

Abstract
Over the past 25 years, substantial advances have been made in our understanding of the cellular and molecular pathways that are essential to maintain a state of health in the mammalian gastrointestinal tract, an organ that is densely colonized with both immune cells and trillions of microbes. Seminal studies in the 1990’s identified that several cytokines, antigen-presentation molecules, and components of the T cell receptor were necessary to prevent the development of spontaneous intestinal inflammation in mice. Subsequent research revealed that these pathways orchestrate beneficial interactions with intestinal microbes, involve complex communication between innate and adaptive immune cells, and can be dysregulated in human inflammatory bowel disease. Here we discuss how these early findings set the stage for numerous other advances and shaped our current knowledge of host-microbiota interactions and intestinal homeostasis in mammals. It is expected that continued investigation of these areas will define novel immunologic mechanisms of tolerance and inflammation in the intestine that can be exploited to benefit human health.
One sentence summary:
This review discusses how seminal findings have shaped our current understanding of immunologic homeostasis and host-microbiota interactions in the mammalian intestine.
Introduction
The anatomical landscape of the mammalian intestine creates a number of considerable challenges to maintain a state of health. The organ system possesses an enormous surface area in which a single layer of intestinal epithelial cells segregates a significant portion of the immune system from trillions of intestinal microorganisms, termed the microbiota, which is comprised of commensal bacteria, fungi, protozoa and viruses (1). Intestinal homeostasis depends on a physical separation of the majority of intestinal microbiota from the mammalian immune system, and this is accomplished through numerous biophysical and biochemical barriers, such as the production of tight junctions, anti-microbial proteins and mucus by the host epithelium. A breakdown in these protective barriers causes aberrant immune responses to the microbiota and is a hallmark of multiple chronic inflammatory diseases, include inflammatory bowel disease (IBD), HIV/AIDS, viral hepatitis, cardiovascular disease, and cancer (2). Despite physical separation, there is a dynamic and reciprocal dialogue between the microbiota, host epithelium, and immune system, which is also essential to regulate a state of intestinal health. The mammalian immune system recognizes and responds to intestinal microbiota, promoting protective innate and adaptive immune pathways that reinforce barrier function, prime protective immune responses to pathogens, and maintain tolerance to beneficial microbiota or food antigens (3). These immune responses also shape the composition of the intestinal microbiota by exerting pressure or tolerance against selective microbes, or by providing nutrient substrates through mucus production or epithelial fucosylation (3). Here we discuss specific immune pathways identified in mice and humans that are essential to regulate intestinal homeostasis, and consider how these findings have shaped our understanding of host-microbe interactions and created a foundation for future therapeutic strategies in inflammatory bowel disease.
T cell- and cytokine-dependent regulation of intestinal health
In 1993, three seminal studies identified that several lines of novel transgenic mice with deficiencies in the immune system develop spontaneous and chronic intestinal inflammation. These included mice deficient in interleukin (IL-)2, IL-10, major histocompatibility complex class II (MHCII), and subunits of the T cell receptor (TCR) (4–6). The results of these studies were surprising and highlighted for the first time an essential role of the immune system in orchestrating intestinal homeostasis.
While it was previously appreciated that IL-10 is a critical immunoregulatory cytokine that limits T cell and macrophage responses (7), the finding that Il10−/− mice develop spontaneous intestinal inflammation solidified an essential role for this pathway in regulating intestinal health. Mice lacking IL-10 develop chronic colitis characterized by extensive mucosal hyperplasia and aberrant inflammatory leukocyte infiltration (4). Additional studies revealed that mice lacking a subunit of the IL-10 receptor, IL-10Rβ, also develop spontaneous intestinal inflammation (8). CD4+ T cells are major mediators of colitis in IL-10- and IL-10R-deficient mice, rather than B cells, and initial studies suggested an association with increased T helper (Th)1 cells and IFNγ production. IL-10 directly inhibits IL-12 production in myeloid cells, and therefore limits Th1 cell differentiation (9, 10). In addition to IL-12, IL-10 also suppresses IL-23 production through inhibition of the shared IL-12p40 subunit at the transcriptional level (10). IL-23 is essential for maintenance and expansion of pathogenic Th17 cells, and therefore IL-10 also limits pathogenic Th17 cell responses by controlling IL-23 induction during mucosal inflammation.
Despite these advances, the exact cellular and molecular pathways by which IL-10 mediates intestinal health were incompletely understood. T cells, B cells, macrophages and dendritic cells (DCs), as well as several non-hematopoietic cells, have been reported as potential cellular sources of IL-10 in the mammalian intestine (10). Cre-/lox technology in mice permitted lineage-specific deletion of IL-10 in specific cell types and revealed that T cell-derived IL-10 was the critical cellular source to regulate mucosal homeostasis (11), and many of these phenotypes could be recapitulated with lineage-specific deletion of IL-10 in FoxP3+ Tregs (12). Expression of IL-10R and STAT3 are also critical in FoxP3+ Tregs to limit spontaneous intestinal inflammation, however over-expression of a dominant negative IL-10R with a CD4-cre was not sufficient to induce spontaneous disease (13, 14). In contrast, expression of IL-10R, but not IL-10, was essential on myeloid cells to condition monocyte-derived anti-inflammatory macrophages and prevent spontaneous intestinal inflammation (15, 16). Thus, Treg-derived IL-10 drives macrophages to execute tolerogenic functions and maintain mucosal homeostasis. Recent studies have clarified that the molecular basis of the anti-inflammatory function of IL-10 in macrophages occurred in part by metabolic reprogramming. In response to inflammatory stimuli, IL-10 regulates cellular metabolism in macrophages by suppressing mTOR activity through the induction of its inhibitor, DDIT4, and preventing glucose uptake and glycolysis, while promoting oxidative phosphorylation (17). In IL-10-deficient mice, dysfunctional mitochondria accumulate in macrophages, resulting in activation of the NLRP3 inflammasome and production of IL-1β. Consistent with this, impairing the inflammasome with caspase-1 deficiency could partially protect IL-10-deficient mice from the development of spontaneous intestinal inflammation (Figure 1).
Figure 1. IL-10-dependent regulation of intestinal health.

T cells, especially Treg cells, are the critical cellular source of IL-10 in the mammalian intestine. The gut microbiota is critical for the induction of intestinal Treg cells and IL-10 production. B. fragilis–derived PSA acts on DCs, resulting in IL-10 expression. In addition, bacteria-stimulated macrophage can also directly secrete IL-10 and support Treg cell development. Thus, DC-derived IL-10 binds to IL-10R expressed on Treg cells, which induces STAT3 activation and the expression of abundant IL-10. IL-10 limits TH1 and TH17 cell differentiation through inhibition of IL-12 and IL-23, respectively. Treg-derived IL-10 drives macrophages to execute tolerogenic functions and maintain intestinal homeostasis. In addition, IL-10 regulates cellular metabolism in macrophages by suppressing mTOR activity, thus reducing glucose uptake and glycolysis while promoting oxidative phosphorylation. Essential pathways that prevent spontaneous inflammation are highlighted in red.
IL-2 deficient mice develop a spontaneous and progressive colonic inflammation with ulceration and bloody diarrhea (6). These results were paradoxical, as prior studies had defined an important role of IL-2 in promoting T cell responses. Similar intestinal inflammation also develops in mice lacking the high-affinity IL-2 receptor alpha subunit, CD25 (18). Elevated levels of both T cells and B cells were characterized in the colonic lamina propria of aged IL-2- or CD25-deficient mice. The infiltrating T cells of such mice are substantially increased in numbers, activation and proliferation (6, 18). Crossing IL-2-deficient mice with Rag2-deficient mice resulted in an absence of colitis, demonstrating an essential role for adaptive immunity in disease progression (19). It was initially hypothesized that the loss of IL-2 producing CD4+ Th1 cells leads to a predominance of IL-4 producing Th2 cells and subsequent hyper-reactivity of mucosal B cells. However, later studies demonstrated that T cells, not B cells, are required for spontaneous intestinal inflammation in IL-2-deficient mice (19). The IL-2-dependent regulation of CD4+ T cells were later refined with the identification of additional heterogeneity of T helper cells, and it is now appreciated that IL-2 promotes the differentiation of Th1 cells, Th2 cells and Treg cells while inhibiting Th17 cells (20). Among these findings, the appreciation of IL-2-dependent Treg cells was one of the most significant advances. Treg cells are vital for preventing autoimmunity, limiting inflammatory responses and maintaining immune homeostasis, particularly within the gastrointestinal tract (21). The development and expansion of inducible Treg cells requires IL-2, and IL-2 sequestration is also a critical mechanism by which Treg cells can suppress effector T cell responses (21). Consistent with this, Treg cells are largely absent in IL-2- or IL-2R-deficient mice, and lineage-specific deletion of the IL-2R on Treg cells was recently found to promote spontaneous intestinal inflammation and other autoimmune diseases (22). The relevant cellular sources of IL-2 in regulating intestinal homeostasis have yet to be defined, but expression has been observed in T cells, DCs, NK cells and innate lymphoid cells (21). In short, these studies define a critical role for T cells as a target of IL-2 and led to a greater understanding of Treg cells in intestinal homeostasis (Figure 2).
Figure 2. IL-2-dependent regulation of intestinal health.

During intestinal homeostasis, IL-2 is produced by activated CD4+ T cells, DCs, and other unknown cells. IL-2 is mainly consumed by regulatory T cells and activated CD4+ T cells. It is now widely appreciated that IL-2 promotes the differentiation of TH1 cells, TH2 cells, and Treg cells while inhibiting TH17 cells. Moreover, Treg cells efficiently sequester IL-2 from effector T cells and use it to maintain high levels of CD25 and Foxp3 expression, enhancing its suppressive capacity. Essential pathways that prevent spontaneous inflammation are highlighted in red.
Beyond IL-2 and IL-10, spontaneous and chronic intestinal inflammation was also reported in mice lacking TCRα, TCRβ, TCRβ and TCRδ, and MHCII (5). The intestinal disease in these mice exhibit chronic diarrhea, wasting syndrome associated with anorectal prolapse. However, athymic or Rag1−/− mice did not exhibit any spontaneous disease, suggesting that dysfunction of αβ T cells, especially MHCII restricted CD4+ T cells, may underlie the pathogenesis of intestinal inflammation (5). A recent study found that absence of MHCII on conventional DCs resulted in chronic intestinal inflammation as well, suggesting that DC-mediated CD4+ T cell priming and subsequent adaptive immune responses, including Treg differentiation, are essential to maintain homeostasis (23) (Figure 3). Collectively, these seminal findings drove a field forward towards extensive investigation into immunoregulatory cytokines, CD4+ T cells, and innate immune populations to better understand intestinal health and disease.
Figure 3. Roles for antigen-presentation in regulating intestinal homeostasis.
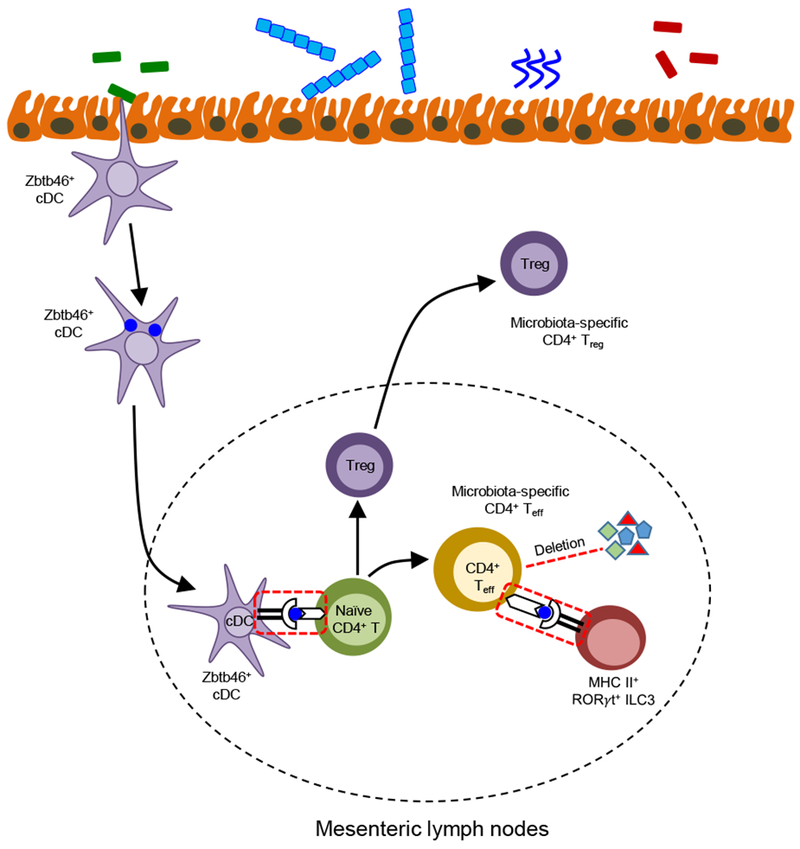
Microbiota are sampled by intestinal Zbtb46+ conventional DCs (cDCs), and bacteria-loaded cDCs then migrate to the draining mesenteric lymph nodes and activate naïve CD4+ T cells by presenting commensal bacteria–derived antigen via MHC-II, resulting in the differentiation of effector and regulatory microbiota–specific CD4+ T cells. MHC-II+ RORγt+ ILC3s contact antigen-experienced T effector cells, resulting in deletion of commensal bacteria–specific CD4+ effector T cells, a process termed intestinal selection. In addition, activated CD4+ T cells partly differentiate into Treg cells, which migrate to lamina propria and regulate intestinal homeostasis. Essential pathways that prevent spontaneous inflammation are highlighted in red.
Reciprocal host-microbiota interactions critically influence intestinal health
Housing conditions can dramatically influence the onset and location of intestinal disease in many of the above-described transgenic mice, and enteric microbes were subsequently found to be necessary for the development of spontaneous intestinal inflammation (1). IL-10-deficient mice re-derived under germ-free conditions failed to develop colonic inflammation, and this could be recapitulated by treatment with broad-spectrum antibiotics before disease onset. Antibiotics also attenuate disease severity in Il10−/− mice after the development of spontaneous colitis, and germ-free mice Il10−/− mice develop chronic colitis after colonization with normal enteric microflora or selected species of microbes derived from healthy wild-type mice (24, 25). It is noteworthy that Helicobacter is indispensable for colonic inflammation in Il10−/− mice, however, Helicobacter alone is insufficient to induce disease, suggesting that alteration in the microbial community or change in bacterial metabolism may affect colitis development (26). Consistent with this, IL-10 deficient mice have reduced probiotic bacteria, such as Lactobacillus, and treating IL-10 deficient mice with different species of Lactobacilli effectively prevents or attenuates colitis (27). A probiotic bacteria mixture containing a combination of eight bacteria treatment could also significantly ameliorate the development of colitis in Il10−/− mice with established disease (28). Thus, IL-10 can influence the colonization of selective bacteria species in the gastrointestinal tract and reciprocally the microflora exerts a crucial role in preventing or controlling disease progression.
Attenuated colitis is also observed in IL-2-, TCRα- or TCRβ-deficient mice housed in a specific pathogen free environment, and no clinical or histologic evidence of colitis in germ-free conditions, although a mild, focal, and non-lethal colonic inflammation was reported in older gnotobiotic IL-2-deificient mice (29, 30). Subsequent studies revealed that abundant Escherichia (E.) coli accumulate in Il2−/− mice, whereas limited E. coli was detected in control mice. Mono-colonization of gnotobiotic Il2−/− mice with the E. coli strain mpk, but not E.coli Nissle, is sufficient to induce colonic inflammation. Also, B. vulgatus mpk protects against E. coli mpk induced colitis in gnotobiotic Il2−/− mice (31, 32). In addition, the absence of MHCII on conventional DCs resulted intestinal inflammation, which could be alleviated by antibiotic treatment and entirely averted under germ-free conditions (23). Taken together, these studies demonstrate that the intestinal microbiota are essential for the development of spontaneous intestinal inflammation in mice, and are likely targets of aberrant immune responses when there is a disruption in immunologic homeostasis. Further, they imply that there are strain-specific differences in microbes that critically influence intestinal disease.
Subsequent seminal studies have revealed that the intestinal microbiota critically influence the composition of lamina propria CD4+ T cell subsets, in particular Th17 and Treg cells. Colonization of mice with segmented filamentous bacteria (SFB) results in a dramatic expansion of Th17 cells. SFB could penetrate the mucus layer in the terminal ileum, and closely interact with the epithelia cells, resulting in Th17 differentiation within the lamina propria through MHCII-dependent antigen presentation of SFB antigens by intestinal dendritic cells and induction serum amyloid A from intestinal epithelial cells (33–35). Recently, it was also found that a number of other intestinal microbes are sufficient to induce Th17 cell responses in the intestine, in part through similar epithelial adhesion mechanisms (36). Gut microbes are also important for intestinal Treg cell differentiation. Colonization of gnotobiotic mice with a complex cocktail of cluster IV and XIVa Clostridia strains increased the development of IL-10 expressing inducible Treg cells, which occurs in part through potent induction of TGFβ (37, 38). B. fragilis derived polysaccharide A (PSA) can also promote accumulation of colonic IL-10 expressing Treg cells via the TLR2-MyD88 signaling pathway (39) (Figure 1). Further, Clostridia species produce short chain fatty acids (SCFAs), such as acetate, propionate and butyrate, resulting from the fermentation of dietary fiber. SCFAs are absorbed by colonic epithelial cells or diffuse into lamina propria, and help to induce colonic Treg cell differentiation through multiple proposed mechanisms (40–42).
Defects in the host immune system reciprocally influences microbial communities and causes intestinal dysbiosis, which in some contexts is sufficient to drive chronic inflammation in the gut. For example, mice lacking both the transcription factor T-bet and Rag2, named TRUC mice, develop spontaneous colonic inflammation in a microbiota dependent manner. Remarkably, this phenomenon was horizontally transmissible to wild-type mice by either co-housing or adoptive transfer of the TRUC mice microbiota. It was proposed that loss of T-bet in the innate immune system leads to overproduction of TNFα in DCs and subsequently induces epithelial cell death (43). Later studies uncovered that Helicobacter typhlonius may drive excess TNFα production and promote colitis in TRUC mice (44). Another example of dysbiosis driven by immunologic defects is observed in mice deficient for the inflammasome component NLRP6. These mice displayed spontaneous colitis that was communicable to wild-type mice after cohousing. Prevotellaceae was recognized as a causative agent of this inflammatory phenotype (45). However, these results have recently been called into question by a recent report housing littermate controls that didn’t observe any dysbiosis or spontaneous disease (46). These findings suggest that disruption of immune responses in the gut can induce microbial dysbiosis, and dysbiosis alone can drive the intestinal inflammation in several contexts.
Innate lymphoid cell regulation of intestinal homeostasis
The IL-10Rβ chain is also utilized by another member of the IL-10 cytokine family, IL-22, which critically influences intestinal health and homeostasis by acting exclusively on non-hematopoietic cells. IL-22 is a pleiotropic cytokine that promotes intestinal tissue repair, protects from intestinal pathogens, and anatomically-restricts select intestinal microbiota (47–49). These functions are in part due to IL-22-mediated induction of mucus or antimicrobial proteins in intestinal epithelial cells, including REGIIIβ, REGIIIγ, S100A8 and S100A9, which limit colonization with commensal bacteria, such as SFB. Conversely, IL-22 also induces fucosylation of intestinal epithelial cells, which promotes colonization with beneficial bacteria and protects from Salmonella typhimurium and Citrobacter rodentium infection (50–52). IL-22 is also context dependent and can promote intestinal inflammation in response to Toxoplasma gondii infection or in models of innate cell-mediated colitis (47).
Although originally thought to be a T cell-derived cytokine, studies of IL-22 contributed to recent revolutionary findings in mice and humans that identified populations of innate lymphocytes, known as innate lymphoid cells (ILCs), and defined these cells as critical regulators of immunity, inflammation and homeostasis in the intestine. ILCs are subdivided into three subgroups based on their transcription factor expression profile, including T-bet+ group 1 ILCs (ILC1), GATA3+ group 2 ILCs (ILC2) and RORγt+ group 3 ILCs (ILC3) (53). Of note, ILC3 contribute to intestinal health through several distinct pathways. ILC3 have a bidirectional relationship with the gut microbes, as the development and activation of ILC3 are regulated by intestinal microbiota-derived signals, and further, the effector function of ILC3 influences microbial composition and intestinal homeostasis (54). In response to myeloid cell derived IL-1β and IL-23 after recognition of microbial signals or microbiota-derived metabolites (e.g., Aryl hydrocarbon receptor ligands), ILC3 produce IL-22, IL-17, IFNγ and GM-CSF (54, 55). Among these cytokines, IL-22 is dominantly produced by ILC3 in the intestine and associated lymphoid tissues. During homeostasis, disruption of the ILC3-IL-22 pathway leads to defects in anatomical containment of selective microbiota that reside in the lymphoid tissue, termed lymphoid tissue-resident commensal bacteria (LRCs) (48). LRCs also induce IL-22 production by ILC3 and myeloid cell-derived IL-10, which collectively enhance LRC colonization and protected mice from lethal intestinal damage (56). IL-22 production by ILC3 is also essential for regulating the composition of intestinal microbes and facilitating colonization resistance. Resistance to intestinal C. rodentium infection in mice is dependent on IL-22 signaling and ILC3 is the dominant source of IL-22 in the first week after infection, then T cell derived IL-22 and B cells contribute largely to resistance and clearance of the infection (49, 57). Other than IL-22, ILC3-derived IL-17, which can act alone or synergistically with IL-22, promotes anti-microbial peptide production from IECs and induces chemokine expression to recruit neutrophils during pathogen infection (47, 49).
ILC3s also regulate intestinal homeostasis or inflammation through direct and indirect interaction with adaptive immune responses. ILC3 secretes lymphotoxin-α3 (LTα3) or express surface lymphotoxin-α1β2 (LTα1β2), which stimulates the T cell-dependent or independent production of IgA, respectively, thereby modulating the composition of the gut microflora (58). ILC3 regulate homeostasis of myeloid cells in the intestine through production of GM-CSF, which promotes Treg cell responses to food antigens and maintains oral tolerance (55) (Figure 1). ILC3 were also found to express high levels of MHCII and directly present antigens to CD4+ T cells. Surprisingly, this interaction results in the inhibition of microbiota-specific CD4+ T cells, and genetic deletion of ILC3-intrisic MHCII leads to the development of spontaneous CD4+ T cell-dependent intestinal inflammation (59). Mechanistically, MHCII+ ILC3 mediated cell death of microbiota-specific CD4+ T cells in the intestine and this has been termed “intestinal selection” given a number of significant similarities to what occurs during thymic selection, and further establishes a paradigm of how antigen presentation regulates intestinal health (60) (Figure 3).
Essential immunologic pathways in human IBD
Human IBD involve a complex interplay between genetic risk factors and extrinsic environmental triggers, such as the microbiota. Genome-wide association studies (GWAS) have identified about 200 susceptibility genes associated with IBD, most of which are involve in regulating intestinal barrier function and host-microbe interactions (61, 62). These analyses helped to provide insight into disease pathobiology and promote advances in diagnostics and therapies. IL-10 and IL-10R signaling were early on recognized as a human IBD risk allele through GWAS (61, 63), and further studies also identified IL-2 and IL-2R (64). HLA polymorphisms also highly segregate IBD cohorts, suggesting a critical role for antigen presentation (62). Genomic loss-of-function mutations in IL10, IL10RA, IL10RB and IL2RA have also been described to cause a unique form of IBD that develops at early stages of life, termed very early onset IBD (VEO-IBD), and is thought to be the result of a monogenic mutations (65). However, these mutations only represent a small population of VEO-IBD patients and advances in whole exome sequencing are rapidly identifying novel variants.
The role of ILCs in human IBD is still in the early stages of investigation, however, several studies have reported that ILC frequencies or responses are altered in the context of Crohn’s disease. An expansion of IL-17-producing and IL-22-producing ILC3 was noted in the inflamed mucosa of patients with Crohn’s disease, and these were dependent upon the fecal stream, suggesting either a pathogenic or compensatory protective role (66, 67). In contrast, other studies have suggested that a reduction in ILC3 occurs in Crohn’s disease, and that human ILC3 may exhibit plasticity, differentiate into pro-inflammatory ILC1 or ex-ILC3, and produce IFNγ (68). Furthermore, the expression of MHCII on ILC3 is lower in patients with Crohn’s disease compare to health controls, and the reduced expression of MHCII is inversely correlated with intestinal Th17 cells, which suggests that ILC3 may limit pathogenic T cells through MHCII in Crohn’s disease as described in mice (59, 60).
Various therapeutic advances have been applied to clinical management of patients with IBD (Figure 4). Among these therapeutic strategies, blockade of TNF has provided substantial therapeutic benefit in some but not all patients, and further patients can go on to loose responsiveness (69). There has been an urgent search for other effective cytokine blockade strategies. The IL-12 p35-p40 and IL-23 p19-p40 are two heterodimeric pro-inflammatory cytokines that are induced in the inflamed mucosa in patients with Crohn’s disease and represent a promising target. Ustekinumab, an anti-p40 monoclonal antibody therapy, was recently approved by the FDA for treatment of moderate to severe Crohn’s disease. However, several cytokine blockade trials have yielded disappointing results. For example, blockade of IL-17 resulted in aggravation of Crohn’s disease and increased susceptibility to fungal infections in a number of patients (70). The reason possibly owing to the protective roles of IL-17 on intestinal epithelial cells, or disrupting ILC3-IL-17 mediated protection from opportunistic fungal infection (54, 71). Other promising therapies that were recently approved, or are in the clinical or pre-clinical trials, include targeting the integrin α4β7 to limit immune cells recruitment to the intestine, and small molecule inhibitors targeting transcriptional regulators or kinases downstream of cytokine receptors (69). In particular, targeting the ILC3 and Th17 cell transcription factor RORγt may hold particular promise givens its ability to selectively target pro-inflammatory responses while preserving tissue protection and innate immunity (72).
Fig. 4. Current and potential therapeutic targets in IBD.
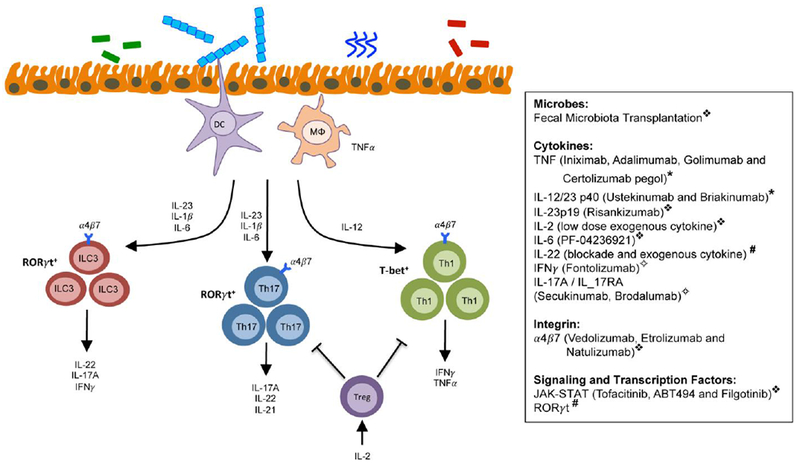
A number of current and potential future targets exist to provide therapeutic benefit in the context of IBD. These targets influence the generation, maintenance, or effector function of innate and adaptive immune cells. Symbol definitions: ❖, efficacy exhibited in clinical trials; *, FDA-approved and providing clinical benefit; #, promising preclinical target in mouse models; ✧, failed or poor efficacy in clinical trials.
The balance of effector and Treg cells in intestinal homeostasis suggest a therapeutic potential of boosting Treg cells in human IBD. Clinical trials and studies have shown that low-dose IL-2 specifically expands and activates Treg cell populations and thus might be a promising strategy for human IBD therapy (73). Further, other strategies aim at delivering Treg cell-inducing microbiota or microbiota-derived products, however the clinical efficacy of probiotic or fecal microbiota transplantation in IBD has thus far demonstrated limited efficacy.
Future directions and perspectives on host-microbiota interactions and intestinal homeostasis
Since the discovery of several transgenic mouse lines that develop spontaneous intestinal inflammation, there has been an astounding number of exciting scientific and technical advances that paved the way for a better understanding of the immune mechanisms that maintain a state of health in the mammalian gastrointestinal tract. Immunologic mediators remain the primary therapeutic targets to limit inflammation in IBD, but there is an urgent need to develop safe and effective strategies that hold preventative or ideally curative potential. Recent appreciation of sophisticated and reciprocal host-microbiota interactions will also allow advanced determination of whether we can harness the microbiota or related byproducts as a tractable treatment or target. These goals can only be achieved by continued investigation of mouse models and innovative exploration of patient oriented translation research that will continually challenge paradigms and advance our scientific understanding of intestinal health towards preventative and curative strategies of intestinal diseases.
Acknowledgements
We thank members of the Sonnenberg laboratory for discussions and critical reading of this manuscript. Research in the Sonnenberg laboratory is supported by the National Institutes of Health (DP5OD012116, R01AI123368, R21DK110262 and U01AI095608), the NIAID Mucosal Immunology Studies Team (MIST), the Crohn’s and Colitis Foundation of America, the Searle Scholars Program, the American Asthma Foundation Scholar Award, and Pilot Project Funding from the Center for Advanced Digestive Care (CADC).
References
- 1.Garrett WS, Gordon JI, Glimcher LH, Homeostasis and inflammation in the intestine. Cell 140, 859 (March 19, 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honda K, Littman DR, The microbiome in infectious disease and inflammation. Annu Rev Immunol 30, 759 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooper LV, Littman DR, Macpherson AJ, Interactions between the microbiota and the immune system. Science 336, 1268 (June 08, 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W, Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263 (October 22, 1993). [DOI] [PubMed] [Google Scholar]
- 5.Mombaerts P et al. , Spontaneous development of inflammatory bowel disease in T cell receptor mutant mice. Cell 75, 274 (October 22, 1993). [DOI] [PubMed] [Google Scholar]
- 6.Sadlack B et al. , Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 75, 253 (October 22, 1993). [DOI] [PubMed] [Google Scholar]
- 7.Moore KW, O’Garra A, de Waal Malefyt R, Vieira P, Mosmann TR, Interleukin-10. Annu Rev Immunol 11, 165 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Spencer SD et al. , The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med 187, 571 (February 16, 1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson NJ et al. , T helper cell 1-type CD4+ T cells, but not B cells, mediate colitis in interleukin 10-deficient mice. J Exp Med 184, 241 (July 01, 1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG, Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol 29, 71 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Roers A et al. , T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med 200, 1289 (November 15, 2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rubtsov YP et al. , Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546 (April, 2008). [DOI] [PubMed] [Google Scholar]
- 13.Chaudhry A et al. , Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566 (April 22, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber S et al. , Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554 (April 22, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shouval DS et al. , Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 40, 706 (May 15, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zigmond E et al. , Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 40, 720 (May 15, 2014). [DOI] [PubMed] [Google Scholar]
- 17.Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R, Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513 (May 05, 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willerford DM et al. , Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3, 521 (October, 1995). [DOI] [PubMed] [Google Scholar]
- 19.Ma A, Datta M, Margosian E, Chen J, Horak I, T cells, but not B cells, are required for bowel inflammation in interleukin 2-deficient mice. J Exp Med 182, 1567 (November 01, 1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boyman O, Sprent J, The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 12, 180 (February 17, 2012). [DOI] [PubMed] [Google Scholar]
- 21.Josefowicz SZ, Lu LF, Rudensky AY, Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol 30, 531 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chinen T et al. , An essential role for the IL-2 receptor in Treg cell function. Nat Immunol 17, 1322 (November, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loschko J et al. , Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J Exp Med 213, 517 (April 04, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madsen KL et al. , Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology 118, 1094 (June, 2000). [DOI] [PubMed] [Google Scholar]
- 25.Sellon RK et al. , Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 66, 5224 (November, 1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dieleman LA et al. , Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect Immun 68, 5107 (September, 2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madsen KL, Doyle JS, Jewell LD, Tavernini MM, Fedorak RN, Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology 116, 1107 (May, 1999). [DOI] [PubMed] [Google Scholar]
- 28.Madsen K et al. , Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 121, 580 (September, 2001). [DOI] [PubMed] [Google Scholar]
- 29.Dianda L et al. , T cell receptor-alpha beta-deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol 150, 91 (January, 1997). [PMC free article] [PubMed] [Google Scholar]
- 30.Schultz M et al. , IL-2-deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am J Physiol 276, G1461 (June, 1999). [DOI] [PubMed] [Google Scholar]
- 31.Schuppler M, Lotzsch K, Waidmann M, Autenrieth IB, An abundance of Escherichia coli is harbored by the mucosa-associated bacterial flora of interleukin-2-deficient mice. Infect Immun 72, 1983 (April, 2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waidmann M et al. , Bacteroides vulgatus protects against Escherichia coli-induced colitis in gnotobiotic interleukin-2-deficient mice. Gastroenterology 125, 162 (July, 2003). [DOI] [PubMed] [Google Scholar]
- 33.Goto Y et al. , Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 40, 594 (April 17, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivanov II et al. , Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485 (October 30, 2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sano T et al. , An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163, 381 (October 08, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atarashi K et al. , Th17 Cell Induction by Adhesion of Microbes to Intestinal Epithelial Cells. Cell 163, 367 (October 08, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Atarashi K et al. , Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232 (August 08, 2013). [DOI] [PubMed] [Google Scholar]
- 38.Atarashi K et al. , Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337 (January 21, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Round JL et al. , The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332, 974 (May 20, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arpaia N et al. , Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451 (December 19, 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furusawa Y et al. , Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446 (December 19, 2013). [DOI] [PubMed] [Google Scholar]
- 42.Smith PM et al. , The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569 (August 02, 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garrett WS et al. , Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33 (October 05, 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powell N et al. , The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 37, 674 (October 19, 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elinav E et al. , NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745 (May 27, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mamantopoulos M et al. , Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 47, 339 (August 15, 2017). [DOI] [PubMed] [Google Scholar]
- 47.Sonnenberg GF, Fouser LA, Artis D, Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 12, 383 (May, 2011).21502992 [Google Scholar]
- 48.Sonnenberg GF et al. , Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336, 1321 (June 08, 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng Y et al. , Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14, 282 (March, 2008). [DOI] [PubMed] [Google Scholar]
- 50.Goto Y et al. , Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 345, 1254009 (September 12, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pham TA et al. , Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe 16, 504 (October 08, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pickard JM et al. , Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature 514, 638 (October 30, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spits H et al. , Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol 13, 145 (February, 2013). [DOI] [PubMed] [Google Scholar]
- 54.Sonnenberg GF, Artis D, Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37, 601 (October 19, 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mortha A et al. , Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 343, 1249288 (March 28, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fung TC et al. , Lymphoid-Tissue-Resident Commensal Bacteria Promote Members of the IL-10 Cytokine Family to Establish Mutualism. Immunity 44, 634 (March 15, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D, CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122 (January 28, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsuji M et al. , Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261 (August 15, 2008). [DOI] [PubMed] [Google Scholar]
- 59.Hepworth MR et al. , Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113 (June 06, 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hepworth MR et al. , Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 348, 1031 (May 29, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khor B, Gardet A, Xavier RJ, Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307 (June 15, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jostins L et al. , Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119 (November 01, 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Glocker EO et al. , Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 361, 2033 (November 19, 2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharfe N, Dadi HK, Shahar M, Roifman CM, Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proc Natl Acad Sci U S A 94, 3168 (April 01, 1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benchimol EI et al. , Incidence, outcomes, and health services burden of very early onset inflammatory bowel disease. Gastroenterology 147, 803 (October, 2014). [DOI] [PubMed] [Google Scholar]
- 66.Geremia A et al. , IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med 208, 1127 (June 06, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Longman RS et al. , CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med 211, 1571 (July 28, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bernink JH et al. , Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14, 221 (March, 2013). [DOI] [PubMed] [Google Scholar]
- 69.Neurath MF, Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol 14, 269 (May, 2017). [DOI] [PubMed] [Google Scholar]
- 70.Hueber W et al. , Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693 (December, 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JS et al. , Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 43, 727 (October 20, 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Withers DR et al. , Transient inhibition of ROR-gammat therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat Med 22, 319 (March, 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Klatzmann D, Abbas AK, The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol 15, 283 (May, 2015). [DOI] [PubMed] [Google Scholar]