

Abstract
Aims
In hypertrophy and heart failure, the proarrhythmic persistent Na+ current (INaL) is enhanced. We aimed to investigate the electrophysiological role of neuronal sodium channel NaV1.8 in human hypertrophied myocardium.
Methods and results
Myocardial tissue of 24 patients suffering from symptomatic severe aortic stenosis and concomitant significant afterload‐induced hypertrophy with preserved ejection fraction was used and compared with 12 healthy controls. We performed quantitative real‐time PCR and western blot and detected a significant up‐regulation of NaV1.8 mRNA (2.34‐fold) and protein expression (1.96‐fold) in human hypertrophied myocardium compared with healthy hearts. Interestingly, NaV1.5 protein expression was significantly reduced in parallel (0.60‐fold). Using whole‐cell patch‐clamp technique, we found that the prominent INaL was significantly reduced after addition of novel NaV1.8‐specific blockers either A‐803467 (30 nM) or PF‐01247324 (1 μM) in human hypertrophic cardiomyocytes. This clearly demonstrates the relevant contribution of NaV1.8 to this proarrhythmic current. We observed a significant action potential duration shortening and performed confocal microscopy, demonstrating a 50% decrease in proarrhythmic diastolic sarcoplasmic reticulum (SR)‐Ca2+ leak and SR‐Ca2+ spark frequency after exposure to both NaV1.8 inhibitors.
Conclusions
We show for the first time that the neuronal sodium channel NaV1.8 is up‐regulated on mRNA and protein level in the human hypertrophied myocardium. Furthermore, inhibition of NaV1.8 reduced augmented INaL, abbreviated the action potential duration, and decreased the SR‐Ca2+ leak. The findings of our study suggest that NaV1.8 could be a promising antiarrhythmic therapeutic target and merits further investigation.
Keywords: Left ventricular hypertrophy, Sodium channels, Late sodium current, HFpEF, Arrhythmias, Calcium, SR‐Ca2+ leak
Introduction
Left ventricular remodelling caused by pressure overload can lead to myocardial hypertrophy. Left ventricular hypertrophy (LVH) is an adaptive response to increased chronic workload and a very common clinical finding.1, 2 It has been reported that LVH can potentially increase the incidents of congestive heart failure (HF) and sudden cardiac death.3, 4 Furthermore, it has been shown that structural modifications due to LVH are associated with HF with preserved ejection fraction (HFpEF).5 LVH is also associated with an increased prevalence of cardiac arrhythmias and constitutes an important risk factor for cardiac morbidity and mortality.6, 7, 8, 9 The pathology of LVH shows not only mechanical but also extensive cellular and molecular remodelling including cardiomyocyte growth changes, dysfunction of excitation–contraction coupling, certain metabolic dysfunctions, and fibrosis.10, 11
Patients with severe aortic stenosis‐dependent symptoms have a tremendous risk of sudden cardiac death if only medically treated.12 In the past few decades, a great progress has been made in underpinning the cellular and molecular mechanisms of remodelling in myocardial hypertrophy. However, the involvement of voltage‐gated sodium channel (NaV) isoforms in HFpEF and/or myocardial hypertrophy with preserved ejection fraction leading to cardiac arrhythmias has not been elucidated comprehensively. Besides the peak sodium current causing the action potential (AP) upstroke, a small persistent sodium current is existing, also known as late sodium current (INaL).13 In case of cardiac pathology, some INaL producing NaV channels reopen or remain active throughout the whole AP. The amplitude of INaL is smaller when compared with peak sodium current but eventually with a larger Na integral due to longer persistence during the course of the AP plateau.14 INaL has been reported to be enhanced in different clinically relevant cardiac pathologies like hypoxia, ischaemia, and HF.15, 16, 17 In severe myocardial hypertrophy, INaL is also enhanced18 and may cause intracellular Na+ overload and prolongation of the action potential duration (APD).19 Consequently, the resulting Na+ accumulation may lead to intracellular Ca2+ overload because of reduced efflux and increased influx of Ca2+ through Na+/Ca2+ exchanger.20, 21
The voltage‐gated sodium channel 1.5 (NaV1.5) is considered the predominant cardiac NaV isoform in the heart. In the past few years, different reports suggested the existence of other non‐cardiac sodium channels including NaV1.8 (SCN10A) in the heart, which like NaV1.5 is tetrodotoxin resistant. In addition, genetic variants of SCN10A have been recently shown to influence cardiac conduction.22, 23 Further evidence about NaV1.8 in cardiac conduction comes from genome‐wide association studies reporting that NaV1.8 could modulate cardiac conduction by effecting PR and QRS intervals.23 However, the expression of NaV1.8 in cardiac tissue and its direct involvement in human cardiac arrhythmias is still poorly understood. In the current study, we investigated the presence and the functional role of NaV1.8 in cardiac hypertrophy with preserved contractility derived exclusively from patients suffering from severe aortic stenosis. To study the role of NaV1.8 channel in the electrophysiological context of human LVH, we used two different NaV1.8 blockers, which are described to be very specific for NaV1.8 (A‐803467 and PF‐01247324). The blocker A‐803467 is over 100‐fold more selective for the NaV1.8 channel vs. other human NaV channels.24 Similarly, the drugPF‐01247324 is a highly specific inhibitor of NaV1.8 over other NaV channels, even when used at very high concentration.25
Materials and methods
Human myocardial tissue
All procedures were conducted in compliance with the local ethics committee. The study conforms to the World Medical Association declaration of Helsinki. Written informed consent was received from all patients prior to inclusion. Myocardial tissue of the hypertrophied left ventricle was obtained from patients (n = 24; Table 1 ) with severe aortic valve stenosis and preserved ejection fraction undergoing an open‐heart surgery for aortic valve replacement as a classic myectomy (Morrow resection). For molecular purposes, we utilized healthy left ventricular myocardium (n = 12) from healthy donor hearts that were not transplanted because of technical reasons.
Table 1.
Patient characteristics: values in the table represent the mean ± standard error of the mean
| Male sex | 50% |
| Age | 66.38 ± 3.66 years |
| Ejection fraction | 56.95 ± 1.81% |
| Dyspnoea | 85.7% |
| Interventricular septum | 15.0 ± 1.7 mm |
| Aortic valve area | 0.8 ± 0.1 cm2 |
| Mean AV pressure gradient | 44 ± 6.4 mm Hg |
| Diabetes | 4.3% |
| ACE‐inhibitors | 17.4% |
| β‐Blockers | 65.2% |
| Diuretics | 76.5% |
| Digoxin | 0.0% |
| Amiodarone | 0.0% |
| AT1 receptor antagonists | 26.1% |
| Aldosterone antagonists | 0.0% |
| Statins | 30.4% |
| Ca channel blockers | 26.1% |
ACE, angiotensin‐converting enzyme; AT1, angiotensin II receptor–type 1; AV, aortic valve.
Western blots
Myocardial tissue samples from patients with LVH (n = 12) compared with healthy control myocardium were homogenized in Tris buffer (pH 7.4) containing Tris–HCl (20 mM), NaCl (200 mM), NaF (20 mM), Na3VO4 (1 mM), dithiothreitol (1 mM), Triton X‐100 (1%), and complete protease and phosphatase inhibitor cocktails (Roche Diagnostics, Germany). Protein concentration was determined by bicinchoninic acid assay (Pierce Biotechnology, United States). Denatured tissue homogenates (10 min, 70°C in 2% beta‐mercaptoethanol) were separated on 7.5% sodium dodecyl sulfate–polyacrylamide gels, then transferred to a nitrocellulose membrane, and incubated with the following primary antibodies: mouse monoclonal anti‐NaV1.8 (1:1000, LSBio, United States, LS‐C109037), rabbit polyclonal anti‐NaV1.5 (1:2000, Alomone Labs, Israel, ASC‐005), and mouse monoclonal anti‐GAPDH (1:20000, Biotrend, Germany, BTMC‐A473‐9) at 4°C overnight. Secondary antibodies included horseradish peroxidase (HRP)‐conjugated goat anti‐rabbit and goat anti‐mouse (1:10000, Jackson ImmunoResearch, United Kingdom, 111‐035‐144 and 115‐035‐062, respectively). The membrane was incubated with secondary antibodies for 1 h at room temperature. Immobilon™ Western Chemiluminescent HRP Substrate (Millipore, Germany) was used for the chemiluminescent detection.
Quantitative real‐time PCR
Human cardiac tissues were snap‐frozen in liquid nitrogen and stored at −80°C. RNA was isolated by use of the SV Total RNA Isolation System (Promega, Germany). DNA standards were generated by serial dilution in diethyl pyrocarbonate water containing yeast tRNA (30 μL/mL) as co‐precipitant and used at varying concentrations between 0.125 and 100 000 fg/μL to ensure exponential growth of DNA amounts in the standard range. One hundred nanogram of RNA was reverse transcribed into cDNA using standard protocols. For quantitative PCR, 10 μL of SYBR Green PCR Master Mix (Thermo Fisher, Germany), 7 μL of nuclease‐free water, 1 μL of forward and 1 μL of reverse primer, and 1 μL of cDNA were mixed. Quantitative PCR was carried out using the iQ5 Multicolor Real‐time PCR Detection System (Bio‐Rad, United States). Forty cycles of 15 s at 95°C followed by 1 min at 60°C were used, and fluorescence was measured after each cycle. After 40 cycles, melt curve analysis was performed to ensure specificity of the products. Thresholds cycles were evaluated and normalized to housekeeping genes and controls. The following primer sequences (5′‐3′) were used for quantitative real‐time PCR analyses: SCN10A, forward TGGCAGATGACCTGGAAGAACC and reverse CGATACGGTAGCAAGTCTTGCG (Origene, Cat. no. HP209444, NM_006514); and GAPDH, forward GTCTCCTCTGACTTCAACAGCG and reverse ACCACCCTGTTGCTGTAGCCAA.
Myocyte isolation
Left ventricular myocardium was rinsed, cut into small pieces, and incubated at 37°C in a spinner flask filled with Joklik‐MEM (JMEM) solution (PAN‐Biotech, Germany) that contained 1.0 mg/mL collagenase (Worthington type 1, 185 U/mg, CellSystems, France) and 13% trypsin (Life Technologies, United States). After 45 min, the supernatant was discarded, and fresh JMEM solution containing collagenase was added. The solution was incubated for 10 min until myocytes were disaggregated. The supernatant that contained disaggregated cells was removed and centrifuged (700 rpm, 5 min).
Fresh JMEM with collagenase was added to the remaining tissue. This procedure was repeated four to five times. After every step, the centrifuged cells were resuspended in JMEM solution that contained bovine calf serum 10%, and pH was adjusted to 7.4 at room temperature. Only cell solutions that contained elongated, not granulated, cardiomyocytes with cross‐striations were selected for experiments, plated on laminin‐coated recording chambers, and left to settle for 30 min.
Whole‐cell patch clamp
INaL measurements
Ruptured‐patch whole‐cell voltage‐clamp was used to measure INaL in human ventricular cardiomyocytes isolated from hypertrophied hearts with microelectrodes (2–3 MΩ). Pipettes were filled with solution containing CsCl (95 mM), Cs‐glutamate (40 mM), NaCl (10 mM), MgCl2 (0.92 mM), Mg‐ATP (5 mM), Li‐GTP (0.3 mM), HEPES (5 mM), niflumic acid (0.03 mM; to block Ca2+‐activated chloride current), nifedipine (0.02 mM; to block Ca2+ current), strophanthidin (0.004 mM; to block Na+/K+‐ATPase), EGTA (1 mM), and CaCl2 (0.36 mM; free [Ca2+]i, 100 nM), and pH was adjusted to 7.2 with CsOH. The bath solution contained NaCl (135 mM), tetramethylammonium chloride (5 mM), CsCl (4 mM), MgCl2 (2 mM), glucose (10 mM), and HEPES (10 mM), and pH was adjusted to 7.4 with CsOH. To minimize contaminating Ca2+ currents during INaL measurements, Ca2+ was omitted from the bath solution. Access resistance was <7 MΩ. Cardiomyocytes were held at −120 mV, and INaL was elicited using a train of pulses to −35 mV (1000 ms duration, 10 pulses, basic cycle length 2 s). Recordings were initiated 3 min after rupture. The measured INaL at −35 mV was leak subtracted before calculation of the INaL integral (between 100 and 500 ms) and was normalized to membrane capacitance of the measured cell. Cardiomyocytes were treated with either A‐803467 (30 nM) or PF‐01247324 (1 μM) for 10 min and compared with the control untreated cells. Measurements were conducted at a temperature of 37.7°C.
Action potential duration measurements
To record the AP from human ventricular cardiomyocytes, the whole‐cell patch‐clamp technique was used to measure membrane potential (current clamp configuration). Microelectrodes (7–8 MΩ) were filled with solution containing K‐Aspartate (92 mM), KCl (48 mM), Mg‐ATP (1 mM), HEPES (10 mM), EGTA (0.02 mM), GTP‐Tris (0.1 mM), and Na2‐ATP (4 mM), and final pH was adjusted at 7.2 with KOH solution. Bath solution contained NaCl (140 mM), KCl (4 mM), MgCl2 (1 mM), CaCl2 (2 mM), glucose (10 mM), and HEPES (10 mM), and final pH was adjusted to 7.4 with NaOH. Action potentials were continuously elicited by square current pulses of 1–2 nA amplitude and 1–5 ms duration at increasing stimulation frequency (0.5–3 Hz). Access resistance was typically ~5–15 MΩ after patch rupture. Fast capacitance was compensated for in a cell‐attached configuration. Recordings were commenced after cell stabilization, which was ~10 min after rupture. Human cell measurements were conducted at a temperature of 37.7°C. Every cell was patched and measured before and after drug application.
In all patch‐clamp experiments, cardiomyocytes were mounted on the stage of a microscope (Nikon T 300). Fast capacitance was compensated in cell‐attached configuration. Membrane capacitance and series resistance were compensated after patch rupture. Signals were filtered with 2.9 and 10 kHz Bessel filters and recorded with an EPC10 amplifier (HEKA Elektronik, Germany). NaV1.8 was inhibited using either A‐803467 (30 nM for 10 min) or PF‐01247324 (1 μM for 5 min). Both drugs were added to the bath solution.
Measurement of Ca2+ sparks
Isolated cardiomyocytes were incubated at room temperature for 30 min with a Fluo‐4 AM loading buffer (10 μM; Molecular Probes, Life Technologies). Experimental solution contained NaCl (136 mM), KCl (4 mM), NaH2PO4 (0.33 mM), NaHCO3 (4 mM), CaCl2 (2 mM), MgCl2 (1.6 mM), HEPES (10 mM), and glucose (10 mM), and final pH was adjusted to 7.4 by addition of NaOH solution at room temperature. NaV1.8 inhibitors A‐803467 (30 nM) and PF‐01247324 (1 μM) were added to their respective groups in the experimental solution.
Cells were continuously superfused during experiments. To wash out the loading buffer and to remove any extracellular dye (as well as to provide enough time for complete de‐esterification of Fluo‐4 AM), cells were superfused with experimental solution for 5 min before the experiments were begun. Ca2+ spark measurements were performed with a laser scanning confocal microscope (LSM 5 Pascal, Zeiss, Germany) using a ×40 oil‐immersion objective. Fluo‐4 was excited by an argon ion laser (488 nm), and emitted fluorescence was collected through a 505 nm long‐pass mission filter. Fluorescence images were recorded in the line scan mode with 512 pixels per line (width of each scan line: 38.4 μm) and a pixel time of 0.64 μs. One image consists of 10 000 unidirectional line scans, which equates to a measurement period of 7.68 s. Experiments were conducted at resting conditions after the sarcoplasmic reticulum (SR) was loaded with Ca2+ by repetitive field stimulation (10 pulses at 1 Hz, 20 V). Ca2+ sparks were analysed with the program SparkMaster for ImageJ. The mean spark frequency of the respective cell resulted from the number of sparks normalized to cell width and scan rate (100 μm−1 s−1).
Results
Regulation of NaV1.5 and NaV1.8 in human left ventricular hypertrophy
To investigate the regulation of NaV1.8 and NaV1.5 protein expression in human LVH compared with healthy control ventricular myocardium tissue homogenates, western blots were performed. We found a significant up‐regulation of NaV1.8 protein expression (1.96‐ ± 0.31‐fold) in LVH compared with healthy control ventricular myocardium (Figure 1 B). In contrast, the expression of NaV1.5 protein was significantly decreased (0.60‐ ± 0.10‐fold) compared with Non‐failing (NF) (Figure 1 B). In accordance to our findings of NaV1.8 expression at the protein level, we recorded a significant up‐regulation of NaV1.8 mRNA levels in LVH (2.34‐ ± 0.64‐fold, n = 5) compared with NF (n = 10) by quantitative PCR. The mRNA expression of NaV1.8 is shown as relative expression to the housekeeping gene GAPDH (Figure 1 C).
Figure 1.
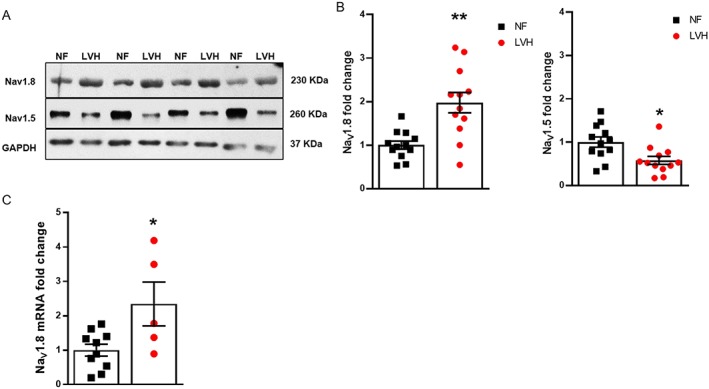
Regulation of NaV1.8 and NaV1.5 expression in hypertrophy. (A) Western blots were performed utilizing left ventricular human tissue homogenates. (B) Densitometry data of NaV1.8 and NaV1.5 show a significant up‐regulation of NaV1.8 and down‐regulation of NaV1.5. GAPDH was used as an internal loading control in all blots [NF: n = 12; and left ventricular hypertrophy (LVH): n = 12]. (C) Real‐time quantitative PCR showing the relative mRNA expression of NaV1.8/GAPDH in left ventricle of human NF (n = 10) and LVH (n = 5). *P ≤ 0.05 and **P ≤ 0.01 vs. NF. Student's t‐test. Data shown as mean ± standard error of the mean and individual values.
NaV1.8 contributes to INaL and action potential duration in human hypertrophy
Given the increased expression of NaV1.8 in human LVH, we investigated the potential contribution to the augmentation of INaL and APD prolongation under hypertrophic condition. We adopted the approach using novel‐specific blockers of NaV1.8 channel A‐803467 and PF‐01247324 to inhibit the activity of this channel in human LVH cardiomyocytes for recording INaL and APDs by patch clamp. Cardiomyocytes were superfused with either A‐803467 or PF‐01247324. Our data show a significant reduction in INaL (Figure 2 ) and also shortening of APD90 (Figure 3 ) when cardiomyocytes were exposed to NaV1.8 inhibitors compared with the control group. Furthermore, both NaV1.8 inhibitors do not exert any effect on the upstroke velocity and amplitude of the AP (Figure 4 A and 4 B). Therefore, it can be concluded that NaV1.8 contributes to INaL in terms of a positive net inward current to APD prolongation in human hypertrophy.
Figure 2.

(A) Original traces and (B) data showing individual and mean values ± standard error of the mean of INaL in human ventricular cardiomyocytes isolated from left ventricular hypertrophy patients (control: n = 8 cells; A‐803467: n = 7 cells; PF‐01247324: n = 7 cells). *P ≤ 0.05. One‐way analysis of variance and Bonferroni's post‐test.
Figure 3.
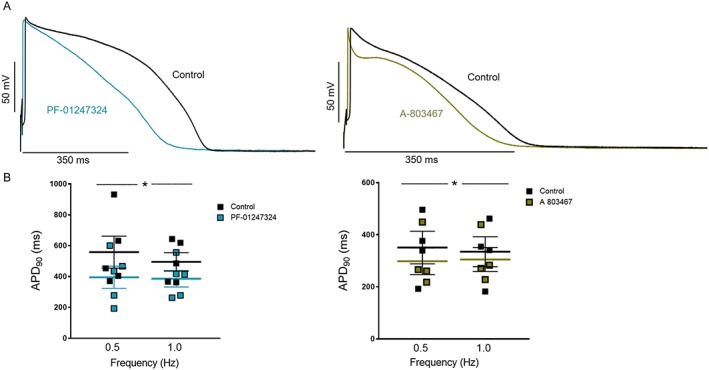
(A) Original action potential recording (0.5 Hz) and (B) data showing individual and mean values ± standard error of the mean of APD90 in left ventricular cardiomyocytes from patients with left ventricular hypertrophy. A‐803467 or PF‐01247324 paced at 0.5 and 1 Hz (n = 5 cells and n = 4 cells, respectively; *P ≤ 0.05 vs. control). Two‐way repeated measures analysis of variance and Bonferroni's post‐test. All the action potential duration (APD) measurements were performed pairwise by wash‐in.
Figure 4.
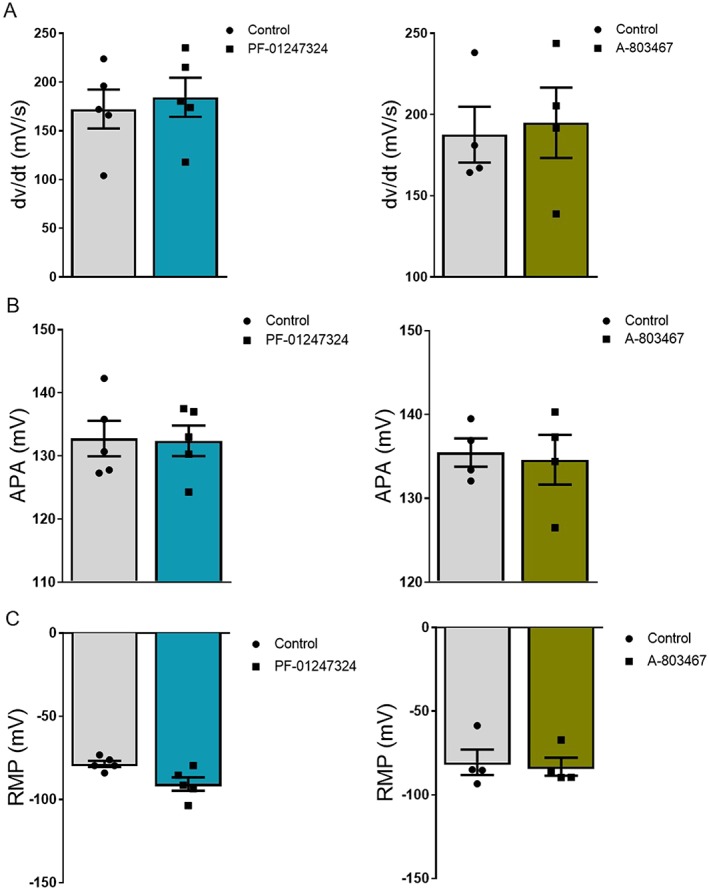
(A) Individual and mean values ± standard error of the mean showing upstroke velocity of action potential in control and drug groups from left ventricular hypertrophy cardiomyocytes. (B) Data showing individual and mean values ± standard error of the mean of action potential amplitude (APA) measurements from isolated human left ventricular hypertrophy cardiomyocytes. (C) Graph shows the resting membrane potential (RMP) of cardiomyocytes measured under control condition and drug treatments (PF‐01247324: n = 5 cells; and A‐804367: n = 4 cells). All measurements were performed pairwise by wash‐in. Paired Student's t‐test was performed.
Contribution of NaV1.8 to proarrhythmic SR‐Ca 2+ leak
During pathological conditions such as HF, an enhanced INaL can lead to disturbed SR‐Ca2+ metabolism and hence potentially proarrhythmic SR‐Ca2+ leakiness. This can give rise to delayed after‐depolarizations and thereby arrhythmias. However, the contribution of different NaV isoforms to SR‐Ca2+ leak in human LVH has not been evaluated. Therefore, we measured SR‐Ca2+ leak in isolated cardiomyocytes from human hypertrophied hearts.
We detected a high incident of spontaneous diastolic Ca2+ release events in hypertrophied cardiomyocytes in the absence of inhibitors (Figure 5 A). When cardiomyocytes were incubated with NaV1.8 inhibitors either A‐803467 or PF‐01247324, a significant decrease in calcium spark frequency (CaSpF) and SR‐Ca2+ leak was observed compared with control group (Figure 5 B and 5 E). In control cells, CaSpF was 0.68 ± 0.12 μm−1 s−1 (n = 145 cells/12 patients), while in A‐806734‐treated myocytes, CaSpF was reduced to 0.27 ± 0.06 μm−1 s−1 (n = 124 cells/11 patients, P ≤ 0.01) and in PF‐01247324‐treated myocytes to 0.26 ± 0.07 μm−1 s−1 (n = 91 cells/9 patients, P ≤ 0.01), respectively (Figure 5 B). NaV1.8 inhibition with PF‐01247324 also resulted in a significant reduction of the Ca2+ spark duration (Figure 5 C), while no differences were recorded for Ca2+ spark amplitude between control and drug treatment groups (Figure 5 D). These data clearly demonstrate the important role of the NaV1.8 channel in SR‐Ca2+ leak regulation in hypertrophy. Taken together, proarrhythmic SR‐Ca2+ release can significantly be decreased by targeting NaV1.8 with specific inhibitors.
Figure 5.
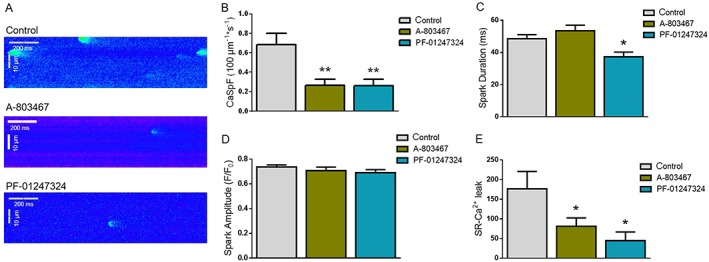
(A) Representative line scan images and (B) calcium spark frequency (CaSpF) in left ventricular cardiomyocytes from patients with left ventricular hypertrophy under control condition and NaV1.8 inhibition. Pre‐incubation with either A‐803467 or PF‐01247324 resulted in a significant decrease of CaSpF in hypertrophy compared with control. (C) Mean values of spark duration and (D) spark amplitude of cardiomyocytes. (E) Calculated full SR‐Ca2+ leak in left ventricular cardiomyocytes from left ventricular hypertrophy patients (control: n = 145; A‐803467: n = 124 cells; PF‐01247324: n = 91 cells). Data shown as mean ± standard error of the mean. One‐way analysis of variance and Bonferroni's post‐test. *P ≤ 0.05, **P ≤ 0.01.
Discussion
LVH is a common clinical finding in daily practice caused, besides some other reason, by hypertension and aortic valve stenosis. At the early stages, myocardial hypertrophy serves as a compensatory mechanism, which can lead to systolic HF at later stages.26 Importantly, patients with symptomatic aortic stenosis have a tremendous risk of sudden cardiac death if only medically treated.12 In the past few decades, a great progress has been made in underpinning the cellular and molecular mechanisms of remodelling during hypertrophy. However, the mechanistic links between dysregulated proteins and in particular arrhythmogenesis, especially in human hypertrophy, still remain elusive. We herein investigated the regulation of neuronal sodium channel NaV1.8 and its role in pathophysiology of human significant hypertrophy. We demonstrate that NaV1.8 expression is up‐regulated during LVH with preserved ejection fraction compared with healthy left ventricles, while NaV1.5 expression is down‐regulated in parallel. To the best of our knowledge, this is the first report of such an ion channel switch in human hypertrophy. Furthermore, inhibition of NaV1.8 by specific blockers causes a significant reduction of the enhanced INaL and consequently leads to abbreviated APDs in LVH. Moreover, we observed a significant decrease of the proarrhythmic SR‐Ca2+ leak by inhibition of NaV1.8 in human hypertrophied cardiomyocytes. Therefore, inhibition of NaV1.8 modulates well‐accepted proarrhythmogenic triggers such as INaL, APD prolongation, and diastolic SR‐Ca2+ leak.
In the current study, we found increased expression of NaV1.8 at both mRNA and protein level in human LVH with preserved contractility when compared with NF. Transcripts of many non‐cardiac NaV channels (NaV1.1, NaV1.3, NaV1.2, and NaV1.6) including NaV1.8 were detected in mouse and dog heart, while there was no previous evidence of NaV1.8 expression in the human ventricle during LVH.27, 28 Moreover, there have been conflicting reports about the involvement of these non‐cardiac NaV isoforms in INaL enhancement in animal models due to species‐specific expression. We already reported that the protein expression of other non‐cardiac NaV channels (NaV1.1 and NaV1.6) was down‐regulated while NaV1.5 was increased showing no significant INaL enhancement in a transverse aortic constriction mouse model of compensated hypertrophy.17 Similar findings were reported in a dog and rat HF model where NaV1.5 protein expression was found to be down‐regulated, whereas a significant increase in INaL was observed.29, 30 These findings from animal models suggest the involvement of other non‐cardiac sodium channels in INaL augmentation and arrhythmogenesis under hypertrophy and HF conditions. However, animal models represent a very artificial variability in hypertrophy, which cannot be compared directly with human hypertrophy with severe aortic stenosis. Moreover, these animal‐derived findings cannot be directly translated into the human because of severe differences in cellular electrophysiology. Therefore, the translational data of our current study add novel knowledge on INaL regulation in hypertrophy and show for the first time that INaL augmentation is also caused by over‐expression of NaV1.8 in human hypertrophy.
Electrophysiological recordings also described that non‐cardiac NaV channels contribute to INaL up to 44% in canine ventricular cardiomyocytes under normal physiological conditions31 and NaV1.8 was suggested to be a major contributor of INaL in healthy mouse ventricular cardiomyocytes.32 However, NaV1.8 was not investigated before in animal models with present heart disease, for example, hypertrophy. It was shown previously by us and other groups that increased INaL is implicated as potential contributor to proarrhythmogenic triggers and thereby to the occurrence of arrhythmias.33, 34, 35 During different pathological conditions associated with heart diseases, over‐expression of these non‐cardiac NaV channels was also shown to be responsible for the prolonged cardiac APD.31 The functional properties of NaV1.8 are characterized by a long AP duration with preserved excitability during sustained stimulation in the dorsal root ganglion.36 In accordance with an enhanced INaL, we observed prolongation of APD in ventricular cardiomyocytes of hypertrophy patients. Inhibition of NaV1.8 with specific blockers showed a significant abbreviation of APD. Up‐regulated NaV1.8 expression in human hypertrophy may cause increased INaL potentially leading to a pathological prolongation of APD in this disease. This prolonged APD can give rise to early after‐depolarizations, thereby posing increased fatal risk for ventricular arrhythmias.13 The partial inhibition of INaL up to 50% not only restored healthy APD in failing cardiomyocytes but also ceased after‐depolarizations.13
Isolated cardiomyocytes from patients with hypertrophic cardiomyopathy showed an augmented INaL with a subsequent increase in intracellular Ca2+ load.18 Enhanced INaL might lead to diastolic Ca2+ overload through the Na+/Ca2+ exchanger operation in reverse mode.37 An increase in diastolic Ca2+ not only results in impaired relaxation but also plays a key role in activating pro‐hypertrophic signalling pathways, leading to increased stiffness of the left ventricle.38, 39 These data suggest that INaL is not only the consequence of hypertrophy but is also involved in inducing diastolic intracellular Ca2+ overload. The resulting Ca2+ load is thought to trigger intracellular spontaneous Ca2+‐release events, leading to cytosolic Ca2+ oscillations, automaticity, and triggered activity. In addition to animal studies, we have previously demonstrated that inhibition of an enhanced INaL with tetrodotoxin or ranolazine reduces the SR‐Ca2+ leak in human diseased cardiomyocytes.37, 40 In the present study, we extend this evidence to human cardiomyocytes with very significant hypertrophy and HFpEF. The highly specific inhibition of NaV1.8 potently suppressed the diastolic SR‐Ca2+ leak in ventricular cardiomyocytes of patients with severe LV hypertrophy. Taken together, the data of our present study define a significant contribution of NaV1.8 in the initiation of proarrhythmic triggers via INaL‐induced SR‐Ca2+ leak and also APD prolongation. The differences between the activation of NaV1.5 and NaV1.8 suggest that the selective inhibition of NaV1.8‐mediated INaL can be antiarrhythmic.
Patients with severe aortic stenosis and concomitant LV hypertrophy including HFpEF are at high risk of sudden cardiac death, and one of the probable causes is lethal arrhythmias. Our current study provides better understanding of electrophysiological disturbances that occur in human severe hypertrophy. We identified a potential new ion channel target (NaV1.8) and provide a respective possible pharmacological antiarrhythmic approach. Therefore, these findings provide basic evidence for in vivo studies.
Conflict of interest
None declared.
Funding
S.A. and S.S. are funded by the Marga und Walter Boll‐Stiftung through a research grant (project no. 220‐12.1‐15). K.S.‐B. is funded by the Bundesministerium für Bildung und Forschung (BMBF) grant e:Bio—Modul II—Verbundprojekt: CaRNAtion (031L0075C to K.S.‐B. and G.H.). S.S. and L.S.M. are funded by the ReForM‐Programme of the University Hospital Regensburg. N.D. and G.H. are funded by the Deutsche Forschungsgemeinschaft (DFG, SFB 1002).
Acknowledgements
We gratefully acknowledge the technical assistance of Timo Schulte, Yvonne Metz, and Johanna Heine.
Ahmad, S. , Tirilomis, P. , Pabel, S. , Dybkova, N. , Hartmann, N. , Molina, C. E. , Tirilomis, T. , Kutschka, I. , Frey, N. , Maier, L. S. , Hasenfuss, G. , Streckfuss‐Bömeke, K. , and Sossalla, S. (2019) The functional consequences of sodium channel NaV1.8 in human left ventricular hypertrophy. ESC Heart Failure, 6: 154–163. 10.1002/ehf2.12378.
References
- 1. Savage DD, Garrison RJ, Kannel WB, Levy D, Anderson SJ, Stokes J III, Feinleib M, Castelli WP. The spectrum of left ventricular hypertrophy in a general population sample: the Framingham Study. Circulation 1987; 75: I26–I33. [PubMed] [Google Scholar]
- 2. Sheridan DJ, Kingsbury MP, Flores NA. Regression of left ventricular hypertrophy; what are appropriate therapeutic objectives? Br J Clin Pharmacol 1999; 47: 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kannel WB. Prevalence and natural history of electrocardiographic left ventricular hypertrophy. Am J Med 1983; 75: 4–11. [DOI] [PubMed] [Google Scholar]
- 4. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med 1990; 322: 1561–1566. [DOI] [PubMed] [Google Scholar]
- 5. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, Gonzalez‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Rev Esp Cardiol (Engl Ed) 2016; 69: 1167. [DOI] [PubMed] [Google Scholar]
- 6. Cooper RS, Simmons BE, Castaner A, Santhanam V, Ghali J, Mar M. Left ventricular hypertrophy is associated with worse survival independent of ventricular function and number of coronary arteries severely narrowed. Am J Cardiol 1990; 65: 441–445. [DOI] [PubMed] [Google Scholar]
- 7. Levy D, Anderson KM, Savage DD, Balkus SA, Kannel WB, Castelli WP. Risk of ventricular arrhythmias in left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol 1987; 60: 560–565. [DOI] [PubMed] [Google Scholar]
- 8. Kurdi M, Booz GW. Three 4‐letter words of hypertension‐related cardiac hypertrophy: TRPC, mTOR, and HDAC. J Mol Cell Cardiol 2011; 50: 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edelmann F. Facts and numbers on epidemiology and pharmacological treatment of heart failure with preserved ejection fraction. ESC Heart Fail 2015; 2: 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kehat I, Molkentin JD. Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 2010; 122: 2727–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DiCarlo LA, Libbus I, Kumar HU, Mittal S, Premchand RK, Amurthur B, KenKnight BH, Ardell JL, Anand IS. Autonomic regulation therapy to enhance myocardial function in heart failure patients: the ANTHEM‐HFpEF study. ESC Heart Fail 2018; 5: 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ross J Jr, Braunwald E. Aortic stenosis. Circulation 1968; 38: 61–67. [DOI] [PubMed] [Google Scholar]
- 13. Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail 2007; 9: 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Antzelevitch C, Nesterenko V, Shryock JC, Rajamani S, Song Y, Belardinelli L. The role of late I Na in development of cardiac arrhythmias. Handb Exp Pharmacol 2014; 221: 137–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002; 105: 2543–2548. [DOI] [PubMed] [Google Scholar]
- 16. Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol 1996; 497: 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, Sag CM, Hund TJ, Mohler PJ, Belardinelli L, Hasenfuss G, Maier LS, Sossalla S. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure‐induced heart disease. J Mol Cell Cardiol 2013; 61: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coppini R, Ferrantini C, Del LM, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Poggesi C, Cerbai E, Mugelli A, Yao L, Fan P, Belardinelli L, Yacoub M, Olivotto I. Response to letter regarding article, “Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy”. Circulation 2013; 128: e157. [DOI] [PubMed] [Google Scholar]
- 19. Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide‐induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther 2006; 318: 214–222. [DOI] [PubMed] [Google Scholar]
- 20. Pieske B, Maier LS, Piacentino V III, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 2002; 106: 447–453. [DOI] [PubMed] [Google Scholar]
- 21. Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schondube FA, Tirilomis T, Tenderich G, Hasenfuss G, Belardinelli L, Maier LS. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts—role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol 2008; 45: 32–43. [DOI] [PubMed] [Google Scholar]
- 22. Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, Navaratnarajah M, Lotlikar A, Sehmi JS, Kooner MK, Deng G, Siedlecka U, Parasramka S, El‐Hamamsy I, Wass MN, Dekker LR, de Jong JS, Sternberg MJ, McKenna W, Severs NJ, de SR, Wilde AA, Anand P, Yacoub M, Scott J, Elliott P, Wood JN, Kooner JS. Genetic variation in SCN10A influences cardiac conduction. Nat Genet 2010; 42: 149–152. [DOI] [PubMed] [Google Scholar]
- 23. Pfeufer A, van NC, Marciante KD, Arking DE, Larson MG, Smith AV, Tarasov KV, Muller M, Sotoodehnia N, Sinner MF, Verwoert GC, Li M, Kao WH, Kottgen A, Coresh J, Bis JC, Psaty BM, Rice K, Rotter JI, Rivadeneira F, Hofman A, Kors JA, Stricker BH, Uitterlinden AG, van Duijn CM, Beckmann BM, Sauter W, Gieger C, Lubitz SA, Newton‐Cheh C, Wang TJ, Magnani JW, Schnabel RB, Chung MK, Barnard J, Smith JD, Van Wagoner DR, Vasan RS, Aspelund T, Eiriksdottir G, Harris TB, Launer LJ, Najjar SS, Lakatta E, Schlessinger D, Uda M, Abecasis GR, Muller‐Myhsok B, Ehret GB, Boerwinkle E, Chakravarti A, Soliman EZ, Lunetta KL, Perz S, Wichmann HE, Meitinger T, Levy D, Gudnason V, Ellinor PT, Sanna S, Kaab S, Witteman JC, Alonso A, Benjamin EJ, Heckbert SR. Genome‐wide association study of PR interval. Nat Genet 2010; 42: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roeloffs R, Zhong C, Mikusa JP, Hernandez G, Gauvin D, Wade C, Zhu C, Pai M, Scanio M, Shi L, Drizin I, Gregg R, Matulenko M, Hakeem A, Gross M, Johnson M, Marsh K, Wagoner PK, Sullivan JP, Faltynek CR, Krafte DS. A‐803467, a potent and selective NaV1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A 2007; 104: 8520–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Payne CE, Brown AR, Theile JW, Loucif AJ, Alexandrou AJ, Fuller MD, Mahoney JH, Antonio BM, Gerlach AC, Printzenhoff DM, Prime RL, Stockbridge G, Kirkup AJ, Bannon AW, England S, Chapman ML, Bagal S, Roeloffs R, Anand U, Anand P, Bungay PJ, Kemp M, Butt RP, Stevens EB. A novel selective and orally bioavailable Nav 1.8 channel blocker, PF‐01247324, attenuates nociception and sensory neuron excitability. Br J Pharmacol 2015; 172: 2654–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaasch WH, Delorey DE, St John Sutton MG, Zile MR. Patterns of structural and functional remodeling of the left ventricle in chronic heart failure. Am J Cardiol 2008; 102: 459–462. [DOI] [PubMed] [Google Scholar]
- 27. Haufe V, Camacho JA, Dumaine R, Gunther B, Bollensdorff C, von Banchet GS, Benndorf K, Zimmer T. Expression pattern of neuronal and skeletal muscle voltage‐gated Na+ channels in the developing mouse heart. J Physiol 2005; 564: 683–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain‐type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A 2002; 99: 4073–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xi Y, Wu G, Yang L, Han K, Du Y, Wang T, Lei X, Bai X, Ma A. Increased late sodium currents are related to transcription of neuronal isoforms in a pressure‐overload model. Eur J Heart Fail 2009; 11: 749–757. [DOI] [PubMed] [Google Scholar]
- 30. Zicha S, Maltsev VA, Nattel S, Sabbah HN, Undrovinas AI. Post‐transcriptional alterations in the expression of cardiac Na+ channel subunits in chronic heart failure. J Mol Cell Cardiol 2004; 37: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Biet M, Barajas‐Martinez H, Ton AT, Delabre JF, Morin N, Dumaine R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non‐cardiac‐type Na+ channels. J Mol Cell Cardiol 2012; 53: 593–598. [DOI] [PubMed] [Google Scholar]
- 32. Yang T, Atack TC, Stroud DM, Zhang W, Hall L, Roden DM. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ Res 2012; 111: 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maltsev VA, Kyle JW, Mishra S, Undrovinas A. Molecular identity of the late sodium current in adult dog cardiomyocytes identified by NaV1.5 antisense inhibition. Am J Physiol HeartCirc Physiol 2008; 295: H667–H676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, Fahrenbach J, Weiss D, Taylor WR, Zafari AM, Dudley SC Jr. Human heart failure is associated with abnormal C‐terminal splicing variants in the cardiac sodium channel. Circ Res 2007; 101: 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin‐dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 2006; 116: 3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patrick HT, Waxman SG. Inactivation properties of sodium channel NaV1.8 maintain action potential amplitude in small DRG neurons in the context of depolarization. Mol Pain 2007; 3: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fischer TH, Herting J, Mason FE, Hartmann N, Watanabe S, Nikolaev VO, Sprenger JU, Fan P, Yao L, Popov AF, Danner BC, Schondube F, Belardinelli L, Hasenfuss G, Maier LS, Sossalla S. Late INa increases diastolic SR‐Ca2+‐leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc Res 2015; 107: 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 2008; 70: 23–49. [DOI] [PubMed] [Google Scholar]
- 39. Dorn GW, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 2005; 115: 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sag CM, Mallwitz A, Wagner S, Hartmann N, Schotola H, Fischer TH, Ungeheuer N, Herting J, Shah AM, Maier LS, Sossalla S, Unsold B. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII‐dependent manner. J Mol Cell Cardiol 2014; 76: 94–105. [DOI] [PubMed] [Google Scholar]