L., Xavier R.J., Root D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z., Herrera-Carrillo E., Berkhout B. Delineation of the Exact Transcription Termination Signal for Type 3 Polymerase III. Mol. Ther. Nucleic Acids. 2018;10:36–44. doi: 10.1016/j.omtn.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.-D., Fernandez J.P., Mis E.K., Khokha M.K., Giraldez A.J. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T., Haniuda K., Moutai T., Matsudaira M., Mizokawa S., Shiratori I., Azuma T., Kitamura D. In-vitro derived germinal centre B cells differentially generate memory B or plasma cells in vivo. Nat. Commun. 2011;2:465. doi: 10.1038/ncomms1475. [DOI] [PubMed] [Google Scholar]
- Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander S., Calado D.P., Srinivasan L., Köchert K., Zhang B., Rosolowski M., Rodig S.J., Holzmann K., Stilgenbauer S., Siebert R. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22:167–179. doi: 10.1016/j.ccr.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Xiao T., Chen C.-H., Li W., Meyer C.A., Wu Q., Wu D., Cong L., Zhang F., Liu J.S. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015;25:1147–1157. doi: 10.1101/gr.191452.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.