

Abstract
Fatty Acid Amide Hydrolase (FAAH) is a serine hydrolase that plays a key role in controlling endogenous levels of endocannabinoids. FAAH inhibition is considered a powerful approach to enhance the endocannabinoid signalling, and therefore it has been largely studied as a potential target for the treatment of neurological disorders such as anxiety or depression, or of inflammatory processes. We present two novel series of amide derivatives of ibuprofen designed as analogues of our reference FAAH inhibitor Ibu-AM5 to further explore its structure-activity relationships. In the new amides, the 2-methylpyridine moiety of Ibu-AM5 was substituted by benzylamino and piperazinoaryl moieties. The obtained benzylamides and piperazinoarylamides showed FAAH inhibition ranging from the low to high micromolar potency. The binding of the new amides in the active site of FAAH, estimated using the induced fit protocol, indicated arylpiperazinoamides binding the ACB channel and the cytosolic port, and benzylamides binding the ACB channel.
Keywords: Ibuprofen amides, FAAH inhibition, fatty acid amide hydrolase, endocannabinoids, induced fit docking
Introduction
N-acylethanolamines (NAE) are endogenous lipid ligands that regulate numerous physiological functions in the body due to activation of cannabinoid receptors, peroxisome proliferator-activated receptor-alpha (PPAR-α), and other targets1. Arachidonoylethanolamide (anandamide, AEA), palmitoylethanolamide, oleoylethanolamide, stearoylethanolamide and linoleoylethanolamide are the principal N-acylethanolamines. Fatty acid amide hydrolase (FAAH) is a serine hydrolase enzyme largely responsible for the hydrolytic degradation of N-acylethanolamines. The FAAH catalytic mechanism exploits an unusual catalytic triad, Ser-Ser-Lys, in which the basic Lys142 activates the nucleophilic Ser241, involving the Ser217 as a “proton shuttle”2. Structurally, FAAH is a homodimer enzyme bound to the membrane3 (Figure 1(a)). Its binding cavity is characterised by a series of separate channels that are crucial for its biological function: (i) the membrane access channel (MAC) that connects the membrane-bound region with the enzyme active site; (ii) the acyl-chain binding channel (ACB) including the catalytic triad and residues involved in the substrate binding; (iii) the cytosolic port (CP), which represents a way out for the hydrophilic product of the substrates hydrolysation4 (Figure 1(b)).
Figure 1.

(a) 3D structure of the homo-dimer rat FAAH (rFAAH) model complexed with Anandamide (AEA). Monomer a and b are shown as green and orange cartoon, respectively. The membrane bilayer is indicated as dashed black line. (b) Details of the rFAAH binding cavity and channels. Key aminoacids of the binding cavity are highlighted as green sticks: Ser217:Ser241:Lys142 (catalytic triad), membrane access channel (MAC), the cytosolic port (CP) and the acyl-chain binding pocket (ACB).
A number of different classes of FAAH inhibitors have been described in the literature, including carbamate derivatives, α-ketoheterocycles, piperazinyl, and piperidinyl ureas and boronic acids5. Inhibition of FAAH increases NAE levels in the brain and other tissues, but does not produce the sorts of behaviours seen with Δ-tetrahydrocannabinol, the main psychoactive ingredient of cannabis6,7 thereby making the enzyme a potentially exciting target for drug development. In humans, most FAAH inhibitors are well-tolerated8–10, the exception being BIA10-2474 which produced its toxic effects by presumed off-target effects11,12. In animal models, FAAH inhibition produces potentially beneficial effects in a variety of animal models of pain, but this has not been translated into the clinic10,13,14. However, other indications remain of great interest, not least in the field of anxiety/depression15,16 and intestinal inflammation17,18.
In 1997, it was reported that the non-steroidal anti-inflammatory drug ibuprofen inhibited FAAH19. Although the potency was modest, the IC50 concentration was in the range that could be achieved in humans. The ability of ibuprofen to inhibit FAAH is shared by other profens such as flurbiprofen20 and carprofen21. In previous studies, our research group has reported the FAAH inhibitory activity of profen amides and showed that the amide of Ibuprofen with 2-amino-3-methylpyridine (Ibu-AM5) (Table 1) was two to three orders of magnitude more potent than ibuprofen itself as a reversible inhibitor of FAAH22,23. The compound has a much lower ulcerogenic potency than ibuprofen24. In other studies, we have explored the SAR of Ibu-AM5 analogues by modifying the 2-aminopyridine moiety25 and the isobutyl moiety26. Here, we present the synthesis, docking studies, and pharmacological characterisation of two new series of Ibuprofen derivatives, the benzylamides, and the piperazinoamides.
Table 1.
Maximum percentage and IC50 values for inhibition of rat brain AEA hydrolysis by compounds 3–16.
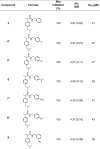 |
|||||||||||
 |
|||||||||||
 |
*Values with ethanol as solvent, taken from25. For the test compounds, the solvent was ethanol except when indicated with †, where DMSO was used. §The inhibition data was better fitted by a curve with a residual activity rather than a curve assuming 100% inhibition. The maximal inhibition is indicated (when it was greater than 50%), and the pI50 and IC50 values refer to the inhibitable portion of the curve. The inability of the compounds to produce a maximal inhibition was not investigated further. #Values for URB-597, as reference, with a preincubation time of 60 min, are taken from35.
Experimental
Materials
Anandamide [ethanolamine-1-3H] (specific activity 2.22 TBq mmol-1) was purchased from American Radiolabeled Chemicals, Inc (St. Louis, MO). All commercially available solvents and reagents were used without further purification and were purchased from Sigma-Aldrich (Milan, Italy).
Chemistry
NMR spectra were recorded on an Inova 500 spectrometer (Varian, Palo Alto, CA). The chemical shifts (δ) are reported in part per million downfield from tetramethylsilane (TMS), which was used as internal standard, and the spectra were recorded in hexadeuteriodimethylsulphoxide (DMSO-d6). Infra-red spectra were recorded on a Vector 22 spectrometer (Bruker, Bremen, Germany) in Nujol mulls. The main bands are given in cm−1. Positive-ion electrospray ionisation (ESI) mass spectra were recorded on a double-focusing MAT 95 instrument (Finnigan, Waltham, MA) with BE geometry. Melting points (mp) were determined on a SMP1 Melting Point apparatus (Stuart Scientific, Stone, UK) and are uncorrected. All products reported showed 1H NMR spectra in agreement with the assigned structures. The purity of the tested compounds was determined by combustion elemental analyses conducted by the Microanalytical Laboratory of the Chemistry Department of the University of Ferrara with a MT-5 CHN recorder elemental analyser (Yanagimoto, Kyoto, Japan) and the values found were within 0.4% of theoretical values.
General procedure for the synthesis of benzylamide derivatives 3–16
A solution of ibuprofen 1 (0.21 g, 1 mmol), 1–(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) (0.19 g, 1.1 mmol) and hydroxybenzotriazole (HOBt) (0.13 g, 1 mmol) in anhydrous acetonitrile (MeCN) (10 ml) was stirred at r.t. for 30 min. Then the appropriate substituted benzylamine 2 (1 mmol) was added. The mixture was then stirred for 24 h at r.t. After the solvent was removed under vacuum, the residue was dissolved in ethyl acetate (AcOEt) (20 ml) and washed sequentially with brine (2 × 5 ml), 10% citric acid (2 × 5 ml), NaHCO3 10% aqueous solution (2 × 5 ml) and water (2 × 5 ml). The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum. The obtained residue was tritured with iPr2O; the precipitate was then filtrated to obtain the compounds 3–16.
N-Benzyl-2–(4-isobutylphenyl)propanamide (3)
Obtained following the general procedure by the condensation between 1 and benzylamine. Yield 78%. m.p. 60–62 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.07–7.30 (m, 9H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3311, 1645, 1546, 1466, 1378, 1230 cm−1. Elemental analysis: calculated for C20H25NO (295.43)% C 81.31; H 8.53; N 4.74; found % C 81.36; H 8.51; N 4.73.
N-(4-Fluorobenzyl)-2–(4-isobutylphenyl)propanamide (4)
Obtained following the general procedure by the condensation between 1 and 4-fluorobenzylamine. Yield 83%. m.p. 58–61 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.04–7.22 (m, 8H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3308, 1638, 1538, 1512, 1463 cm−1. Elemental analysis: calculated for C20H24FNO (313.42)% C 76.60; H 7.72; N 4.47; found % C 76.70; H 7.70; N 4.45.
N-(4-Chlorobenzyl)-2–(4-isobutylphenyl)propanamide (5)
Obtained following the general procedure by the condensation between 1 and 4-chlorobenzylamine. Yield 82%. m.p. 65–68 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.3 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.07–7.30 (m, 8H, Ar), 8.42 (t, J = 5.0 Hz, 1H, NH). IR (Nujol) 3270, 3084, 1904, 1709, 1646, 1560, 1463, 1422 cm−1. Elemental analysis: calculated for C20H24ClNO (329.87)% C 72.82; H 7.33; N 4.25; found % C 72.88; H 7.32; N 4.24.
N-(4-(tert-Butyl)benzyl)-2–(4-isobutylphenyl)propanamide (6)
Obtained following the general procedure by the condensation between 1 and 4-(tertbuthyl)benzylamine. Yield 80%. Oil. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.23 (s, 9H, CH3) 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (m, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.07–7.26 (m, 8H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Film) 3291, 1649, 1547, 1514 cm−1. Elemental analysis: calculated for C24H33NO (351.53)% C 82.00; H 9.46; N 3.98; found % C 82.07; H 9.44; N 3.97.
2–(4-Isobutylphenyl)-N-(4-(trifluoromethyl)benzyl)propanamide (7)
Obtained following the general procedure by the condensation between 1 and 4-(trifluoromethyl)benzylamine. Yield 82%. m.p. 62–64 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.07–7.60 (m, 8H, Ar), 8.49 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3332, 3274, 1639, 1541, 1462 cm−1. Elemental analysis: calculated for C21H24F3NO (363.42)% C 69.40; H 6.66; N 3.85; found % C 69.48; H 6.64; N 3.83.
2–(4-Isobutylphenyl)-N-(4-methoxybenzyl)propanamide (8)
Obtained following the general procedure by the condensation between 1 and 4-methoxybenzylamine. Yield 81%. m.p. 78–80 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 3.68 (s, 3H, OCH3) 4.23 (d, J = 5.5 Hz, 2H, CH2) 6.08–7.23 (m, 8H, Ar), 8.31 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3284, 2360, 1710, 1648, 1462 cm−1. Elemental analysis: calculated for C21H27NO2 (325,45)% C 77.50; H 8.36; N 4.30; found % C 77.56; H 8.34; N 4.28.
2–(4-Isobutylphenyl)-N-(3-(trifluoromethyl)benzyl)propanamide (9)
Obtained following the general procedure by the condensation between 1 and 3-(trifluoromethyl)benzylamine. Yield 83%. m.p. 53–55 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.04–7.41 (m, 8H, Ar), 8.35 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3288, 3073, 1651, 1584, 1452, 1329, 1165 cm−1. Elemental analysis: calculated for C21H24F3NO (363.42)% C 69.40; H 6.66; N 3.85; found % C 69.47; H 6.64; N 3.84.
2–(4-Isobutylphenyl)-N-(2-methoxybenzyl)propanamide (10)
Obtained following the general procedure by the condensation between 1 and 2-methoxybenzylamine. Yield 82%. m.p. 61–63 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 3.68 (s, 3H, OCH3) 4.23 (d, J = 5.5 Hz, 2H, CH2) 6.75–7.24 (m, 8H, Ar), 8.15 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3275, 1777, 1641, 1564, 1462, cm−1. Elemental analysis: Calculated for C21H27NO2 (325.45)% C 77.50; H 8.36; N 4.30; found % C 77.57; H 8.34; N 4.28.
N-(2-Chlorobenzyl)-2–(4-isobutylphenyl)propanamide (11)
Obtained following the general procedure by the condensation between 1 and 2-chlorobenzylamine. Yield 85%. m.p. 60–63 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.08–7.04 (m, 8H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Nujol): 3270, 1710, 1666, 1641, 1562 cm−1. Elemental analysis: calculated for C20H24ClNO (329.87)% C 72.82; H 7.33; N 4.25; found % C 72.88; H 7.32; N 4.24.
N-(3-Hydroxy-4-methoxybenzyl)-2–(4-isobutylphenyl)propanamide (12)
Obtained following the general procedure by the condensation between 1 and 3-hydroxy-4-methoxybenzylamine. Yield 80%. m.p. 82–85 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 3.72 (s, 3H, OCH3) 4.23 (d, J = 5.5 Hz, 2H, CH2) 6.62–7.21 (m, 7H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH) 9.45 (s, 1H, OH). IR (Nujol) 3334, 3276, 1642, 1564, 1462 cm−1. Elemental analysis: calculated for C21H27NO3 (341.45)% C 73.87; H 7.97; N 4.10; found % C 73.90; H 7.95; N 4.08.
N-(3,4-Dichlorobenzyl)-2–(4-isobutylphenyl)propanamide (13)
Obtained following the general procedure to by the condensation between 1 and 3,4-dichlorobenzylamine. Yield 83%. m.p. 78–82 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2), 7.07–7.60 (m, 7H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3268, 3072, 1647, 1549, 1428 cm−1. Elemental analysis: calculated for C20H23Cl2NO (364.31)% C 65.94; H 6.36; N 3.84; found % C 66.03; H 6.35; N 3.82.
N-(2,4-Dichlorobenzyl)-2–(4-isobutylphenyl)propanamide (14)
Obtained following the general procedure by the condensation between 1 and 2,4-dichlorobenzylamine. Yield 79%. m.p. 73–75 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.07–7.60 (m, 7H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3274, 3083, 1646, 1557 cm−1. Elemental analysis: calculated for C20H23Cl2NO (364.31)% C 65.94; H 6.36; N 3.84; found % C 66.03; H 6.35; N 3.82.
N-(2,5-Dichlorobenzyl)-2–(4-isobutylphenyl)propanamide (15)
Obtained following the general procedure by the condensation between 1 and 2,5-dichlorobenzylamine. Yield 82%. m.p. 93–96 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.3 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.06–7.40 (m, 7H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3268, 3072, 1647, 1549, 1428 cm−1. Elemental Analysis: calculated for C20H23Cl2NO (363.42)% C 65.94; H 6.36; N 3.84; found % C 66.03; H 6.35; N 3.82.
N-(2,6-Dichlorobenzyl)-2–(4-isobutylphenyl)propanamide (16)
Obtained following the general procedure by the condensation between 1 and 2,6-dichlorobenzylamine. Yield 82%. m.p. 130–135 °C. 1H NMR (DMSO-d6) δ 0.85 (d, J = 6.5 Hz, 6H, CH3), 1.34 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 6.5 Hz, 2H, CH2), 3.62 (q, J = 7.0 Hz, 1H, CH), 4.23 (d, J = 5.5 Hz, 2H, CH2) 7.04–7.45 (m, 7H, Ar), 8.39 (t, J = 5.5 Hz, 1H, NH). IR (Nujol) 3310, 1641, 1534, 1437 cm−1. Elemental analysis: calculated for C20H23Cl2NO (363.42)% C 65.94; H 6.36; N 3.84; found % C 66.00; H 6.35; N 3.82.
General procedure for the synthesis of phenylpiperazine derivatives 18–27
A solution of 1 (0.21 g, 1 mmol), EDC (0.19 g, 1.1 mmol) and HOBt (0.13 g, 1 mmol) in anhydrous MeCN (10 ml) was stirred at r.t. for 30 min, then the appropriate arylpiperazine 17 (1 mmol) was added. The mixture was then stirred for 12 h at r.t. After the solvent was removed under vacuum, the residue was dissolved in AcOEt (20 ml) and washed sequentially with brine (2 × 5 ml), 10% citric acid (2 × 5 ml), NaHCO3 10% aqueous solution (2 × 5 ml) and water (2 × 5 ml). The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum. The obtained residue was tritured with iPr2O; the precipitate was then filtrated to obtain the compounds 18–27.
2–(4-Isobutylphenyl)-1–(4-phenylpiperazin-1-yl)propan-1-one (18)
Obtained following the general procedure by the condensation between 1 and 1-phenylpiperazine. Yield 97%. m.p. 75–80 °C. 1H NMR (DMSO-d6) δ 0.83 (d, J = 7.0 Hz, 6H, CH3), 1.30 (d, J = 7.0 Hz, 3H, CH3), 1.80 (hept, J = 7.0 Hz, 1H, CH), 2.42 (d, J = 7.0 Hz, 2H, CH2), 3.16 (m, 2H, CH2), 3.20 (m, 2H, CH2), 3.40 (m, 1H, CH), 3.48–3.65 (m, 4H, CH2), 6.81 (m, 1H Ar), 7.04–7.45 (m, 6H, Ar), 7.53 (m, 1H Ar), 7.59 (m, 1H Ar). IR (Nujol) 3273, 1741, 1631, 1600, 1508, 1465 cm−1. Elemental analysis: calculated for C23H30N2O (350.51)% C 78.82; H 8.63; N 7.99; found % C 78.89; H 8.67; N 7.85.
1–(4-(3-Chlorophenyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (19)
Obtained following the general procedure by the condensation between 1 and 1–(3-chlorophenyl)piperazine. Yield 95%. Oil. 1H NMR (DMSO-d6) δ 0.84 (d, J = 6.5 Hz, 6H, CH3), 1.32 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.43 (d, J = 7.0 Hz, 2H, CH2), 2.89 (m, 2H, CH2), 3.23 (m, 2H, CH2), 3.41 (q, J = 7.0 Hz, 1H, CH), 3.45–3.68 (m, 4H, CH2), 6.70 (m, 1H Ar), 7.06–7.43 (m, 6H, Ar), 7.50 (m, 1H Ar). IR (Film) 3437, 1732, 1646, 1594, 1486, 1463, 1384, 1231 cm−1. Elemental analysis: calculated for C23H29ClN2O (384.95)% C 71.76; H 7.59; N 7.28; found % C 71.75; H 7.60; N 7.35.
1–(4-(4-Chlorophenyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (20)
Obtained following the general procedure by the condensation between 1 and 1–(4-chlorophenyl)piperazine. Yield 83%. Oil. 1H NMR (DMSO-d6) δ 0.94 (d, J = 7.0 Hz, 6H, CH3), 1.32 (d, J = 6.5 Hz, 3H, CH3), 1.81 (hept, J = 7.0 Hz, 1H, CH), 2.66 (m, 2H, CH2), 2.95 (m, 2H, CH2), 3.11 (m, 2H, CH2), 3.50–3.68 (m, 4H, CH2), 4.17 (q, J = 6.5 Hz, 1H, CH), 6.89 (m, 1H, Ar), 7.03 (m, 1H, Ar), 7.16–7.21 (m, 5H, Ar), 7.30 (m, 1H, Ar). IR (Film) 3421, 2955, 1731, 1645, 1497, 1463, 1384 cm−1. Elemental analysis: calculated for C23H29ClN2O (384.95)% C 71.76; H 7.59; N 7.28; found % C 71.70; H 7.65; N 7.30.
1–(4-(3,4-Dichlorophenyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (21)
Obtained following the general procedure by the condensation between 1 and 1–(3,4-dichlorophenyl)piperazine. Yield 90%. Oil. 1H NMR (DMSO-d6) δ 0.86 (d, J = 6.5 Hz, 6H, CH3), 1.33 (d, J = 7.0 Hz, 3H, CH3), 1.80 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.41 (d, J = 7.0 Hz, 2H, CH2), 2.92 (m, 2H, CH2), 3.17 (m, 2H, CH2), 3.22 (m, 1H, CH), 3.40–3.71 (m, 4H, CH2), 6.88 (m, 1H Ar), 7.06–7.43 (m, 5H, Ar), 7.53 (m, 1H Ar). IR (Film) 3433, 1728, 1645, 1594, 1555, 1484, 1230 cm−1. Elemental analysis: calculated for C23H28Cl2N2O (419.39)% C 64.87; H 6.73; N 6.68; found % C 64.78; H 6.68; N 6.59.
1–(4-(4-Fluorophenyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (22)
Obtained following the general procedure by the condensation between 1 and 1–(4-fluorophenyl)piperazine. Yield 97%. m.p. 45–50 °C. 1H NMR (DMSO-d6) δ 0.90 (d, J = 7.0 Hz, 6H, CH3), 1.36 (d, J = 6.5 Hz, 3H, CH3), 1.87 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.61 (m, 2H, CH2), 2.99 (m, 2H, CH2), 3.06 (m, 2H, CH2), 3.44–3.61 (m, 4H, CH2), 4.19 (q, J = 6.5 Hz, 1H, CH), 6.97 (m, 1H, Ar), 7.02 (m, 1H, Ar), 7.10–7.27 (m, 5H, Ar), 7.34 (m, 1H, Ar). IR (Nujol) 3445, 2955, 2929, 1644, 1510, 1441, 1230 cm−1. Elemental analysis: calculated for C23H29FN2O (368.23)% C 74.97; H 7.93; N 7.60; found % C 75.01; H 7.90; N 7.55.
2–(4-Isobutylphenyl)-1–(4-(4-methoxyphenyl)piperazin-1-yl)propan-1-one (23)
Obtained following the general procedure by the condensation between 1 and 1–(4-methoxyphenyl)piperazine. Yield 95%. Oil. 1H NMR (DMSO-d6) δ 0.95 (d, J = 7.0 Hz, 6H, CH3), 1.30 (d, J = 6.5 Hz, 3H, CH3), 1.83 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.62 (m, 2H, CH2), 3.03 (m, 2H, CH2), 3.09 (m, 2H, CH2), 3.40–3.59 (m, 4H, CH2), 3.66 (s, 3H, CH3), 4.22 (q, J = 6.5 Hz, 1H, CH), 7.01 (m, 1H, Ar), 7.04 (m, 1H, Ar), 7.12–7.23 (m, 5H, Ar), 7.38 (m, 1H, Ar). IR (Film) 3440, 2954, 2930, 1732, 1644, 1464, 1442, 1246 cm−1. Elemental analysis: calculated for C24H32N2O2 (380.53)% C 75.75; H 8.48; N 7.36; found % C 75.80; H 8.53; N 7.30.
2–(4-Isobutylphenyl)-1–(4-(3-methoxyphenyl)piperazin-1-yl)propan-1-one (24)
Obtained following the general procedure by the condensation between 1 and 1–(3-methoxyphenyl)piperazine. Yield 92%. Oil. 1H NMR (DMSO-d6) δ 0.82 (d, J = 7.0 Hz, 6H, CH3), 1.28 (d, J = 6.0 Hz, 3H, CH3), 1.79 (hept, J = 6.0–7.0 Hz, 1H, CH), 2.87 (m, 2H, CH2), 3.01 (m, 2H, CH2), 3.13 (m, 2H, CH2), 3.44–3.54 (m, 4H, CH2), 3.68 (s, 3H, CH3), 4.08 (q, J = 6.5 Hz, 1H, CH), 6.34 (m, 2H, Ar), 6.42 (m, 1H, Ar), 7.05–7.10 (m, 3H, Ar), 7.16 (m, 2H, Ar). IR (Film) 2956, 2928, 1734, 1647, 16071460,1203 cm−1. Elemental analysis: calculated for C24H32N2O2 (380.53)% C 75.75; H 8.48; N 7.36; found % C 75.70; H 8.50; N 7.34.
2–(4-Isobutylphenyl)-1–(4-(m-tolyl)piperazin-1-yl)propan-1-one (25)
Obtained following the general procedure by the condensation between 1 and 1–(3-methylphenyl)piperazine. Yield 91%. Oil. 1H NMR (DMSO-d6) δ 0.81 (d, J = 7.0 Hz, 6H, CH3), 1.27 (d, J = 6.0 Hz, 3H, CH3), 1.78 (hept, J = 6.0–7.0 Hz, 1H, CH), 2.20 (s, 3H, CH3), 2.50 (d, J = 7.0 Hz, 2H, CH2), 2.83 (m, 1H, CH2), 2.98 (m, 1H, CH2), 3.13 (m, 1H, CH2), 3.46–3.53 (m, 4H, CH2), 3.73 (m, 1H, CH2), 4.09 (q, J = 6.5 Hz, 1H, CH), 6.58–6.64 (m, 3H, Ar), 7.05 (m, 1H, Ar), 7.09 (d, J = 8.0, 2H, Ar), 7.16 (d, J = 8.0 Hz, 2H, Ar). IR (Film) 3483, 2955, 1926, 1644, 1602, 1494, 1434, 1233,1185 cm−1. Elemental analysis: calculated for C24H32N2O (364.25)% C 79.08; H 8.85; N 7.68; found % C 79.15; H 8.92; N 7.60.
1–(4-(2,3-Dimethylphenyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (26)
Obtained following the general procedure by the condensation between 1 and 1–(2,3-dimethylphenyl)piperazine. Yield 90%. Oil. 1H NMR (DMSO-d6) δ 0.83 (d, J = 7.0 Hz, 6H, CH3), 1.30 (d, J = 7.0 Hz, 3H, CH3), 1.78 (hept, J = 7.0 Hz, 1H, CH), 2.12 (m, 3H, CH3), 2.18 (m, 3H, CH3), 2.40 (m, 2H, CH2) 2.12 (m, 2H, CH2), 2.50 (m, 2H, CH2), 2.72 (m, 2H, CH2), 3.56 (m, 2H, CH2), 4.09 (q, J = 7 Hz, 1H, CH), 6.76 (m, 1H, Ar), 6.69 (m, 1H, Ar), 6.99 (m, 1H, Ar), 7.11 (m, 2H, Ar), 7.17 (m, 2H, Ar). IR (Film) 2959, 1731, 1644, 15111471, 1367, 1235 cm−1. Elemental analysis: calculated for C25H34N2O (378.56)% C 79.32; H 9.05; N 7.40; found % C 79.28; H 9.02; N 7.50
2–(4-Isobutylphenyl)-1–(4-(o-tolyl)piperazin-1-yl)propan-1-one (27)
Obtained following the general procedure by the condensation between 1 and 1–(2-methylphenyl)piperazine. Yield 91%. Oil. 1H NMR (DMSO-d6) δ 0.83 (d, J = 6.5 Hz, 6H, CH3), 1.29 (d, J = 7.0 Hz, 3H, CH3), 1.81 (hept, J = 6.5–7.0 Hz, 1H, CH), 2.21 (s, 3H, CH3), 2.24 (m, 1H, CH2), 2.41 (d, J = 6.0 Hz, 2H, CH2), 2.63 (m, 2H, CH2), 2.76 (m, 1H, CH2), 3.43 (m, 1H, CH2), 3.56 (m, 2H, CH2), 3.69 (m, 1H, CH2), 4.10 (q, J = 7.0 Hz, 1H, CH), 6.82 (m, 1H, Ar), 6.93 (m, 1H, Ar), 7.07–7.14 (m, 4H, Ar), 7.42 (d, J = 7.5, 2H, Ar). IR (Film) 2923, 1639, 1600, 1494, 14631377,1224, 1147 cm−1. Elemental analysis: calculated for C25H32N2O (364.25)% C 79.32; H 8.85; N 7.68; found % C 79.27; H 9.00; N 7.59.
2–(4-Isobutylphenyl)-1-(piperazin-1-yl)propan-1-one trifluoroacetate (29)
Compound 1 (2.06 g, 10 mmol), EDC (2.09 g, 11 mmol) and HOBt (1.35 g, 10 mmol) were dissolved in MeCN (10 ml). The mixture was stirred at r.t. for 30 min, then BOC-piperazine (28) (1.86 g, 10 mmol) was added. The mixture was stirred at r.t. for 12 h. After the solvent was removed under vacuum. The residue was dissolved in AcOEt (20 ml) and washed sequentially with brine (2 × 5 ml), 10% citric acid (2 × 5 ml), saturated NaHCO3 aqueous solution (2 × 5 ml) and water (2 × 5 ml). The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum. The obtained residue was dissolved in dichloromethane, without further purification, added trifluoroacetic acid (TFA) (20 ml) and stirred at r.t. for 24 h to obtain the BOC de-protected compound. Then the solvent was removed under vacuum and to the obtained residue diethyl ether (Et2O) (20 ml) was added leading to formation of a solid that was filtered to give the title compound. Yield 97%. Oil. 1H NMR (DMSO-d6) δ 0.84 (d, J = 5.0 Hz, 6H, CH3), 1.28 (d, J = 5.0 Hz, 3H, CH3), 1.81 (m, 1H, CH), 2.41 (d, J = 6.0 Hz, 2H, CH2), 2.56 (m, 1H, CH2), 3.00 (m, 3H, CH2), 3.34 (m, 1H, CH2), 3.58 (m, 1H, CH2), 3.74 (m, 2H, CH2), 4.09 (m, 1H, CH), 7.11 (m, 2H, Ar), 7.18 (d, 2H, Ar), 8.88 (s, 1H, NH). IR (Film) 2957, 2925, 2854, 1674, 1636, 1461, 1442, 1367, 1199, 1082 cm−1. Elemental analysis: calculated for C19H27F3N2O3 (388.42)% C 58.75; H 7.01; N 7.21; found % C 58.80; H 6.99; N 7.17.
General procedure for the synthesis of benzylpiperazine 30–34
To a solution of compound 29 (0.39 g, 1 mmol) in dichloromethane (CH2Cl2) (10 ml) the appropriate arylaldehyde (1.6 mmol), sodium sodium hydrogen carbonate (NaHCO3) (0.10 g, 1.2 mmol) and sodium triacetoxyborohydride (NaBHAc3) (0.32 g, 1.5 mmol) were added; the mixture was then stirred at r.t. for 24 h. After the mixture was basified to pH 10 with a solution of NaOH 0.1 N, then extracted with CH2Cl2 (3 × 20 ml). The organic phases were collected, dried over sodium Na2SO4, filtrated and the solvent removed to obtain the desired compound.
1–(4-Benzyl-1-yl)-2–(4-isobutylphenyl)propan-1-one (30)
Obtained following the general procedure by the reductive alkylation between 29 and benzaldehyde. Yield 91%. Oil. 1H NMR (DMSO-d6) δ 0.85 (d, J = 7.0 Hz, 6H, CH3), 1.25 (d, J = 7.0 Hz, 3H, CH3), 1.78 (m, 1H, CH2), 1.81 (q, J = 6.0 Hz, 1H, CH), 2.21 (m, 1H, CH2), 2.34 (m, 1H, CH2), 2.41 (d, J = 7.5 Hz, 2H, CH2), 2.50 (s, 1H, CH2), 3.36 (m, 3H, CH2), 3.38 (s, 2H, CH2), 3.60 (m, 1H, CH2), 4.03 (m, 1H, CH), 7.08 (m, 2H, Ar), 7.13 (d, 2H, Ar), 7.23 (m, 3H, Ar), 7.29 (d, 2H, Ar). IR (Film) 3448, 2954, 2929, 2645, 1462, 1230, 1032, 1000 cm−1. Elemental analysis: calculated for C24H32N2O (364.25)% C 79.08; H 8.85; N 7.68; found % C 79.15; H 8.80; N 7.65.
1–(4-(2-Chlorobenzyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (31)
Obtained following the general procedure by the reductive alkylation between 29 and 2-chlorobenzaldehyde. Yield 87%. Oil. 1H NMR (DMSO-d6) δ 0.83 (d, J = 7.0 Hz, 6H, CH3), 1.23 (d, J = 7.0 Hz, 3H, CH3), 1.79 (q, J = 6.0 Hz, 1H, CH), 2.40 (d, J = 7.5 Hz, 2H, CH2), 3.32–3.75 (m, 8H, CH2), 4.00 (m, 1H, CH), 4.56 (s, 2H, CH2), 6.99–7.56 (m, 8H, Ar). IR (Film) 3416, 2955, 2927, 1628, 1060, 1033 cm−1. Elemental analysis: calculated for C24H31ClN2O (398.98)% C 72.25; H 7.83; N 7.02; found % C 72.30; H 7.81; N 7.08.
1–(4-(3-Chlorobenzyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (32)
Obtained following the general procedure by the reductive alkylation between 29 and 3-chlorobenzaldehyde. Yield 90%. Oil. 1H NMR (DMSO-d6) δ 0.96 (d, J = 6.5 Hz, 6H, CH3), 1.36 (d, J = 7.5 Hz, 3H, CH3), 1.94 (q, J = 6.5–7.5 Hz, 1H, CH), 2.52 (d, J = 7.5 Hz, 2H, CH2), 3.37–3.85 (m, 8H, CH2), 4.14 (m, 1H, CH), 4.62 (s, 2H, CH2), 7.13–7.55 (m, 8H, Ar). IR (Film) 3414, 2955, 2928,1702, 1631, 1464, 1434, 1228, 1196 cm−1. Elemental analysis: calculated for C24H31ClN2O (398.98)% C 72.25; H 7.83; N 7.02; found % C 72.38; H 7.85; N 7.00.
2–(4-Isobutylphenyl)-1–(4-(4-(trifluoromethyl)benzyl)piperazin-1-yl)propan-1-one (33)
Obtained following the general procedure by the reductive alkylation between 29 and 4-(trifluoromethyl)benzaldehyde. Yield 50%. Oil. 1H NMR (DMSO-d6) δ 0.96 (d, J = 6.5 Hz, 6H, CH3), 1.37 (d, J = 6.5 Hz, 3H, CH3), 1.90 (q, J = 6.5 Hz, 1H, CH), 2.53 (s, 2H, CH2), 2.61 (d, J = 6.5 Hz, 2H, CH2), 3.43–3.70 (m, 8H, CH2), 4.12 (m, 1H, CH), 7.13–7.27 (m, 8H, Ar). IR (Film) 3332, 2955, 32868, 1644, 1510, 1462, 1367, 1228, 1164, 1125, 1066 cm−1. Elemental analysis: calculated for C25H31F3N2O (432.53)% C 69.42; H 7.22; N 6.48; found % C 69.48; H 7.21; N 6.53.
1–(4-(3-Fluorobenzyl)piperazin-1-yl)-2–(4-isobutylphenyl)propan-1-one (34)
Obtained following the general procedure by the reductive alkylation between 29 and 3-fluorobenzaldehyde. Yield 49%. Oil. 1H NMR (DMSO-d6) δ 0.96 (d, J = 6.5 Hz, 6H, CH3), 1.36 (d, J = 6.5 Hz, 3H, CH3), 1.89 (q, J = 6.5 Hz, 1H, CH), 2.51 (s, 2H, CH2), 2.61 (d, J = 6.5 Hz, 2H, CH2), 3.32–3.52 (m, 8H, CH2), 4.12 (m, 1H, CH), 7.15–7.25 (m, 8H, Ar). IR (Film) 3407, 2955, 2926, 1713, 1696, 1631, 1591, 1254, 1001 cm−1. Elemental analysis: calculated for C24H31FN2O (382.52)% C 75.36; H 8.17; N 7.32; found % C 75.40; H 8.05; N 7.40.
Computational studies
FAAH receptor and ibuprofen amides preparation
The crystal structure of the fatty acid amide hydrolase (FAAH) (PDB ID: 3QK5)27 has been downloaded from the Protein Data Bank website. Both monomers A and B were treated with the Protein Preparation Wizard28 tool implemented in Maestro ver. 11.129 in order to add all the hydrogen atoms and assign the correct bond orders. Moreover, the co-crystallised ligand (QK5), as well as all the crystallographic water molecules, were removed. Residue Lys142 was considered in its deprotonated form, according to the proposed catalytic mechanism2,3,30. The 3 D structure of amides described above was built using the Graphical User Interface of Maestro ver. 11.129, as (S)-enantiomers in view of our data with (S)-Ibu-AM5, which was ten-fold more potent inhibitor than the (R)-enantiomer23. The protonation state of these amides at pH 7.4 in water has been calculated using the Epik module31, revealing the protonation of the nitrogen atom of the piperazine ring only of compounds 30–34 characterised by a methylene bridge between the aryl and the piperazine rings. Finally, each compound was then minimised with the OPLS_2005 force field using the Polak-Ribiere Conjugate Gradient (PRCG) algorithm and 2500 iteration steps.
Induced-Fit docking protocol
Only the monomer A of the rat FAAH (rFAAH) receptor was considered for the induced-fit docking (IFD)32,33. The IFD protocol has three stages. In the first stage, ligands were docked to rigid protein using initial Glide softened potential (van der Waals radii scaling). The top 20 poses for each ligand were retained. In the second stage, a Prime side-chain prediction for each protein/ligand complex on residues within a default distance of 5 Å was performed followed by a Prime minimisation of the same set of residues and protein/ligand complexes. In the third stage, a Glide re-docking of each protein/ligand complex within a specified default lowest-energy structure (30 kcal/mol) was carried out. In this step, each ligand is rigorously docked using the default Glide settings, into the induced-fit receptor structure. At the end of the final stage, two methods were used for estimation of the binding energy for each output complex pose (IFDScore and Glide_emodel). Glide SP (Standard Precision) was used for all docking calculations. Docking poses were ranked on the basis of Glide_emodel energy. A preliminary validation of the computational protocol was performed by reproducing with IFD the crystallographic binding mode of QK5 ligand complexed in the model PDB ID: 3QK5 (QK5) (Supplementary Figure S2-A)27 and that of the (S)-Ibu-AM5 found after Molecular Dynamics simulations23 (Supplementary Figure S2-B).
Pharmacology
FAAH assay
Frozen (−80 °C) brains (minus cerebella) from adult Wistar or Sprague-Dawley rats were thawed and homogenised in 20 mM HEPES, 1 mM MgCl2, pH 7.0. and thereafter centrifuged at ∼35000 × g for 20 min at 4 °C. Homogenates were washed (by centrifugation at ∼35000 × g for 20 min at 4 °C followed by resuspension in the buffer) twice and incubated at 37 °C for 15 min in order to hydrolyse all endogenous FAAH substrates. After a further centrifugation, pellets were resuspended in 50 mM Tris-HCl buffer, pH 7.4, containing 1 mM EDTA and 3 mM MgCl2, and frozen at −80 °C in aliquots until used for the assay. For the FAAH assay34 test compounds, homogenates (usually 0.5–0.8 μg protein per assay, diluted with 10 mM Tris-HCl, 1 mM EDTA pH 7.4) and 25 μL of [3H]AEA in 10 mM Tris- HCl, 1 mM EDTA, pH 7.4, containing 1% w/v fatty acid-free bovine serum albumin, final substrate concentration of 0.5 μM) were incubated for 10 min at 37 °C (final assay volume 200 μL). Reactions were stopped by placing the tubes on ice. Final assay concentrations of the solvents used for the compounds (ethanol or DMSO) were in the range 1–5%. Activated charcoal (80 μL + 320 μL 0.5 M HCl) was added and the samples were mixed and left at room temperature for about 30 min. Following centrifugation at 2500 rpm for 10 min, aliquots (200 μL) of the supernatants were analysed for tritium content by liquid scintillation spectroscopy with quench correction. Blank values were obtained by the use of buffer rather than homogenate.
In general, FAAH assays upon three homogenates were undertaken using separate inhibitor dilution series (from a stock solution), with 6 concentrations of inhibitor in half-log concentrations (i.e. 1, 3, 10 µM etc) ranging from 0.3–100 µM. Data were expressed as % of vehicle control and analysed using the algorithm log(inhibitor) vs. response – variable slope (four parameters) built into the GraphPad Prism computer programme for the Macintosh (GraphPad Software Inc., San Diego, CA). Two different curve fits were chosen: one where the top (uninhibited) value was set to 100 and the bottom (maximum inhibition) was set to 0, and one where the top was set to 100 and the bottom allowed to float. The best model (when the bottom value returned >0) was chosen by Akaike’s informative criteria. Since the programme uses log10 inhibitor concentrations, the IC50 values for the inhibitable fraction are derived from the corresponding − log10(IC50) (pI50) values. Hence the SE values are for the pI50 values rather than the IC50 values. In consequence, we report both pI50 and IC50 values to indicate the SE values.
Results and discussion
The target amides of ibuprofen were synthesised according to the Schemes 1–3. The benzylamides were obtained by coupling ibuprofen (1) with substituted benzylamines (2) in the presence of EDC and HOBt in MeCN solution. This synthetic pathway was found to be clean and high yielding.
Scheme 1.
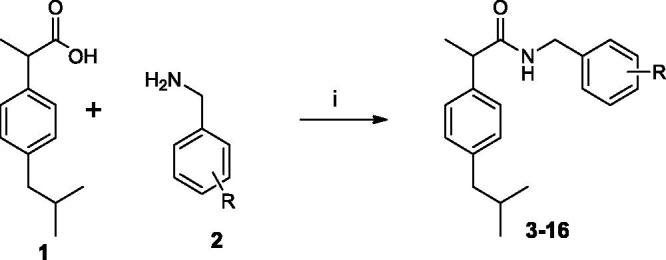
Synthesis of Ibuprofen amides 3–16. (i) EDC, OH-Bt, MeCN, r.t. 24 h.
Scheme 2.
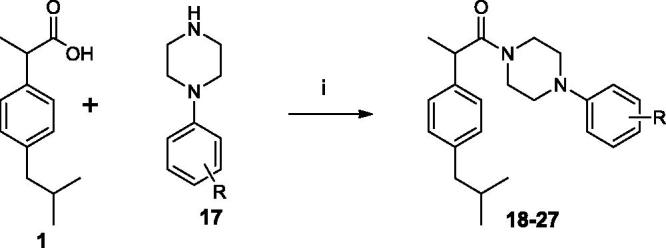
Synthesis of Ibuprofen amides 18–27. (i) EDC, OH-Bt, MeCN, r.t. 12 h.
Scheme 3.

Synthesis of Ibuprofen amides 30–34. (i) EDC, OH-Bt, MeCN, r.t. 12 h, (ii) TFA, CH2Cl2, r.t., 24 h.; (iii) NaBHAc3, NaHCO3, ArCHO, r.t., 24 h.
The benzylamides 3–16, along with the reference compound Ibu-AM5, were evaluated for their ability to inhibit FAAH. The inhibition assays were performed using 0.5 μM [3H]AEA as substrate and rat brain homogenates as the enzyme source. For comparison, the % inhibition produced by URB597 (which shows time-dependent inhibition) using this assay in our laboratory but following a 60 min preincubation, was 8, 38, 98 and 100 at concentrations of 0.1, 1, 10 and 100 nM, respectively35. The results of these primary assays are shown in Table 1. The replacement of the Ibu-AM5 2-methylpyridine moiety with a benzyl moiety to give benzylamides 3–16 led to a reduction in inhibitory activity. In general, benzylamide derivatives showed IC50 values ranging 18–51 µM with the exception of 11, and 15. Compounds 11 and 15 showed FAAH inhibitory activity with IC50 values of 4.1 and 4.4 µM, respectively (Table 1). As shown in Table 1, the position and the kind of substituent on benzyl moiety affect the inhibitory activity. The presence of 4-substituent is not favourable for the inhibitory activity. Conversely, the presence of substituents at 2-position improved the activity as compared with the unsubstituted amide 3, as showed by the 2-chloro (11) and 2,5-dichloro (15) derivatives. Moreover, the moving of chlorine atom from 5-position of amide 15 to 4-position to give the analogue 14 led to a reduction in activity (IC50 21 µM). While the moving of a chlorine atom from 5-position to 6-position (amide 16) led to a complete loss of activity.
We also studied the binding of the newly designed compounds in the active site of FAAH by using the Induced fit protocol (IFD) implemented in Maestro (Schrödinger), that takes into consideration not only ligand flexibility, but also protein rearrangements upon ligand binding. Calculations were carried out in the monomer A of the rat FAAH (rFAAH) (PDB ID: 3QK5). Taking into considerations previous results on stereoselectivity of Ibu-AM5, demonstrating (S)-IbuAM5 being 10-fold more active than (R)-Ibu-AM523, we performed and discussed docking calculations on the (S)-enantiomers of selected compounds in the series. However, for the sale of completeness, docking results for the (R)-enantiomers were reported as Supplementary information. A preliminary validation of the computational protocol was performed on (S)-Ibu-AM5 and QK5 ligand. Results highlighted that the pose with the lowest Glide-emodel and IFDScore reproduced closely the binding mode of (S)-Ibu-AM5 and QK5 (RMSD: 1.34 Å and 0.58 Å respectively; (Supplementary Figure S1). We have compared the binding mode of amides 3, 11 and 15. Best poses of compounds 3 (Figure 2(a)), 11 and 15 (Figure 2(b)) were positioned within the ACB channel with the benzylamide moiety oriented toward the membrane, and the isobutyl moiety pointing toward the catalytic triad, but differed for the position along the ACB channel, being 3 slightly shifted toward the catalytic triad. The more active compounds of the series, 11 and 15 showed high similarity to the binding mode predicted for Ibu-AM5 (Supplementary Figure S2). All the compounds engaged one hydrogen bond (H-bond) interaction of the carbonyl with the Thr488 side chain, but differed for the positioning of the rest of the molecule (Figure 2). In particular, the isobutyl well matching the hydrophobic region formed by Ile491, Phe244, Ile238, and Leu192 in the case of 3, or Phe381, Ala377, Phe432, and Leu380 in the case of amide 11, and Leu192, Ile491, and Leu404 in the case of amide 15. The binding mode adopted by amides 11 and 15 positioned the substituted benzyl ring in a similar position within the hydrophobic region at the gorge of MA channel and appeared favoured with respect to amide 3, since they showed an additional H-bond interaction with Trp531 (amide 11) or Asp403 (amide 15).
Figure 2.

The lowest emodel binding modes of (S)-benzylamides : (a) 3, (b) 11 and 15. Key interacting residues of FAAH are displayed as green, cyan and orange lines relatively to benzylamides 3, 11, and 15 that are represented as green, cyan and orange sticks, respectively. Hydrogen bond interactions detected by Maestro 11.1 are shown as dashed black lines.
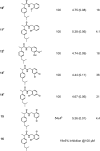
With the aim to increase the hydrophilicity of the amides and to reduce the flexibility of the amide chain a new series of arylpiperazinoamides was designed. As indicated in Scheme 2, amides 18–27 were prepared by condensation between Ibuprofen (1) and aryl-substituted piperazines (17) using the EDC method. In general, all the arylpiperazinoamides displayed FAAH inhibitory activity better than benzylamide series. As shown in Table 2 the N-phenylpiperazinoamide 18 displayed inhibitory activity comparable to benzylamides 11 and 15. The introduction of 4-methoxy group on the phenyl ring (amide 23) did not change the activity, while the displacement of the methoxy group into the 3-position (amide 24) led to a drop in activity. The replacement of 4-methoxy group with fluorine atom produced a reduction in activity (compound 22, IC50 17 μM). In contrast, the introduction in the same position of chlorine atom to give compound 20 improved ten-fold the inhibitory activity. Moving the chlorine from 4- to 3-position (amide 19) did not affect the activity. However, the 3,4-dichlorophenyl substituted amide 21 that, contrary to expectation, showed inhibitory activity an order of magnitude lower potency than analogue 20. The substitution of 3-chlorine of amide 19 with the methyl group to afford compound 25 caused a clear reduction in activity. The introduction of the second methyl group in 2-position (amide 26) restored the activity, while the displacement of 3-methyl into 2-position produced loss of activity (amide 27).
Table 2.
Maximum percentage and IC50 values for inhibition of rat brain AEA hydrolysis by compounds 18–27.
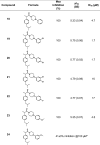 |
|||||||||||
 |
*The inhibition data was better fitted by a curve with a residual activity rather than a curve assuming 100% inhibition. The maximal inhibition is indicated (when it was greater than 50%), and the pI50 and IC50 values refer to the inhibitable portion of the curve. The inability of the compounds to produce a maximal inhibition was not investigated further.
Induced-Fit docking at the enzyme active site revealed that all the arylpiperazinoamide derivatives adopted a different binding mode when compared to benzylamides or to the lead Ibu-AM5, binding deeper in the ACB, establishing, in most cases, direct interactions with at least one residue of the catalytic triad through the carbonyl of the amide, and the arylpiperazinoamide moiety entering the cytosolic port (CP). In order to clarify the SARs of this interesting novel series of ibuprofen derivatives, we analysed the binding mode of 19, 20, 21, 25, and 26. Figure 3(a) represents the binding mode of amide 19, showing two H-bonds with the catalytic triad residues Ser241 and Ser217, and a clear interaction of the chlorine with the NH of Cys269. Accordingly, the introduction of a second chlorine in 4-position, would clash with the side chain of Val270, thus forcing the 3,4-dichlorophenyl ring of compound 21 to move toward Phe381 (Figure 3(a)). This movement also induces a rearrangement of the isobutylphenyl moiety determining the disruption of the hydrophobic network that could account for the 10-fold loss of activity. Interestingly compound 20, which showed the same FAAH inhibitory activity of analogue 19, showed the same position as the isobutylphenyl moiety, albeit the 4-chlorophenyl ring assumed an intermediate conformation with respect to amides 19 and 21. We also studied the binding of amides 25, and 26 differing for the number and the position of methyl groups on the piperazine phenyl ring (Figure 3(b)). Docking results did not allow to give a clear explanation of how these subtle structural differences gave rise to the observed striking differences in activity. The two compounds indeed occupied the same binding pocket, and all established a H-bond with Ser241. Nevertheless, the introduction of the 2-methyl forced the phenyl ring to assume a perpendicular conformation with respect to the piperazine ring as an effect of steric hindrance with piperazine ring hydrogens. This led to different binding modes for 2-methylphenyl (compound 26) and 3-methylphenyl analogue (25), that resulted in a different ability in engaging T-shape π-π stacking interactions with Phe381, only observed for 2-methyl derivative (Figure 3(b)). Albeit docking results gave interesting suggestions about some aspects of SAR of the benzyl- and the piperazine phenyl derivatives, the lack of correlation between IFDscores (Data not shown) and IC50 suggests some caution in interpreting the results.
Figure 3.

Superposition between the lowest emodel binding mode of (S)-piperazinoarylamides: (a) 19, 20 and 21 depicted as yellow, purple and dark-green sticks, respectively; (b) 25 and 26 dysplayed as brown and violet sticks, respectively. rFAAH key residues involved in ligand interactions are displayed as lines coloured relatively as the interacting ligand. Hydrogen bond interactions and T-shaped π-π stacking interaction detected by Maestro 11.1 are shown as dashed black lines. Globally, the substitution of a chorine atom to the phenyl ring (compound 19) allows an Hbond interaction with Cys269. The addition of a one more methyl group on the aromatic ring (amide 26) does not affect the binding mode of the two ligands, except for the phenyl ring itself that in the case of amide 26 is slightly oriented toward the Phe381 residue engaging one T-shape π-π stacking interaction.
As further modification, a methylene bridge was inserted between the piperazine and the aromatic ring. The amides 30–34 were prepared by condensation of ibuprofen (1) with N-BOC-piperazine (29), using the same EDC method described above. After BOC-deprotection, a benzyl group was added by reductive alkylation, treating the intermediate 6 with the suitable substituted arylaldehyde in presence of NaHCO3, NaBHAc3 in CH2Cl2 solution (Scheme 3). Unfortunately, this modification afforded the poor active analogues 30–34 (Table 3). This loss of activity could be attributed to the protonation of the nitrogen atom of the piperazine ring that is unfavourable in the hydrophobic environment of the FAAH channels. The induced fit docking of 30 and 32 confirmed this hypothesis, yielding binding modes characterised by the loss of interactions with Ser241, and by a closed conformation (Supplementary Figure S3), due largely to an intramolecular cation-π interaction, thus explaining the loss in the inhibition activity.
Table 3.
Maximum percentage and IC50 values for inhibition of rat brain AEA hydrolysis by compounds 30–34.
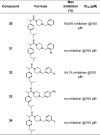 |
In conclusion, the present study has further explored the pharmacophore of Ibu-AM5 with respect to its interaction with FAAH. Although the benzyl- and the piperazinoamides were logical areas to explore, neither set of derivatives improved upon the inhibitory potency of Ibu-AM5. Nevertheless, the arylpiperazinoamide derivatives showed a binding mode involving residues from the cytosolic port, which has been poorly explored as potential binding site of FAAH inhibitors and could, therefore, be considered interesting leads for the design of novel and more potent FAAH inhibitors.
Supplementary Material
Funding Statement
This work was supported by the Regione Autonoma della Sardegna Project L.R. 7/2007 under grant no. 2012_CRP-59473 to VO; the University of Cagliari [grant FIR 2016–17] to VO and by the Swedish Research Council under the grant no. 12158, medicine, and the Research Funds of Umeå University Medical Faculty to CJF.
Acknowledgements
The authors thank Mona Svensson for excellent technical help. BC acknowledges the SCoPE Datacenter of the University of Napoli for the access to HPC infrastructures.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Di Marzo V. New approaches and challenges to targeting the endocannabinoid system. Nat Rev Drug Discov 2018;17:623–39. [DOI] [PubMed] [Google Scholar]
- 2.Palermo G, Rothlisberger U, Cavalli A, De Vivo M. Computational insights into function and inhibition of fatty acid amide hydrolase. Eur J Med Chem 2015;91:15–26. [DOI] [PubMed] [Google Scholar]
- 3.Bracey M, Hanson M, Masuda K, et al. Structural adaptions in a membrane enzyme that terminates endocannabinoid signaling. Science 2002;298:1793–6. [DOI] [PubMed] [Google Scholar]
- 4.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem 2005;74:411–32. [DOI] [PubMed] [Google Scholar]
- 5.Tuo W, Leleu-Chavain N, Spencer J, et al. Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and n-acylethanolamine acid amidase inhibitors. J Med Chem 2017;60:4–46. [DOI] [PubMed] [Google Scholar]
- 6.Kathuria S, Gaetani S, Fegley D, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 2003;9:76–81. [DOI] [PubMed] [Google Scholar]
- 7.Justinova Z, Mangieri R, Bortolato M, et al. Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiat 2008;64:930–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li GL, Winter H, Arends R, et al. Assessment of the pharmacology and tolerability of pf-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects. Br J Clin Pharmacol 2012;73:706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pawsey S, Wood M, Browne H, et al. Safety, tolerability and pharmacokinetics of FAAH inhibitor v158866: a double-blind, randomised, placebo-controlled phase I study in healthy volunteers. Drugs R D 2016;16:181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagenlehner FME, van Till JWO, Houbiers JGA, et al. Fatty acid amide hydrolase inhibitor treatment in men with chronic prostatitis/chronic pelvic pain syndrome: an adaptive double-blind, randomized controlled trial. Urology 2017;103:191–7. [DOI] [PubMed] [Google Scholar]
- 11.van Esbroeck ACM, Janssen APA, Cognetta AB III, et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474. Science 2017;356:1084–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.US Food and Drug Administration FDA works with regulatory partners to understand French-based Biotrial phase 1 clinical study. 22 January 2016. Retrieved 23 January 2016. https://www.fda.gov/Drugs/DrugSafety/ucm482740.htm
- 13.Huggins JP, Smart TS, Langman S, et al. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor pf-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 2012;153:1837–46. [DOI] [PubMed] [Google Scholar]
- 14.Vernalis P. Vernalis plc Completes Investment in its NCE Development [press release]. 2015. Retrieved from http://www.vernalis.com/media-centre/latest-releases/708-vernalis-plc-completes-investment-in-its-nce-development-pipeline (URL checked 28 May 2018).
- 15.Fowler CJ. The potential of inhibitors of endocannabinoid metabolism as anxiolytic and antidepressive drugs-A practical view. Eur Neuropsychopharmacol 2015;25:749–62. [DOI] [PubMed] [Google Scholar]
- 16.Patel S, Hill MN, Cheer JF, et al. The endocannabinoid system as a target for novel anxiolytic drugs. Neurosci Biobehav Rev 2017;76:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storr M, Keenan C, Emmerdinger D, et al. Targeting endocannabinoid degradation protects against experimental colitis in mice: involvement of CB1 and CB2 receptors. J Mol Med 2008;86:925–36. [DOI] [PubMed] [Google Scholar]
- 18.Sałaga M, Mokrowiecka A, Zakrzewski PK, et al. Experimental colitis in mice is attenuated by changes in the levels of endocannabinoid metabolites induced by selective inhibition of fatty acid amide hydrolase (FAAH). J Crohns Colitis 2014;8:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fowler CJ, Tiger G, Stenström A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure-activity relationship. J Pharmacol Exp Ther 1997;283:729–34. [PubMed] [Google Scholar]
- 20.Fowler CJ, Janson U, Johnson RM, et al. Inhibition of anandamide hydrolysis by the enantiomers of ibuprofen, ketorolac, and flurbiprofen. Arch Biochem Biophys 1999;362:191–6. [DOI] [PubMed] [Google Scholar]
- 21.Favia AD, Habrant D, Scarpelli R, et al. Identification and characterization of carprofen as a multitarget fatty acid amide hydrolase/cyclooxygenase inhibitor. J Med Chem 2012;55:8807–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt S, Paylor B, Boldrup L, et al. Inhibition of fatty acid amide hydrolase, a key endocannabinoid metabolizing enzyme, by analogues of ibuprofen and indomethacin. Eur J Pharmacol 2007;565:26–36. [DOI] [PubMed] [Google Scholar]
- 23.Karlsson J, Morgillo CM, Deplano A, et al. Interaction of the N-(3-Methylpyridin-2-yl)amide derivatives of flurbiprofen and ibuprofen with FAAH: enantiomeric selectivity and binding mode. PLoS One 2015;10:e0142711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cocco M, Congiu C, Onnis V, et al. Synthesis of ibuprofen heterocyclic amides and investigation of their analgesic and toxicological properties. Eur J Med Chem 2003;38:513–18. [DOI] [PubMed] [Google Scholar]
- 25.Fowler CJ, Björklund E, Lichtman AH, et al. Inhibitory properties of ibuprofen and its amide analogues towards the hydrolysis and cyclooxygenation of the endocannabinoid anandamide. J Enzyme Inhib Med Chem 2013;28:172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deplano A, Morgillo CM, Demurtas M, et al. Novel propanamides as fatty acid amide hydrolase inhibitors. Eur J Med Chem 2017;136:523–42. [DOI] [PubMed] [Google Scholar]
- 27.Gustin DJ, Ma Z, Min X, et al. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg Med Chem Lett 2011;21:2492–6. [DOI] [PubMed] [Google Scholar]
- 28.Sastry GM, Adzhigirey M, Day T, et al. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aid Mol Des 2013;27:221–34. [DOI] [PubMed] [Google Scholar]
- 29.Schrödinger Release 2017-1: Maestro, Schrödinger, LLC, New York, NY, 2017. [Google Scholar]
- 30.Lodola A, Castelli R, Mor M, Rivara S. Fatty acid amide hydrolase inhibitors: a patent review (2009–2014). Expert Opin Ther Pat 2015;25:1247–66. [DOI] [PubMed] [Google Scholar]
- 31.Shelley JC, Cholleti A, Frye L, et al. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 2007;21:681–91. [DOI] [PubMed] [Google Scholar]
- 32.Sherman W, Day T, Jacobson MP, et al. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006;49:534–53. [DOI] [PubMed] [Google Scholar]
- 33.Schrödinger Suite 2017-1 Induced Fit Docking protocol; Glide, Schrödinger, LLC, New York, NY, 2017; Prime, Schrödinger, LLC, New York, NY, 2017. [Google Scholar]
- 34.Boldrup L, Wilson SJ, Barbier AJ, Fowler CJ. A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J Biochem Biophys Methods 2004;60:171–7. [DOI] [PubMed] [Google Scholar]
- 35.Wilson AA, Hicks JW, Sadovski O, et al. Carbonyl‐labeled carbamates as fatty acid amide hydrolase radiotracers for positron emission tomography. J Med Chem 2013;56:201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.