

Abstract
Tafamidis meglumine, a transthyretin (TTR) stabilizer, is effective in delaying the progression of neuropathy in TTR amyloidosis with Val30Met mutations. However, its efficacy in TTR amyloid cardiomyopathy is not fully elucidated. Herein, we report a 73‐year‐old Japanese man with a diagnosis of TTR amyloid cardiomyopathy with Val30Met mutation treated with tafamidis. To evaluate treatment response, cardiac magnetic resonance imaging was performed before and after 12 months of tafamidis treatment. Native T1, extracellular volume, and left ventricular mass showed no obvious worsening, and findings of other diagnostic studies also supported the efficacy of tafamidis to delay the progression of amyloid cardiomyopathy. Our case suggests that serial native T1 and extracellular volume may be novel non‐invasive imaging methods to monitor the treatment response to TTR stabilizers in cardiac amyloidosis and also that tafamidis may be effective in suppressing cardiac progression in TTR amyloid cardiomyopathy with Val30Met mutation.
Keywords: Transthyretin, Amyloidosis, Tafamidis, Cardiac magnetic resonance, Native T1, Extracellular volume
Introduction
It has been demonstrated that tafamidis meglumine (Vyndaqel®; Pfizer Inc., New York, NY, USA), a transthyretin (TTR) stabilizer, is effective against peripheral neurological impairment in TTR amyloidosis with Val30Met mutations.1, 2 However, its efficacy in TTR amyloid cardiomyopathy with Val30Met mutations is not fully elucidated. Cardiac magnetic resonance (CMR) imaging has emerged as an imaging technique in cardiac amyloidosis that provides detailed information about the presence, location, and distribution of hypertrophy, as well as visualization and measurement of cardiac amyloid infiltration. Both myocardial native T1 and extracellular volume (ECV) using CMR imaging have been validated as surrogate markers of infiltration and have been shown to correlate with disease burden with high diagnostic and prognostic value in TTR cardiac amyloidosis.3 Herein, we report a case of TTR amyloidosis with Val30Met mutation, with serial native T1 and ECV assessments to evaluate the treatment response to tafamidis.
Case report
A 73‐year‐old Japanese man was transferred to our hospital with dyspnoea. He had no medical or family history of note. Blood pressure was 140/75 mmHg, heart rate 137 beats/min, and respiratory rate 24/min with oxygen saturation of 92% on room air. Electrocardiogram showed atrial fibrillation, ST depression, and inverted T waves in leads V3–V6. QRS width was 112 ms. Echocardiography showed mild left ventricular (LV) concentric hypertrophy (interventricular septum/posterior wall thickness of 11/11 mm, LV end‐diastolic/end‐systolic dimension of 46/25 mm, and LV mass of 180.6 g), LV ejection fraction of 60%, dilated left atrium (LA) with LA diameter of 50 mm and LA volume of 77 mL/m2, diastolic function at the upper limit of normal range (E/e′ 14.6), and preserved right ventricular (RV) systolic function (tricuspid annular plane systolic excursion of 20 mm and RV fractional area change of 44%). On laboratory examination, troponin T was 0.025 ng/mL, and B‐type natriuretic peptide was 252.4 pg/mL. Spirometry showed an obstructive pattern [vital capacity of 3.28 L (103.7%) and forced expiratory volume in 1 second of 1.88 L (59.7%)] with normal diffusing capacity of the lung for carbon monoxide of 13.24 mL/min/mmHg (88.3%). Coronary angiography showed no significant coronary artery stenosis.
Right ventricular endomyocardial biopsy revealed diffuse interstitial amyloid deposits in the myocardium, with positive immunohistochemical staining for TTR (Figure 1 A–C). Genetic analysis revealed Val30Met mutation (GTG → ATG), indicating a diagnosis of hereditary TTR amyloidosis. The patient showed vitreous opacities and chronic glaucoma, suggesting ocular amyloidosis, without peripheral/autonomic neuropathy or carpal tunnel syndrome. The patient was started on tafamidis 20 mg per day for prophylaxis against progression of neuropathy.
Figure 1.
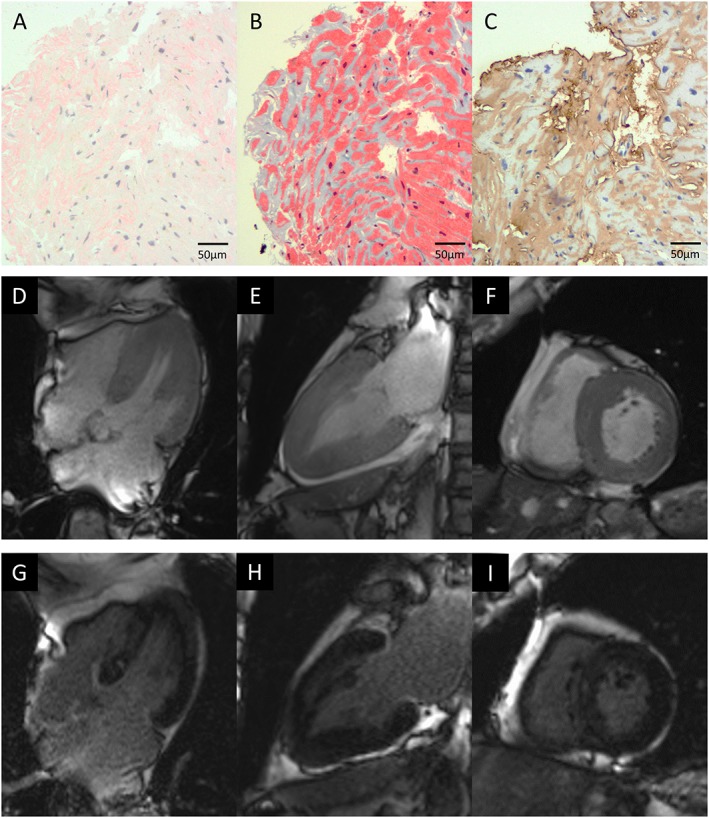
Endomyocardial biopsy and cardiac magnetic resonance imaging. (A) Congo red staining and (B) Masson's trichrome staining revealed diffuse amyloid deposits in the myocardial interstitium, which were (C) immunopositive for transthyretin. Cardiac magnetic resonance imaging prior to tafamidis showed diffuse left ventricular hypertrophy by cine imaging ((D) four‐chamber view; (E) two‐chamber view; (F) short‐axis view) and diffuse late gadolinium enhancement of the left ventricle ((G) four‐chamber view; (H) two‐chamber view; (I) short‐axis view).
To evaluate the efficacy of tafamidis treatment on cardiac lesions, the patient underwent 3 tesla CMR evaluations before and after 12 months of tafamidis treatment. CMR imaging before tafamidis treatment showed diffuse LV hypertrophy (Figure 1 D–F) and diffuse myocardial late gadolinium enhancement (Figure 1 G–I), which showed no obvious progression at follow‐up (LV mass by CMR: 115.0 g before and 111.3 g at 12 months). As shown in Figure 2 , prolonged native T1 time (1387 ms) and increased ECV (42.0%) were observed before tafamidis treatment, and no obvious worsening was found at 12 months follow‐up (native T1 time of 1356 ms and ECV of 40.4%). Troponin T and B‐type natriuretic peptide levels were unchanged at 0.016 ng/mL and 271.5 pg/mL, respectively, after 12 months of tafamidis. Echocardiography showed no significant progression in LV morphological features (interventricular septum/posterior wall thickness of 12/12 mm, LV end‐diastolic/end‐systolic dimension of 43/25 mm, and LV mass of 184.1 g), with LV ejection fraction of 60%, as well as no obvious changes in LA size, diastolic function, or RV systolic function (LA diameter of 47 mm, LA volume of 73 mL/m2, E/e′ of 14.4, tricuspid annular plane systolic excursion of 18 mm, and RV fractional area change of 46%). Spirometry after tafamidis also showed no obvious change [vital capacity of 3.20 L (102.2%), forced expiratory volume in 1 s of 1.84 L (59.6%), and diffusing capacity of the lung for carbon monoxide of 12.46 mL/min/mmHg (89.2%)]. Comparison of electrocardiograms before and after tafamidis is shown in Figure 3 , which also showed no obvious change in QRS width or inverted T waves.
Figure 2.
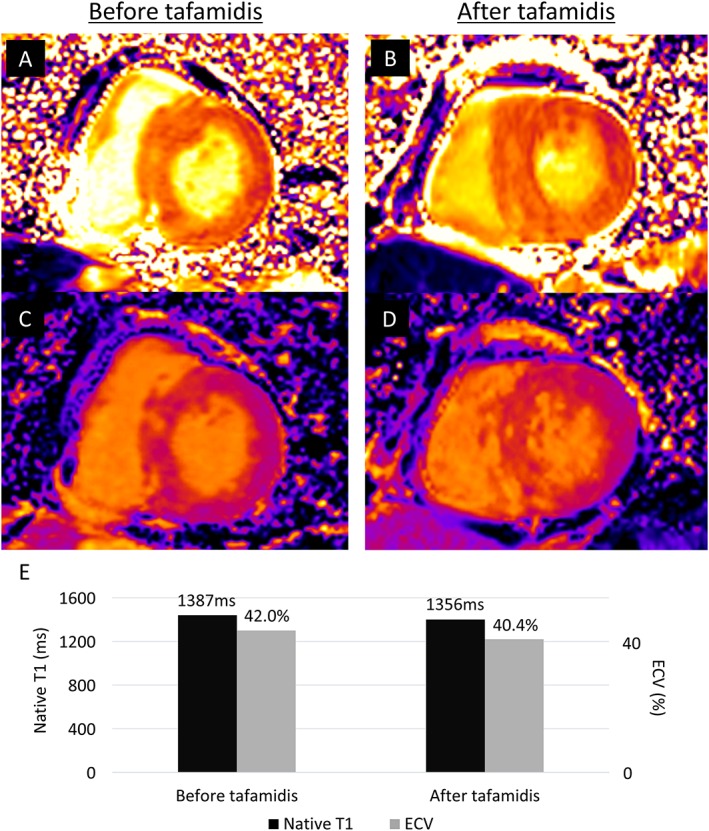
Native T1 and extracellular volume (ECV) before and after 12 months of tafamidis treatment. Three tesla cardiac magnetic resonance imaging showing native T1 mapping (A) before and (B) after tafamidis. There was no obvious worsening of native T1 value (1387 to 1356 ms). ECV (C) before and (D) after tafamidis also showed no significant worsening (42.0 to 40.4%). The serial changes are summarized in (E).
Figure 3.
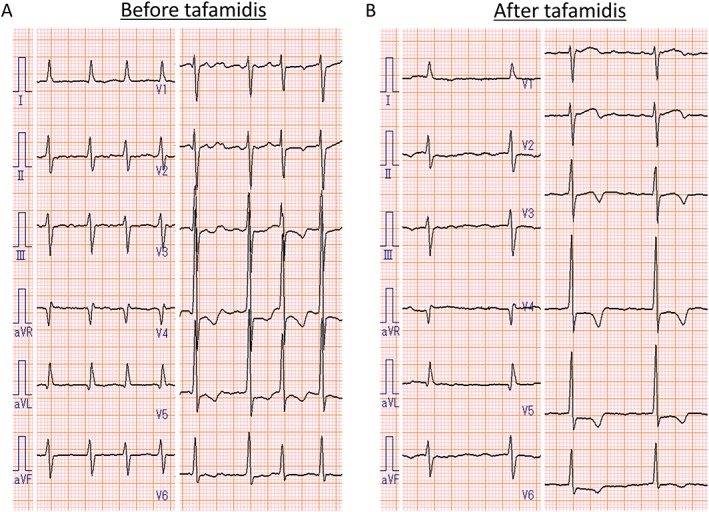
Electrocardiograms (A) before and (B) after 12 months of tafamidis treatment. No obvious change in QRS width or inverted T waves was observed.
Discussion
We report a case of TTR amyloid cardiomyopathy with Val30Met mutation, with serial native T1 and ECV evaluation using CMR imaging to evaluate the treatment response to tafamidis on cardiac lesions. Native T1 and ECV showed no significant worsening after 12 months of tafamidis treatment, suggesting that tafamidis may have suppressed cardiac progression. We also found no significant progression of LV mass at 12 months (2% increase by echocardiography and 3% decrease by CMR imaging), in contrast to the rapid progression reported in TTR amyloid cardiomyopathy (LV mass increase in 12 months: 9% by CMR imaging and 22% by echocardiography) and changes in LV morphological features associated with a poor prognosis.4, 5 Findings of other diagnostic studies, including no obvious change in LV diastolic function,6 also supported the efficacy of tafamidis to delay the progression of amyloid cardiomyopathy. To the best of our knowledge, our case is the first published report of a serial evaluation of tafamidis treatment using native T1 and ECV in TTR amyloid cardiomyopathy.
The efficacy of tafamidis treatment in TTR amyloid cardiomyopathy is not fully elucidated. In TTR amyloidosis with non‐Val30Met mutations, recent reports including the Phase 3 ATTR‐ACT Trial (Tafamidis in Transthyretin Cardiomyopathy Clinical Trial) have reported the potential of TTR stabilizers (diflunisal or tafamidis) to slow cardiac progression and improve survival7, 8, 9, 10, 11; however, the efficacy of tafamidis in TTR amyloid cardiomyopathy with Val30Met mutation has not been elucidated. Our case suggests that tafamidis may be effective in suppressing cardiac progression in TTR amyloid cardiomyopathy with Val30Met mutation, based on our serial CMR evaluations. Further studies are required to evaluate whether the efficacy of tafamidis on the myocardial substrate differs between early‐onset and late‐onset Val30Met cases12, 13, 14 or among TTR amyloid cardiomyopathies with other mutations.15
In conclusion, serial native T1 and ECV evaluation using CMR imaging may be novel non‐invasive imaging methods to monitor the treatment response to TTR stabilizers in cardiac amyloidosis. Our case also suggests that tafamidis may suppress cardiac progression in TTR amyloid cardiomyopathy with Val30Met mutations.
Conflict of interest
Yoshiki Sekijima has received royalties from Pfizer related to tafamidis patents and has received speaker honoraria from Pfizer. All other authors declare that they have no conflict of interest.
Shintani, Y. , Okada, A. , Morita, Y. , Hamatani, Y. , Amano, M. , Takahama, H. , Amaki, M. , Hasegawa, T. , Ohta‐Ogo, K. , Kanzaki, H. , Ishibashi‐Ueda, H. , Yasuda, S. , Shimazaki, C. , Yoshinaga, T. , Yazaki, M. , Sekijima, Y. , and Izumi, C. (2019) Monitoring treatment response to tafamidis by serial native T1 and extracellular volume in transthyretin amyloid cardiomyopathy. ESC Heart Failure, 6: 232–236. 10.1002/ehf2.12382.
References
- 1. Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté‐Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceição IM, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Wilson A, Grogan DR. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012; 79: 785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ando Y, Sekijima Y, Obayashi K, Yamashita T, Ueda M, Misumi Y, Morita H, Machii K, Ohta M, Takata A, Ikeda S. Effects of tafamidis treatment on transthyretin (TTR) stabilization, efficacy, and safety in Japanese patients with familial amyloid polyneuropathy (TTR‐FAP) with Val30Met and non‐Val30Met: a phase III, open‐label study. J Neurol Sci 2016; 362: 266–271. [DOI] [PubMed] [Google Scholar]
- 3. Martinez‐Naharro A, Treibel TA, Abdel‐Gadir A, Bulluck H, Zumbo G, Knight DS, Kotecha T, Francis R, Hutt DF, Rezk T, Rosmini S, Quarta CC, Whelan CJ, Kellman P, Gillmore JD, Moon JC, Hawkins PN, Fontana M. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol 2017; 70: 466–477. [DOI] [PubMed] [Google Scholar]
- 4. Benson MD, Teague SD, Kovacs R, Feigenbaum H, Jung J, Kincaid JC. Rate of progression of transthyretin amyloidosis. Am J Cardiol 2011; 108: 285–289. [DOI] [PubMed] [Google Scholar]
- 5. Amano M, Izumi C, Nishimura S, Kuroda M, Sakamoto J, Tamaki Y, Enomoto S, Miyake M, Tamura T, Kondo H, Nakagawa Y. Predictors of prognosis in light‐chain amyloidosis and chronological changes in cardiac morphology and function. Am J Cardiol 2017; 120: 2041–2048. [DOI] [PubMed] [Google Scholar]
- 6. Fritschka M, Schlegl M, Borges A, Werner M, Gebker R, Pieske B, Kelle S. Unusual case of ATTR amyloidosis with cardiac manifestation and situs inversus totalis. Clin Res Cardiol 2017; 106: 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merlini G, Planté‐Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, Packman J, Tripp T, Grogan DR. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non‐Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res 2013; 6: 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Damy T, Judge DP, Kristen AV, Berthet K, Li H, Aarts J. Cardiac findings and events observed in an open‐label clinical trial of tafamidis in patients with non‐Val30Met and non‐Val122Ile hereditary transthyretin amyloidosis. J Cardiovasc Transl Res 2015; 8: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, Quyyumi AA, Aarts J, Falk RH. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail 2015; 8: 519–526. [DOI] [PubMed] [Google Scholar]
- 10. Rosenblum H, Castano A, Alvarez J, Goldsmith J, Helmke S, Maurer MS. TTR (transthyretin) stabilizers are associated with improved survival in patients with TTR cardiac amyloidosis. Circ Heart Fail 2018; 11: e004769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington‐Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018; 379: 1007–1016. [DOI] [PubMed] [Google Scholar]
- 12. Koike H, Ando Y, Ueda M, Kawagashira Y, Iijima M, Fujitake J, Hayashi M, Yamamoto M, Mukai E, Nakamura T, Katsuno M, Hattori N, Sobue G. Distinct characteristics of amyloid deposits in early‐ and late‐onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci 2009; 287: 178–184. [DOI] [PubMed] [Google Scholar]
- 13. Sekijima Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther 2014; 39: 225–233. [DOI] [PubMed] [Google Scholar]
- 14. Izumiya Y, Takashio S, Oda S, Yamashita Y, Tsujita K. Recent advances in diagnosis and treatment of cardiac amyloidosis. J Cardiol 2018; 71: 135–143. [DOI] [PubMed] [Google Scholar]
- 15. Fujita T, Inomata T, Kaida T, Iida Y, Ikeda Y, Nabeta T, Ishii S, Maekawa E, Naruke T, Koitabashi T, Kitamura E, Sekijima Y, Ako J. Tafamidis for the treatment of hereditary transthyretin amyloid cardiomyopathy: a case report. Cardiology 2017; 137: 74–77. [DOI] [PubMed] [Google Scholar]