

Abstract
Background
Cystic fibrosis (CF) is the commonest inherited life‐shortening illness in white populations, caused by a mutation in the gene that codes for the cystic fibrosis transmembrane regulator protein (CFTR), which functions as a salt transporter. This mutation mainly affects the airways where excess salt absorption dehydrates the airway lining leading to impaired mucociliary clearance. Consequently, thick, sticky mucus accumulates making the airway prone to chronic infection and progressive inflammation; respiratory failure often ensues. Other complications include malnutrition, diabetes and subfertility.
Increased understanding of the condition has allowed pharmaceutical companies to design mutation‐specific therapies targeting the underlying molecular defect. CFTR potentiators target mutation classes III and IV and aim to normalise airway surface liquid and mucociliary clearance, which in turn impacts on the chronic infection and inflammation. This is an update of a previously published review.
Objectives
To evaluate the effects of CFTR potentiators on clinically important outcomes in children and adults with CF.
Search methods
We searched the Cochrane Cystic Fibrosis Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the reference lists of relevant articles, reviews and online clinical trial registries. Last search: 21 November 2018.
Selection criteria
Randomised controlled trials (RCTs) of parallel design comparing CFTR potentiators to placebo in people with CF. A separate review examines trials combining CFTR potentiators with other mutation‐specific therapies.
Data collection and analysis
The authors independently extracted data, assessed the risk of bias in included trials and used GRADE to assess evidence quality. Trial authors were contacted for additional data.
Main results
We included five RCTs (447 participants with different mutations) lasting from 28 days to 48 weeks, all assessing the CFTR potentiator ivacaftor. The quality of the evidence was moderate to low, mainly due to risk of bias (incomplete outcome data and selective reporting) and imprecision of results, particularly where few individuals experienced adverse events. Trial design was generally well‐documented. All trials were industry‐sponsored and supported by other non‐pharmaceutical funding bodies.
F508del (class II) (140 participants)
One 16‐week trial reported no deaths, or changes in quality of life (QoL) or lung function (either relative or absolute change in forced expiratory volume in one second (FEV1) (moderate‐quality evidence). Pulmonary exacerbations and cough were the most reported adverse events in ivacaftor and placebo groups, but there was no difference between groups (low‐quality evidence); there was also no difference between groups in participants interrupting or discontinuing treatment (low‐quality evidence). Number of days until the first exacerbation was not reported, but there was no difference between groups in how many participants developed pulmonary exacerbations. There was also no difference in weight. Sweat chloride concentration decreased, mean difference (MD) ‐2.90 mmol/L (95% confidence interval (CI) ‐5.60 to ‐0.20).
G551D (class III) (238 participants)
The 28‐day phase 2 trial (19 participants) and two 48‐week phase 3 trials (adult trial (167 adults), paediatric trial (52 children)) reported no deaths. QoL scores (respiratory domain) were higher with ivacaftor in the adult trial at 24 weeks, MD 8.10 (95% CI 4.77 to 11.43) and 48 weeks, MD 8.60 (95% CI 5.27 to 11.93 (moderate‐quality evidence). The adult trial reported a higher relative change in FEV1 with ivacaftor at 24 weeks, MD 16.90% (95% CI 13.60 to 20.20) and 48 weeks, MD 16.80% (95% CI 13.50 to 20.10); the paediatric trial reported this at 24 weeks, MD 17.4% (P < 0.0001)) (moderate‐quality evidence). These trials demonstrated absolute improvements in FEV1 (% predicted) at 24 weeks, MD 10.80% (95% CI 8.91 to 12.69) and 48 weeks, MD 10.44% (95% CI 8.56 to 12.32). The phase 3 trials reported increased cough, odds ratio (OR) 0.57 (95% CI 0.33 to 1.00) and episodes of decreased pulmonary function, OR 0.29 (95% CI 0.10 to 0.82) in the placebo group; ivacaftor led to increased dizziness in adults, OR 10.55 (95% CI 1.32 to 84.47). There was no difference between groups in participants interrupting or discontinuing treatment (low‐quality evidence). Fewer participants taking ivacaftor developed serious pulmonary exacerbations; adults taking ivacaftor developed fewer exacerbations (serious or not), OR 0.54 (95% CI 0.29 to 1.01). A higher proportion of participants were exacerbation‐free at 24 weeks with ivacaftor (moderate‐quality evidence). Ivacaftor led to a greater absolute change from baseline in FEV1 (% predicted) at 24 weeks, MD 10.80% (95% CI 8.91 to 12.69) and 48 weeks, MD 10.44% (95% CI 8.56 to 12.32); weight also increased at 24 weeks, MD 2.37 kg (95% CI 1.68 to 3.06) and 48 weeks, MD 2.75 kg (95% CI 1.74 to 3.75). Sweat chloride concentration decreased at 24 weeks, MD ‐48.98 mmol/L (95% CI ‐52.07 to ‐45.89) and 48 weeks, MD ‐49.03 mmol/L (95% CI ‐52.11 to ‐45.94).
R117H (class IV) (69 participants)
One 24‐week trial reported no deaths. QoL scores (respiratory domain) were higher with ivacaftor at 24 weeks, MD 8.40 (95% CI 2.17 to 14.63), but no relative changes in lung function were reported (moderate‐quality evidence). Pulmonary exacerbations and cough were the most reported adverse events in both groups, but there was no difference between groups; there was no difference between groups in participants interrupting or discontinuing treatment (low‐quality evidence). Number of days until the first exacerbation was not reported, but there was no difference between groups in how many participants developed pulmonary exacerbations. No changes in absolute change in FEV1 or weight were reported. Sweat chloride concentration decreased, MD ‐24.00 mmol/L (CI 95% ‐24.69 to ‐23.31).
Authors' conclusions
There is no evidence supporting the use of ivacaftor in people with the F508del mutation. Both G551D phase 3 trials demonstrated a clinically relevant impact of ivacaftor on outcomes at 24 and 48 weeks in adults and children (over six years of age) with CF. The R117H trial demonstrated an improvement in the respiratory QoL score, but no improvement in respiratory function.
As new mutation‐specific therapies emerge, it is important that trials examine outcomes relevant to people with CF and their families and that adverse events are reported robustly and consistently. Post‐market surveillance is essential and ongoing health economic evaluations are required.
Plain language summary
Ivacaftor (marketed as Kalydeco®), a new specific therapy for cystic fibrosis
Review question
How does ivacaftor affect clinical outcomes (survival, quality of life and lung function) in people with cystic fibrosis (CF)?
Background
In people with CF, airway surfaces do not have enough water because of an abnormal protein; this makes it difficult to clear thick and sticky mucus, which leads to lung infections. Ivacaftor works on the abnormal protein in people with some mutations to allow the airways retain more water and better clear mucus, so fewer lung infections develop.
Ivacaftor was aimed at people with class III and IV mutations, and has been studied in people with G551D (class III), R117H (class IV) and F508del (class II) mutations.
Trial characteristics
We included five trials (447 participants) comparing ivacaftor to placebo (dummy treatment with no active medication) lasting between four and 48 weeks. Three trials enrolled a total of 238 people with at least one copy of the G551D mutation, one trial enrolled 140 people with two copies of the F508del mutation and one enrolled 69 people with at least one copy of the R117H mutation. The evidence is up to date as of 21 November 2018.
Key results
F508del mutation
The trial did not report any deaths or show improvements in lung function, quality of life scores or weight. Cough and pulmonary exacerbations (flare ups of lung disease) were the most reported adverse events when taking both ivacaftor and placebo; there were a similar number of flare ups for both groups. Sweat chloride concentrations were reduced with ivacaftor.
G551D mutation
No deaths were reported. Both children and adults taking ivacaftor showed improvements in lung function, but only adults reported higher quality of life scores. People given placebo reported more coughing and experienced more episodes of decreased lung function; more adults taking ivacaftor reported episodes of dizziness. Similar numbers of people taking ivacaftor and placebo delayed the course of medication, or withdrew from the trial altogether, due to side effects (e.g. psychological issues, liver disease, severe breathing problems). There were more serious pulmonary exacerbations whilst taking placebo compared to ivacaftor. Adults taking ivacaftor were admitted to hospital less often and had fewer courses of intravenous antibiotics for exacerbations. Both children and adults and children increased their weight with ivacaftor. There was a drop in sweat chloride concentrations with ivacaftor.
R117H mutation
No deaths occurred in this trial. While quality of life scores improved with ivacaftor, lung function did not. Cough and pulmonary exacerbations (flare ups of lung disease) were the most reported adverse events when taking both ivacaftor and placebo; there were a similar number of flare ups for both groups. There was no difference in weight; but as for other mutations there was a reduction in sweat chloride concentration with ivacaftor. Evidence suggests that ivacaftor is an effective treatment for people (over six years of age) with cystic fibrosis and the G551D mutation, but not for those with the F508del or R117H mutations.
Quality of the evidence
In most trials, individuals were put into different treatment groups at random with equal chances of being given either placebo or ivacaftor; no one could work out which treatment the next person would receive, so that healthier people did not receive ivacaftor and make the results seem better. We were not sure whether anyone involved in the trial knew who was receiving which treatment and how this might affect results. No trials reported all results clearly; sometimes they did not report them in a way that we could use in the review and sometimes they did not report the data at all. This affected our certainty regarding the overall results. Information about some side effects was limited as not many people experienced them, therefore, it is difficult to judge whether there was a difference between treatment groups
We judged the evidence in this review to be moderate to low quality.
Trial funding sources
All trials were sponsored by Vertex Pharmaceuticals Incorporated. The National Institute of Health (NIH), the Cystic Fibrosis Foundation (CFF) and other non‐pharmaceutical funding bodies also supported the trials.
Summary of findings
Summary of findings for the main comparison. Summary of findings ‐ ivacaftor compared with placebo for cystic fibrosis with the F508del CFTR mutation.
| Ivacaftor compared with placebo for cystic fibrosis with the F508del CFTR mutation | ||||||
| Patient or population: adults and children with cystic fibrosis and with the F508del CFTR mutation Settings: outpatients Intervention: ivacaftor Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Ivacaftor | |||||
|
Survival Follow‐up: 16 weeks |
No deaths reported. | No deaths reported. | NA |
140 (1 study) |
NA | |
|
QoL Total score Follow‐up: NA |
Not reported. | NA | NA | NA | ||
|
QoL (CFQ‐R) Respiratory domain Follow‐up: 16 weeks |
See comment.1 | See comment.1 | NA | 140 (1 study) | ⊕⊕⊕⊝ moderate3 | There was no significant difference between groups at 16 weeks. |
|
FEV1 % predicted Relative change from baseline Follow‐up:16 weeks |
Not reported.2 | The mean FEV1 (% predicted) was 2.4% higher (0.95% lower to 5.75% higher) in the ivacaftor group. | NA | 140 (1 study) | ⊕⊕⊕⊝ moderate3 | There was no significant difference between groups at 16 weeks. |
|
FEV1 % predicted Absolute change from baseline Follow‐up: 16 weeks |
Not reported.2 | The mean FEV1 (% predicted) was 1.7% higher (0.65% lower to 4.05% higher) in the ivacaftor group. | NA | 140 (1 study) | ⊕⊕⊕⊝ moderate3 | There was no significant difference between groups at 16 weeks. |
|
Adverse events Follow‐up:16 weeks |
The most commonly reported adverse events in the placebo group were: pulmonary exacerbation, cough, oropharyngeal pain and fatigue. | The most commonly reported adverse events in the ivacaftor group were: cough, pulmonary exacerbation, upper respiratory tract infection and nasal congestion. | NA | 140 (1 study) | ⊕⊕⊝⊝ low3,4 | There was no significant difference between groups in terms of any other adverse events. |
|
Time to first pulmonary exacerbation Follow‐up: 16 weeks |
Not reported. | NA | NA | NA | ||
| *The basis for the assumed risk is the mean placebo group risk across studies, unless otherwise stated. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CFTR: cystic fibrosis transmembrane regulator; CI: confidence interval; FEV1: forced expiratory volume at 1 second; HR: hazard ratio; MD: mean difference; QoL: quality of life. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Presentation of the data prevented including data in analysis therefore results are presented narratively. 2. Only differences between placebo and ivacaftor groups were presented, results within the placebo group were not presented therefore an assumed risk cannot be calculated. 3. Downgraded once due to risk of bias from incomplete outcome data and/or selective reporting in all of the included studies. 4. Downgraded once due to imprecision: few events occurred therefore CIs for occurrence of specific events are very wide (also see Analysis 1.2)
Summary of findings 2. Summary of findings ‐ ivacaftor compared with placebo for cystic fibrosis with at least one G551D CFTR mutation.
| Ivacaftor compared with placebo for cystic fibrosis with at least one G551D CFTR mutation | ||||||
| Patient or population: adults and children with cystic fibrosis and with at least one G551D CFTR mutation Settings: outpatients Intervention: ivacaftor Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Ivacaftor | |||||
|
Survival Follow‐up: 2 ‐ 48 weeks |
No deaths reported. | No deaths reported. | NA | 238 (3 studies) | NA | |
|
QoL Total score Follow‐up: NA |
Not reported. | NA | NA | NA | ||
|
QoL (CFQ‐R) Respiratory domain Follow‐up: 2 ‐ 48 weeks |
See comment.1 | See comment.1 | NA |
222 (3 studies) |
⊕⊕⊕⊝ moderate2 | A small phase 2 trial (n = 19) showed no significant difference at 2 or 4 weeks. A phase 3 trial (n = 151) in adults showed significantly higher quality of life in the ivacaftor group compared to placebo at 48 weeks A phase 3 trial (n = 52) showed no significant difference at 24 or 48 weeks in the child version of the CFQ‐R. The same trial showed a significantly higher quality of life in the ivacaftor group compared to placebo at 48 weeks in the parent / caregiver version at 24 weeks but not at 48 weeks.5 |
|
FEV1 % predicted Relative change from baseline Follow‐up: 2 ‐ 48 weeks |
See comment.1 | See comment.1 | NA |
232 (3 studies) |
⊕⊕⊕⊝ moderate2 | A small phase 2 trial (n = 19) showed no significant difference at 2 or 4 weeks. Two phase 3 trials (n = 213) showed a significant improvement in FEV1 at 24 weeks (MD ranged between 16.9% to 17.4%) One phase 3 trial (n = 161) showed a significant improvement in FEV1 at 48 weeks (MD 16.8%). 6 |
|
FEV1 L and % predicted Absolute change from baseline Follow‐up: 2 to 48 weeks |
See comment.1 | See comment.1 | NA |
232 (3 studies) |
⊕⊕⊝⊝ low2,3 | A small phase 2 trial (n = 19) showed no significant difference at 2 or 4 weeks. Two phase 3 trials (n = 213) showed a significant improvement in FEV1 (L and % predicted) at 24 weeks and 48 weeks.7 |
|
Adverse events Follow‐up: 2 ‐ 48 weeks |
The most commonly reported adverse events in the placebo group were: pulmonary exacerbation, cough, oropharyngeal pain and headache. | The most commonly reported adverse events in the ivacaftor group were: cough, pulmonary exacerbation, upper respiratory tract infection and headache. | NA |
232 (3 studies) |
⊕⊕⊝⊝ low2,4 | Pulmonary exacerbations were significantly more common in the placebo group. There was no significant difference between groups in terms of any other adverse events.8 |
|
Time to first pulmonary exacerbation Follow‐up: 48 weeks |
41% of the placebo group were exacerbation free at 48 weeks. | 67% of the ivacaftor group were exacerbation free at 48 weeks. | HR 0.46 (95% CI 0.29 to 0.73) |
161 (1 study) |
⊕⊕⊕⊝ moderate2 | There was also a statistically significant difference between groups at 24 weeks; 78% and 51% of the ivacaftor and placebo groups were exacerbation free respectively; HR 0.46 (95% CI 0.28 to 0.76). |
| *The basis for the assumed risk is the mean placebo group risk across studies, unless otherwise stated. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CFTR: cystic fibrosis transmembrane regulator; CI: confidence interval; FEV1: forced expiratory volume at 1 second; HR: hazard ratio; MD: mean difference; QoL: quality of life. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1. Presentation of the data prevented pooling the data from all three studies therefore results are summarised narratively. 2. Downgraded once due to risk of bias from incomplete outcome data and/or selective reporting in all of the included studies. 3. Downgraded once due to unexplained heterogeneity. 4. Downgraded once due to imprecision: few events occurred therefore CIs for occurrence of specific events are very wide (also see Analysis 2.5, Analysis 2.6 and Analysis 2.8). 5. See analyses for numerical results (Analysis 2.1;Analysis 2.2; Analysis 2.3). 6. See analysis for numerical results (Analysis 2.4). 7. See analyses for numerical results (Analysis 2.14; Analysis 2.15). 8. See analyses for numerical results (Analysis 2.5; Analysis 2.6; Analysis 2.8).
Summary of findings 3. Summary of findings ‐ ivacaftor compared with placebo for cystic fibrosis with at least one R117H CFTR mutation.
| Ivacaftor compared with placebo for cystic fibrosis with at least one R117H CFTR mutation | ||||||
|
Patient or population: adults and children with cystic fibrosis and with at least one R117H CFTR mutation Settings: outpatients Intervention: ivacaftor Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Ivacaftor | |||||
|
Survival Follow‐up: 2 ‐ 24 weeks |
No deaths reported. | No deaths reported. | NA |
69 (1 study) |
NA | |
|
QoL Total score Follow‐up: NA |
Not reported. | NA | NA | NA | ||
|
QoL (CFQ‐R) Respiratory domain Follow‐up: 2 ‐ 24 weeks |
Not reported. | The mean adjusted CFQ‐R respiratory domain score was 8.40% higher (2.17% higher to 14.63% higher) in the ivacaftor group. | NA |
69 (1 study) |
⊕⊕⊕⊝ moderate2 | There was a significant improvement in CFQ‐R respiratory domain scores at 24 weeks.3 |
|
FEV1 % predicted Relative change from baseline Follow‐up: 2 ‐ 24 weeks |
See comment.1 | The mean adjusted FEV1 (% predicted) was 5.00% higher (0.24% lower to 10.24% higher) in the ivacaftor group. | NA |
69 (1 study) |
⊕⊕⊕⊝ moderate2 | There was no significant difference between groups at 24 weeks.3 |
|
FEV1 L and % predicted Absolute change from baseline Follow‐up: 2 ‐ 24 weeks |
See comment.1 | The mean adjusted FEV1 (% predicted) was 2.10% higher (1.13% lower to 5.33% higher) in the ivacaftor group. | NA |
69 (1 study) |
⊕⊕⊕⊝ moderate2 | There was no significant difference between groups at 24 weeks.3 |
|
Adverse events Follow‐up: 2 ‐ 24 weeks |
The most commonly reported adverse events in the placebo group were: pulmonary exacerbation and cough. | The most commonly reported adverse events in the ivacaftor group were: pulmonary exacerbation, cough, headache, nasal congestion, oropharyngeal pain, diarrhoea and increased sputum. | NA |
69 (1 study) |
⊕⊕⊝⊝ low2,4 | There was no significant difference between groups in terms of any other adverse events. |
|
Time to first pulmonary exacerbation Follow‐up: 24 weeks |
63% of the placebo group were exacerbation free at 24 weeks. | 68% of the ivacaftor group were exacerbation free at 24 weeks. | HR 0.93 (95% CI 0.42 to 2.08) |
69 (1 study) |
⊕⊕⊕⊝ moderate2 | There was no significant difference between groups at 24 weeks. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CFQ‐R: Cystic Fibrosis Questionnaire‐Revised; CFTR: cystic fibrosis transmembrane regulator; CI: confidence interval; FEV1: forced expiratory volume at 1 second; HR: hazard ratio; MD: mean difference; QoL: quality of life. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1. Presentation of the data prevented including data in analysis therefore results are presented narratively. 2. Downgraded once due to risk of bias from selective reporting. 3. Treatment effect was adjusted for baseline values of age and % predicted FEV1. 4. Downgraded once due to imprecision: few events occurred therefore CIs for occurrence of specific events are very wide (also see Analysis 3.4)
Background
A glossary of terms specific to this review can be found in the appendices (Appendix 1); a more general glossary of terms used in Cochrane Reviews can be accessed at Cochrane Glossary.
Description of the condition
Cystic fibrosis (CF) is the most common inherited life‐shortening illness in white populations, with a prevalence of 1 in 2000 at birth in Europeans (Bobadilla 2002) and varying prevalence in North American populations depending on ethnic composition (Hamosh 1998). According to the data report from the UK CF Registry which was published in July 2014, there were 10,338 people with a confirmed diagnosis of CF in the UK (UK CF Registry Report 2013). The clinical features of CF arise from abnormalities in a protein called the cystic fibrosis transmembrane conductance regulator (CFTR) (Riordan 1989; Southern 1997). Normally, CFTR protein is transported to the outer cell membrane, where it has a role in co‐ordinating the transport of the salt ions, sodium (Na+) and chloride (Cl‐), in and out of the cell. This salt transport role is particularly important in the airways, pancreas, sweat gland, and vas deferens.
In the lungs of people with CF, defective salt transport leads to a reduction in airway surface liquid volume. This, in turn, leads to compromised mucociliary clearance, which initiates a cycle of infection, inflammation and progressive lung damage, eventually causing respiratory failure and premature death. Other consequences of CFTR dysfunction, including those related to abnormalities in the inflammatory response, are probably important but not as well characterized. In addition, CFTR‐related ion transport abnormalities can lead to other systemic complications. These include malnutrition and diabetes (through pancreatic damage, salt depletion through excess loss in sweat) and subfertility (in men and women) caused by various factors.
Over 1900 mutations have been identified in the CFTR gene, which can be categorised according to the impact they have on CFTR synthesis, processing, or function (CFMD 2012). These categories have been described (Rowntree 2003; Southern 2007); and a summary is presented in the additional tables (Table 4). The most common mutation is the delta F508 (F508del) mutation (class II mutation) and is present on at least one allele in 7990 (90.8%) people with CF in the UK. The second most common mutation in the UK is the G551D mutation (class III mutation). This mutation exists in 514 (5.84%) people with CF in the UK. The R117H mutation (class IV mutation) is the third most common mutation, present in 398 (4.52%) people with CF in the UK (UK CF Registry Report 2013).
1. Classes of mutations affecting CFTR production, structure, and function.
| Class | Example mutation | Impact on CFTR structure and function |
| I | G542X | Synthesis of CFTR is critically impaired, and no functional protein is produced. This is due to the presence of a premature stop codon in the nucleotide sequence. Individuals have minimal CFTR function. |
| II | ΔF508 | A full length of CFTR is produced, but this is structurally abnormal and destroyed by the cell before it reaches the cell membrane. This is called a defect in the intracellular trafficking pathway. Minimal amounts of CFTR can escape being destroyed and be transported to the cell membrane. However here, class II CFTR products display defective ion transport. Individuals have minimal CFTR function. |
| III | G551D | CFTR is produced and embedded in the cell membrane, but the chloride channel does not respond (‘switch on’) to normal stimulation from the cell. This means there is no significant ion transport across the protein. Individuals have some residual CFTR function. |
| IV | R347P | CFTR is transported to the outer cell membrane, and responds to normal stimulation, but functions at a low level because chloride ions do not cross the channel appropriately. Individuals have some residual CFTR function. |
| V | A455E | Normal CFTR is produced, but the amount of protein is reduced. Individuals have some residual CFTR function. |
CFTR: cystic fibrosis transmembrane conductance regulator
Description of the intervention
Understanding how the mutations, which are described in the additional tables (Table 4), affect the production, structure, and function of CFTR has led to the concept of mutation‐specific therapies. One strategy, relating mainly to mutations classes III to V but also to class II, is to improve the function of CFTR that has reached the cell membrane, but whose function is compromised (McKone 2004). Drugs that improve CFTR function in the cell membrane are called potentiators and have the potential to correct the abnormal salt transport that characterises CF. Potentiators can be administered orally, or as an inhaled preparation.
Two distinct approaches have resulted in the recognition of candidate drugs with this mode of action (Amaral 2007):
testing of compounds known to affect CFTR or other ion‐channels (either pharmaceutical drugs or chemicals which occur naturally in plants, herbs, fruits or food components);
high throughput screening, which involves testing large numbers of diverse chemicals, on laboratory cell lines, to identify which of these may stimulate CFTR.
In addition to potentiators, other drugs which aim to correct defects of the CFTR are also under investigation in clinical trials. These include correctors (which increase the amount of CFTR in the cell membrane by preventing early degradation ‐ class II mutations) and 'stop‐codon therapies' which act to prevent structural abnormalities of CFTR that occur when premature stop codons terminate protein synthesis too early ‐ class I mutations). Cochrane Reviews of trials assessing correctors and one assessing stop codon therapies have been published (Aslam 2017; Southern 2018).
How the intervention might work
The CFTR potentiator ivacaftor was identified through high‐throughput screening by Vertex Pharmaceuticals Incorporated. In cell and animal models, it was able to restore CFTR function for class III mutations and correct the abnormal transmembrane salt transport (Van Goor 2009). However, the precise mechanism of action of ivacaftor is unclear, owing to the limited amount of data.
The correction of the basic defect in CF in the airways of the lung could lead to the normalization of airway surface liquid; the resulting re‐establishment of mucociliary clearance, would then have a beneficial impact on the chronic infection and inflammation that characterizes CF lung disease.
Why it is important to do this review
Since CFTR potentiators are novel therapies, it is important that randomised controlled trials (RCTs) testing these agents are critically appraised. This will enable examination of the evidence relating to the benefits and harms of CFTR potentiators. It is important that funding bodies have a clear evidence base on which to assess new therapies for CF that aim to correct the basic defect, particularly given the large prevalence of people with mutations that might be impacted by CFTR potentiators. It is likely that these therapies will represent a significant healthcare resource. In addition, critical appraisal of included trials will help inform future trial design.
This review aims to collate evidence from RCTs that have evaluated the benefits and harms of CFTR potentiators in people with CF. This is an update of a previously published version (Patel 2012; Patel 2015).
Objectives
To evaluate the effects of CFTR potentiators on clinically important outcomes in children and adults with CF.
Methods
Criteria for considering studies for this review
Types of studies
RCTs of parallel design (published or unpublished). Cross‐over trials were considered inappropriate given the potential longer‐term impact of this therapy on outcomes.
Types of participants
We have included trials involving children or adults with CF, as confirmed either by the presence of two disease‐causing mutations, or by a combination of a positive sweat test and recognised clinical features of CF. We included trials involving people with any level of disease severity and any relevant mutation class, where CFTR has been demonstrated to successfully embed within the cell membrane and display defective function.
Types of interventions
In a post hoc change we have only included trials comparing CFTR potentiators to placebo or another intervention. Trials where CFTR potentiators are used in combination with other CFTR function modulators were excluded. There are many ongoing trials that use a CFTR potentiator alongside a CFTR corrector and we believe it would be more appropriate to conduct a separate systematic review for these trials (Southern 2018).
Types of outcome measures
Primary outcomes
Survival (in a post hoc change mortality data were also considered)
-
Quality of life (QoL) (measured using validated quantitative scales or scores (e.g. Cystic Fibrosis Questionnaire‐Revised (CFQ‐R) (Quittner 2009))
total QoL score
different sub‐domains which may be reported
Forced expiratory flow rate at one second (FEV1) (relative change from baseline)
Secondary outcomes
-
Adverse effects
graded by review authors as mild (therapy does not need to be discontinued)
graded by review authors as moderate (therapy is discontinued, and the adverse effect ceases)
graded by review authors as severe (life‐threatening or debilitating, or which persists even after treatment is discontinued)
other adverse effects of therapy (of any severity) that are not classifiable according to these categories, including pulmonary exacerbation (protocol defined or physician defined) and time‐to‐exacerbation data
-
Hospitalisation
number of days
number of episodes
School or work attendance (i.e. number of days missed)
-
Other physiological measures of lung function (litres or per cent (%) predicted for age, sex and height)
FEV1 absolute values (rather than "relative change from baseline", which is specified as primary outcome)
forced vital capacity (FVC) (absolute values and change from baseline)
-
Extra courses of antibiotics (measured as time‐to the next course of antibiotics and the total number of courses of antibiotics)
oral
intravenous
inhaled
-
Radiological measures of lung disease (assessed using any scoring system)
chest radiograph scores
computerised tomogram (CT) score
-
Acquisition of respiratory pathogens
Pseudomonas aeruginosa
Staphylococcus aureus
Haemophilus influenzae
other significant pathogen
-
Eradication of respiratory pathogens (as defined by trial authors)
P aeruginosa
S aureus
H influenzae
other significant pathogen
-
Nutrition and growth (measured as relative change from baseline) (including z scores or centiles)
weight
body mass index (BMI)
height
Sweat chloride (change from baseline) as a measure of CFTR function
Cost of treatment
Search methods for identification of studies
There are no restrictions regarding language or publication status.
Electronic searches
We identified relevant trials from the Group's Cystic Fibrosis Trials Register using the term: 'drugs that augment function of abnormal CFTR protein in the cell membrane'. Relevant trials have been tagged with this term for indexing purposes in the Group's Cystic Fibrosis Trials Register.
This is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of the Cochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the handsearching of two journals – Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work was identified by searching through the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Group's website.
Date of latest search of the Group's Cystic Fibrosis Trials Register: 21 November 2018.
We also searched online clinical trial registries (Appendix 1). Date of last search of clinical trial registries: 21 November 2018.
Searching other resources
We screened references of included trials to identify other potentially relevant trials. We also contacted authors of included trials, leaders in the field, and companies known to be developing and investigating CFTR potentiators, to identify any trials that may have been missed by these searches.
Data collection and analysis
Selection of studies
For the original review, two authors (SP and IS) independently assessed the suitability of each trial identified by the search. From the 2018 update onwards MS and IS independently assessed the suitability of each trial and if disagreement arose regarding suitability for inclusion, we attempted to reach a consensus by discussion, failing which a third author (KWS) arbitrated.
Data extraction and management
For the updated review, two authors (MS and IS) independently extracted relevant data from each included trial using a standardised data extraction form. If disagreement arose on data extraction, we attempted to reach a consensus by discussion, failing which a fourth author (KWS) arbitrated.
We intended to report on our primary outcome 'survival' as survival to time X (time‐to‐event data). However, these data were not available, so we considered mortality data (number of deaths) as an alternative outcome (post hoc change). For lung function, where possible we reported the relative change from baseline in FEV1, since this way of presenting FEV1 is very important in clinical practice. If this was not possible, we reported absolute change from baseline in FEV1 as a secondary outcome. We extracted QoL as the absolute change from baseline.
With regards to the secondary outcome 'Adverse effects', we extracted the total number of participants who experienced adverse effects or required trial drug interruption or termination. When extracting data on pulmonary exacerbations, we noted whether they were protocol‐defined or physician‐defined. We reported the number of participants who experienced episodes of pulmonary exacerbation or reported time‐to‐exacerbation data. We extracted the number of participants who required hospitalisations. With regards to 'Extra courses of antibiotics', we reported on the total number of courses of antibiotics.
For the secondary outcomes 'Change from baseline in weight' and 'Change from baseline in sweat chloride concentration', we extracted the absolute change from baseline results.
For continuous outcomes, where standard deviations (SDs) were not provided, we calculated the standard error of the mean (SEM) from the 95% confidence intervals (CIs) and inserted the results into a generic inverse variance (GIV) analysis.
If the trial author presented non‐parametric data, we reported results in the written text and not in the analysis.
We have reported data at 4 weeks, 16 weeks, 24 weeks and 48 weeks.
Assessment of risk of bias in included studies
Two authors (MS and IS) assessed the risk of bias for each trial using the Cochrane risk of bias tool (Higgins 2011a). This includes assessment of the following methodological aspects of the included trials:
procedure for randomisation (selection bias);
allocation concealment (selection bias);
masking (blinding) of the intervention from participants, clinicians, and trial personnel evaluating outcomes (performance bias);
missing outcome data (attrition bias);
selective outcome reporting (reporting bias);
other sources of bias.
We also assessed whether all participants were included in an intention‐to‐treat analysis, regardless of whether they completed the treatment schedule or not. If disagreement arose on the assessment of risk of bias of a trial, we attempted to reach a consensus by discussion, failing which a third author (KWS) arbitrated.
Measures of treatment effect
For binary outcomes, we calculated a pooled estimate of the treatment effect for each outcome using the pooled odds ratio (OR) and 95% CIs or 99% confidence intervals for analysis of separate adverse events. If calculating a pooled OR was not appropriate, we calculated an estimate of the treatment effect for each outcome using the OR and 95% CIs.
For continuous outcomes, we calculated the mean change from baseline for each group or the mean post‐intervention values and 95% CIs for each group. We produced a pooled estimate of treatment effect by calculating the mean difference (MD) and 95% CIs. For QoL, CFQ‐R was the most frequently used questionnaire and so we calculated the MD and 95% CIs. No other questionnaire was used.
For time‐to‐event outcomes, such as 'Time to first pulmonary exacerbation', we used measures of survival analysis, and calculated hazard ratios (HR) and 95% CIs between different arms of the trial.
In future updates of this review, if different trials present data for the same outcomes in different forms (e.g. absolute values of lung function measures, or change in these measures from a baseline), we will combine these in a meta‐analysis where appropriate.
Where the trials did not report change data, but instead presented absolute post‐treatment data without baseline data (so it was not possible to calculate change data), we planned to use absolute post‐treatment data instead of change from baseline. However, if the report presented baseline and post‐treatment data for any outcome, we calculated SDs for the change from baseline, for example if the CI was available. If there was not enough information available to calculate the SDs for the changes, we planned to impute them from other trials in the review, where data were available and trials were similar (i.e. when they used the same measurement scale, had the same degree of measurement error, had the same time periods between baseline and final value measurement and had conducted the randomisation process appropriately). If neither of these methods were possible, we planned to calculate a change‐from‐baseline SD, making use of an imputed correlation coefficient (methods described in chapter 16 in the Cochrane Handbook of Systematic Reviews of Interventions (Higgins 2011b)).
Unit of analysis issues
Within this review, we have only included results from RCTs of parallel design in which individual trial participants were randomised. We excluded cross‐over trials, because they are not appropriate for evaluating therapies that potentially correct the underlying defect (Higgins 2011b). We did not identify any cluster RCTs. Further updates of this review will include any eligible cluster RCTs identified and these will be included and analysed as described in the Cochrane Handbook for Systematic Reviews of Inteventions (Higgins 2011b).
Where trials with multiple intervention groups reported dichotomous data i.e. adverse effects, we pooled the data to form one intervention group and compared data to the placebo group as recommended in chapter 16 of the Cochrane Handbook for Systematic Reviews of Inteventions (Higgins 2011b).
Dealing with missing data
In order to allow an intention‐to‐treat analysis, we extracted data on the number of participants with each outcome event, by allocated treated group, irrespective of compliance and whether or not the participant was later thought to be ineligible or otherwise excluded from treatment or follow‐up. We calculated the number of participants with outcome data and checked if this was consistent with the number of originally randomised participants. We checked that this finding was consistent with findings on the online ongoing trials database (clinicaltrials.gov). If there were any discrepancies or any uncertainty, we contacted the primary investigators for clarification. For more detail about what we requested and what information we were provided with, see the risk of bias section below (Incomplete outcome data (attrition bias)).
Assessment of heterogeneity
We assessed heterogeneity through a visual examination of the forest plots, and by considering the I² statistic (Higgins 2003) together with the Chi² test (P < 0.1) (Deeks 2011). The I² statistic reflects the likelihood that the variation of results across trials is due to heterogeneity rather than chance, and we interpreted this statistic using the following classification:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
If we had identified heterogeneity between trials, we would have conducted a sensitivity analysis including only homogenous trials to determine the effect of heterogeneity on the overall treatment effect of the intervention.
Assessment of reporting biases
In order to identify selective outcome reporting, where possible we compared outcomes described in the trial protocol with those reported in the publication(s). We requested protocols for specific trials from the primary investigators, corresponding author, or relevant pharmaceutical company when they were not available. We have recorded the proportion of protocols that were available to us. If the protocol was not available, we checked published information on the trial registry databases. We also compared outcomes listed in the 'Methods' section of the final paper with those presented in the 'Results' section. If the published papers partially reported negative findings (i.e. P > 0.05), we contacted the primary investigators for these data.
We planned to assess publication bias by constructing and assessing the symmetry of a funnel plot. This would have been possible if we had included more than 10 trials in the review.
Data synthesis
We used a fixed‐effect model to analyse data from trials which we did not consider to be heterogeneous (see classifications above). If substantial or considerable heterogeneity had been present (I² greater than 50%), we used a random‐effects model to analyse data.
Subgroup analysis and investigation of heterogeneity
We planned to investigate any heterogeneity that we identified using subgroup analyses of potential confounding factors. For this review, we planned that these confounding factors would be:
age (children (defined as younger than 18 years of age) versus adults);
gender;
different mutation classes (Table 4).
As we did not seek individual patient data from trial investigators, we did not undertake a subgroup analysis on the basis of disease severity. Such an analysis may be incorporated in future updates of this review.
Sensitivity analysis
If we had been able to combine a sufficient number of trials (at least 10), we planned to examine the impact of risk of bias on the results examined by comparing meta‐analyses including and excluding trials with concerns of high risk of selection or reporting bias due to issues relating to randomisation, allocation concealment, or masking of interventions from participants or trial personnel.
Summary of findings and quality of the evidence (GRADE)
In a post hoc change from protocol, we have presented three summary of findings tables (Table 1; Table 2; Table 3) under the comparison of 'Ivacaftor compared to placebo' according to mutation class (see Table 4).
The following outcomes were reported in all tables (chosen based on relevance to clinicians and consumers):
survival;
QoL (total score);
QoL (respiratory domain);
FEV1 (relative and absolute change);
adverse events; and
time to first pulmonary exacerbation.
For clarity in the tables, adverse events are not presented according to the subdomains in Effects of interventions; instead the authors have inserted a general statement about the summary of findings for these outcomes and the evidence is graded based on all of the subdomains combined.
We determined the quality of the evidence using the GRADE approach; and downgraded evidence in the presence of a high risk of bias in at least one trial, indirectness of the evidence, unexplained heterogeneity or inconsistency, imprecision of results, high probability of publication bias. We evidence by one level if they considered the limitation to be serious and by two levels if very serious.
Results
Description of studies
Results of the search
The search of the Group's Cystic Fibrosis Trials Register identified a total of 201 publications (abstracts and full papers) representing 67 trials. A further 91 trials were identified from ongoing trials databases. Five trials (67 references) were included and 62 trials (115 references) were excluded. The two trials (two references) awaiting classification have both been published as conference abstracts; further details below (Kazani 2016; Uttamsingh 2016). In a post hoc change (2018), references to corrector and potentiator combination therapy trials have not been listed in the review, meaning 79 trials (110 references) identified have not been listed in the reference section of this review. The results of the search are displayed in the PRISMA diagram below (Figure 1).
1.
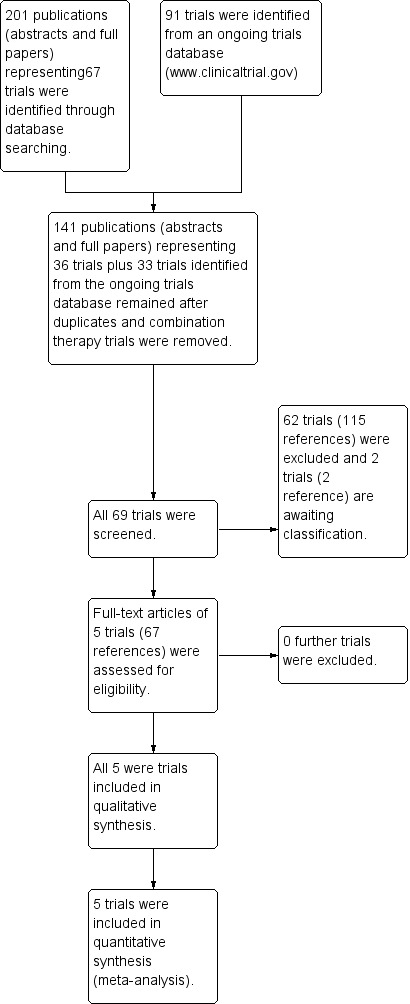
Study flow diagram.
No additional trials were identified by screening references of included trials or by contacting authors of included trials, leaders in the field, and companies known to be developing and investigating CFTR potentiators.
Included studies
We included five trials (67 references) with 447 participants in this review (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). All trials were available as full texts.
Trial design
All five included trials were RCTs of parallel design. The responsible funding body in all included trials was Vertex Pharmaceuticals Incorporated. The National Institute of Health (NIH), the Cystic Fibrosis Foundation (CFF) and other non‐pharmaceutical funding bodies were also involved in supporting the trials (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011).
The F508del trial (n = 140) was a phase 2 trial that lasted 16 weeks (DISCOVER 2011). Participants who met pre‐defined eligibility criteria and completed Part A of the trial were continued onto an open‐label extension phase (Part B) lasting 96 weeks (n = 48); data from this extension were not eligible for inclusion in this review (DISCOVER 2011). There were three trials of people with the G551D mutation; the phase 2 trial had three arms and lasted 28 days (Accurso 2010); the two phase 3 trials lasted 48 weeks (ENVISION 2013; STRIVE 2011). Participants in these trials who completed 48 weeks of treatment (n = 194) were entered into an open‐label extension phase, which lasted up to 96 weeks; participants from this trial were not eligible for inclusion in this review because they were not re‐randomised to treatment or control (PERSIST 2014). The phase 3 R117H trial (n = 69) lasted for 24 weeks (KONDUCT 2015). Participants that completed 24 weeks of treatment were eligible to enrol into an open‐label extension phase which lasted up to 104 weeks with an interim analysis at 12 weeks (KONTINUE 2017). Data from KONTINUE were not eligible for inclusion as participants were not re‐randomised (KONTINUE 2017).
All trials were conducted at multiple centres (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). The F508del trial was run across 34 sites in North America (DISCOVER 2011). Although this trial recruited participants with a class II mutation, we included it in the initial version of the review on the basis that the trial evaluated ivacaftor and its effect of the minimal amount of CFTR reaching the cell surface in this mutation. The phase 2 G551D trial recruited participants from 13 sites in North America and Europe (Accurso 2010). The paediatric G551D trial (65 sites) and the adult G551D trial (29 sites) were conducted in North America, Europe and Australia (ENVISION 2013; STRIVE 2011). The phase 3 R117H trial was conducted at 31 sites across Europe and North America (KONDUCT 2015).
Outcome data were reported at time frames ranging from 2 to 48 weeks (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011).
Participants
All 447 participants in the included trials had a confirmed diagnosis of CF. The F508del trial examined the effect of ivacaftor on people homozygous for the F508del mutation (class II mutation) (DISCOVER 2011). In the three G551D trials (class III mutation), participants were required to possess at least one G551D‐CFTR allele (Accurso 2010; ENVISION 2013; STRIVE 2011). The R117H trial required participants to have at least one R117H‐CFTR allele (KONDUCT 2015).
Two trials recruited recruited participants aged 12 years and older (DISCOVER 2011; STRIVE 2011); participants in the F508del trial had a mean age of 25.5 years (DISCOVER 2011) and participants in the adult G551D trial had a mean age of 23.2 years (STRIVE 2011). The phase 2 G551D trial recruited participants aged 18 years and over and participants had a median 21 age of years (Accurso 2010). The paediatric phase 3 G551D trial enrolled participants aged 6 to 11 years of age and participants had a mean age of 8.9 years (ENVISION 2013). The R117H trial recruited those over 6 years of age and participants had a mean age of 31 years (KONDUCT 2015).
In the F508del trial 140 participants were allocated in a 4:1 ratio to either intervention or placebo (DISCOVER 2011). Sample sizes in the G551D trials ranged between 19 participants (Accurso 2010) and 167 participants (STRIVE 2011). The phase 2 G551D trial allocated 19 participants in a 2:2:1 ratio to either one of two intervention groups or a placebo group respectively (Accurso 2010). In the phase 3 G551D trials, 52 children and 167 adults were allocated in a 1:1 ratio to either intervention or placebo (ENVISION 2013; STRIVE 2011). During randomisation, the adults were stratified according to age (less than 18 years versus 18 years and over) and pulmonary function (less than 70% versus 70% and over of the predicted FEV1) (STRIVE 2011). Similarly, a 1:1 ratio was used to allocate participants to intervention or placebo groups in the R117H trial; participants were stratified by age (6 to 11 years, 12 to 17 years, and 18 years and over) and % predicted FEV1 (< 70%, ≥ 70% to ≤ 90% and > 90%) during randomisation (KONDUCT 2015).
All participants had a baseline FEV1 reading of 40% or over for age, sex and height and where baseline information was available, similar characteristics were seen (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011).
Interventions
Ivacaftor was the intervention drug in all included trials (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). Four trials compared 150 mg of ivacaftor every 12 hours to placebo (ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). The phase 2 G551D trial compared 150 mg and 250 mg of ivacaftor every 12 hours to placebo (Accurso 2010).
To determine the doses to be used, two G551D trials (the phase 2 trial and the paediatric phase 3 trial) conducted earlier investigations (Accurso 2010; ENVISION 2013). Ivacaftor dose levels used in the phase 2 G551D trial were determined based on pharmacokinetic modelling of data from a previous cross‐over trial (Accurso 2010). Pre‐trial single‐dose pharmacokinetic analysis and phase 2a data were used to determine doses in the paediatric phase 3 G551D trial (ENVISION 2013).
Participants in four of the five included trials continued on prescribed medications, that were approved for CF, during the trial period (Accurso 2010; ENVISION 2013; DISCOVER 2011; STRIVE 2011). No information was provided for the R117H trial regarding how drug dosage was determined or whether participants continued taking prescribed medications (KONDUCT 2015).
Outcomes
The primary end points in the trials were safety (Accurso 2010) or absolute change in FEV1 (ENVISION 2013; KONDUCT 2015; STRIVE 2011) or both (DISCOVER 2011).
All trials employed the CFQ‐R respiratory domain to measure QoL (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). One trial reported data for all domains (Accurso 2010). In the adult phase 3 G551D trial, data for other CFQ‐R domains were reported where improvements were seen in the ivacaftor group (STRIVE 2011). Relative change from baseline in FEV1 was reported in the full text by four trials (Accurso 2010; DISCOVER 2011; KONDUCT 2015; STRIVE 2011) and in a conference abstract by the fifth trial (24‐week interim data only) (ENVISION 2013).
All trials reported on the safety profile of ivacaftor and the total number of participants who developed pulmonary exacerbations (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). The adult G551D trial and R117H trial also reported data for hospitalisation and number of days of hospitalisation (KONDUCT 2015; STRIVE 2011). Three trials reported on the number of participants who required extra courses of antibiotics (DISCOVER 2011; KONDUCT 2015; STRIVE 2011).
All trials examined the effect of ivacaftor on absolute change in FEV1 (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). Two trials reported on absolute change from baseline in FEV1 for subgroups, according to pulmonary function, geographic region and gender (ENVISION 2013; STRIVE 2011).
Three trials reported on weight (change from baseline), BMI and BMI as z score for age (ENVISION 2013; DISCOVER 2011; STRIVE 2011) and one trial reported change in BMI from baseline (KONDUCT 2015). Two trials reported on height z scores (ENVISION 2013; STRIVE 2011).
All included trials reported on change from baseline in sweat chloride concentration (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011).
Nasal potential difference, a primary outcome in the Accurso trial, was not included in this review, as it is not yet a validated outcome measure (Accurso 2010).
Excluded studies
A total of 62 trials (115 references) were listed as excluded. We excluded 12 trials which were of cross‐over design (Altes 2011; Berkers 2017; Edgeworth 2017; Davies 2012; KONNECTION 2013; EudraCT Number: 2016‐001619‐19; McGarry 2015; NCT01685801; NCT01784419; NCT02709109; NCT02742519; NCT03068312) and one trial which carried out secondary analysis on trials of cross‐over design (Accurso 2013). Eleven trials were excluded as they used treatments to correct the molecular defect and were not potentiators (ALBATROSS 2017; Chadwick 1998; Clancy 2012; FLAMINGO 2017; Horsley 2018; McCarty 2002; NCT03474042; NCT02323100; Rubenstein 1998; Rubenstein 2006; Zeitlin 2002). Six trials were excluded as they examined stop codon therapies for class I mutations (Kerem 2014; Pradal 2002; Romano 2000; Sermet‐Gaudelus 2010; Wilschanski 2003; Wilschanski 2008). We excluded 12 trials due to an observational study design (EudraCT Number: 2016‐001440‐18; Hubert 2018; NCT01549314; NCT01863238; NCT02039986; NCT02141464; NCT02311140; NCT02445053; NCT02722057; NCT03390985; NCT03652090; Seliger 2015) and a further 12 trials were excluded as they had a single interventional arm with no comparator (ARRIVAL 2018; Davies 2016; EudraCT Number: 2014‐000817‐30; NCT01946412; NCT02310789; NCT02690519; NCT02707562; NCT02934698; NCT03256799; NCT03256968; NCT03277196; PERSIST 2014). Three trials used treatments to amplify or modify the molecular defect rather than potentiate it (NCT02718495; NCT02724527; NCT03258424). Four trials did not use mutation‐specific treatments e.g. antibiotic therapy (EUudraCT Number: 2016‐001785‐29; NCT02443688; NCT02759562; RIO‐CF 2017). One trial excluded people with CF (TOPIC 2018).
We excluded 79 trials (110 references) comparing potentiator and corrector combination therapy to placebo, but these have not been referenced within the list of excluded studies as a separate systematic review has been conducted which focuses on combination therapy (Southern 2018).
Studies awaiting classification
Two trials are listed as awaiting classification (Kazani 2016; Uttamsingh 2016), one of which has been terminated according to the record on clinicaltrials.gov (Kazani 2016).
Trial design
Both trials are double‐blind RCTs of parallel design, but one is a phase 1 trial (Uttamsingh 2016) and the second is a phase 2 trial (Kazani 2016). The phase 1 trial lasted for seven days with no additional follow‐up (Uttamsingh 2016). The phase 2 trial had a duration of 14 days with a follow‐up to 42 days (Kazani 2016).
Participants
In the phase 1 trial, investigators enrolled 40 participants with a confirmed diagnosis of CF, but no details of participants' baseline characteristics are available (Uttamsingh 2016).
In the three‐arm phase 2 trial, only the third phase of the trial was eligible which randomised 49 adults with CF heterozygous with one allele represented as any CFTR mutation and the other allele must represent a class III, IV, V, VI CFTR mutation (Kazani 2016). In the phase 2 trial the mean age of participants was 31.7 years. With regards to gender split the trial randomised more males (n = 30) than females (n = 19).
Intervention
Investigators enrolled 40 participants and allocated them in a 4:1 ratio to either intervention or placebo (Uttamsingh 2016). CTP‐656 (a deuterated analogue of ivacaftor) was used as the intervention drug. Participants received either 75mg, 150mg, or 225mg of CTP‐656. Dose escalation was initiated only after safety and tolerability were found to support proceeding to the higher dose. All participants were dosed under fed conditions; a high‐fat breakfast was provided on Day 1 through Day 7 approximately 30 minutes prior to dosing (Uttamsingh 2016). (Since this trial was undertaken CTP‐656 has been sold by its developers (Concert Pharmaceuticals Inc.) to Vertex and has been renamed VX‐561.)
The phase 2 trial compared QBW251 (a membrane‐associated wild‐type CFTR potentiator) at two doses ‐ 150 mg twice daily and 450 mg twice daily ‐ to placebo. The 450 mg daily dose group was split into participants with a number of different mutations (n = 12) and a second group who were homozygous for F508del (n = 19) (Kazani 2016).
Outcomes
The phase 1 trial's main outcome was the pharmacokinetic (PK) profile and PK blood samples were collected at several time points post‐dose on Day 1 and Day 7, and at 12 and 24 hours post‐dose on Day 2 through Day 6. The plasma concentrations of CTP‐656 and its metabolites, (D‐M1 and D‐M6) were analysed by a combination of liquid chromatography with mass spectrometry for all three dose cohorts (Uttamsingh 2016).
In the phase 2 trial, the primary outcomes were the change in LCI and adverse events; secondary outcomes included the change in FEV1, the change in CFQR reported outcomes and the change in sweat chloride. Some limited results have been published on clinicaltrials.gov (Kazani 2016).
Risk of bias in included studies
A summary of the risk of bias judgements can be found in the figures (Figure 2; Figure 3).
2.
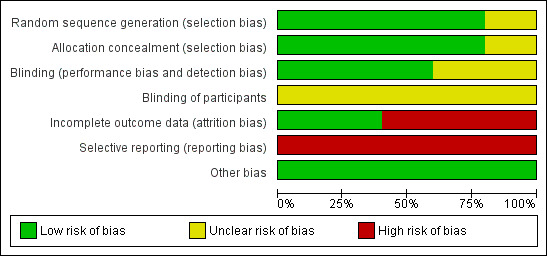
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies. This Figure demonstrates the high proportion of trials that were judged to have a high risk of attrition bias (incomplete outcome data addressed) and selective reporting bias.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Sequence generation
Four trials reported on random sequence generation; all four employed an unblinded statistician (not otherwise associated with the trial) to produce the final randomisation list using a code produced by the trial sponsor and we judged these to have a low risk of bias for sequence generation (Accurso 2010; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). One phase 3 G551D trial did not report on how the children were randomised and we judged this to have an unclear risk of bias (ENVISION 2013).
Allocation concealment
Four trials reported on allocation concealment (Accurso 2010; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). Participants were allocated to groups using an interactive voice response system (IVRS) according to the concealed randomisation list and we judged these to have a low risk of bias with regards to allocation concealment (Accurso 2010; DISCOVER 2011; KONDUCT 2015; STRIVE 2011). The paediatric phase 3 G551D trial did not report on allocation concealment and we judged this to have an unclear risk of bias (ENVISION 2013).
Blinding
Three trials reported on the blinding of trial personnel (Accurso 2010; DISCOVER 2011; STRIVE 2011). All trial personnel were blinded to participant treatment assignments. Tasks requiring unblinded personnel were conducted by people not otherwise involved in the trials; hence we judged these three trials to have a low risk of bias for blinding of personnel (Accurso 2010; DISCOVER 2011; STRIVE 2011). Two trials stated that they were 'double‐blinded' but did not provide sufficient information to allow a clear judgement giving an unclear risk of bias (ENVISION 2013; KONDUCT 2015).
With regards to the blinding of participants, none of the included trials reported details of the oral tablets such as colour, size, consistency or taste; therefore we judge them all to have an unclear risk of bias with regards to the blinding of participants (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
Incomplete outcome data
There was low attrition (less than 15%) in all included trials (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
In the phase 2 G551D trial, one participant withdrew consent prior to dosing, but all 19 remaining participants completed the trial and were included in the analysis of reported outcomes; therefore, we judged this trial to have a low risk of attrition bias (Accurso 2010). In the R117H trial, two participants from the ivacaftor group withdrew after dosing due to one participant becoming pregnant and one for non‐compliance. All 69 participants who received at least one dose of the trial drug were included in the analysis. We judged this trial to also have a low risk of attrition bias (KONDUCT 2015).
In the remaining three trials, some participant data were excluded from the analysis and these trials were judged to have a high risk of attrition bias (DISCOVER 2011; ENVISION 2013; STRIVE 2011).
A total of 10 participants (7.1%) withdrew from the F508del trial (DISCOVER 2011); eight out of 112 (7.1%) participants withdrew from the ivacaftor group (three due to adverse events, two for non‐compliance, one refused medication, one was lost to follow up; and one due to "early termination per sponsor decision") and two participants out of 28 (7.1%) withdrew from the placebo group due to adverse events (DISCOVER 2011). Participant data were excluded from the analysis of the change from baseline in sweat chloride concentration (one participant excluded from the ivacaftor group) and the absolute change from baseline in FEV1 (one participant excluded from the ivacaftor group). The author was approached for information on these missing data (DISCOVER 2011).
In the paediatric phase 3 G551D trial, 4 out of 52 children (7.7%) withdrew from the placebo group, one for each of the following reasons: adverse events; refusal of medication; withdrawal of consent; and wrong genotype. Participant data were excluded from the analysis for absolute change from baseline in % predicted FEV1 (one child excluded from placebo group), change from baseline in sweat chloride concentration (three children excluded from ivacaftor group and three children excluded from the placebo group) and CFQ‐R respiratory domain paediatric version (one child excluded from placebo group). The author was approached for information on these missing data and revealed that a modified intention‐to‐treat analysis (not per protocol) was employed, where data were excluded from the analysis (ENVISION 2013).
In the adult phase 3 G551D trial, 10 out of 167 participants (6.0%) withdrew from the placebo group; four due to adverse events, one on the physician's decision, two required prohibited medication; one withdrew consent; one was the wrong genotype; and one had "Increased Lab Draws/Difficult Lab Stick". Six out of 167 participants (3.6%) withdrew from the ivacaftor group; one due to adverse events; two for non‐compliance; one became pregnant; one refused medication; and one withdrew consent. After these 16 randomised participants (9.6%) withdrew, a total of 161 participants (78 in the placebo group and 83 in the ivacaftor group) were included in the analysis. However, in the analysis of change from baseline in CFQ‐R respiratory domain scores, data from 10 participants (seven participants from the placebo group and three participants from the ivacaftor group) were excluded; and data from nine participants (four participants from the placebo group and five participants from the ivacaftor group) were excluded from the analysis of the change from baseline in sweat chloride concentration. The author was approached for information on these missing data (STRIVE 2011).
Selective reporting
Trial protocols were available for the phase 2 G551D and the adult phase 3 G551D trials (Accurso 2010; STRIVE 2011). In the phase 2 trial, weight was measured at days 1, 3, 14, 21 and 28, but not reported. In such a trial, the change in weight of participants from baseline is a key outcome and by not reporting this outcome, the trial is at high risk of selective reporting bias (Accurso 2010). In the adult trial, the following tertiary outcomes were not reported in the full text: change from baseline in oxygen saturation; change from baseline in EuroQol Questionnaire (EQ‐5D); and outpatient sick visits to the clinic or hospital for CF–related complications. Also CFQ‐R domain scores were reported for domains where improvements were seen in the ivacaftor group only. Hence, this trial was also judged to have a high risk of selective reporting bias (STRIVE 2011).
Full protocols were not available, either online or upon request, for the remaining three trials (DISCOVER 2011; ENVISION 2013; KONDUCT 2015). In the paediatric phase 3 G551D trial, FVC was measured at days 1 and 15, then at weeks 8, 16, 24, 32, 40 and 48; but results were not reported in the full text; also, relative change in FEV1 from baseline was reported at 24 weeks, but not at any other time point (ENVISION 2013). In such trials, the change from baseline in FVC and relative change from baseline in FEV1 are key outcomes and by not reporting these outcomes, the trial is at high risk of selective reporting (ENVISION 2013). In the F508del trial, there were missing data for change from baseline in FEV1 score (at day 15 and week 8) and in change from baseline in FVC or mid‐forced expiratory flow (FEF25‐75%) measured throughout the trial. Also, one withdrawal from the ivacaftor group was due to "early termination per sponsor decision" and attempts to illicit further information about this were unsuccessful; we therefore found the risk of selective reporting in this trial to be high (DISCOVER 2011). In the R117H trial, a protocol synopsis was made available by the trial sponsor which reported a number of tertiary outcomes; however, no results were made available for these outcomes. We therefore judged the trial to be at high risk of selective reporting bias (KONDUCT 2015).
Other potential sources of bias
The baseline characteristics of participants were similar in all included trials (Accurso 2010; ENVISION 2013; DISCOVER 2011; KONDUCT 2015; STRIVE 2011).
In the phase 2 G551D trial, drug dosing was monitored by clinical staff via returned dosage units and dosing diaries; an adherence rate of 100% (range 92.6 to 100) was achieved (Accurso 2010). In the paediatric phase 3 G551D trial, reviewing dosing with participants and caregivers ensured correct doses were taken; mean rates of compliance of 94.4% in the ivacaftor group and of 95.7% in the placebo group were reported (ENVISION 2013). Two trials ensured adherence by drug accountability (DISCOVER 2011; STRIVE 2011). No information was provided regarding drug adherence for the R117H trial (KONDUCT 2015).
In the R117H trial additional age subgroup analyses were performed for participants aged 6 to 11 years and ≥ 18 years, however, there were only two participants between 12 to 17 years of age so no statistical analysis was performed for this group (KONDUCT 2015). No reason was provided for the low numbers in 12 to 17 age group (KONDUCT 2015).
All trials were funded by a single company, Vertex Pharmaceuticals Incorporated (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
Effects of interventions
See: Table 1; Table 2; Table 3
In the summary of findings tables, the quality of the evidence has been graded for pre‐defined outcomes (see above) and definitions of these gradings provided.
Ivacaftor versus placebo
Primary outcomes
1. Survival
No survival data or deaths were reported by any of the included trials (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
2. QoL
a. total QoL score
None of the trials reported on total QoL scores (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
b. different sub‐domains
All five trials reported the CFQ‐R respiratory domain scores (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011). The phase 2 G551D trial also reported scores for all other CFQ‐R domains (Accurso 2010) and the adult phase 3 G551D trial reported scores for CFQ‐R domains where improvements were reported in the ivacaftor group (STRIVE 2011).
In the F508del trial, investigators reported lower scores on the CFQ‐R respiratory domain score in the ivacaftor group (MD ‐1.3) at 16 weeks, but this difference was not significant (DISCOVER 2011) (moderate‐quality evidence). Insufficient data for this outcome were reported for inclusion in the analysis.
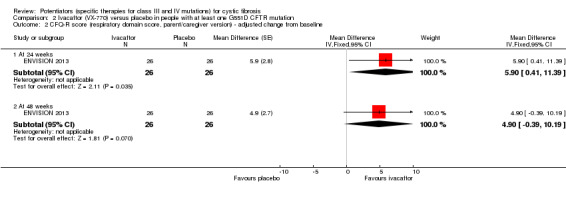
Data from both phase 3 G551D trials have not been combined as the paediatric trial reported individual change from baseline respiratory domain scores, for both children and parents or caregivers (ENVISION 2013), but the adult trial pooled the CFQ‐R respiratory domain scores from adult and child questionnaires (STRIVE 2011). In the paediatric trial, at 24 weeks children in the ivacaftor group reported higher CFQ‐R respiratory domain scores, MD 6.10 (95% CI ‐1.35 to 13.55), but the difference between groups was not significant (ENVISION 2013) (Analysis 2.1) (moderate‐quality evidence). In the same trial at 24 weeks, the parents or caregivers of children in the ivacaftor group reported significantly higher CFQ‐R respiratory domain scores, MD 5.90 (95% CI 0.41 to 11.39) (Analysis 2.2) (moderate‐quality evidence). In the adult trial, at 24 weeks participants reported significantly higher CFQ‐R respiratory domain scores in the ivacaftor group, MD 8.10 (95% CI 4.77 to 11.43) (STRIVE 2011) (Analysis 2.3) (moderate‐quality evidence).
2.1. Analysis.
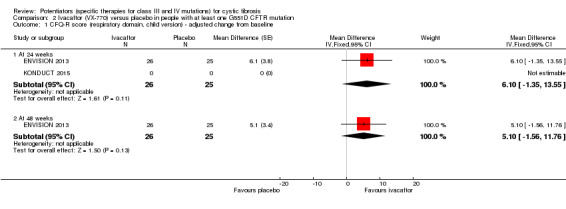
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 1 CFQ‐R score (respiratory domain, child version) ‐ adjusted change from baseline.
2.2. Analysis.
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 2 CFQ‐R score (respiratory domain score, parent/caregiver version) ‐ adjusted change from baseline.
2.3. Analysis.
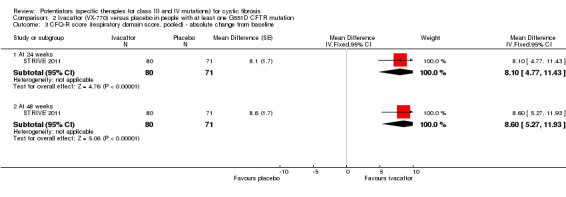
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 3 CFQ‐R score (respiratory domain score, pooled) ‐ absolute change from baseline.
At 48 weeks, both children and and parents or caregivers reported higher CFQ‐R respiratory domain scores in the ivacaftor group, MD 5.10 (95% CI ‐1.56 to 11.76) and MD 4.90 (95% CI ‐0.39 to 10.19) respectively (Analysis 2.1; Analysis 2.2) (moderate‐quality evidence); in neither case were treatment differences significant (ENVISION 2013). In the adult trial at 48 weeks, investigators reported significantly higher CFQ‐R respiratory domain scores in the ivacaftor group, MD 8.60 (95% CI 5.27 to 11.93) (STRIVE 2011) (Analysis 2.3) (moderate‐quality evidence). At the same time‐point, significantly higher CRQ‐R scores in the physical functioning scale (P < 0.001), social functioning scale (P = 0.0026), eating disturbances scale (P = 0.0021) and treatment burden scale (P = 0.0419) were observed (STRIVE 2011). Higher scores for the body, emotion and digestive scales were also seen but the treatment differences between groups for these domains were not significant (STRIVE 2011).
Accurso reported on improvement in the respiratory domain score at two weeks and four weeks using medians and ranges, precluding the analysis of these data in RevMan (Accurso 2010). At two weeks, a median (range) increase of 5.6 (0.0 to 16.7) points was achieved in the 150 mg ivacaftor group and 5.6 (‐11.1 to 11.1) in the 250 mg ivacaftor group. The median (range) change of 2.8 (‐5.6 to 11.1) in the placebo group meant the difference between the two treatment and the placebo groups was not significant. At four weeks, there were no significant differences between treatment groups and the placebo group; the 150 mg group reported a median (range) improvement of 8.3 (0.0 to 16.7) points, the 250 mg group reported a median (range) improvement of 11.1 (‐5.6 to 33.3) points and the placebo group reported a median (range) improvement of 2.8 (‐5.6 to 11.1) points (Accurso 2010). Accurso also presented median (range) scores for all other CFQ‐R domains which are shown in the additional tables (Table 5). None of the improvements were significant (Accurso 2010) (moderate‐quality evidence).
2. CFQ‐R domain scores in the phase 2 G551D study at 4 weeks (median (range)).
| Domain | Day 14 | Day 28 | ||||
| Placebo (n = 4) | VX‐770 150 mg (n = 8) | VX‐770 250 mg (n = 7) | Placebo (n = 4) | VX‐770 150 mg (n = 8) | VX‐770 250 mg (n = 7) | |
| Body image | 0 (‐11.1 to 22.2) | 0 (‐22.2 to 0) | 0 (‐11.1 to 22.2) | ‐5.6 (‐11.1 to 22.2) | 0 (‐22.2 to 11.1) |
0 (‐11.1 to 44.4) |
| Digestive symptoms | ‐5.6 (‐22.2 to 0) | 0 (0 to 22.2) | 0 (‐11.1 to 22.2) | 0 (‐22.0 to 0) | 5.6 (0 to 22.2) |
0 (‐11.1 to 33.3) |
| Eating disturbances | 0 (‐11.1 to 11.1) | 0 (0 to 11.1) | 0 (‐11.1 to 0) | ‐5.6 (‐11.1 to 0) | 0 (0 to 0) |
0 (‐11.1 to 0) |
| Emotional functioning | 13.3 (‐6.7 to 20.0) | 0 (‐26.7 to 0) | 0 (‐20.0 to 26.7) | 3.3 (0 to 20.0) | 0 (‐6.7 to 6.7) | 6.7 (‐6.7 to 20.0) |
| Health perceptions | 5.6 (‐11.1 to 11.1) | 0 (‐22.2 to 22.2) | 0 (‐33.3 to 11.1) | 0 (‐11.1 to 11.1) | 0 (‐22.2 to 22.2) |
0 (‐11.1 to 11.1) |
| Physical functioning | 0 (0 to 4.2) | 2.1 (‐4.2 to 16.7) | 4.2 (0 to 8.3) | 2.1 (0 to 4.2) |
4.2 (‐8.3 to 25.0) |
0 (0 to 12.5) |
| Respiratory symptoms | 2.8 (‐5.6 to 11.1) | 5.6 (0 to 16.7) | 5.6 (‐11.1 to 11.1) | 2.8 (‐5.6 to 11.1) | 8.3 (0 to 16.7) | 11.1 (‐5.6 to 33.3) |
| Role | 0 (0 to 8.3) | 0 (‐8.3 to 8.3) | 0 (0 to 8.3) | 0 (0 to 0) | 0 (‐8.3 to 8.3) |
0 (‐8.3 to 8.3) |
| Social | 2.8 (‐5.6 to 5.6) | 0 (‐44.4 to 11.1) | ‐5.6 (‐16.7 to 5.6) | 2.8 (‐11.1 to 5.6) | 0 (‐11.1 to 5.6) |
0 (‐16.7 to 11.1) |
| Treatment burden | 0 (‐11.1 to 11.1) | ‐5.6 (‐22.2 to 11.1) | 0 (‐22.2 to 22.2) | 0 (0 to 0) | ‐5.6 (‐22.2 to 11.1) | 0 (‐22.2 to 11.1) |
| Vitality | 4.2 (‐16.7 to 8.3) | 4.2 (‐8.3 to 16.7) | 0 (‐8.3 to 16.7) | ‐8.3 (‐16.7 to 0) | 0 (‐16.7 to 25.0) |
0 (0 to 16.7) |
| Weight | 0 (0 to 33.3) | 0 (0 to 33.3) | 0 (‐33.3 to 33.3) | 0 (0 to 33.3) |
0 (0 to 33.3) |
0 (‐33.3 to 33.3) |
CFQ‐R: Cystic Fibrosis Questionnaire‐Revised
In the R117H trial, investigators reported a significant improvement in the CFQ‐R respiratory domain scores in the ivacaftor group, MD 8.40 (95% CI 2.17 to 14.63) (KONDUCT 2015) (Analysis 3.1) (moderate‐quality evidence). The treatment effect was adjusted for continuous baseline values of age and % predicted FEV1 (KONDUCT 2015). The R117H trial conducted a further subgroup analysis by age, which only showed a significant improvement in the respiratory domain score for adults in the ivacaftor group and not children (Table 6).
3.1. Analysis.
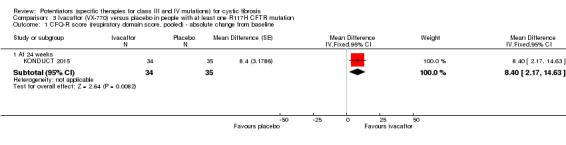
Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 1 CFQ‐R score (respiratory domain score, pooled) ‐ absolute change from baseline.
3. Outcomes for age subgroups in R117H study at 24 weeks.
| Overall | 6 ‐ 11 years | ≥18 years | ||||
| Treatment difference | P value | Treatment difference | P value | Treatment difference | P value | |
| Quality of life (CFQ‐R) ‐ respiratory domain (pooled) | 8.4 | 0.009 | ‐6.1 | 0.19 | 12.6 | 0.002 |
| % predicted FEV1 ‐ absolute change from baseline | 2.1 | 0.20 | ‐6.3 | 0.03 | 5.0 | 0.01 |
| % predicted FEV1 ‐ relative change from baseline | 5.0 | 0.06 | ‐6.8 | 0.04 | 0.2 | 0.008 |
| BMI ‐ absolute change from baseline | 0.26 | 0.78 | ‐0.18 | 0.87 | 0.31 | 0.78 |
| Sweat chloride ‐ absolute change from baseline | ‐24.0 | <0.0001 | ‐27.6 | <0.001 | ‐21.9 | <0.001 |
Treatment difference was adjusted for age and % predicted FEV1
BMI: body mass index CFQ‐R: cystic fibrosis questionnaire ‐ revised FEV1: forced expiratory volume in one second
3. FEV1 (relative change from baseline)
All included trials reported this outcome (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
At 16 weeks investigators reported no significant difference in the relative change from baseline in FEV1 between groups in the F508del trial, MD 2.40% (95% CI ‐0.95 to 5.75) (DISCOVER 2011) (Analysis 1.1) (moderate‐quality evidence).
1.1. Analysis.

Comparison 1 Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation, Outcome 1 FEV1 ‐ relative change from baseline.
At 24 weeks in the phase 3 G551D trials, both children and adults in the ivacaftor group reported significant improvements in the relative change from baseline in FEV1 (ENVISION 2013; STRIVE 2011). For the paediatric trial, only interim data were reported in a conference abstract which were insufficient for inclusion into the meta‐analysis, MD 17.4% (P < 0.0001) (ENVISION 2013); but for the adult trial investigators reported MD 16.90% (95% CI 13.60 to 20.20) (Analysis 2.4). Data at 48 weeks were not reported in the paediatric trial (ENVISION 2013); however, the adult trial reported a significant improvement in the relative change from baseline in FEV1 in the ivacaftor group at 48 weeks, MD 16.80% (95% CI 13.50 to 20.10) (STRIVE 2011) (Analysis 2.4) (moderate‐quality evidence).
2.4. Analysis.
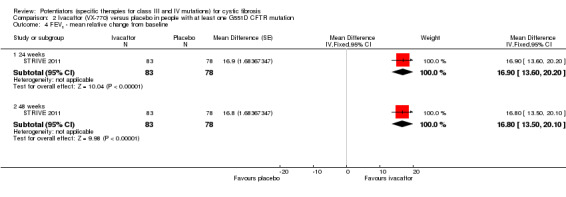
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 4 FEV1 ‐ mean relative change from baseline.
Again Accurso reported medians (range) from the phase 2 G551D trial; at four weeks there was a relative improvement from baseline in % predicted FEV1 of 8.7% (2.3 to 31.3) in the 150 mg group, 4.4% (0.0 to 18.3) in the 250 mg group and 7.3% (5.2 to 8.2) in the placebo group (Accurso 2010). Treatment differences between ivacaftor arms and the placebo group were not significant (Accurso 2010) (moderate‐quality evidence).
In the R117H trial, investigators included the relative change from baseline in FEV1 data in a results table; however, the absolute change in FEV1 was a primary outcome and this was the focus of the text (KONDUCT 2015). At 24 weeks investigators reported no significant difference in the relative change from baseline in FEV1 between treatment groups MD 5.00% (95% CI ‐0.24 to 10.24) (Analysis 3.2) (moderate‐quality evidence). Treatment effect was adjusted for continuous baseline values of age and % predicted FEV1 (KONDUCT 2015).
3.2. Analysis.
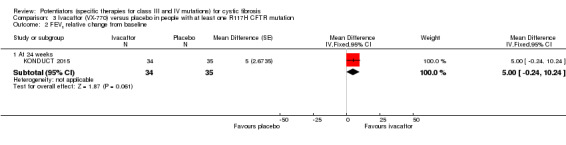
Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 2 FEV1 relative change from baseline.
Secondary outcomes
1. Adverse effects
All included trials reported the number of participants who experienced adverse effects (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
The F508del trial reported the number of participants who experienced adverse effects that occurred in at least 5% of participants taking ivacaftor (DISCOVER 2011) (Analysis 1.2). Accurso reported the number of participants who experienced any adverse effect in the phase 2 G551D trial (Accurso 2010); we have pooled data for the 150 mg group and 250 mg group and compared these to the placebo group (Analysis 2.6). Both phase 3 G551D trials reported the number of participants who experienced any serious adverse effect and the number of participants who experienced all adverse effects that occurred in more than 5% of participants in either treatment arm (ENVISION 2013; STRIVE 2011). We have combined the data from both trials (Analysis 2.5). Serious adverse effects were defined as life‐threatening or debilitating, which is consistent with our definition of severe. The R117H trial reported the number of participants with any adverse event, serious adverse events (the authors' definition of a serious adverse event was not present in the text), adverse events leading to discontinuation and adverse events occurring in 15% of participants in any ivacaftor treatment group (KONDUCT 2015) (Analysis 3.3; Analysis 3.4).
1.2. Analysis.

Comparison 1 Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation, Outcome 2 Adverse effects.
2.6. Analysis.
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 6 All adverse events.
2.5. Analysis.
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 5 Adverse events occurring in greater than or equal to 5% of trial participants.
3.3. Analysis.

Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 3 Adverse events.
3.4. Analysis.
Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 4 Adverse events in > 15% of participants.
For the F508del trial, phase 3 G551D trials and the R117H trial, we have reported on adverse effects that occurred more frequently in either the ivacaftor or placebo groups where there was greater or equal to 5% incidental difference between groups in the number of participants affected (DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011). Given the small sample size in the phase 2 G551D trial by Accurso (n = 19), we have not reported on the most common adverse effects from this trial (Accurso 2010). For all trials we have present below the results that were statistically significant in trial reports. Where numerical data were not provided, we have discussed the results narratively.
a. mild (therapy does not need to be discontinued)
In the F508del trial, episodes of cough, nausea, rash and contact dermatitis were experienced by more participants assigned to ivacaftor compared to those assigned to placebo. Pulmonary exacerbations were more common in participants in the placebo group (DISCOVER 2011) (Analysis 1.2) (low‐quality evidence). In the adult phase 3 G551D trial, more adults in the placebo group experienced episodes of pulmonary exacerbation, cough, haemoptysis, and decreased pulmonary function in comparison to participants in the ivacaftor group. Adverse effects that occurred in more adults in the ivacaftor group were headache, upper respiratory tract infection, nasal congestion, rash, and dizziness (STRIVE 2011) (Analysis 2.5) (low‐quality evidence). In the paediatric phase 3 G551D trial, episodes of cough, productive cough, vomiting, rales, and decreased pulmonary function were experienced by more children in the placebo group in comparison to children in the ivacaftor group. Episodes of oropharyngeal pain, headache, nasopharyngitis, upper respiratory tract infection, otitis media, diarrhoea, and an increased blood eosinophil count were reported in more children in the ivacaftor group (ENVISION 2013) (Analysis 2.5) (low‐quality evidence). In the R117H trial more episodes of nasal congestion, oropharyngeal pain, abdominal pain, wheeze and CF lung pathogen colonisation were experienced in the ivacaftor group compared to the placebo group (KONDUCT 2015) (Analysis 3.4) (low‐quality evidence).
There were no statistically significant differences at the 1% significance level to allow for multiple analyses related to adverse events in any of the included trials (Analysis 2.5; Analysis 2.6, Analysis 1.2; Analysis 3.4).
b. moderate (therapy is discontinued and the adverse effect ceases)
The F508del trial and both phase 3 G551D trials reported the number of participants who experienced adverse effects that required interruption of the intervention (DISCOVER 2011; ENVISION 2013; STRIVE 2011). The R117H trial did not report any participants requiring interruption of the intervention (KONDUCT 2015).
In the F508del trial, two participants in the ivacaftor group (1.8%) stopped receiving the intervention due to elevated liver enzyme values; but the intervention was subsequently restarted as both cases were found to be caused by concurrent pathology (DISCOVER 2011). No trial drug interruption occurred in the placebo group. The difference between groups in number of participants who required trial interruption was not significant, OR 1.29 (95% CI 0.06 to 27.62) (Analysis 1.3) (low‐quality evidence). In the paediatric phase 3 G551D trial, three participants from the placebo group experienced adverse effects requiring interruption of the intervention compared to one participant in the ivacaftor group; but the details of these adverse events were not reported (ENVISION 2013) (low‐quality evidence). In the adult trial, 11 adverse events in the ivacaftor group (13.3%) compared to four in the placebo group (5.1%) required trial drug interruption. Adverse events in the ivacaftor group were: haemoptysis (n = 1); migraine (n = 1); upper respiratory tract infection (n = 1); pulmonary exacerbation (n = 1); increased hepatic enzyme (n = 2); pulmonary exacerbation and anaphylactic shock (n = 1); lymph node pain and gynaecomastia (n = 1); myalgia and diarrhoea (n = 1); decreased weight and pulmonary exacerbation (n = 1); and vulvovaginal mycotic infection, oral candidiasis and two episodes of pulmonary exacerbation (n = 1). Adverse events in the placebo group were: migraine (n = 1); pulmonary exacerbation (n = 1); increased blood lactate dehydrogenase and increased hepatic enzymes (n = 1); and rash, nephrolithiasis and renal colic (n = 1). Investigators reported that five participants in the placebo group required drug interruption; however, one of these participants subsequently withdrew from the trial and will be discussed under 'severe' adverse events (STRIVE 2011) (low‐quality evidence). Combined data for both phase 3 G551D trials showed no significant difference between treatment arms, OR 1.18 (95% CI 0.14 to 9.92) (Analysis 2.7) (low‐quality evidence). Heterogeneity was substantial (I² = 64%) which is possibly due to the difference in ages between the adult and paediatric trials.
1.3. Analysis.

Comparison 1 Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation, Outcome 3 Severity of adverse effects of therapy with regards to study drug interruption (moderate) or discontinuation (severe).
2.7. Analysis.
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 7 Severity of adverse effects of therapy with regards to study drug interruption (moderate) or discontinuation (severe).
c. severe (life‐threatening or debilitating, or which persists even after treatment is discontinued)
All included trials reported the number of participants who developed adverse effects that required discontinuation of the intervention (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
In the F508del trial, three participants in the ivacaftor group (2.7%) discontinued due to asthenia, fatigue and headache (n = 1), arthritis (n = 1) and myopathy (n = 1); a further participant in this group developed fatigue, depression and suicidal ideation that was considered to be life‐threatening (DISCOVER 2011). In comparison, two participants in the placebo group (7.1%) discontinued medication because of abnormal feelings of cognitive disorder (n = 1) and elevated liver enzymes and increased lactate dehydrogenase levels (n = 1). The abnormal liver enzyme profile was considered to be caused by concomitant use of an anabolic steroid like a nutritional supplement (DISCOVER 2011). The number of participants requiring trial drug discontinuation between treatment arms was not significant, OR 0.36 (95% CI 0.06 to 2.25) (Analysis 1.3) (low‐quality evidence).
In the paediatric phase 3 G551D trial, one participant from the placebo group withdrew due to anxiety and psychological issues (ENVISION 2013). In the adult trial, four participants from the placebo group withdrew from the trial; one each due to increased hepatic enzyme levels, atrioventricular block, panic attack and respiratory failure. There was one dropout from the ivacaftor group due to increased hepatic enzymes (STRIVE 2011). Combined data for the phase 3 G551D trials showed no significant difference between treatment arms in the number of participants discontinuing the trial drug, OR 0.25 (95% CI 0.04 to 1.57) (Analysis 2.7) (low‐quality evidence). Likewise, combined data from the two trials for serious adverse effects, did not show an increase with ivacaftor compared with placebo (Analysis 2.8), with the exception of pulmonary exacerbation which will be discussed under other adverse effects of therapy (below).
2.8. Analysis.
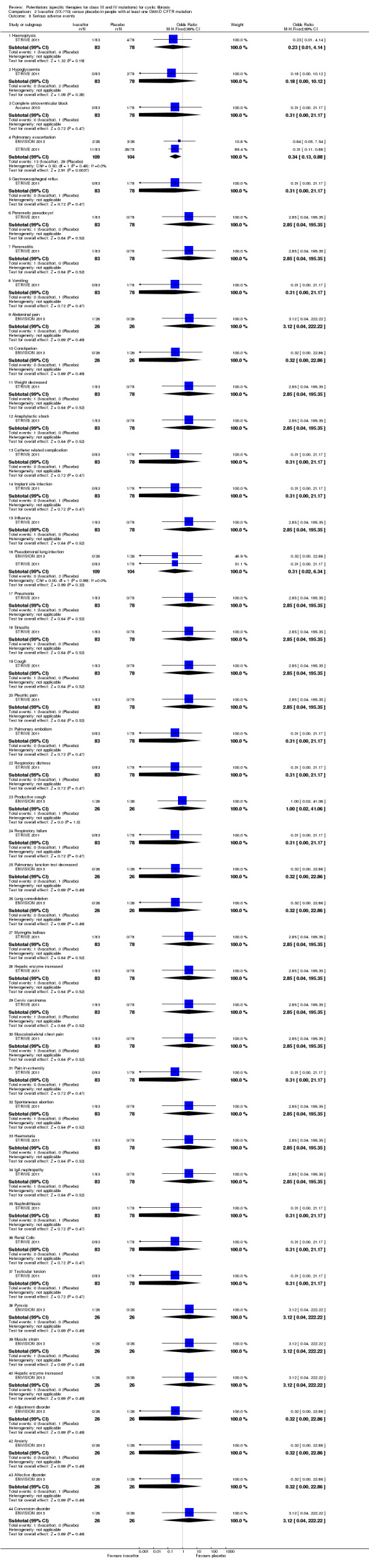
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 8 Serious adverse events.
No severe adverse effects were reported in any of the participants in the phase 2 G551D trial and all adverse events resolved without discontinuation of the intervention (Accurso 2010) (low‐quality evidence).
The R117H trial reported six (17%) severe adverse events in the placebo group and four (12%) in the ivacaftor group with no participants discontinuing the intervention due to adverse events (KONDUCT 2015). There was no significant difference in the number of serious adverse events between treatment arms, OR 0.64 (95% CI 0.16 to 2.52) (Analysis 3.3) (low‐quality evidence).
d. other adverse effects of therapy (of any severity) that are not classifiable according to these categories
The number of participants who experienced episodes of pulmonary exacerbation was reported by all trials (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011) (low‐quality evidence). In all cases pulmonary exacerbations were protocol‐defined; both phase 3 G551D trials and also the F508del trial employed the same definition (DISCOVER 2011; ENVISION 2013; STRIVE 2011), but this was different to the definition employed by the phase 2 G551D trial (Accurso 2010). The R117H trial reported protocol‐defined pulmonary exacerbations, but the full protocol was not available to determine this definition (KONDUCT 2015). Data for all G551D participants in this review are shown separately (Analysis 2.9). In the adult phase 3 G551D trial, there were minor differences in the number of participants experiencing episodes of pulmonary exacerbations between the full text, supplementary appendix and an online trials database (clinicaltrials.gov). Data were extracted from the online trials database where data for serious and other cases of pulmonary events were reported.
2.9. Analysis.
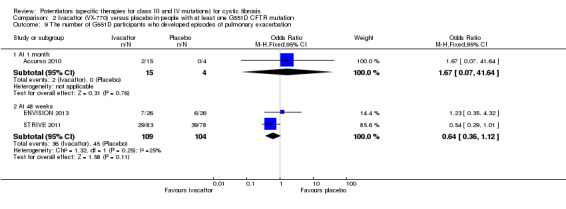
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 9 The number of G551D participants who developed episodes of pulmonary exacerbation.
In the phase 2 G551D trial, one participant from each of the treatment arms experienced an exacerbation compared to none in the placebo group (Accurso 2010); when analysed, there was no significant difference in the number of participants who experienced pulmonary exacerbations between the ivacaftor group (combined data from 150 mg and 250 mg groups) and the placebo group at one month, OR 1.67 (95% CI 0.07 to 41.64) (Analysis 2.9). In the phase 3 G551D trials, at 48 weeks significantly fewer participants in the ivacaftor group experienced serious episodes of pulmonary exacerbations compared to participants in the placebo group, OR 0.34 (99% CI 0.13 to 0.88) (Analysis 2.8). Combined data for all exacerbations in these trials was not significant between groups at this time‐point, OR 0.64; 95% CI 0.36 to 1.12) (Analysis 2.9). In the adult trial, the meta‐analysis of all exacerbations shows significantly fewer participants in the ivacaftor group experienced exacerbations than in the placebo group, OR 0.54 (95% CI 0.29 to 1.01) (STRIVE 2011) (Analysis 2.9). In the F508del trial, investigators reported a non‐significant lower frequency of pulmonary exacerbations in the ivacaftor group, OR 0.44 (99% CI 0.14 to 1.41) (Analysis 1.2).
The adult phase 3 G551D trial reported data for the time‐to‐first pulmonary exacerbation (STRIVE 2011). At 24 weeks, a significantly greater proportion of participants in the ivacaftor group (78%) were exacerbation‐free in comparison to participants in the placebo group (51%). This corresponds to a hazard ratio (HR) of 0.46 (95% CI 0.28 to 0.76) (P = 0.002) so within the first 24 weeks, adults receiving ivacaftor were 54% less likely to experience an exacerbation than those assigned to placebo (Analysis 2.10). At 48 weeks, 67% of participants in the ivacaftor group and 41% of participants in the placebo group were exacerbation‐free, HR 0.46 (95% CI 0.29 to 0.73), and the difference between groups remained significant (P < 0.001) (Analysis 2.10) (moderate‐quality evidence). Therefore during the 48‐week trial period, participants receiving ivacaftor were 54% less likely to experience an exacerbation than those assigned to placebo (STRIVE 2011).
2.10. Analysis.
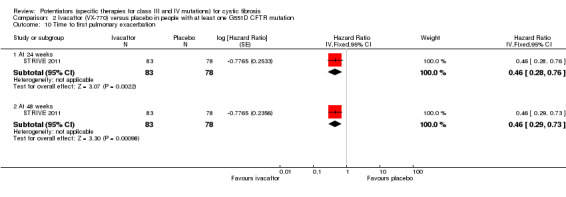
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 10 Time to first pulmonary exacerbation.
The R117H trial reported that at 24 weeks there was no significant difference between treatment groups for the time‐to‐first pulmonary exacerbation, HR 0.93 (95% CI 0.42 to 2.08) (KONDUCT 2015) (Analysis 3.6) (moderate‐quality evidence). Additionally, there was no significant difference between the ivacaftor and placebo groups in the number of participants experiencing a pulmonary exacerbation, OR 0.81 (95% CI 0.30 to 2.19) (Analysis 3.5).
3.6. Analysis.
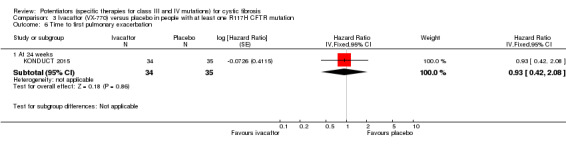
Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 6 Time to first pulmonary exacerbation.
3.5. Analysis.
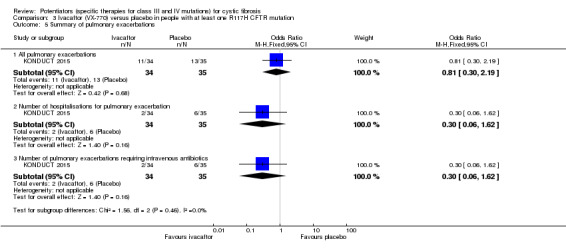
Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 5 Summary of pulmonary exacerbations.
There was no significant difference in the hepatic enzyme profile between groups in either phase 3 G551D trial (Analysis 2.5; Analysis 2.11). The majority of participants in the F508del trial had aspartate aminotransferase (AST), alanine aminotransferase (ALT) and bilirubin values less than or equal to two times ULN (DISCOVER 2011).
2.11. Analysis.

Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 11 Maximum liver function test abnormalities in adolescents/adults at 48 weeks.
2. Hospitalisation
The adult phase 3 G551D trial (n = 167) reported on the number of hospitalisations for episodes of pulmonary exacerbation and also on the number of days admitted (STRIVE 2011). The R117H trial reported the number of hospital admissions for episodes of pulmonary exacerbation (KONDUCT 2015).
a. number of days
In the G551D trial, of the episodes of pulmonary exacerbation that required hospitalisations, those occurring in participants in the ivacaftor group required less time in hospital, MD ‐0.23 days (95% CI ‐3.74 to 3.28) (Analysis 2.12). We note this non‐significant summary statistic conflicts with the trial report. The Supplementary table 2B of the supplementary appendix reports a P value of 0.0275 for this difference; however, this has been calculated from a Wilcoxon rank‐sum test, stratified by baseline % predicted FEV1 severity and age group (STRIVE 2011). This is a non‐parametric test used when data are skewed and where values are usually given as medians and ranges. In this trial, however, mean and SDs have been reported. As stated above, after entering these data into Review Manager, we have calculated a different P value of 0.90 which was not significant (Analysis 2.12).
2.12. Analysis.
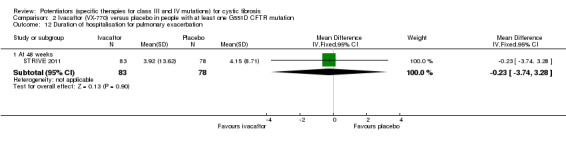
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 12 Duration of hospitalisation for pulmonary exacerbation.
b. number of episodes
Similarly, in the G551D trial significantly fewer episodes of pulmonary exacerbation required hospitalisation in participants in the ivacaftor group, OR 0.37 (95% CI 0.16 to 0.81) (Analysis 2.13) (STRIVE 2011). The R117H trial did not show a significant difference between treatment groups in the number of participants requiring hospital admission for pulmonary exacerbations (Analysis 3.5) (KONDUCT 2015).
2.13. Analysis.

Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 13 Number of hospitalisations for pulmonary exacerbations.
3. School or work attendance
No individual data for school or work attendance was reported by any trial (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
4. Other physiological measures of lung function
a. FEV1 (absolute values)
All included trials reported on the absolute change from baseline in FEV1 (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011). For the four phase three trials, this outcome was a primary endpoint (DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011). The data in the paediatric phase 3 G551D trial were adjusted based on the continuous baseline value of % of predicted FEV1 (ENVISION 2013). Data in the R117H trial were adjusted for continuous baseline values of age and % predicted FEV1 (KONDUCT 2015).
Investigators reported no significant differences between groups in the F508del trial for change from baseline of % predicted FEV1 at 16 weeks, MD 1.70% (95% CI ‐0.65 to 4.05) (DISCOVER 2011) (Analysis 1.4) (moderate‐quality evidence).
1.4. Analysis.
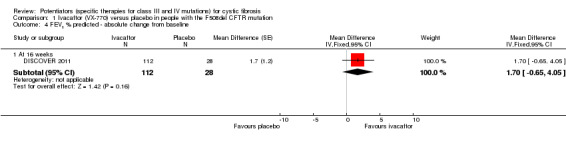
Comparison 1 Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation, Outcome 4 FEV1 % predicted ‐ absolute change from baseline.
Both phase 3 G551D trials reported results early (day 15), but we are unable to include these results in our analysis (ENVISION 2013; STRIVE 2011). At this time point, an estimated MD of 13.0% points can be deduced from the graph (Figure 2 in the full paper) for the paediatric trial (ENVISION 2013) and the adult trial reported a significant MD of 9.2% points (P < 0.001) (STRIVE 2011). At 24 weeks combined data from both of these trials show a significant increase in FEV1 (% predicted) from baseline in the ivacaftor group, MD 10.80% (95% CI 8.91 to 12.69) (P < 0.00001) with no heterogeneity (I² = 0%) (Analysis 2.14) (low‐quality evidence). Combined data at 48 weeks still demonstrate a significant change from baseline in FEV1 (% predicted), MD 10.44% (95% CI 8.56 to 12.32), again with no heterogeneity (I² = 0%) (Analysis 2.14) (low‐quality evidence). For the change from baseline in FEV1 (L), combined data at 24 weeks show a significant increase in in the ivacaftor group, MD 0.33 L (95% CI 0.17 to 0.49), but with considerable heterogeneity (I² = 85%) (Analysis 2.15) (low‐quality evidence). At 48 weeks the combined data still demonstrate a significant improvement, MD 0.31 L (95% CI 0.11 to 0.50), but again with considerable heterogeneity (I² = 90%) (Analysis 2.15) (low‐quality evidence). We do not know why the I² values are different between the meta‐analyses of % predicted FEV1 and FEV1 (L); we are not able to infer anything from it as there are only two trials in the analysis (ENVISION 2013; STRIVE 2011).
2.14. Analysis.

Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 14 FEV1 (% predicted) ‐ mean absolute change from baseline.
2.15. Analysis.
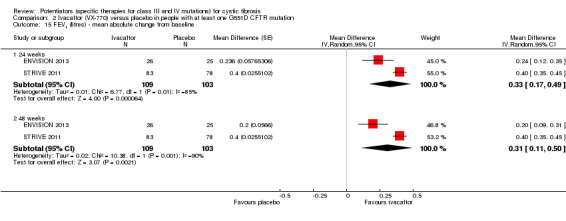
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 15 FEV1 (litres) ‐ mean absolute change from baseline.
At four weeks, in the phase 2 G551D trial, investigators reported a median (range) change from baseline in FEV1 of 0.25 L (0.05 to 0.75) in the 150 mg group, 0.17 L (0 to 0.37) in the 250 mg group and 0.20 L (0.12 to 0.33) in the placebo group (low‐quality evidence). Treatment differences between the ivacaftor arms and the placebo arm were not significant (Accurso 2010).
Subgroup analysis of the G551D trials
Improvements in the absolute change from baseline in FEV1 amongst pre‐defined subgroups were reported at 48 weeks by the phase 3 G551D trials (ENVISION 2013; STRIVE 2011).
In the paediatric trial at 48 weeks, children in three subgroups demonstrated significant improvements in the change from baseline in % predicted FEV1 in the ivacaftor group compared to the placebo group:
a baseline % predicted FEV1 below 90%, MD 14.90 (95% CI 7.30 to 22.50);
were from Europe, MD 24.60 (95% CI 6.40 to 42.80); and
were female, MD 13.80 (95% CI 4.20 to 23.40) (ENVISION 2013).
However, there was no significant difference in the change from baseline in % predicted FEV1 between treatment and placebo in the following subgroups:
baseline % predicted FEV1 over 90%, MD 6.90% (95% CI ‐3.80 to 17.60);
from North America, MD 5.80% (95% CI ‐2.60 to 14.20);
from Australia, MD 4.20% (95% CI ‐3.70 to 12.10); and
male, MD 5.20% (95% CI ‐2.20 to 12.60) (ENVISION 2013) (Analysis 2.16).
2.16. Analysis.
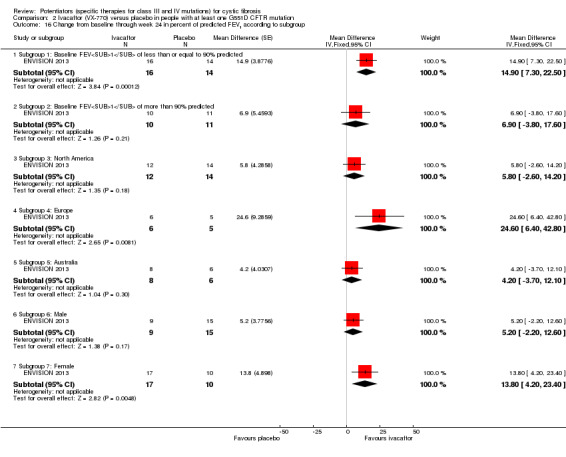
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 16 Change from baseline through week 24 in percent of predicted FEV1 according to subgroup.
The adult phase 3 G551D trial reported significant improvements in FEV1 % predicted favouring ivacaftor over placebo in all subgroups (Table 7); insufficient data precludes inclusion of these results in a meta‐analysis (STRIVE 2011).
4. Absolute change in FEV1 % predicted amongst pre‐defined subgroups in the adult phase 3 G551D study at 48 weeks.
| Subgroup | Treatment effect | P value |
| Baseline percent of predicted FEV1 of less than 70% | 10.6 | < 0.001 |
| Baseline percent of predicted FEV1 of greater or equal to 70% | 10.3 | < 0.001 |
| Participants from North America | 9.0 | < 0.001 |
| Participants from Europe | 9.9 | < 0.001 |
| Participants from Australia | 11.9 | 0.008 |
| Male | 11.0 | < 0.001 |
| Female | 11.6 | < 0.001 |
| Less than 18 years | 11.4 | 0.005 |
| Greater than or equal to 18 years | 9.9 | < 0.001 |
The treatment effect represents the difference between the ivacaftor group and the placebo group with respect to the absolute change from baseline through week 48.
FEV1: forced expiratory volume at one second
The R117H trial reported that at 24 weeks there was no significant treatment differences in absolute % predicted FEV1 between the ivacaftor and placebo groups, MD 2.10% (95% CI ‐1.13 to 5.33) (Analysis 3.7) (KONDUCT 2015) (low‐quality evidence).
3.7. Analysis.
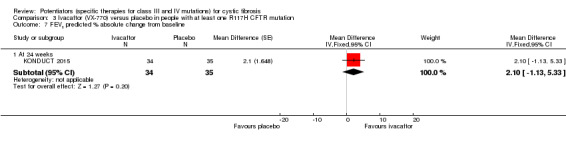
Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 7 FEV1 predicted % absolute change from baseline.
b. FVC (absolute values and change from baseline)
Three trials reported on the change from baseline in FVC (Accurso 2010; DISCOVER 2011; STRIVE 2011). None of the data were included in a meta‐analysis as they were either non‐parametric data (reported as median and ranges) (Accurso 2010) or insufficient data were provided (DISCOVER 2011; STRIVE 2011).
The F508del trial reported no significant differences between groups in FVC, but no numerical data were provided (DISCOVER 2011).
The phase 2 G551D trial reported a relative improvement from baseline FVC at two weeks in all three groups; there was a 9.0% improvement in the 150 mg ivacaftor group, a 7.4% improvement in the 250 mg ivacaftor group and a 1.0% improvement in the placebo group (Accurso 2010). Treatment differences between the ivacaftor groups and the placebo group were not significant. At four weeks a median (range) change from baseline of 0.18 L (‐0.29 to 0.48) in the 150 mg group, 0.09 L (‐0.09 to 0.43) in the 250 mg group and 0.10 L in the placebo group were reported. These values corresponded to relative changes from baseline of 4.6% in the 150 mg group, 2.6% in the 250 mg group and 2.3% in the placebo group. Treatment differences between the ivacaftor groups and the placebo group were not significant (Accurso 2010).
The adult phase 3 G551D trial reported on the absolute change from baseline in FVC in the form of a graph (supplemental figure 4B of the supplementary appendix) (STRIVE 2011). From the graph, an estimated MD between intervention and placebo arms of 11.5% at 24 weeks and 7.0% at 48 weeks can be deduced. These results should be interpreted with caution, as they are not accurate (STRIVE 2011).
5. Extra courses of antibiotics
Three trials reported on extra courses of antibiotics required by participants (n = 376) (DISCOVER 2011; KONDUCT 2015; STRIVE 2011).
The F508del trial reported on the number of new sinopulmonary signs or symptom events that required new or changed antibiotic therapy; however, the route of administration was not stated (DISCOVER 2011). Investigators reported proportionately fewer episodes of new sinopulmonary signs or symptom events requiring antibiotic treatment in the ivacaftor group, OR 0.56 (95% CI 0.24 to 1.30); however, the difference was not significant (Analysis 1.5).
1.5. Analysis.

Comparison 1 Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation, Outcome 5 Antibiotic treatment of sinopulmonary signs or symptoms.
The adult phase 3 G551D trial reported firstly the number of episodes of pulmonary exacerbation that required intravenous antibiotics and secondly the number of days on intravenous antibiotics to treat pulmonary exacerbations (see below) (STRIVE 2011). The R117H trial reported the number of episodes of pulmonary exacerbation requiring intravenous antibiotics (KONDUCT 2015).
a. oral
This outcome was not reported in any trial.
b. intravenous
In the adult phase 3 G551D trial, Ramsey reported significantly fewer episodes of pulmonary exacerbations requiring intravenous antibiotics occurred in participants in the ivacaftor group, OR 0.34 (95% CI 0.18 to 0.64) (P = 0.0009) (Analysis 2.17). In addition, more days of intravenous antibiotic administration were required for exacerbations occurring in participants in the placebo group, MD ‐4.35 (95% CI ‐10.51 to 1.81) (Analysis 2.18). Supplementary table 2B of the supplementary appendix reports a P value of 0.0183 for this difference; however, this has been calculated from a Wilcoxon rank‐sum test, stratified by baseline % predicted FEV1 severity and age group (STRIVE 2011). This is a non‐parametric test used when data are skewed and where values are usually given as medians and ranges. In this trial however, mean and SDs have been reported. After entering these data into Review Manager, we have calculated a different P value of 0.17 which was not significant (Analysis 2.18).
2.17. Analysis.
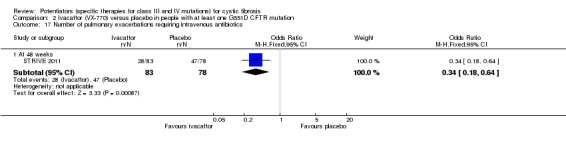
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 17 Number of pulmonary exacerbations requiring intravenous antibiotics.
2.18. Analysis.
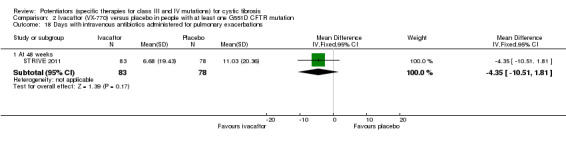
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 18 Days with intravenous antibiotics administered for pulmonary exacerbations.
In the R117H trial, investigators reported fewer episodes of pulmonary exacerbation requiring intravenous antibiotics in the ivacaftor group, OR 0.30 (95% CI 0.06 to 1.62), however, this result was not significant (P = 0.16) (Analysis 3.5) (KONDUCT 2015).
c. inhaled
This outcome was not reported in either trial.
6. Radiological measures of lung disease
No trials reported chest radiograph scores or CT scores.
7. Acquisition of respiratory pathogen
No trial reported on the acquisition of any respiratory pathogens (e.g. P aeruginosa, S aureus, H influenzae).
8. Eradication of respiratory pathogens
No trial reported on the eradication of any respiratory pathogens (e.g. P aeruginosa, S aureus, H influenzae).
9. Nutrition and growth
a. weight
Three trials (n = 359) reported the absolute change from baseline in weight (kg) (DISCOVER 2011; ENVISION 2013; STRIVE 2011); one of these additionally reported on weight‐for‐age z score (DISCOVER 2011).
The F508del trial, reported no significant difference in weight gain between the ivacaftor group and the placebo group at 16 weeks, MD ‐0.20 kg (95% CI ‐1.18 to 0.78) (Analysis 1.6). There was also no significance difference between groups in weight‐for‐age z scores (MD 0.43), but insufficient data prevented inclusion of this outcome in the analysis (DISCOVER 2011).
1.6. Analysis.
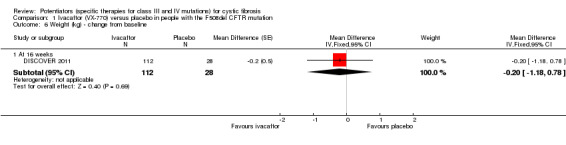
Comparison 1 Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation, Outcome 6 Weight (kg) ‐ change from baseline.
Combined data from the phase 3 G551D trials at 24 weeks demonstrated significant weight gain in the ivacaftor group, MD 2.37 kg (95% CI 1.68 to 3.06) with moderate heterogeneity (I² = 38%) (ENVISION 2013; STRIVE 2011) (Analysis 2.19). Combined data from the same trials at 48 weeks also demonstrated significant weight gain in the ivacaftor group compared to placebo, MD 2.75 kg (95% CI 1.74 to 3.75), but with no heterogeneity (I² = 0%) (ENVISION 2013; STRIVE 2011) (Analysis 2.19).
2.19. Analysis.
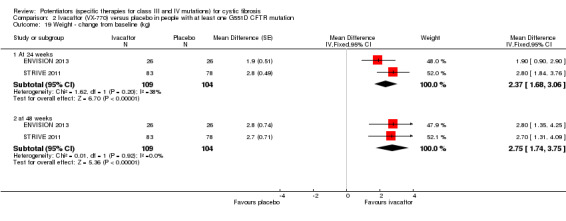
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 19 Weight ‐ change from baseline (kg).
b. BMI
Four trials reported on the change from baseline in BMI (DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011) and three additionally reported BMI for age z score (DISCOVER 2011; ENVISION 2013; STRIVE 2011), but none of them provided sufficient information for inclusion into a meta‐analysis (DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
In the F508del trial, a lower BMI score was reported in the ivacaftor group in comparison to the placebo group at 16 weeks, MD ‐0.04 mg/m², but the difference between groups was not significant (DISCOVER 2011). In the phase 3 G551D trials, both children and adults in the ivacaftor group scored significantly higher BMI values at 48 weeks, MD 1.09 kg/m² (P = 0.0003) and MD 0.93 kg/m² (P < 0.0001) respectively (ENVISION 2013; STRIVE 2011).
In the F508del trial, at 16 weeks there was no significant difference between the ivacaftor group and the placebo group in BMI‐for‐age z score amongst a subgroup of participants aged 20 years or below, MD 0.75 (DISCOVER 2011). The paediatric phase 3 G551D trial reported significantly higher BMI‐for‐age‐z scores in the ivacaftor group compared to placebo at 24 weeks, MD 0.34 (P ≤ 0.001), and 48 weeks, MD 0.45 (P < 0.001) (ENVISION 2013). At 48 weeks, the adult phase 3 G551D trial reported significantly higher BMI‐for‐age‐z scores in the ivacaftor group amongst a subgroup of 47 participants aged 12 to 20 years, MD 0.33 (P = 0.0490) (STRIVE 2011).
The R117H trial reported a higher BMI score in the ivacaftor group compared to placebo at 24 weeks, MD 0.26 kg/m² (95% CI ‐1.57 to 2.09), but this was not significant (P = 0.1) (KONDUCT 2015) (Analysis 3.8). Treatment effect was adjusted for age and categorical % predicted FEV1 at baseline (KONDUCT 2015).
3.8. Analysis.

Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 8 BMI absolute change from baseline.
c. height
Two trials reported on height z scores (ENVISION 2013; STRIVE 2011). The paediatric phase 3 G551D trial reported improvements in the ivacaftor group at both 24 weeks (MD 0.06) and 48 weeks (MD 0.12) (ENVISION 2013). In the adult phase 3 G551D trial reported improvements in the ivacaftor group at 24 weeks (MD 0.05) and 48 weeks (MD 0.06) amongst a subset of 47 participants aged 12 to 20 years (STRIVE 2011).
10. Sweat chloride (change from baseline)
The change from baseline in sweat chloride concentration was reported by all trials (n = 447) (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
In the F508del trial, investigators reported significant reductions in sweat chloride concentration at 16 weeks in the ivacaftor group compared to the placebo group, MD ‐2.90 mmol/L (95% CI ‐5.60 to ‐0.20) (DISCOVER 2011) (Analysis 1.7).
1.7. Analysis.

Comparison 1 Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation, Outcome 7 Sweat chloride concentration ‐ change from baseline.
In the phase 2 G551D trial, at four weeks there was a significant difference in the median (range) change from baseline in sweat chloride concentration between the 150 mg group (within group change ‐59.5 mmol/L (‐66.0 to ‐19.0) and the placebo group (within group change 5.0 mmol/L (‐2.0 to 11.0) (P = 0.02); a similar significant difference was seen between the 250 mg group (within group change ‐38.0 mmol/L (‐47.0 to ‐10.5) and placebo group (within group change 5.0 mmol/L (‐2.0 to 11.0) (P = 0.03) (Accurso 2010). In the phase 3 G551D trials, reduced sweat chloride concentrations were reported early (day 15) in children, MD ‐50 mmol/L (estimated from a graph) (ENVISION 2013) and in adults, MD ‐47.1 mmol/L (P < 0.001) (STRIVE 2011). Combined data from these trials demonstrated significant reductions in sweat chloride concentration at 24 weeks, MD ‐50.19 mmol/L (95% CI ‐56.20 to ‐44.18) with a moderate level of heterogeneity (I² = 57%) (Analysis 2.20), and at 48 weeks, MD ‐49.74 mmol/L (95% CI ‐54.61 to ‐44.87) with a moderate level of heterogeneity (I² = 40%) (Analysis 2.20). Secondary analysis of combined data from these trials reported that the number needed to treat (NNT) for a reduction of 20 mmol/L in sweat chloride concentration was 1.03. Data from each phase 3 G551D trial demonstrated that significant changes in sweat chloride were reported regardless of whether participants were FEV1 responders (5% point improvement) or minimal responders (less than 5% point improvement) (ENVISION 2013; STRIVE 2011) (Table 8).
2.20. Analysis.
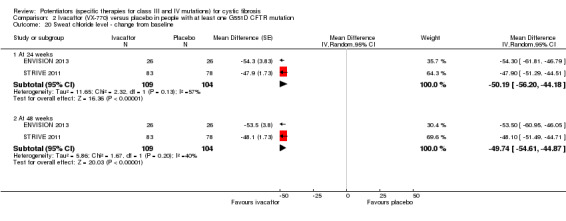
Comparison 2 Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation, Outcome 20 Sweat chloride level ‐ change from baseline.
5. Change from baseline in sweat chloride concentration in FEV1 responders and FEV1 non‐responders in the phase 3 G551D studies.
| Paediatric phase 3 G551D study(ENVISION 2013) | Adult phase 3 G551D study(STRIVE 2011) | |||
| Treatment difference (mmol/L) | P value | Treatment difference (mmol/L) | P value | |
| Minimal responders (less than 5% point improvement) | ‐55.8 | <0.0001 | ‐46.1 | < 0.0001 |
| FEV1 responders (5% point improvement) | ‐53.9 | <0.0001 | ‐49.7 | < 0.0001 |
FEV1: forced expiratory volume at one second
In the R117H trial, at 24 weeks a significant reduction in sweat chloride concentrations was reported in the ivacaftor group compared to placebo group, MD ‐24.00 mmol/L (95% CI ‐28.01 to ‐19.99) (P = <0.0001) (KONDUCT 2015) (Analysis 3.9). Treatment effect was adjusted for continuous baseline values of age and % predicted FEV1 (KONDUCT 2015). In addition, age subgroup analysis showed significant reductions in sweat chloride concentrations for both participants aged 6 to 11 years and 18 years and over (Table 6).
3.9. Analysis.

Comparison 3 Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation, Outcome 9 Sweat chloride concentration ‐ change from baseline.
11. Cost of treatment
No trials reported on the cost of ivacaftor treatment.
Discussion
Summary of main results
The CFTR protein product of a class III mutation is transported to the cell membrane, but displays defective chloride ion gating and dysregulation of sodium channels resulting in a severe CF phenotype; characterised by chronic airway infection and inflammation and pancreatic insufficiency. Agents that repair the underlying gating defects (CFTR potentiators) restore CFTR function for class III mutations and correct the abnormal transmembrane salt transport in cell and animal models (Van Goor 2009). These CFTR potentiators may also have a role in repairing the underlying gating defect in the minimal amounts of class II CFTR that are transported to the cell membrane and for the restrictive movement of chloride ions in class IV CFTR.
This review examines whether CFTR potentiators improve clinically relevant endpoints in CF. Five trials with 447 participants met the inclusion criteria for this review; one compared 150 mg and 250 mg doses of ivacaftor (a potentiator) to placebo (Accurso 2010) and four compared 150 mg of ivacaftor to placebo (DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011). Three trials examined the impact of ivacaftor on people with a class III mutation (G551D) (Accurso 2010; ENVISION 2013; STRIVE 2011). One trial enrolled participants with a class II mutation (F508del) (DISCOVER 2011) and one enrolled participants with a class IV mutation (R117H) (KONDUCT 2015).
The trial that evaluated ivacaftor for people with F508del enrolled participants aged 12 years or over (n = 140) for a duration of 16 weeks (DISCOVER 2011). The first G551D trial was a phase 2 trial enrolling participants 18 years and over and measuring a number of outcomes at 14 and 28 days (Accurso 2010). This was followed by two phase 3 trials, the first enrolling participants aged 12 years and older (n = 167) (STRIVE 2011) and the second enrolled participants aged 6 to 11 years (n = 52) (ENVISION 2013). These trials reported outcomes at 24 and 48 weeks. The R117H trial enrolled participants over six years of age (n = 69) and measured outcomes at 24 weeks (KONDUCT 2015).
The primary end points for these trials were safety (Accurso 2010) or change in FEV1 (ENVISION 2013; KONDUCT 2015; STRIVE 2011) or both (DISCOVER 2011). With respect to this review's primary outcomes, there were no deaths reported in any of the trials and the length and size of the trials precluded valid assessment of the impact of ivacaftor on survival. For CFQ‐R scores, no significant difference was reported in the F508del trial (DISCOVER 2011). The adults randomised to ivacaftor in the phase 2 G551D trial did not report significantly higher CFQ‐R respiratory domain scores 28 days (Accurso 2010). However, adults in the phase 3 G551D trial reported significantly higher CFQ‐R respiratory scores at 24 weeks, MD 8.10 (95% CI 4.63 to 11.57) and at 48 weeks, MD 8.70 (95% CI 5.51 to 11.89) (Analysis 2.3). This finding was not reproduced in the paediatric participants (ENVISION 2013). The R117H trial reported significantly higher CFQ‐R scores at 24 weeks in participants randomised to ivacaftor, MD 8.40 (95% CI 2.17 to 14.63) (Analysis 3.1) (KONDUCT 2015). Again, for relative change in FEV1, no significant improvement was reported in the F508del trial (DISCOVER 2011). The phase 2 G551D trial did not report treatment with ivacaftor resulted in a significant improvement in relative change in FEV1 (Accurso 2010). However, in the adult G551D phase 3 trial, significant improvements in relative change in FEV1 were seen early (after 15 days) and maintained through to 48 weeks, MD 16.80% (95% CI 13.50 to 20.10) (STRIVE 2011). Significant improvements in this outcome were also seen at 24 weeks in the paediatric trial, MD 17.4% (P < 0.0001), but results at 48 weeks were not published (ENVISION 2013). The R117H did not report any significant improvement in relative change in FEV1 (KONDUCT 2015).
We compared the number of participants experiencing adverse effects between the ivacaftor group and the placebo group. Combined data from both G551D phase 3 trials demonstrated a reduced reporting of cough, OR 0.57 (95% CI 0.33 to 1.00) and reduced episodes of decreased pulmonary function, OR 0.29 (95% CI 0.10 to 0.82) in the ivacaftor group (Analysis 2.5). Increased reports of dizziness were recorded in participants receiving ivacaftor in the adult G551D trial (STRIVE 2011), but this adverse effect was not reported by any other trial (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015). Data from the R117H trial showed increased reporting of adverse effects in the ivacaftor group except for pulmonary exacerbation, OR 0.93 (95% CI 0.35 to 2.44), however, these results were not significant (KONDUCT 2015) (Analysis 3.4). No trial reported a significant increase in adverse effects leading to interruption or termination of the trial drug (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
Combined data from the phase 3 G551D trials demonstrated significantly fewer participants experienced 'serious' cases of pulmonary exacerbation in the ivacaftor group, OR 0.34 (95% CI 0.17 to 0.70) (Analysis 2.13). When considering all data for exacerbations, fewer adults in the ivacaftor group reported exacerbations in the adult phase 3 G551D trial, OR 0.54 (95% CI 0.29 to 1.01) (Analysis 2.9); also, more participants in the placebo group required hospitalisation and IV antibiotics for pulmonary exacerbations and a greater proportion of participants in the ivacaftor group were exacerbation‐free at the 24 and 48 week time points (Analysis 2.10) (STRIVE 2011). In the R117H trial fewer participants in the ivacaftor group experienced pulmonary exacerbations, hospitalisation and IV antibiotics for the exacerbation, but results were not significant (Analysis 3.5) (KONDUCT 2015). This effect was not reported by the remaining three trials (Accurso 2010; DISCOVER 2011; ENVISION 2013).
The adverse effects profile of ivacaftor should be interpreted with caution as it was based on small numbers of participants. Furthermore, none of the included trials were powered to assess this outcome (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011).
No significant weight gain or change in BMI scores were reported in the F508del or R117H trials (DISCOVER 2011; KONDUCT 2015). Participants in both phase 3 G551D trials demonstrated significant weight gain on ivacaftor at 24 weeks, MD 2.37 kg (95% CI 1.68 to 3.06) and 48 weeks, MD 2.75 kg (95% CI 1.74 to 3.75) (Analysis 2.19). These two trials also reported that significantly higher BMI scores were achieved in the ivacaftor group; for children, MD 1.09 kg/m² (P = 0.0003) and for adults, MD 0.93 kg/m² (P < 0.0001). Significantly higher BMI for age z scores were reported for children in the ivacaftor group at 24 weeks, MD 0.34 (P ≤ 0.001) and at 48 weeks, MD 0.45 (P < 0.001) (ENVISION 2013). In a subset of participants aged 12 to 20 years old from the adult trial, a higher BMI for age Z score was reported at 48 weeks, MD 0.33 (P = 0.0490) (STRIVE 2011).
All trials reported significant reductions in sweat chloride concentrations (Accurso 2010; DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011). The F508del trial reported a MD of ‐2.90 mmol/L (95% CI ‐5.60 to ‐0.20) (DISCOVER 2011). The phase 2 G551D trial reported median values at 28 days; in the 150 mg ivacaftor group, median ‐64.5 mmol/L (P = 0.02) and in the 250 mg ivacaftor group, median ‐43.0 mmol/L (P = 0.03) (Accurso 2010). Combined data from the phase 3 G551D trials demonstrated significant reductions in sweat chloride concentration in participants on ivacaftor at 24 weeks, MD ‐50.19 mmol/L (95% CI ‐56.20 to ‐44.18) and 48 weeks, MD ‐49.74 mmol/L (95% CI ‐54.61 to ‐44.87) (Analysis 2.20). Similarly, the ivacaftor group in the R117H trial reported a significant reduction in sweat chloride concentration at 24 weeks, MD ‐24.00 mmol/L (95% CI ‐28.01 to ‐19.99) (Analysis 3.9).
Overall completeness and applicability of evidence
The DISCOVER trial recruited 140 participants homozygous for the F508del mutation, who were a representative sample for people with this mutation aged from 12 years upwards. This trial was included because it evaluated ivacaftor, although on a population with class II mutations in which a minimal amount of CFTR is able to reach the cell surface. In light of the results that failed to reach statistical significance in this population, trials with younger participants with F508del are not appropriate (DISCOVER 2011).
Three trials recruited 238 participants with the G551D mutation representing a significant proportion of all people with this mutation in the countries where the trials were undertaken (Accurso 2010; ENVISION 2013; STRIVE 2011). It can be assumed that the results are applicable to those with the G551D mutation not included in the trials; however, current trials have not enrolled children under six years of age or pregnant women. There are insufficient data for some outcomes, which we considered to be important for this review: school or work attendance; radiological measures of lung disease; acquisition of respiratory pathogens; and cost of treatment. One G551D trial reported on hospitalisations and extra courses of antibiotics (STRIVE 2011).
The R117H trial recruited 69 participants with the R117H mutation. Results are not applicable to individuals with other class IV mutations and the small sample size reduces the applicability of this evidence to people with cystic fibrosis who carry the R117H mutation (KONDUCT 2015).
There is no current evidence to support the use of CFTR potentiators in people with other class III to V mutations (not G551D). While there are trials recruiting people with other class III to V mutations, these have not be included in this review due to their cross‐over trial design (KONNECTION 2013; NCT01685801; NCT01784419).
Quality of the evidence
All the included trials were RCTs which represent the highest quality with regards to trial design.
The overall risk of bias across the included trials was judged to be moderate. Random sequence generation, allocation concealment and blinding of trial personnel were well‐documented. Blinding of participants was less clear, as details of ivacaftor and placebo tablets (such as taste, colour and consistency) were not provided. Participant data were excluded from the analysis in four trials (DISCOVER 2011; ENVISION 2013; KONDUCT 2015; STRIVE 2011). The risk of selective reporting was generally high as trials did not always report on outcomes outlined in the protocol, or present data for all outcomes and report on outcomes considered important by the review authors.
The quality of the evidence in this review is judged to be moderate to low, mainly due to risk of bias from incomplete outcome data and selective reporting and imprecision of results, particularly where few individuals have experienced adverse events (Table 1; Table 2; Table 3).
Potential biases in the review process
The review authors conducted a comprehensive literature search of online journal databases using the Cystic Fibrosis and Genetic Disorders Review Group's Cystic Fibrosis Trials Register and online trials databases (Appendix 1) and also of manual searching of journal conference abstracts. Two authors independently applied the inclusion and exclusion criteria to the resultant literature and extracted relevant trials. The authors independently extracted data and assessed the risk of bias of the included trials. If they failed to reach a consensus on the risk of bias in a trial, a third author (KWS) arbitrated. The initial analyses were undertaken by one review author (SP) and checked for appropriateness by the review statistician (KD). MS completed the analysis for the update of the review. This methodological approach ensured that risks of bias in the review process were kept to a minimum.
This review has assessed all available published trial data. Trial authors were contacted for relevant unpublished information and individual participant data. To date, a protocol synopsis and summary of product characteristics have been provided with regards to information requested for the R117H trial (KONDUCT 2015). We are not aware of any unpublished trials, although we have highlighted issues with selective reporting in the included trials.
Agreements and disagreements with other studies or reviews
We are aware of one other published review of ivacaftor for people with CF which included the phase 3 G551D trials and data from the open‐label extensions to these trials (Whiting 2014). In line with this Cochrane Review, the Whiting review also concluded that ivacaftor has a clinically relevant impact on people with CF with the G551D mutation. We identified disagreements between the two reviews concerning the methodological quality and risk of bias assessments for the phase 3 G551D trials, although both reviews have used the Cochrane risk of bias tool. In the Whiting review, the authors judged the risk of bias for all methodological aspects of the adult phase 3 G551D trial to be low; whereas we judged this trial to have an unclear risk of detection bias and high risks of attrition bias and selective reporting bias (STRIVE 2011). With regards to the paediatric phase 3 G551D trial, disagreements were identified for participant and outcome assessor blinding, incomplete outcome data and selective reporting. The Whiting review judged this trial to have a low risk of detection bias, performance bias and selective outcome reporting bias and an unclear risk of attrition bias; while we judged the trial to have an unclear risk of detection and performance bias and a high risk of attrition bias and selective outcome reporting bias (ENVISION 2013). The overall methodological quality of the phase 3 G551D trials was assessed to be higher in the Whiting review than in this review.
Authors' conclusions
Implications for practice.
The F508del trial demonstrated no evidence to support the use of ivacaftor in those with the F508del mutation (DISCOVER 2011). The two G551D phase 3 trials demonstrated a clinically relevant impact of ivacaftor on outcomes at 24 and 48 weeks in children (over six years of age) and adults with cystic fibrosis (CF) and the G551D mutation (ENVISION 2013; STRIVE 2011). The R117H trial demonstrated an improvement in the respiratory domain of the CFQ‐R but no improvement in respiratory function (KONDUCT 2015). These trials were judged to have a moderate risk of bias.
Implications for research.
Ivacaftor is the first intervention that corrects the underlying molecular defect in CF. It has demonstrated effectiveness in people with a class III mutation (G551D) and some effectiveness with class IV mutations (R117H) and has the potential to be used for other class III and IV mutations. Phase 3 trials examining ivacaftor in combination with a CFTR corrector (lumacaftor) in class II mutations (F508del) have been assessed in a separate Cochrane Review (Southern 2018).
As new mutation‐specific therapies emerge, it is important that the lessons learnt from this review are taken on board, in particular with respect to trial design (cross‐over trials are not appropriate given the potential for the therapies under consideration to fundamentally change the natural history of the condition), selective reporting and the details of participant blinding. It is important that these trials examine valid outcomes that are relevant to people with CF and their families.
With novel therapies and approaches, such as ivacaftor, the reporting of adverse events is critical and this should be undertaken in a robust and consistent manner. Valid post‐market surveillance is also essential.
In view of the cost implications for this therapy, ongoing health economic evaluations are required.
What's new
| Date | Event | Description |
|---|---|---|
| 18 December 2018 | New citation required and conclusions have changed | One new trial was included (KONDUCT 2015). An improvement in the respiratory domain of the CFQ‐R was demonstrated but no improvement in respiratory function. Three authors have stepped down from the review (Kerry Dwan, Carlos Echevarria and Michael Schechter) and two new authors have joined the team (Mica Skilton and Ashma Krishan). |
| 18 December 2018 | New search has been performed | One trial has been included from the previous ongoing studies section (KONDUCT 2015). A search of the Cochrane Cystic Fibrosis and Genetic Disorders Review Group's Cystic Fibrosis Register identified 99 new references (45 trials) which were potentially eligible for inclusion in this review. There were 14 references to already included trials: two references were added to the Accurso trial (Accurso 2010); six references were added to the Ramsey trial (STRIVE 2011); three references were added to the Davies trial (ENVISION 2013); and three references which are linked to both the Davies and Ramsey trials were also added (ENVISION 2013; STRIVE 2011). A further four references were identified and added to already excluded trials: three references to the Kerem trial (Kerem 2014); one reference to the Davies trial (Davies 2012). There were 29 references to 10 new trials identified, all were excluded (ALBATROSS 2017; Berkers 2017; Davies 2016; Edgeworth 2017; FLAMINGO 2017; Horsley 2018; McGarry 2015; NCT02323100; RIO‐CF 2017; Seliger 2015). Two new references were added to studies awaiting classification (Kazani 2016; Uttamsingh 2016). A search of two clinical trials registries identified 33 new relevant trials, all of which were excluded. In a post hoc change, corrector and potentiator combination therapy trials have not been referenced in the review, meaning the remaining 52 references identified have not been listed. |
Acknowledgements
The review authors would like to acknowledge help and support of the Cystic Fibrosis and Genetics Disorders Review Group in particular Nikki Jahnke and Tracey Remmington for their help and support. They would also like to acknowledge the contribution of Dr Kerry Dwan, Dr Carlos Echevarria, Professor Michael Schechter and Dr Sarah Nevitt on earlier versions of this protocol and review.
We would also like to acknowledge the trial investigators; Dr Jane Davies, Professor Bonnie Ramsey, Professor Patrick Flume and Dr Frank Accurso for their attempts to retrieve requested information from the trial sponsor and Vertex Pharmaceuticals for providing additional information.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Glossary
| Term | Explanation |
| CFTR (cystic fibrosis transmembrane regulator) | A protein which is in the outer membrane of cells. It works by regulating transport of salt in and out of the cell. Problems with the amount of CFTR in the cell membrane, its structure, or the manner in which it functions can lead to altered transport of salt across the cell membrane. In the lungs of people with CF, these problems cause thick airway secretions. Abnormalities of CFTR can also lead to problems in other organs. |
| CFTR correctors | Drugs or chemicals which work by increasing the amount of CFTR in the cell membrane. |
| CFTR potentiators | Drugs or chemicals which increase the effectiveness of CFTR at transporting salt across the cell membrane. |
| CFTR stop‐codon therapies | Drugs or chemicals which help the cell to synthesise CFTR. They specifically work in people with CF in whom the basic defect happens because CFTR which is made is too short, and so functions abnormally. |
Appendix 2. Electronic search strategies
| Database / Resource | Search terms | Date last searched |
| European Medicines Agency (www.clinicaltrialsregister.eu/) |
cystic fibrosis AND ivacaftor | 13 April 2018 |
| US National Institute of Health trials database (clinicaltrials.gov/) |
cystic fibrosis AND ivacaftor | 13 April 2018 |
| WHO ICTRP (apps.who.int/trialsearch/) |
cystic fibrosis AND ivacaftor | 13 April 2018 |
Data and analyses
Comparison 1. Ivacaftor (VX‐770) versus placebo in people with the F508del CFTR mutation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 FEV1 ‐ relative change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 1.1 At 16 weeks | 1 | 140 | Mean Difference (Fixed, 95% CI) | 2.4 [‐0.95, 5.75] |
| 2 Adverse effects | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Pulmonary exacerbation | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.14, 1.41] |
| 2.2 Upper respiratory tract infection | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.18, 11.10] |
| 2.3 Cough | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.62 [0.59, 11.59] |
| 2.4 Productive cough | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.36 [0.15, 37.71] |
| 2.5 Headache | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.18, 11.10] |
| 2.6 Nasal congestion | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.71 [0.22, 13.09] |
| 2.7 Sinusitis | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.08 [0.13, 33.75] |
| 2.8 Oropharyngeal pain | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.14, 4.90] |
| 2.9 Upper abdominal pain | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.8 [0.11, 29.87] |
| 2.10 Nausea | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.65 [0.17, 41.76] |
| 2.11 Diarrhea | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.74 [0.08, 6.49] |
| 2.12 Contact dermatitis | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.48 [0.08, 158.51] |
| 2.13 Rash | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.23 [0.12, 228.54] |
| 2.14 Fatigue | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.12, 4.45] |
| 2.15 Pyrexia | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.14, 9.20] |
| 2.16 Increase in C‐reactive protein | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.53 [0.09, 26.08] |
| 3 Severity of adverse effects of therapy with regards to study drug interruption (moderate) or discontinuation (severe) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Adverse effect requiring study drug interruption (moderate) | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.06, 27.62] |
| 3.2 Adverse effect requiring study drug discontinuation (severe) | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.06, 2.25] |
| 4 FEV1 % predicted ‐ absolute change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 4.1 At 16 weeks | 1 | 140 | Mean Difference (Fixed, 95% CI) | 1.70 [‐0.65, 4.05] |
| 5 Antibiotic treatment of sinopulmonary signs or symptoms | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 At 16 weeks | 1 | 140 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.24, 1.30] |
| 6 Weight (kg) ‐ change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 6.1 At 16 weeks | 1 | 140 | Mean Difference (Fixed, 95% CI) | ‐0.2 [‐1.18, 0.78] |
| 7 Sweat chloride concentration ‐ change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 7.1 At 16 weeks | 1 | 140 | Mean Difference (Fixed, 95% CI) | ‐2.9 [‐5.60, ‐0.20] |
Comparison 2. Ivacaftor (VX‐770) versus placebo in people with at least one G551D CFTR mutation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 CFQ‐R score (respiratory domain, child version) ‐ adjusted change from baseline | 2 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 1.1 At 24 weeks | 2 | 51 | Mean Difference (Fixed, 95% CI) | 6.1 [‐1.35, 13.55] |
| 1.2 At 48 weeks | 1 | 51 | Mean Difference (Fixed, 95% CI) | 5.1 [‐1.56, 11.76] |
| 2 CFQ‐R score (respiratory domain score, parent/caregiver version) ‐ adjusted change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 2.1 At 24 weeks | 1 | 52 | Mean Difference (Fixed, 95% CI) | 5.9 [0.41, 11.39] |
| 2.2 At 48 weeks | 1 | 52 | Mean Difference (Fixed, 95% CI) | 4.9 [‐0.39, 10.19] |
| 3 CFQ‐R score (respiratory domain score, pooled) ‐ absolute change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 3.1 At 24 weeks | 1 | 151 | Mean Difference (Fixed, 95% CI) | 8.1 [4.77, 11.43] |
| 3.2 At 48 weeks | 1 | 151 | Mean Difference (Fixed, 95% CI) | 8.6 [5.27, 11.93] |
| 4 FEV1 ‐ mean relative change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 4.1 24 weeks | 1 | 161 | Mean Difference (Fixed, 95% CI) | 16.9 [13.60, 20.20] |
| 4.2 48 weeks | 1 | 161 | Mean Difference (Fixed, 95% CI) | 16.8 [13.50, 20.10] |
| 5 Adverse events occurring in greater than or equal to 5% of trial participants | 2 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 5.1 Pulmonary exacerbation | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.64 [0.30, 1.33] |
| 5.2 Cough | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.57 [0.27, 1.19] |
| 5.3 Productive Cough | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.81 [0.29, 2.24] |
| 5.4 Pulmonary function test decreased | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.29 [0.07, 1.14] |
| 5.5 Wheezing | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.12 [0.28, 4.52] |
| 5.6 Rales | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.86 [0.28, 2.69] |
| 5.7 Oropharyngeal pain | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.26 [0.52, 3.05] |
| 5.8 Nasal congestion | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.39 [0.55, 3.53] |
| 5.9 Nasopharyngitis | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.32 [0.46, 3.76] |
| 5.10 Upper respiratory tract infection | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.91 [0.74, 4.90] |
| 5.11 Bronchitis | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.57 [0.13, 18.48] |
| 5.12 Otitis media | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 4.55 [0.23, 89.19] |
| 5.13 Rhinorrhoea | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.65 [0.17, 2.45] |
| 5.14 Sinusitis | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.75 [0.21, 2.68] |
| 5.15 Ear Infection | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.18 [0.00, 10.67] |
| 5.16 Pharyngitis streptococcal | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.17 Rhinitis | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.86 [0.40, 8.70] |
| 5.18 Viral Infection | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.36 [0.04, 3.24] |
| 5.19 Sinus Headache | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.44 [0.26, 8.01] |
| 5.20 Headache | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.60 [0.66, 3.92] |
| 5.21 Dizziness | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 10.55 [0.69, 162.40] |
| 5.22 Pyrexia | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.96 [0.35, 2.60] |
| 5.23 Fatigue | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.83 [0.23, 3.07] |
| 5.24 Abdominal pain upper | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.87 [0.26, 2.91] |
| 5.25 Abdominal pain | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.29 [0.46, 3.59] |
| 5.26 Constipation | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.34 [0.06, 2.05] |
| 5.27 Diarrhea | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.36 [0.45, 4.11] |
| 5.28 Vomiting | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.58 [0.20, 1.68] |
| 5.29 Nausea | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.13 [0.37, 3.40] |
| 5.30 Gastrooesophageal reflux disease | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.31 Weight decreased | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.46 [0.05, 4.42] |
| 5.32 Asparate aminotransferase increased | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.02 [0.40, 10.31] |
| 5.33 Alanine aminotransferase increased | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.83 [0.21, 3.31] |
| 5.34 Eosinophil count increased | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.26 [0.15, 69.97] |
| 5.35 C‐reactive protein increased | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.74 [0.12, 4.37] |
| 5.36 Blood glucose increased | 1 | 166 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.71 [0.25, 11.72] |
| 5.37 Hypoglycaemia | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.69 [0.09, 5.18] |
| 5.38 white blood cell count decreased | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.39 Bacteria sputum Identified | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.02 [0.39, 10.32] |
| 5.40 Joint Sprain | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.10 [0.00, 4.71] |
| 5.41 Breath Sounds abnormal | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.35 [0.05, 2.49] |
| 5.42 Arthralgia | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.34 [0.28, 6.44] |
| 5.43 Myalgia | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.44 Back pain | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.94 [0.17, 5.03] |
| 5.45 Neck pain | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.46 Haemoptysis | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.51 [0.16, 1.65] |
| 5.47 Respiratory tract congestion | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.71 [0.18, 2.86] |
| 5.48 Sinus Congestion | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.97 [0.39, 10.10] |
| 5.49 Dyspnoea | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.36 [0.04, 3.24] |
| 5.50 Pleutiric pain | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.44 [0.27, 21.87] |
| 5.51 Respiration abnormal | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.46 [0.05, 4.42] |
| 5.52 Pharyngeal erythema | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.53 Culture throat positive | 1 | 51 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.17 [0.09, 55.58] |
| 5.54 Rash | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.03 [0.58, 7.06] |
| 5.55 Acne | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.95 [0.30, 12.62] |
| 5.56 Lymphadenopathy | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.48 [0.02, 12.25] |
| 5.57 Excoriation | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.48 [0.02, 12.25] |
| 5.58 Neutrophil count decreased | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.59 Forced expiratory volume decreased | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 5.60 Seasonal allergy | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.48 [0.02, 12.25] |
| 5.61 Allergic rhinitis | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 5.41 [0.09, 312.04] |
| 6 All adverse events | 1 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 6.1 Cardiac Murmur | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.2 Chest discomfort | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.60] |
| 6.3 Pulmonary exacerbation | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.4 Upper respiratory tract infection | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.5 Respiratory tract congestion | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.6 Cough | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.09 [0.04, 30.57] |
| 6.7 Cough decreased | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.8 Productive cough | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.21 [0.00, 11.63] |
| 6.9 Sputum abnormal | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.10 Sputum decreased | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.11 Sputum discolored | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.12 Increased upper airway secretion | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.13 Increased bronchial secretion | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.14 Rhonchi | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.15 Headache | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.16 Tension headache | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.17 Sinus headache | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.18 Sinusitis | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.19 Chronic sinusitis | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.60] |
| 6.20 Sinus congestion | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.21 Nasal congestion | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.22 Nasal oedma | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.23 Nasal dryness | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.24 Nasal turbinate abnormality | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.25 Nasal septum disorder | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.26 Mucosal erosion | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.27 Epistaxis | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.28 Oral candidiasis | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.29 Pharyngeal oedema | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.30 Pharyngeal erythema | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.31 Pharyngolaryngeal pain | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.32 Tonsillar hypertrophy | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.60] |
| 6.33 Abdominal distension | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.60] |
| 6.34 Upper abdominal pain | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.35 Decreased appetite | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.60] |
| 6.36 Nausea | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.37 Vomiting | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.38 Diarrhoea | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.46 [0.01, 16.23] |
| 6.39 Flatulence | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.60] |
| 6.40 Pyuria | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.41 White blood cells in urine | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.42 Rash | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.67 [0.02, 114.48] |
| 6.43 Contact dermatitis | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.08 [0.00, 6.60] |
| 6.44 Erythema | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.45 Excoriation | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.46 Pruritis | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.47 Genital pruritis (female) | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.48 Vulvovaginal mycotic infection | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.49 Photosensitivity reaction | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.50 Allergy to arthropod bite | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.51 Skin laceration | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.52 Injection site oedema | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.53 Fatigue | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.54 Somnolence | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.55 Joint crepitation | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.56 Lymphadenopathy | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 6.57 Blood creatinine increased | 1 | 19 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.93 [0.01, 78.14] |
| 7 Severity of adverse effects of therapy with regards to study drug interruption (moderate) or discontinuation (severe) | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Adverse effect requiring study drug interruption (moderate) | 2 | 213 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.14, 9.92] |
| 7.2 Adverse effect requiring study drug discontinuation (severe) | 3 | 232 | Odds Ratio (M‐H, Random, 95% CI) | 0.25 [0.04, 1.57] |
| 8 Serious adverse events | 3 | Odds Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 8.1 Haemoptysis | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.23 [0.01, 4.14] |
| 8.2 Hypoglycaemia | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.18 [0.00, 10.12] |
| 8.3 Complete atrioventricular block | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.4 Pulmonary exacerbation | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.34 [0.13, 0.88] |
| 8.5 Gastrooesophageal reflux | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.6 Pancreatic pseudocyst | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.7 Pancreatitis | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.8 Vomiting | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.9 Abdominal pain | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.12 [0.04, 222.22] |
| 8.10 Constipation | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.00, 22.86] |
| 8.11 Weight decreased | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.12 Anaphylactic shock | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.13 Catheter related complication | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.14 Implant site infection | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.15 Influenza | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.16 Pseudomonal lung infection | 2 | 213 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.02, 6.34] |
| 8.17 Pneumonia | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.18 Sinusitis | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.19 Cough | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.20 Pleuritic pain | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.21 Pulmonary embolism | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.22 Respiratory distress | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.23 Productive cough | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 1.0 [0.02, 41.06] |
| 8.24 Respiratory failure | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.25 Pulmonary function test decreased | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.00, 22.86] |
| 8.26 Lung consolidation | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.00, 22.86] |
| 8.27 Myringitis bullous | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.28 Hepatic enzyme increased | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.29 Cervix carcinoma | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.30 Musculoskeletal chest pain | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.31 Pain in extremity | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.32 Spontaneous abortion | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.33 Haematuria | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.34 IgA nephropathy | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 2.85 [0.04, 195.35] |
| 8.35 Nephrolithiasis | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.36 Renal Colic | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.37 Testicular torsion | 1 | 161 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.31 [0.00, 21.17] |
| 8.38 Pyrexia | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.12 [0.04, 222.22] |
| 8.39 Muscle strain | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.12 [0.04, 222.22] |
| 8.40 Hepatic enzyme increased | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.12 [0.04, 222.22] |
| 8.41 Adjustment disorder | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.00, 22.86] |
| 8.42 Anxiety | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.00, 22.86] |
| 8.43 Affective disorder | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 0.32 [0.00, 22.86] |
| 8.44 Conversion disorder | 1 | 52 | Odds Ratio (M‐H, Fixed, 99% CI) | 3.12 [0.04, 222.22] |
| 9 The number of G551D participants who developed episodes of pulmonary exacerbation | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 At 1 month | 1 | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.67 [0.07, 41.64] |
| 9.2 At 48 weeks | 2 | 213 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.36, 1.12] |
| 10 Time to first pulmonary exacerbation | 1 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 10.1 At 24 weeks | 1 | 161 | Hazard Ratio (Fixed, 95% CI) | 0.46 [0.28, 0.76] |
| 10.2 At 48 weeks | 1 | 161 | Hazard Ratio (Fixed, 95% CI) | 0.46 [0.29, 0.73] |
| 11 Maximum liver function test abnormalities in adolescents/adults at 48 weeks | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 2x to 3x ULN AST | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.97 [0.57, 6.84] |
| 11.2 2x to 3x ULN ALT | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.23, 2.63] |
| 11.3 2x to 3x ULN Bilirubin | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.90 [0.17, 21.39] |
| 11.4 3x to 5x ULN AST | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.46 [0.04, 5.21] |
| 11.5 3x to 5x ULN ALT | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.01, 3.88] |
| 11.6 3x to 5x ULN Bilirubin | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.7 5x to 8x ULN AST | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.85 [0.11, 71.12] |
| 11.8 5x to 8x ULN ALT | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.31 [0.01, 7.71] |
| 11.9 5x to 8x ULN Bilirubin | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 11.10 8x AST | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.06, 15.28] |
| 11.11 8x ALT | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.83 [0.35, 134.32] |
| 11.12 8x Bilirubin | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 12 Duration of hospitalisation for pulmonary exacerbation | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 12.1 At 48 weeks | 1 | 161 | Mean Difference (IV, Fixed, 95% CI) | ‐0.23 [‐3.74, 3.28] |
| 13 Number of hospitalisations for pulmonary exacerbations | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 At 48 weeks | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.16, 0.81] |
| 14 FEV1 (% predicted) ‐ mean absolute change from baseline | 2 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 14.1 24 weeks | 2 | 213 | Mean Difference (Fixed, 95% CI) | 10.80 [8.91, 12.69] |
| 14.2 48 weeks | 2 | 213 | Mean Difference (Fixed, 95% CI) | 10.44 [8.56, 12.32] |
| 15 FEV1 (litres) ‐ mean absolute change from baseline | 2 | Mean Difference (Random, 95% CI) | Subtotals only | |
| 15.1 24 weeks | 2 | 212 | Mean Difference (Random, 95% CI) | 0.33 [0.17, 0.49] |
| 15.2 48 weeks | 2 | 212 | Mean Difference (Random, 95% CI) | 0.31 [0.11, 0.50] |
| 16 Change from baseline through week 24 in percent of predicted FEV1 according to subgroup | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 16.1 Subgroup 1: Baseline FEV1 of less than or equal to 90% predicted | 1 | 30 | Mean Difference (Fixed, 95% CI) | 14.9 [7.30, 22.50] |
| 16.2 Subgroup 2: Baseline FEV1 of more than 90% predicted | 1 | 21 | Mean Difference (Fixed, 95% CI) | 6.9 [‐3.80, 17.60] |
| 16.3 Subgroup 3: North America | 1 | 26 | Mean Difference (Fixed, 95% CI) | 5.8 [‐2.60, 14.20] |
| 16.4 Subgroup 4: Europe | 1 | 11 | Mean Difference (Fixed, 95% CI) | 24.60 [6.40, 42.80] |
| 16.5 Subgroup 5: Australia | 1 | 14 | Mean Difference (Fixed, 95% CI) | 4.2 [‐3.70, 12.10] |
| 16.6 Subgroup 6: Male | 1 | 24 | Mean Difference (Fixed, 95% CI) | 5.2 [‐2.20, 12.60] |
| 16.7 Subgroup 7: Female | 1 | 27 | Mean Difference (Fixed, 95% CI) | 13.8 [4.20, 23.40] |
| 17 Number of pulmonary exacerbations requiring intravenous antibiotics | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 At 48 weeks | 1 | 161 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.18, 0.64] |
| 18 Days with intravenous antibiotics administered for pulmonary exacerbations | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 18.1 At 48 weeks | 1 | 161 | Mean Difference (IV, Fixed, 95% CI) | ‐4.35 [‐10.51, 1.81] |
| 19 Weight ‐ change from baseline (kg) | 2 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 19.1 At 24 weeks | 2 | 213 | Mean Difference (Fixed, 95% CI) | 2.37 [1.68, 3.06] |
| 19.2 at 48 weeks | 2 | 213 | Mean Difference (Fixed, 95% CI) | 2.75 [1.74, 3.75] |
| 20 Sweat chloride level ‐ change from baseline | 2 | Mean Difference (Random, 95% CI) | Subtotals only | |
| 20.1 At 24 weeks | 2 | 213 | Mean Difference (Random, 95% CI) | ‐50.19 [‐56.20, ‐44.18] |
| 20.2 At 48 weeks | 2 | 213 | Mean Difference (Random, 95% CI) | ‐49.74 [‐54.61, ‐44.87] |
Comparison 3. Ivacaftor (VX‐770) versus placebo in people with at least one R117H CFTR mutation.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 CFQ‐R score (respiratory domain score, pooled) ‐ absolute change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 1.1 At 24 weeks | 1 | 69 | Mean Difference (Fixed, 95% CI) | 8.4 [2.17, 14.63] |
| 2 FEV1 relative change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 2.1 At 24 weeks | 1 | 69 | Mean Difference (Fixed, 95% CI) | 5.0 [‐0.24, 10.24] |
| 3 Adverse events | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Any adverse event | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.18 [0.01, 3.96] |
| 3.2 Serious adverse event | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.16, 2.52] |
| 3.3 Adverse event leading to discontinuation | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Adverse events in > 15% of participants | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Pulmonary exacerbation | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.35, 2.44] |
| 4.2 Cough | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.42, 3.47] |
| 4.3 Headache | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.29 [0.35, 4.69] |
| 4.4 Increased sputum | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.33, 5.47] |
| 4.5 Nasal congestion | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.33, 5.47] |
| 4.6 Oropharyhgeal pain | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.84 [0.51, 15.79] |
| 4.7 Diarrhoea | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.33, 5.47] |
| 4.8 Abdominal pain | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 10.48 [0.54, 202.47] |
| 4.9 Wheezing | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.53 [0.48, 42.82] |
| 4.10 Cystic fibrosis lung pathogen colonisation | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.29 [0.32, 33.31] |
| 5 Summary of pulmonary exacerbations | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 All pulmonary exacerbations | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.81 [0.30, 2.19] |
| 5.2 Number of hospitalisations for pulmonary exacerbation | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.06, 1.62] |
| 5.3 Number of pulmonary exacerbations requiring intravenous antibiotics | 1 | 69 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.30 [0.06, 1.62] |
| 6 Time to first pulmonary exacerbation | 1 | Hazard Ratio (Fixed, 95% CI) | Subtotals only | |
| 6.1 At 24 weeks | 1 | 69 | Hazard Ratio (Fixed, 95% CI) | 0.93 [0.42, 2.08] |
| 7 FEV1 predicted % absolute change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 7.1 At 24 weeks | 1 | 69 | Mean Difference (Fixed, 95% CI) | 2.1 [‐1.13, 5.33] |
| 8 BMI absolute change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 8.1 At 24 weeks | 1 | 69 | Mean Difference (Fixed, 95% CI) | 0.26 [‐1.57, 2.09] |
| 9 Sweat chloride concentration ‐ change from baseline | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 9.1 At 24 weeks | 1 | 69 | Mean Difference (Fixed, 95% CI) | ‐22.00 [‐28.01, ‐19.99] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Accurso 2010.
| Methods | 3‐arm RCT of parallel design. Multicentre: 13 centres reported. Duration: 28 days. |
|
| Participants | Adults with CF with a G551D‐CFTR mutation on at least 1 allele. Total of 19 participants were enrolled in Part 2 of this trial. Gender n (%) Total cohort: 10 (53) males; Treatment group 1: 3 (38) males; Treatment group 2: 4 (57) males; Control group: 3 (75) males. White race n (%) Total cohort: 19 (100); Treatment group 1: 8 (100); Treatment group 2: 7 (100); Control group: 4 (100). Age median (range) Total cohort: 21 (18 ‐ 42) years; Treatment group 1: 23 (18 ‐ 40) years; Treatment group 2: 21 (20 ‐ 38) years; Control group: 24 (18 ‐ 42) years. BMI median (range) Total cohort: 22 (20 ‐ 25); Treatment group 1: 22 (20 ‐ 23); Treatment group 2: 23 (20 ‐ 25); Control group: 22 (21 ‐ 23). CFTR Genotype n (%) G551D/F508del Total cohort: 16 (84); Treatment group 1: 7 (88); Treatment group 2: 5 (71); Control group: 4 (100). G551D/3849+10kbC→T Total cohort: 1 (5); Treatment group 1: 0 (0); Treatment group 2: 1 (14) Control group: 0 (0). G551D/621+1G→T Total cohort: 1 (5); Treatment group 1: 1 (12); Treatment group 2: 0 (0); Control group: 0 (0). G551D/G542X Total cohort: 1 (5); Treatment group 1: 0 (0); Treatment group 2: 1 (14); Control group: 0 (0). FEV1 % predicted Median (range) Total cohort: 69 (40 ‐ 122); Treatment group 1: 65 (42 ‐ 122); Treatment group 2: 76 (40 ‐ 106); Control group: 77 (53 ‐ 112). 40% to <70% n (%) Total cohort: 10 (53); Treatment group 1: 5 (62); Treatment group 2: 3 (43); Control group: 2 (50). 70% to <90% n (%) Total cohort: 5 (26); Treatment group 1: 2 (25); Treatment group 2: 3 (43); Control group: 0 (0). ≥90% value n (%) Total cohort: 4 (21); Treatment group 1: 1 (12); Treatment group 2: 1 (14); Control group: 2 (50). Sweat chloride (mmol/L) mean (range) Total cohort: 95.5 (84.8 ‐ 115.8); Treatment group 1: 100.1 (86.8 ‐ 112.5); Treatment group 2: 97.3 (84.8 ‐ 115.8); Control group: 93.8 (88.0 ‐ 109.5). CFQ‐R respiratory domain (points) mean (range) Total cohort: 72.2 (16.7 ‐ 88.9); Treatment group 1: 69.4 (16.7 ‐ 88.9); Treatment group 2: 72.2 (61.1 ‐ 83.3); Control group: 80.6 (38.9 ‐ 83.3). |
|
| Interventions | Treatment Group 1: VX‐770 150 mg 2x daily (n = 8). Treatment Group 2; VX‐770 250 mg 2x daily (n = 7). Control: placebo 2x daily (n = 4). |
|
| Outcomes |
Primary outcome: 1. Safety and adverse effects* Secondary outcomes: 1. CFTR ion channel function. 2. Change in QoL from baseline* 3. Relative change from baseline in FEV1* 4. Relative change from baseline in FVC* and relative change from baseline in FEF25‐75 5. Change from baseline in sweat chloride concentration.* |
|
| Notes | This was a 2‐part trial; part 1 was of cross‐over design and so not included in this review. We only present details of the second part which was of parallel design. The responsible funding body was Vertex Pharmaceuticals Incorporated. * These outcomes are presented in the review. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list generated by a statistician not otherwise associated with the trial. |
| Allocation concealment (selection bias) | Low risk | Participants assigned to a treatment arm using an Interactive Voice Response system according to a concealed randomisation list. This ensured the outcome assessors and participants were unaware of their allocation. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | All trial personnel were blinded to participants' treatment. Exceptions to this were the laboratory staff and clinical pharmacist who prepared the medication. These trial personnel were not otherwise involved in the trial. |
| Blinding of participants | Unclear risk | All participants received the same number of tablets to maintain trial and personnel blinding. However, there is no report on details of the tablets (e.g. colour, size, taste). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All together, 20 participants were randomised. 1 of 5 placebo participants withdrew due to withdrawal of consent prior to dosing (5% of total participants). All other participants were accounted for in the analysis. |
| Selective reporting (reporting bias) | High risk | The change from baseline in weight, although not reported as an outcome, was measured at day 1, 3, 14, 21 and 28. It was not reported in the publication. |
| Other bias | Low risk | Similar baseline characteristics and high adherence rate (100% [range, 92.6 ‐ 100]). |
DISCOVER 2011.
| Methods | 2‐part RCT, both parts of parallel design. Participants who met set out criteria by the end of part A were eligible to proceed to part B of the trial. In part A, participants were randomly assigned to the intervention or control group in a 4:1 ratio respectively. Multicentre: 34 sites. Duration: part A lasted 16 weeks with part B, an open label extension, lasting up to 96 weeks. |
|
| Participants | 140 participants randomised to treatment or placebo in a ratio of 4:1 (treatment group n = 112; placebo n = 28). Gender n (%) Treatment group: 58 (52) males; Placebo group: 16 (57) males. Age years, mean (SD)/median (range) Treatment group: 22.8 (10.3)/19.5 (12 ‐ 52); Placebo group: 25.0 (8.4)/24.0 (12 ‐ 39). CFTR Genotype All homozygous for the F508del‐CFTR mutation. Height cm, median (range) Treatment group: 164.0 (139.0 ‐ 189.6); Placebo group: 170.7 (145.6 ‐ 184.0). Weight kg, median (range) Treatment group: 55.9 (35.1 ‐ 99.8); Placebo group: 64.9 (44.2 ‐ 100.3). BMI kg/m², median (range) Treatment group: 20.4 (15.7 ‐ 31.6); Placebo group: 21.5 (17.9 ‐ 40.8). Sweat chloride mmol/L, mean (SD)/median (range) Treatment group: 101.4 (10.3)/101.0 (79.5 ‐ 135.5); Placebo group: 102.4 (7.9)/101.5 (91.0 ‐ 122.0). FEV1 % predicted, mean (SD)/median (range) Treatment group: 79.7 (22.7)/79.0 (40 ‐ 129); Placebo group: 74.8 (24.1)/67.0 (43 ‐ 127). |
|
| Interventions |
Part A Treatment group: 150 mg of VX‐770 (ivacaftor) 2x daily (n = 112). Control group: placebo (n = 28). Part B Treatment group: 150 mg of VX‐770 2x daily. Control group: placebo. |
|
| Outcomes |
Primary outcomes 1. Adverse events, clinical laboratory values, ECGs, vital signs and physical examination.* 2. Absolute change in FEV1 % predicted from baseline through 16 weeks.* Secondary outcomes 1. Change from baseline through week 16 in sweat chloride concentration.* 2. Change from baseline through week 16 in weight.* 3. Participant reported health using the disease‐specific CFQ‐R through week 16.* |
|
| Notes | The responsible funding body was Vertex Pharmaceuticals Incorporated. * These outcomes are presented in the review. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Eligible participants were randomised to a treatment group (ivacaftor or placebo) in a 1:1 ratio using a randomisation code produced by Vertex. |
| Allocation concealment (selection bias) | Low risk | 2 statisticians were involved. The trial statistician was blinded whereas the other was unblinded and not associated with the trial. The unblinded statistician generated the final randomisation list that was provided to the Interactive Voice Response/Interactive Web Response System. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | All site personnel including the investigator, the trial monitor, and the Vertex trial team were blinded. Laboratory personnel or their designee were unblinded to the bio‐analysis results, but remained blinded to treatment assignment. A clinical pharmacologist not involved in the conduct of the trial reviewed the bio‐analysis results on an ongoing basis, but remained blinded to the participant’s identity number and treatment assignment. |
| Blinding of participants | Unclear risk | The participants were 'blinded.' There is no report on details of the tablets (e.g. colour, size, taste). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 8 withdrawals were reported in the ivacaftor group and 2 in the placebo group. Analyses were performed on all randomised participants who received at least 1 dose of the trial drug (ivacaftor n = 112 and placebo n = 28), as shown in table 3 of the full paper. However, when assessing the results on clinicaltrials.gov, the number in the ivacaftor group is presented as 111 instead of 112 for the outcomes change from baseline in sweat chloride and absolute change from baseline in FEV1, suggesting an ITT analysis was not conducted. |
| Selective reporting (reporting bias) | High risk | The protocol for this trial was not available. There are no data for the change from baseline in FEV1 at day 15 or week 8. In addition no data were reported for the outcome measures FVC or FEF25‐75 at 16 weeks. 1 withdrawal from the trial was due to "early termination per sponsor decision" and no additional information was provided. The author was approached for information on these missing data. |
| Other bias | Low risk | Similar baseline characteristics. |
ENVISION 2013.
| Methods | 2‐arm RCT of parallel design. Participants were randomly assigned in a 1:1 ratio. Multicentre: 29 centres reported. Duration: 48 weeks. |
|
| Participants | 52 children enrolled onto this trial (control n = 26, intervention n = 26). Children aged 6 to 11 years were eligible for inclusion Gender n (%) Total cohort: 25 (48) males; Treatment group: 9 (35) males; Control group: 16 (62) males. Age years, mean (range) Total cohort: 8.9 (range 6 ‐ 12); Treatment group: 8.9 (range 6 ‐ 12); Control group: 8.9 (range 6 ‐ 12). CFTR Genotype NB Participants with at least one G551D allele were eligible. 44 participants had the G551D/F508del genotype. 8 participants had the G551D‐CFTR mutation on one allele and a non‐F508del‐CFTR mutation on the second allele Height cm, mean (range) Total cohort: 133.8 (110.5 ‐ 168.6); Treatment group: 134.9 (115.0 ‐ 168.6); Control group: 132.6 (110.5 ‐ 155.8). Weight kg, mean (range) Total cohort: 30.9 (17.8 ‐ 62.6); Treatment group: 31.8 (18.8 ‐ 62.6); Control group: 30.0 (17.8 ‐ 46.3). FEV1 % predicted Participants with baseline FEV1 40% ‐ 105% of the predicted value for persons of their age, sex and height were eligible. Mean (range) Total cohort: 84.2 (44.0 ‐ 133.8); Treatment group: 84.7 (52.4 ‐ 133.8); Control group: 83.7 (44.0 ‐ 116.3). < 70%, n (%) Total cohort: 12 (23); Treatment group: 4 (15); Control group: 8 (31). > 70% to < 90%, n (%) Total cohort: 18 (35); Treatment group: 12 (46); Control group: 6 (23). > 90%, n (%) Total cohort: 22 (42); Treatment group: 10 (38); Control group: 12 (46). Sweat chloride mmol/L, mean (range) Total cohort: 104.6 (54.0 ‐ 128.0); Treatment group: 104.3 (54.0 ‐ 128.0); Control group: 104.8 (92.0 ‐ 121.0). CFQ‐R score for respiratory domain, mean (range) Child version Total cohort: 79 (25.0 ‐ 100.0); Treatment group: 78 (33.3 ‐ 100.0); Control group: 80 (25.0 ‐ 100.0). Parent/caregiver version Total cohort: 81 (33.3 ‐ 100.0); Treatment group: 81 (33.3 ‐ 100.0); Control group: 81 (38.9 ‐ 100.0). |
|
| Interventions |
48‐week RCT Treatment: VVX‐770 also known as ivacaftor 150 mg 2x daily (n = 26). Control: placebo (n = 26). Open‐label extension Treatment: VVX‐770 also known as ivacaftor 150 mg 2x daily. Control: placebo. |
|
| Outcomes |
Primary Outcome 1. Absolute change from baseline through week 24 in % predicted FEV1* Secondary outcomes Absolute changes from baseline in: 1. % predicted FEV1 (through week 48);* 2. weight (at weeks 24 and 48);* 3. concentrations of sweat chloride (a measure of CFTR function) (at weeks 24 and 48);* 4. participant‐reported respiratory symptoms as assessed by the respiratory domain of the child version of the CFQ‐R (at weeks 24 and 48);* 5. safety.* |
|
| Notes | The responsible funding body was Vertex Pharmaceuticals Incorporated. * These outcomes are presented in the review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Participants randomised in a 1:1 ratio, however method not reported. |
| Allocation concealment (selection bias) | Unclear risk | No description of allocation concealment. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double blind. It is not stated how attending researchers were blinded. |
| Blinding of participants | Unclear risk | Described as double blind. It is not stated how participants were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | In the paediatric phase 3 G551D trial 4/52 participants (7.7%) withdrew from the total trial population. All 4 participants withdrew from the placebo group. In the analysis of the following outcomes participant data were excluded:
A modified intention‐to‐treat analysis (not per protocol) was employed, where data were excluded from the analysis (ENVISION 2013). |
| Selective reporting (reporting bias) | High risk | At each visit (days 1, 15 and weeks 8, 16, 24, 32, 40 and 48) participant FVC measurements were recorded. These results were not published in the full text, nor made available upon request. Data for the relative change from baseline in FEV1 (an important outcome in such a trial) were not reported at 48 weeks. |
| Other bias | Low risk | Similar baseline characteristics and high adherence rate. |
KONDUCT 2015.
| Methods | Phase 3, double‐blind, RCT of parallel design. Multicentre: 31 centres reported. Duration: 24 weeks with an open‐label extension phase for up to 104 weeks. Participants were not re‐randomised for the extension phase. |
|
| Participants | 69 participants aged 6 years or older with CF with at least 1 allele of the R117H‐CFTR mutation were enrolled. Total: control n = 35, intervention n = 34; 6 ‐ 11 years control n = 8, intervention n = 9; ≥ 18 years control n = 26, intervention n = 24. Age years, mean (SD) Overall: treatment group: 29.2 (16.6); placebo group: 32.7 (17.4). 6 ‐ 11 years: treatment group 8.8 (1.9); placebo group 9 (1.6). ≥ 18 years: treatment group 37.5 (12.1); placebo group 40.6 (12.6). Gender n (%) Overall: treatment group 19 (56%) females; placebo group 20 (57%) females. 6 ‐ 11 years: treatment group 5 (56%) females; placebo group 3 (38%) females. ≥ 18 years: treatment group 13 (54%); placebo group 16 (62%). Weight kg, mean (SD) Overall: treatment group: 66.1 (25.5); placebo group: 62.8 (25.4). 6 ‐ 11 years: treatment group 32.9 (13.3); placebo group 34.0 (9.1). ≥ 18 years: treatment group 77.9 (16.7); placebo group 71.7 (22.5). BMI kg/m2, mean (SD) Overall: treatment group: 24.5 (6.3); placebo group: 23.1 (6). 6 ‐ 11 years: treatment group 17.6 (3.3); placebo group 17.1 (2.4). ≥ 18 years: treatment group 26.9 (5.2); placebo group 24.9 (5.7). FEV1 % predicted, mean (SD) Participants with baseline FEV1 40% to 90% (for participants aged 12 years or older) or 40% to 105% (for participants aged 6 to 11 years) of the predicted value for age, sex, and height were eligible. Overall: treatment group: 75.7 (19.3); placebo group: 70.2 (18.9). 6 ‐ 11 years: treatment group 97.5 (8.6); placebo group 94.0 (8.4). ≥ 18 years: treatment group 67.0 (15.4); placebo group 62.3 (14.4). FEV1 % predicted, n (%) <70% Overall: treatment group: 13 (38%); placebo group: 15 (43%). 6 ‐ 11 years: treatment group 0; placebo group 0. ≥ 18 years: treatment group 13 (54%); placebo group 15 (58%). ≥ 70 to ≤ 90% Overall: treatment group: 14 (41%); placebo group: 14 (40%). 6 ‐ 11 years: treatment group 3 (33%); placebo group 2 (25%). ≥ 18 years: treatment group 10 (42%); placebo group 11 (42%). > 90% Overall: treatment group: 7 (21%); placebo group: 6 (17%). 6 ‐ 11 years: treatment group 6 (67%); placebo group 6 (75%). ≥ 18 years: treatment group 1 (4%); placebo group 0. Sweat chloride n/mmol/l, mean (SD) Overall: treatment group: 32/67.3 (23.5); placebo group: 35/73.4 (19.7). 6 ‐ 11 years: treatment group 8/64.2 (22.6); placebo group 8/74.7 (28.6). ≥ 18 years: treatment group 23/69.3 (24.1); placebo group 26/73.0 (17.3). CFQ‐R score for respiratory domain, n/score, mean (SD) Overall: treatment group: 33/75.3 (20.1); placebo group: 34/66.4 (24.4). 6 ‐ 11 years: treatment group 8/92.7 (7.0); placebo group 7/91.7 (6.8). ≥ 18 years: treatment group 24/68.4 (19.1); placebo group 26/59.2 (23.2). CFTR genotype (Arg117His/Phe508del), mean (SD) Overall: treatment group: 28 (82%); placebo group: 25 (71%). 6 ‐ 11 years: treatment group 8 (89%); placebo group 6 (75%). ≥ 18 years: treatment group 19 (79%); placebo group 19 (73%). |
|
| Interventions | Ivacaftor 150 mg orally 2x daily for 24 weeks. | |
| Outcomes |
Primary outcome: 1. absolute change from baseline in % predicted FEV1 (through 24 weeks). Secondary outcomes: 1. change from baseline in BMI (through 24 weeks); 2. change from baseline in sweat chloride (through 24 weeks); 3. change from baseline in the respiratory domain of the CFQ‐R (through 24 weeks); 4. time to first pulmonary exacerbation (through 24 weeks); 5. number of participants with adverse events and serious adverse events (through 24 weeks). |
|
| Notes | The responsible funding body was Vertex Pharmaceuticals Incorporated. * These outcomes are presented in the review. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were assigned to receive placebo or ivacaftor (1:1) via a randomisation specification and randomisation code. The code list was sent to the IVRS. |
| Allocation concealment (selection bias) | Low risk | A masked biostatistician created the randomisation specification and dummy randomisation code, this was then approved by an unmasked biostatistician not associated with the trial, prior to generating the final randomisation list. An unblinded quality check biostatistician reviewed and approved final randomisation list. The biostatistician not associated with the trial provided the final list to IVRS. |
| Blinding (performance bias and detection bias) All outcomes | Unclear risk | Described as double blinded. Other than the biostatisticians, it is not stated which trial personnel were blinded or how they were blinded. |
| Blinding of participants | Unclear risk | Described as double blinded. There is no report on details of the tablets (e.g. colour, size, taste). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | In total, 69 participants were randomised. 2 withdrew from the intervention group with reasons given. A full analysis set included all randomised participants who received at least 1 dose of trial drug. |
| Selective reporting (reporting bias) | High risk | No protocol was available online to compare outcomes in the protocol with those published. The author was approached for further information and a protocol synopsis provided. This showed the following tertiary outcomes which were reported in the protocol but no with no published results. 1. PK parameter estimates of ivacaftor and metabolites, M1 and M6, derived from plasma concentration‐time data 2. Change from baseline in nonrespiratory domains of the CFQ‐R 3. Change from baseline in weight 4. Change from baseline in height 5. CF‐related complications (pancreatitis or distal ileal obstruction syndrome 6. Change from baseline in inflammatory mediators 7. Change from baseline in qualitative microbiological cultures 8. Change from baseline in IRT 9. Change from baseline in fecal elastase‐1 |
| Other bias | Low risk | Similar baseline characteristics. There were only 2 participants between 12 ‐ 17 years of age so no age subgroup statistical analysis were performed for this group. They were included in overall analysis. No comment was made regarding the reason for low enrolment in this age group. |
STRIVE 2011.
| Methods | 2‐arm RCT of parallel design. Multicentre international trial with participants from Europe, North America and Australia. Duration: up to 48 weeks. |
|
| Participants | Total of 167 participants recruited: treatment group n = 84; control group n = 83. Participants (at least 12 years of age) were stratified according to age (< 18 years versus ≥18 years) and pulmonary function (under 70% versus 70% and over of the predicted FEV1). Gender, n (%) Total cohort: 77 (48) males; Treatment group: 39 (47) males; Control group: 38 (49) males. Non‐Hispanic or white, n (%) Total cohort: 158 (98); Treatment group: 81 (98); Control group: 77 (99). Geographic distribution, n (%) North America Total cohort: 100 (62); Treatment group: 50 (60); Control group: 50 (64). Europe Total cohort: 42 (26); Treatment group: 23 (28); Control group: 19 (24). Australia Total cohort: 19 (12); Treatment group: 10 (12); Control group: 9 (12). Age years, mean (range) Total cohort: 25.5 (12 ‐ 53); Treatment group: 26.2 (12 ‐ 53); Control group: 24.7(12 ‐ 53). Age distribution, n (%) Under 18 years Total cohort: 36 (22); Treatment group: 19 (23); Control group: 17 (22). 18 years and over Total cohort: 125 (78); Treatment group: 64 (77); Control group: 61 (78). Height cm, mean (range) Total cohort: 167.1 (142.2 ‐ 189.8); Treatment group: 167.7 (142.8 ‐ 185.0); Control group: 166.5 (142.2 ‐ 189.8). Weight kg, mean (range) Total cohort: 61.5 (30.2 ‐ 109.9); Treatment group: 61.7(30.2 ‐ 107.2); Control group: 61.2(31.9 ‐ 109.9). BMI, mean (range) Total cohort: 21.8 (14.8 ‐ 38.9); Treatment group: 21.7 (14.8 ‐ 38.9); Control group: 21.9 (15.2 ‐ 38.6). Positive for Pseudomonas aeruginosa, n (%) Total cohort: 122 (76); Treatment group: 64 (77); Control group: 57 (73). CFTR Genotype Non–G551D‐CFTR allele n (%),NB Only participants with at least 1 G551D allele were eligible. Class I Total cohort: 21 (13); Treatment group: 10 (12); Control group: 11 (14). Class II Total cohort: 132 (82); Treatment group: 70 (84); Control group: 62 (79). Class III Total cohort: 1 (1); Treatment group: 0; Control group: 1 (1). Class IV Total cohort: 4 (2); Treatment group: 2 (2); Control group: 2 (3). Class V Total cohort: 1 (1); Treatment group: 0; Control group: 1 (1). Unknown Total cohort: 2(1); Treatment group: 1(1); Control group: 1(1). FEV1 % predicted, mean (range) NB Only participants with baseline FEV1 40% ‐ 90% of the predicted value for persons of their age, sex and height were eligible. Total cohort: 63.6 (31.6 ‐ 98.2); Treatment group: 63.5 (37.3 ‐ ‐98.2); Control group: 63.7 (31.6 ‐ 97.1). FEV1 distribution, n (%) Under 70% of predicted value Total cohort: 94 (58); Treatment group: 49 (59); Control group: 45 (58). 70% and over of predicted value Total cohort: 67 (42); Treatment group: 34 (41); Control group: 33 (42). Sweat chloride mmol/L, mean (range) Total cohort: 100.2 (58.0 ‐ 128.0); Treatment group: 100.4 (74.5 ‐ 128.0); Control group: 100.1 (58.0 ‐ 121.5). |
|
| Interventions |
48‐week RCT Treatment: VX‐770 also known as ivacaftor 150 mg 2x daily (n = 83). Control: placebo (n = 78). Open‐label extension Treatment: VVX‐770 also known as ivacaftor 150 mg 2x daily. Control: placebo. |
|
| Outcomes |
Primary outcome 1. Absolute change from baseline through week 24 in % predicted FEV1.* Secondary outcomes 1. Change from baseline through week 48 in % predicted FEV1.* 2. Time to the first pulmonary exacerbation through week 24 and week 48. * 3. Participant‐reported health using the disease‐specific CFQ‐R.* 4. Change in weight from baseline to week 24 and week 48. * 5. Change from baseline in the concentration of sweat chloride through week 24 and week 48.* Tertiary endpoints 1. Number and duration of pulmonary exacerbations.* 2. Total number of days of hospitalisation for pulmonary exacerbations.* 3. Need for antibiotic therapy for sinopulmonary signs or symptoms.* |
|
| Notes | * These outcomes are presented in the review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Eligible participants were randomised to a treatment group (ivacaftor or placebo) in a 1:1 ratio using a randomisation code produced by Vertex. |
| Allocation concealment (selection bias) | Low risk | 2 biostatisticians involved in randomisation process; 1 blinded trial statistician and 1 unblinded statistician not involved in the trial. The statistician not associated with the trial generated the final randomisation list that was provided to an Interactive Voice Response System. |
| Blinding (performance bias and detection bias) All outcomes | Low risk | All site personnel including the investigator, the trial monitor, and the Vertex trial team were blinded. Laboratory personnel or their designee were unblinded to the bio‐analysis results, but remained blinded to treatment assignment. A clinical pharmacologist not involved in the conduct of the trial reviewed the bio‐analysis results on an ongoing basis, but remained blinded to the participant’s identity number and treatment assignment. |
| Blinding of participants | Unclear risk | All participants were stated to have been blinded, however, details of the tablets have not been provided (e.g. colour, size, taste). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 167 participants were randomised ‐ 83 participants to the placebo group and 84 participants to the ivacaftor group. 10 participants withdrew from the placebo group and 6 participants withdrew from the ivacaftor group. 78 participants in the placebo group and 83 participants in the ivacaftor group (total n = 161) were included in the analysis. The review authors found a minor discrepancy in the number of participants analysed for the outcomes measures: change from baseline in CFQ‐R respiratory domain scores (total analysed n = 151) and the change from baseline in sweat chloride concentration (total analysed n = 152) (STRIVE 2011). |
| Selective reporting (reporting bias) | High risk | Protocol provided. The following tertiary outcomes were reported in the protocol, but results for these outcomes were not published. 1. Change from baseline in oxygen saturation through weeks 24 and 48. 2. Change from baseline in EuroQol Questionnaire (EQ‐5D) through weeks 24 and 48. 3. Outpatient sick visits to the clinic or hospital for CF–related complications through weeks 24 and 48. In addition to this, results were only reported for CFQ‐R domains where improvements were seen in the ivacaftor group. |
| Other bias | Low risk | Similar baseline characteristics. The responsible funding body was Vertex Pharmaceuticals Incorporated. |
ALT: alanine transaminase AST: aspartate transaminase BMI: body mass index CF: cystic fibrosis CFQ‐R: cystic fibrosis questionnaire ‐ revised CFTR: cystic fibrosis transmembrane regulator ECG: electro‐cardiogram FEF25‐75: forced expiratory flow at 25‐75% of the pulmonary volume FEV1: forced expiratory volume at one second FVC: forced vital capacity IRT: immunoreactive trypsinogen ITT: intention to treat IVRS: interactive voice response system MRI: magnetic resonance image PK: pharmacokinetic QoL: quality of life RCT: randomised controlled trial ULN: upper limit of normal
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Accurso 2013 | Secondary analysis on trials of cross‐over design. |
| ALBATROSS 2017 | Intervention to correct molecular defect, not a potentiator. |
| Altes 2011 | Trial of cross‐over design. |
| ARRIVAL 2018 | Single‐arm interventional study, no comparison to placebo. |
| Berkers 2017 | Trial of cross‐over design. |
| Chadwick 1998 | Intervention to correct molecular defect, not a potentiator. |
| Clancy 2012 | Intervention to correct molecular defect, not a potentiator. |
| Davies 2012 | Trial of cross‐over design. |
| Davies 2016 | Single‐arm interventional trial. |
| Edgeworth 2017 | Trial of cross‐over design. |
| EudraCT Number: 2014‐000817‐30 | Single‐arm interventional study. Intervention was not with CFTR potentiator. |
| EudraCT Number: 2016‐001440‐18 | Observational study. Intervention was not with CFTR potentiator. |
| EudraCT Number: 2016‐001619‐19 | Trial of cross‐over design. Intervention was not CFTR potentiator. |
| EUudraCT Number: 2016‐001785‐29 | Intervention used antibiotic therapy rather than CFTR potentiator. |
| FLAMINGO 2017 | Intervention to correct molecular defect, not a potentiator. |
| Horsley 2018 | Intervention to correct molecular defect, not a potentiator. |
| Hubert 2018 | Retrospective cohort design. |
| Kerem 2014 | Intervention for stop codon mutations. |
| KONNECTION 2013 | Trial of cross‐over design. |
| McCarty 2002 | Intervention to correct molecular defect, not a potentiator. |
| McGarry 2015 | Trial of cross‐over design. |
| NCT01549314 | Prospective cohort study design. |
| NCT01685801 | Trial of cross‐over design. |
| NCT01784419 | Trial of cross‐over design. |
| NCT01863238 | Prospective cohort study design. |
| NCT01946412 | Single‐arm interventional study. |
| NCT02039986 | Prospective cohort study design. |
| NCT02141464 | Prospective cohort study design. |
| NCT02310789 | Single‐arm interventional study. Enrolled healthy participants only. |
| NCT02311140 | Prospective cohort study design. |
| NCT02323100 | Intervention was glycerol phenylbutyrate corrector therapy, not a potentiator. |
| NCT02443688 | Intervention was with anti‐inflammatory drug rather than CFTR potentiator. |
| NCT02445053 | Prospective cohort study design. |
| NCT02690519 | Single‐arm interventional trial design. |
| NCT02707562 | Single‐arm interventional trial design. |
| NCT02709109 | Trial of cross‐over design. |
| NCT02718495 | Intervention was with a CFTR amplifier versus placebo. |
| NCT02722057 | Cohort study design. |
| NCT02724527 | Compared another mutation‐specific therapy (corrector) with placebo. |
| NCT02742519 | Trial of cross‐over design. |
| NCT02759562 | Intervention was with a monoclonal antibody rather than CFTR potentiator. |
| NCT02934698 | Single‐arm interventional study. |
| NCT03068312 | Trial of cross‐over design. |
| NCT03256799 | Single‐arm interventional study. Intervention was not CFTR potentiator. |
| NCT03256968 | Single‐arm interventional study. Intervention was not CFTR potentiator. |
| NCT03258424 | Intervention was with a CFTR amplifier versus placebo. |
| NCT03277196 | Non‐randomised, intervention compared to observational arm, not placebo. |
| NCT03390985 | Prospective cohort study design. |
| NCT03474042 | Intervention was with CFTR corrector versus placebo. |
| NCT03652090 | Cohort study design. Intervention was a cell sampling procedure. |
| PERSIST 2014 | Single‐arm interventional study. |
| Pradal 2002 | Intervention for stop codon mutations. |
| RIO‐CF 2017 | Intervention was a soluble guanylase cyclase stimulator, not a potentiator. |
| Romano 2000 | Intervention for stop codon mutations. |
| Rubenstein 1998 | Intervention to correct molecular defect, not a potentiator. |
| Rubenstein 2006 | Intervention to correct molecular defect, not a potentiator. |
| Seliger 2015 | Observational trial. |
| Sermet‐Gaudelus 2010 | Intervention for stop codon mutations. |
| TOPIC 2018 | Participants with CF were excluded. |
| Wilschanski 2003 | Intervention for stop codon mutations. |
| Wilschanski 2008 | Intervention for stop codon mutations. |
| Zeitlin 2002 | Intervention to correct molecular defect, not a potentiator. |
CFTR: cystic fibrosis transmembrane regulator
Characteristics of studies awaiting assessment [ordered by study ID]
Kazani 2016.
| Methods | RCT, double‐blind. Phase 2. Parallel design. Duration: 2 weeks (with follow‐up to Day 42). |
| Participants |
Eligibility criteria (Part 3) Confirmed diagnosis of CF. Heterozygous with one allele represented as any CFTR mutation and the other allele must represent a class III, IV, V, VI CFTR mutation. Aged over 18 years. BMI: 15 ‐ 35 kg/m². FEV1 40% ‐ 100% . Oxygen saturation > 90% on room air. 2016 abstract describes an interim analysis of 40 participants who were enrolled into Part 3 of the trial. According to the clinicaltrials.gov record, 49 participants with CF with a class III, IV, V, or VI mutation on one allele and any other CFTR mutation on the other allele were randomised and all completed the trial. Randomised: overall n = 49; intervention 1 n = 6; intervention 2 (multiple mutations) n = 12, intervention 2 (homozygous for F508del) n = 19; control group n = 12. Age, mean (SD): intervention 1 ‐ 39.3 (5.47) years; intervention 2 (multiple mutations) ‐ 32.7 (13.77) years, intervention 2 (homozygous for F508del) ‐ 27 (5.44) years; control ‐ 27.9 (6.37) years. Gender split: overall ‐ 30 males, 19 females; intervention 1 ‐ 5 males, 1 female; intervention 2 (multiple mutations) – 7 males, 5 females, intervention 2 (homozygous for F508del) ‐ 10 males, 9 females; control ‐ 8 males, 4 females. |
| Interventions | Intervention 1 (n = 4): QBW251 150 mg twice daily (oral capsule). Intervention 2 (n = 12): QBW251 450 mg twice daily (oral capsule). Control (n = 10): placebo (oral capsule). |
| Outcomes |
Primary outcomes 1. Change in LCI 2. Adverse events Secondary outcomes 1. Change in FEV1 2. Change in CFQR reported outcomes 3. Change in sweat chloride |
| Notes | Only part 3 of trial which includes people with CF is eligible to be included in review. Parts 1 and 2 enrolled healthy participants. The trial has been terminated, but some results are posted on clinicaltrials.gov. Sponsor is Novartis. |
Uttamsingh 2016.
| Methods | RCT, double‐blind. Phase 1. Parallel design. Duration: 7 days. |
| Participants | 40 participants (each cohort enrolled 10 new participants; 8 participants received active drug and 2 received placebo (4:1 randomisation). |
| Interventions | Intervention 1: CTP‐656 75 mg once daily. Intervention 2: CTP‐656 150 mg once daily. Intervention 3: CTP‐656 225 mg once daily. Control: placebo. All participants were dosed under fed conditions; a high‐fat breakfast was provided on Day 1 through Day 7 approximately 30 minutes prior to dosing. Dose escalation was initiated only after safety and tolerability were found to support proceeding to the higher dose. |
| Outcomes | PK blood samples were collected at several time points post‐dose on Day 1 and Day 7, and at 12 and 24 hours post‐dose on Day 2 through Day 6. The plasma concentrations of CTP‐656 and its metabolites, D‐M1 and D‐M6 were analysed by LC‐MS/MS. The steady‐state PK profiles for CTP‐656, D‐M1 and D‐M6 were evaluated for the 3 dose cohorts. |
| Notes | The author was approached for further information. Drug initially developed by Concert Pharmaceuticals Inc. before being sold to Vertex for further development in 2017 and being re‐named VX‐561. |
BMI: body mass index CF: cystic fibrosis CFQ‐R: cystic fibrosis questionnaire‐revised CFTR: cystic fibrosis transmembrane regulator FEV1: forced expiratory volume at one second LCI: lung clearance index LC‐MS/MS: liquid chromatography with mass spectrometry PK: pharmacokinetic RCT: randomised controlled trial
Differences between protocol and review
Full review 2016
We have made several changes since the protocol stage.
The title has been changed from 'Mutation‐specific therapies that potentiate cystic fibrosis transmembrane conductance regulator (CFTR) function in cystic fibrosis' to the current title of this review.
We decided to only include trials that compare a CFTR potentiator to a placebo or other intervention. Trials comparing a CFTR potentiator alongside another CFTR modulator will be published in a different review.
For the primary outcome of survival, in a post hoc change, mortality data were also considered.
We have decided to add cost of treatment as a secondary outcome of interest.
In a post hoc change, we have edited the methods section that 99% confidence intervals will be used to analyse separate adverse events. This is the most appropriate statistical approach for considering adverse events individually.
We have added a Summary of Findings table based on the most clinically important outcomes, in line with the outcomes to be reported in the ongoing review of CFTR correctors (Southern 2018).
Update 2018
In a post hoc change, corrector and potentiator combination therapy trials have not been referenced in the review, meaning the 39 references to combination therapy identified in the 2018 searches have not been listed as excluded studies
Contributions of authors
| Roles and responsibilities | |
| TASK | WHO WILL UNDERTAKE THE TASK? |
| Protocol stage: draft the protocol | IS with comments from all |
| Review stage: select which trials to include (2 + 1 arbiter) | IS, SP, MS |
| Review stage: extract data from trials (at least 2 people) | IS, SP, CE |
| Review stage: enter data into RevMan | SP, KD |
| Review stage: carry out the analysis | SP, KD |
| Review stage: interpret the analysis | IS, SP, KD |
| Review stage: draft the final review | SP with comments from all |
| Update stage: update the review | MS, IS, SP, AK |
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Mica Skilton declares no potential conflict of interest.
Ian Sinha declares no potential conflict of interest.
Sanjay Patel declares no potential conflict of interest.
Kevin Southern declares no potential conflict of interest.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Accurso 2010 {published data only}
- Accurso F, Rowe SM, Durie PR, Konstan MW, Dunitz J, Hornick D, et al. Improvement in sweat chloride concentration by the CFTR potentiator VX‐770 in subjects with cystic fibrosis and the G551D‐CFTR mutation. Pediatric Pulmonology 2009;44(S32):296. [Abstract no.: 240; CFGD Register: BD165c; MEDLINE: ] [Google Scholar]
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation. New England Journal of Medicine 2010;363(21):1991‐2003. [CFGD Register: BD165i; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Online supplementary appendix to "Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation" [online]. New England Journal of Medicine 2010;363(21):1‐33 online. [CFGD Register: BD165j; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Online supplementary material (disclosure forms) to "Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation" [online]. New England Journal of Medicine 2010;363(21):1991‐2003 online. [CFGD Register: BD165k; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Online supplementary material (protocol) to "Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation" [online]. New England Journal of Medicine 2010;363(21):1‐121 online. [CFGD Register: BD165l; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso FJ, Rowe SM, Durie PR, Konstan MW, Dunitz J, Hornick DB, et al. Final results of a 14‐and 28‐day study of VX‐770 in subjects with CF. Journal of Cystic Fibrosis 2009;8 Suppl 2:S25. [Abstract no.: 97; CFGD Register: BD165d; MEDLINE: ] [Google Scholar]
- Accurso FJ, Rowe SM, Durie PR, Konstan MW, Dunitz J, Hornick DB, et al. Interim results of phase 2a study of VX‐770 to evaluate safety, pharmacokinetics, and biomarkers of CFTR activity in cystic fibrosis subjects with G551D. Pediatric Pulmonology 2008;43(S1):295. [Abstract no.: 267; CFGD Register: BD165b; MEDLINE: ] [Google Scholar]
- Accurso FJ, Goor F, Zha J, Stone AJ, Dong Q, Ordonez CL, et al. Online Data Supplement to "Sweat chloride as a biomarker of CFTR activity: Proof of concept and ivacaftor clinical trial data". Journal of Cystic Fibrosis 2014;13(2):139‐147 online. [CENTRAL: 1000304; CFGD Register: BD165r; CRS: 5500131000000025] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso FJ, Goor F, Zha J, Stone AJ, Dong Q, Ordonez CL, et al. Sweat chloride as a biomarker of CFTR activity: Proof of concept and ivacaftor clinical trial data. Journal of Cystic Fibrosis 2014;13(2):139‐47. [CENTRAL: 978403; CFGD Register: BD165q; CRS: 5500050000000097; EMBASE: 2014086288] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniu SA. Cystic fibrosis transmembrane regulator potentiators as promising cystic fibrosis therapies. Evaluation of: Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation. New England Journal of Medicine 2010;363(21):1991‐2003. Expert Opinion on Investigational Drugs 2011;20(3):423‐5. [CFGD Register: BD165m; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Boyle M, Clancy JP, Rowe SM, Durie P, Dunitz J, Konstan MW, et al. Effect of VX‐770, a CFTR potentiator, on spirometry and QOL assessment in subjects with CF and the G551D‐CFTR mutation. Pediatric Pulmonology 2009;44(S32):287. [Abstract no.: 217; CFGD Register: BD165f] [Google Scholar]
- Clancy JP, Rowe SM, Durie P, Freedman S, Dong Q, Ordonez C, et al. NPD evaluation of ion transport in G551D CF patients treated with a CFTR potentiator. Pediatric Pulmonology 2009;44(S32):288. [Abstract no.: 222; CFGD Register: BD165e; MEDLINE: ] [Google Scholar]
- Donaldson S, Accurso F, Rowe S, Clancy J, Boyle M, Dunitz J, et al. Improved CFTR and lung function with VX‐770, a novel investigational potentiator of CFTR, in subjects with the G551D‐CFTR mutation. European Respiratory Society Annual Congress; 2010 Sep 18‐22; Barcelona, Spain. 2010:34s. [Abstract no.: 359; CENTRAL: 777402; CFGD Register: BD165s; CRS: 5500050000000276]
- Konstan MW, Accurso FJ, Boyle MP, Clancy JP, Ordonez CL, Zha Jetal. Relationship between Pulmonary outcomes, biomarkers Of CF Disease, And Serum drug Levels in Subjects with The G551D‐CFTR Mutation Treated With VX‐770, An Investigational Oral Potentiator Of CFTR. American Journal of Respiratory and Critical Care Medicine 2010;181(Meeting Abstracts):A2336. [CENTRAL: 758845; CFGD Register: BD165t; CRS: 5500050000000280] [Google Scholar]
- Rowe SM, Clancy JP, Boyle M, Goor F, Ordonez C, Dong Q, et al. Parallel effects of VX‐770 on transepithelial potential difference in vitro and in vivo. Journal of Cystic Fibrosis 2010;9 Suppl 1:S20. [Abstract no.: 74; CFGD Register: BD165h; MEDLINE: ] [Google Scholar]
- Rowe SM, Liu B, Hill A, Hathorne H, Cohen M, Beamer JR, et al. Optimizing nasal potential difference analysis for CFTR modulator development: assessment of ivacaftor in CF subjects with the G551D‐CFTR mutation. PloS One 2013;8(7):e66955. [CFGD Register: BD165o; ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe SM, Goor F, Clancy JJP, Durie PR, Konstan MW, Dunitz J, et al. Corresponding effects of VW‐770 on NPD in vivo & human bronchial epithelial (HBE) cells in vitro. Pediatric Pulmonology 2010;45 Suppl 33:319. [Abstract no.: 281; CFGD Register: BD165g] [Google Scholar]
- Goor F, Hadida S, Negulescu P, Clancy JP, Accurso FJ, Ashlock MA, et al. Relationship of VX‐770 activity on CFTR function in a cell culture system to the activity observed in a clinical study of VX‐770. Pediatric Pulmonology 2008;43(S31):314.. [Abstract no.: 319; CFGD Register: BD165a; MEDLINE: ] [Google Scholar]
DISCOVER 2011 {published data only}
- Flume PA, Borowitz D, Liou T, Li H, Yen K, Ordonez C, et al. VX‐770 in subjects with CF and homozygous for the F508del‐CFTR mutation. Pediatric Pulmonology 2011;46 Suppl 34:284. [Abstract no.: 206; CFGD Register: BD168b; MEDLINE: ] [Google Scholar]
- Flume PA, Borowitz DS, Liou TG, Li H, Yen K, Ordonez CL, et al. VX‐770 in subjects with cystic fibrosis who are homozygous for the F508del‐CFTR mutation. Journal of Cystic Fibrosis 2011;10 Suppl 1:S16. [Abstract no.: 61; CFGD Register: BD168a; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordonez CL, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del‐CFTR mutation. Chest 2012;142(3):718‐24. [CFGD Register: BD168c; ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordonez CL, et al. Ivacaftor in subjects with cystic fibrosis who are homozygous for the homozygous for the F508del‐CFTR mutation. Chest 2012;142(3):718‐24 online. [CFGD Register: BD168d] [DOI] [PMC free article] [PubMed] [Google Scholar]
ENVISION 2013 {published data only}
- Aherns R, Rodriguez S, Yen K, Davies JC. VX‐770 in subjects 6 to 11 years with cystic fibrosis and the G551D‐CFTR mutation. Pediatric Pulmonology 2011;46(S34):283. [Abstract no.: 203; CFGD Register: BD171a; MEDLINE: ] [Google Scholar]
- Borowitz D, Lubarsky B, Wilschanski M, Munck A, Gelfond D, Bodewes F, et al. Nutritional status improved in cystic fibrosis patients with the G551D mutation. Digestive Diseases and Sciences 2016;61(1):198‐207. [CFGD Register: BD170u // BD171p] [DOI] [PubMed] [Google Scholar]
- Borowitz D, Ramsey B, Dong Q, Yen K, Elborn JS. Measures of nutritional status in two Phase 3 trials of ivacaftor in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S13. [Abstract no.: WS6.3; CFGD Register: BD171c//BD170d; ] [Google Scholar]
- Borowitz D, Ramsey B, Rodriguez S, Yen K, Elborn JS. Nutritional status measures among persons with CF carrying the G551D‐CFTR mutation who received ivacaftor or placebo in phase 3 clinical trials. Pediatric Pulmonology 2012;47(S35):298. [Abstract no.: 214; CFGD Register: BD171f//BD170i; ] [Google Scholar]
- Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. American Journal of Respiratory and Critical Care Medicine 2013;187(11):1219‐25. [CFGD Register: BD171g; ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Online Supplement to "Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation" [online]. American Journal of Respiratory and Critical Care Medicine 2013;187(11):1219‐1225 online. [CFGD Register: BD171h; ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KC, Li H, Yen K, Ahrens R, on behalf of theVX08‐770‐103SG. Ivacaftor in subjects 6 to 11 years of age with cystic fibrosis and the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S13. [Abstract no.: WS6.5; CFGD Register: BD171b; ] [Google Scholar]
- Elborn JS, Rodriguez S, Lubarsky B, Gilmartin G, Bell S. Effect of ivacaftor in patients with cystic fibrosis and the G551D‐CFTR mutation who have baseline FEV >90% of predicted. Pediatric Pulmonology 2013;48 Suppl 36:298. [Abstract no.: 257; CENTRAL: 921689; CFGD Register: BD171k // BD188e // BD170m; CRS: 5500125000000401] [Google Scholar]
- Elborn S, Plant B, Konstan M, Aherns R, Rodriguez S, Munck A, et al. Lung function, weight, and sweat chloride responses in patients with cystic fibrosis and the G551D‐CFTR mutation treated with ivacaftor: A secondary analysis. European Respiratory Journal 2013;42:1073s. [Abstract no.: 5059; CENTRAL: 1099910; CFGD Register: BD170o // BD171n ; CRS: 5500050000000278; EMBASE: 71843034] [Google Scholar]
- Flume P, Wainwright CE, Tullis E, Rodriguez S, Davies J, Wagener J. Pulmonary exacerbations in CF patients with the G551D‐CFTR mutation treated with ivacaftor. Journal of Cystic Fibrosis 2013;12 Suppl 1:S63. [Abstract no.: 58; CFGD Register: BD171j//BD170j; ] [DOI] [PubMed] [Google Scholar]
- Konstan MW, Plant BJ, Elborn JS, Rodriguez S, Munck A, Ahrens R, et al. Efficacy response in CF patients treated with ivacaftor: post‐hoc analysis. Pediatric Pulmonology 2015;50(5):447‐55. [CFGD Register: BD170v // BD171r] [DOI] [PubMed] [Google Scholar]
- McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long‐term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp‐CFTR mutation: a phase 3, open‐label extension study (PERSIST). The Lancet. Respiratory Medicine 2014;2:902‐10. [CENTRAL: 1017318; CFGD Register: BD171l // BD170n; CRS: 5500131000000246; JID:: 101605555; PUBMED: 25311995] [DOI] [PubMed] [Google Scholar]
- Plant BJ, Konstan M, Aherns R, Rodriguez S, Munck A, Elborn JS, et al. Lung function, weight, and sweat chloride responses in patients with cystic fibrosis and the G551D‐CFTR mutation treated with ivacaftor: A secondary analysis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S62. [Abstract no.: 53; CFGD Register: BD171i//BD170k; ] [Google Scholar]
- Quittner A, Ramsey B, Rodriguez S, Yen K, Elborn JS. Patient‐reported outcomes in phase 3 trials of ivacaftor in subjects with CF who have the G551D‐CFTR mutation. Pediatric Pulmonology 2012;47 Suppl 35:297. [Abstract no.: 212; CFGD Register: BD171e//BD170g; ] [Google Scholar]
- Quittner AL, Ramsey B, Dong Q, Yen K, Elborn JS. Patient‐reported outcomes in Phase 3 trials of ivacaftor in subjects with CF who have the G551D‐CFTR mutation [abstract]. Journal of Cystic Fibrosis 2012;11 Suppl 1:S67, Abstract no: 43. [CFGD Register: BD171d//BD170e; ] [Google Scholar]
- Stalvey MS, Niknian M, Higgins M, Tarn V, Heltshe SL, Rowe SM. Ivacaftor improves linear growth in children with cystic fibrosis (CF) and a G551D‐CTFR mutation: data from the ENVISION study. Journal of Cystic Fibrosis 2016;15 Suppl:S101. [Abstract no.: 197; CFGD Register: BD171o] [Google Scholar]
- Stalvey MS, Niknian M, Higgins M, Tarn VE, Heltshe SL, Rowe SM. Ivacaftor improves linear growth in G551D cystic fibrosis children: results of a multicenter, placebo‐controlled study. Pediatric Pulmonology 2015;50 Suppl 41:402. [Abstract no.: 554; CENTRAL: 1092205; CFGD Register: BD171m; CRS: 5500135000001394] [Google Scholar]
- Stalvey MS, Pace J, Niknian M, Higgins MN, Tarn V, Davis J, et al. Growth in prepubertal children with cystic fibrosis treated with ivacaftor. Pediatrics 2017;139(2):e20162522. [CFGD Register: BD171q] [DOI] [PMC free article] [PubMed] [Google Scholar]
KONDUCT 2015 {published data only}
- Moss R. Study of ivacaftor in subjects with cystic fibrosis who have the R117H‐CFTR mutation (KONDUCT). Www.clinicaltrials.gov (www.clinicaltrials.gov) (accessed 22 Oct 2014) 2014. [CENTRAL: 1012721; CFGD Register: BD204b; CRS: 5500131000000199; NCT01614457]
- Moss R, Flume PA, Elborn J, Cooke J, Rowe SM, McColley SA, et al. Effects of ivacaftor in CF patients with R117H‐CFTR. Pediatric Pulmonology 2014;49 Suppl 38:221. [Abstract no.: 17; CENTRAL: 1012722; CFGD Register: BD204c; CRS: 5500131000000201] [Google Scholar]
- Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His‐CFTR mutation: a double‐blind, randomised controlled trial. The Lancet. Respiratory Medicine 2015;3(7):524‐33. [CFGD Register: BD204d] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, et al. Ivacaftor treatment in patients with cystic fibrosis who have an R117H‐CFTR mutation, the KONDUCT study. Journal of Cystic Fibrosis 2014;13 Suppl 2:S44. [Abstract no.: WS23.6; CENTRAL: 996577; CFGD Register: BD204a; CRS: 5500129000000012] [Google Scholar]
STRIVE 2011 {published data only}
- Borowitz D, Lubarsky B, Wilschanski M, Munck A, Gelfond D, Bodewes F, et al. Nutritional status improved in cystic fibrosis patients with the G551D mutation. Digestive Diseases and Sciences 2016;61(1):198‐207. [CFGD Register: BD170u // BD171p] [DOI] [PubMed] [Google Scholar]
- Borowitz D, Ramsey B, Dong Q, Yen K, Elborn JS. Measures of nutritional status in two Phase 3 trials of ivacaftor in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S13. [Abstract no.: WS6.3; CFGD Register: BD170d//BD171c; ] [Google Scholar]
- Borowitz D, Ramsey B, Rodriguez S, Yen K, Elborn JS. Nutritional status measures among persons with CF carrying the G551D‐CFTR mutation who received ivacaftor or placebo in phase 3 clinical trials. Pediatric Pulmonology 2012;47 Suppl 35:298. [Abstract no.: 214; CFGD Register: BD170i//BD171f; ] [Google Scholar]
- Elborn JS, Rodriguez S, Lubarsky B, Gilmartin G, Bell S. Effect of ivacaftor in patients with cystic fibrosis and the G551D‐CFTR mutation who have baseline FEV >90% of predicted. Pediatric Pulmonology 2013;48 Suppl 36:298. [Abstract no.: 257; CENTRAL: 921689; CFGD Register: BD170m // BD171k // BD188e ; CRS: 5500125000000401] [Google Scholar]
- Elborn S, Plant B, Konstan M, Aherns R, Rodriguez S, Munck A, et al. Lung function, weight, and sweat chloride responses in patients with cystic fibrosis and the G551D‐CFTR mutation treated with ivacaftor: A secondary analysis. European Respiratory Journal 2013;42:1073s. [Abstract no.: 5059; CENTRAL: 1099910; CFGD Register: BD170o // BD171n ; CRS: 5500050000000278; EMBASE: 71843034] [Google Scholar]
- Flume P, Wainwright CE, Tullis E, Rodriguez S, Davies J, Wagener J. Pulmonary exacerbations in CF patients with the G551D‐CFTR mutation treated with ivacaftor. Journal of Cystic Fibrosis 2013;12 Suppl 1:S63. [Abstract no.: 58; CFGD Register: BD170j//BD171j; ] [DOI] [PubMed] [Google Scholar]
- Flume PA, Wainwright CE, Elizabeth Tullis D, Rodriguez S, Niknian M, Higgins M, et al. Recovery of lung function following a pulmonary exacerbation in patients with cystic fibrosis and the G551D‐CFTR mutation treated with ivacaftor. Journal of Cystic Fibrosis 2018;17(1):83‐8. [CFGD Register: BD170w] [DOI] [PubMed] [Google Scholar]
- Griese M, Ramsey B, Rodriguez S, Yen K, Elborn JS, on behalf of the VX08‐770‐102 Study Group. Pulmonary exacerbations in a Phase 3 trial of ivacaftor in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S67. [Abstract no.: 44; CFGD Register: BD170f; ] [Google Scholar]
- Konstan MW, Plant BJ, Elborn JS, Rodriguez S, Munck A, Ahrens R, et al. Efficacy response in CF patients treated with ivacaftor: post‐hoc analysis. Pediatric Pulmonology 2015;50(5):447‐55. [CFGD Register: BD170v // BD171r] [DOI] [PubMed] [Google Scholar]
- McKone E, Li H, Yen K, Davies JC, on behalf of the VX08‐770‐105 study group. Long‐term safety and efficacy of ivacaftor in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S13. [Abstract no.: WS6.4; CFGD Register: BD170h; ] [Google Scholar]
- McKone E, Sawicki G, Millar S, Pasta D, Rubin J, Konstan M, et al. Improved rate of decline in percent predicted FEV1 (ppfev1) is not associated with acute improvement in ppfev1 in patients with cystic fibrosis (CF) treated with ivacaftor. Journal of Cystic Fibrosis 2016;15 Suppl 1:S42. [Abstract no.: ePS03.4; CFGD Register: BD170s] [Google Scholar]
- McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long‐term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp‐CFTR mutation: a phase 3, open‐label extension study (PERSIST). The Lancet. Respiratory Medicine 2014;2:902‐10. [CENTRAL: 1017318; CFGD Register: BD170n // BD171l; CRS: 5500131000000246; JID:: 101605555; PUBMED: 25311995] [DOI] [PubMed] [Google Scholar]
- Plant BJ, Konstan M, Aherns R, Rodriguez S, Munck A, Elborn JS, et al. Lung function, weight, and sweat chloride responses in patients with cystic fibrosis and the G551D‐CFTR mutation treated with ivacaftor: A secondary analysis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S62. [Abstract no.: 53; CFGD Register: BD170k//BD171i; ] [Google Scholar]
- Quittner A, Ramsey B, Rodriguez S, Yen K, Elborn JS. Patient‐reported outcomes in phase 3 trials of ivacaftor in subjects with CF who have the G551D‐CFTR mutation. Pediatric Pulmonology 2012;47 Suppl 35:297. [Abstract no.: 212; CFGD Register: BD170g//BD171e; ] [Google Scholar]
- Quittner A, Suthoff E, Rendas‐Baum R, Bayliss MS, Sermet‐Gaudelus I, Castiglione B, et al. Effect of ivacaftor treatment in patients with cystic fibrosis and the G551D‐CFTR mutation: patient‐reported outcomes in the STRIVE randomized, controlled trial. Health and Quality of Life Outcomes 2015;13:93. [CENTRAL: 1108956; CFGD Register: BD170r; CRS: 5500050000000282; PUBMED: 26135562] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quittner AL, Ramsey B, Dong Q, Yen K, Elborn JS. Patient‐reported outcomes in Phase 3 trials of ivacaftor in subjects with CF who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S67. [Abstract no.: 43; CFGD Register: BD170e//BD171d; ] [Google Scholar]
- Ramsey B, Dong Q, Yen K, Elborn J. Efficacy and safety of VX‐770 in subjects with cystic fibrosis and the G551D‐CFTR mutation. Pediatric Pulmonology 2011;46(S34):286. [Abstract no.: 211; CFGD Register: BD170a; MEDLINE: ] [Google Scholar]
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. New England Journal of Medicine 2011;365(18):1663‐72. [CFGD Register: BD170b; ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. Online Supplementary Appendix to "A CFTR potentiator in patients with cystic fibrosis and the G551D mutation" [online]. New England Journal of Medicine 2011;365(18):1663‐1672 online. [CFGD Register: BD170c; ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seliger VI, Accurso FJ, Konstan MW, Dong Q, Lubarsky B, Mueller P. Effect of ivacaftor on circulating inflammatory indices in CF patients with the G551D‐CFTR mutation. Pediatric Pulmonology 2013;48 Suppl 36:298. [Abstract no.: 259; CENTRAL: 921687; CFGD Register: BD170l; CRS: 5500125000000399] [Google Scholar]
- Solem CT, Vera‐Llonch M, Liu S, Botteman M, Castiglione B. Impact of pulmonary exacerbations and lung function on generic health‐related quality of life in patients with cystic fibrosis. Health and Quality of Life Outcomes 2016;14:63. [CFGD Register: BD170t] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solem CT, Vera‐Llonch M, Liu S, Botteman M, Lin FJ, Castiglione B. Impact of pulmonary exacerbations on EQ‐5D measures in patients with cystic fibrosis. Value in Health 2014;17(7):A535. [Abstract no.: PSY77; CENTRAL: 1055060; CFGD Register: BD170q; CRS: 5500050000000277; EMBASE: 71673604] [DOI] [PubMed] [Google Scholar]
- Solem CT, Vera‐Llonch M, Liu S, Botteman MF, Lasch K, Rodriguez S, et al. Responsiveness of the EQ‐5D index and visual analog scale to changes in lung function in patients with cystic fibrosis. Value in Health 2014;17(3):A178. [Abstract no.: PRS52; CENTRAL: 1061016; CFGD Register: BD170p; CRS: 5500050000000281; EMBASE: 71488485] [Google Scholar]
References to studies excluded from this review
Accurso 2013 {published data only}
- Accurso FJ, Ratjen F, Altes T, Lubarsky B, Dong Q, Kang L, et al. Effect of withdrawal of ivacaftor therapy on CFTR channel activity and lung function in patients with cystic fibrosis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S62. [Abstract no.: 56; CFGD Register: BD189//BD188c; ] [Google Scholar]
ALBATROSS 2017 {published data only}
- Bell S, Boeck K, Drevinek P, Plant B, Barry P, Elborn S, et al. GLPG2222 in subjects with cystic fibrosis and the F508del/Class III mutation on stable treatment with ivacaftor: results from a phase II study (ALBATROSS). Journal of cystic fibrosis 2018;17(Suppl 3):S2. [CFGD Register: BD247a; clinicaltrials.gov: NCT03045523] [Google Scholar]
- Bell SC, Boeck K, Drevinek P, Plant BJ, Elborn J, Kock H, et al. Results from a phase ii study ‐ albatross ‐ evaluation of glpg2222 in subjects with cf and the f508del/class iii mutation on stable treatment with ivacaftor. Pediatric Pulmonology 2018;53(S2):249. [CFGD Register: BD247b; clinicaltrials.gov: NCT03045523] [Google Scholar]
Altes 2011 {published data only}
- Altes T, Johnson M, Higgins M, Fidler M, Botfield M, Mugler III JP, et al. The effect of ivacaftor treatment on lung ventilation defects, as measured by hyperpolarized helium‐3 MRI, on patients with cystic fibrosis and a G551D‐CFTR mutation. Journal of Cystic Fibrosis 2014;13 Suppl 2:S6. [Abstract no.: WS3.2; CENTRAL: 1000056; CFGD Register: BD172d; CRS: 5500131000000009] [Google Scholar]
- Altes T, Johnson M, Higgins M, Fidler M, Botfield M, Tustison N, Leiva‐Salinas C. Use of hyperpolarized helium‐3 MRI to assess response to ivacaftor treatment in patients with cystic fibrosis. Journal of Cystic Fibrosis 2017;16:267‐274. [DOI] [PubMed] [Google Scholar]
- Altes T, Johnson MA, Miller GW, Mugler JP, Flors L, Mata J, et al. Hyperpolarized Gas MRI of ivacaftor therapy in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S67. [Abstract no.: 46; CFGD Register: BD172b; ] [Google Scholar]
- Altes T, Johnson MA, Miller GW, Mugler JP, Flors L, Mata J, et al. Hyperpolarized gas MRI of ivacaftor therapy in persons with cystic fibrosis and the G551D‐CFTR mutation. Pediatric Pulmonology 2012;47 Suppl 35:291. [Abstract no.: 196; CFGD Register: BD172c; ] [Google Scholar]
- Altes T, Johnson MA, Miller GW, Mugler JP, Flors L, Mata J, et al. Hyperpolarized helium‐3 magnetic resonance imaging of CFTR potentiator therapy in subjects with cystic fibrosis and the G551D mutation. Pediatric Pulmonology 2011;46(S34):284. [Abstract no.: 205; CFGD Register: BD172a; MEDLINE: ] [Google Scholar]
ARRIVAL 2018 {published data only}
- Rosenfeld M, Wainwright CE, Higgins M, Wang LT, McKee C, Campbell D, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single‐arm study. The Lancet. Respiratory Medicine 2018;6(7):545‐53. [PUBMED: 29886024] [DOI] [PMC free article] [PubMed] [Google Scholar]
Berkers 2017 {published data only}
- Berkers G, Mourik P, Dekkers JF, Kruisselbrink E, Vonk AM, Heida‐Michel S, et al. Correlation between individual clinical responses and forskolin‐induced swelling of paired intestinal organoids upon CFTR modulator treatment. Pediatric Pulmonology 2017;52 Suppl 47:295. [CFGD Register: BD242] [Google Scholar]
Chadwick 1998 {published data only}
- Chadwick S, Browning JE, Stern M, Cheng SH, Gruenert DC, Geddes DM, et al. Nasal application of glycerol in DF508 cystic fibrosis patients. Pediatric Pulmonology 1998;26 Suppl 17:278. [CFGD Register: BD147] [Google Scholar]
Clancy 2012 {published data only}
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX‐809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del‐CFTR mutation. Thorax 2012;67(1):12‐8. [CFGD Register: BD166c; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JP, Rowe SM, Accurso FJ, Ballmann M, Boyle MP, DeBoeck C, et al. A phase II, randomized, placebo‐controlled, clinical trial of four doses of VX‐809 in CF patients homozygous for the F508del CFTR mutation. Pediatric Pulmonology 2010;45 Suppl 33:298. [Abstract no.: 224; CFGD Register: BD166b; MEDLINE: ] [Google Scholar]
- Clancy JP, Rowe SM, Liu B, Hathorne H, Dong Q, Wisseh S, et al. Variability of nasal potential difference measurements in clinical testing of CFTR modulators [abstract]. Pediatric Pulmonology 2011;46 Suppl 34:283, Abstract no: 202. [CFGD Register: BD165n, BD166d; MEDLINE: ] [Google Scholar]
- Clancy JP, Spencer‐Green G, for the VX‐809‐101 Study Group. Clinical evaluation of VX‐809, a novel investigational oral F508del‐CFTR corrector, in subjects with cystic fibrosis homozygous for the F508del‐CFTR mutation. Journal of Cystic Fibrosis 2010;9 Suppl 1:S20. [Abstract no.: 73; CFGD Register: BD166a; MEDLINE: ] [Google Scholar]
Davies 2012 {published data only}
- Accurso FJ, Ratjen F, Altes T, Lubarsky B, Dong Q, Kang L, et al. Effect of withdrawal of ivacaftor therapy on CFTR channel activity and lung function in patients with cystic fibrosis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S62. [Abstract no.: 56; CFGD Register: BD189//BD190b//BD188c; ] [Google Scholar]
- Davies J, Sheridan H, Bell N, Cunningham S, Davis SD, Elborn JS, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D‐CFTR mutation and preserved spirometry: a randomised controlled trial. The Lancet. Respiratory Medicine 2013;1(8):630‐8. [CFGD Register: BD188f] [DOI] [PubMed] [Google Scholar]
- Davies JC, Sheridan H, Lee P, Song T, Stone A, Ratjen F. Lung clearance index to evaluate the effect of ivacaftor on lung function in subjects with CF who have the G551D‐CFTR mutation and mild lung disease. Pediatric Pulmonology 2012;47 Suppl 35:311. [Abstract no.: 249; CFGD Register: BD188b; ] [Google Scholar]
- Davies JC, Sheridan H, Lee PS, Song T, Stone A, Ratjen F, et al. Effect of ivacaftor on lung function in subjects with CF who have the G551D‐CFTR mutation and mild lung disease: a comparison of lung clearance index (LCI) vs. spirometry. Journal of Cystic Fibrosis 2012;11 Suppl 1:S15. [Abstract no.: WS7.6; CFGD Register: BD188a; ] [Google Scholar]
- Elborn JS, Rodriguez S, Lubarsky B, Gilmartin G, Bell S. Effect of ivacaftor in patients with cystic fibrosis and the G551D‐CFTR mutation who have baseline FEV >90% of predicted. Pediatric Pulmonology 2013;48 Suppl 36:298. [Abstract no.: 257; CENTRAL: 921689; CFGD Register: BD188e // BD170m // BD171k; CRS: 5500125000000401] [Google Scholar]
- Ratjen F, Sheridan H, Lee P, Song T, Stone A, Davies JC. Lung clearance index as an outcome measure in cystic fibrosis clinical trials. Pediatric Pulmonology 2011;46 Suppl 34:282. [Abstract no.: 201; CENTRAL: 921614; CFGD Register: BD188d; CRS: 5500125000000389] [Google Scholar]
- Ratjen FA, Sheridan H, Lee P‐S, Song T, Stone A, Davies J. Lung clearance index as an endpoint in a multicenter randomized control trial of ivacaftor in subjects with cystic fibrosis who have mild lung disease. American Journal of Respiratory and Critical Care Medicine 2012;185:A2819. [CENTRAL: 1107525; CFGD Register: BD188g; CRS: 5500050000000279; EMBASE: 71987408] [Google Scholar]
Davies 2016 {published data only}
- Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2‐5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open‐label, single‐arm study. The Lancet. Respiratory medicine 2016;4(2):107‐15. [PUBMED: 26803277] [DOI] [PMC free article] [PubMed] [Google Scholar]
Edgeworth 2017 {published data only}
- Button BM, Edgeworth D, Finlayson F, Fantidis M, Wilson L, Talbot A, et al. Effect of ivacaftor on wellness, quality of life and cognitive function in adults with cystic fibrosis and G551D mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S18. [Abstract no.: WS09.5; CENTRAL: 1081475; CFGD Register: BD222a; CRS: 5500135000000012] [Google Scholar]
- Button BM, Edgeworth D, Wilson LM, Sayer J, Tierney A, Finlayson F, et al. Ivacaftor improves wellness, quality of life and cognitive function in G551D cystic fibrosis. Pediatric Pulmonology 2015;50 Suppl 41:437. [Abstract no.: 643; CENTRAL: 1092211; CFGD Register: BD222e; CRS: 5500135000001400] [Google Scholar]
- Edgeworth D, Keating D, Ellis M, Button B, Williams E, Clark D, et al. Improvement in exercise duration, lung function and well‐being in G551D‐cystic fibrosis patients: a double‐blind, placebo‐controlled, randomized, cross‐over study with ivacaftor treatment. Clinical Science 2017;131(15):2037‐45. [CFGD Register: BD222f] [DOI] [PubMed] [Google Scholar]
- Edgeworth D, Keating D, Williams E, Clark D, Button B, Tierney A, et al. Exercise improvements in ivacaftor treated G551D cystic fibrosis patients are not solely related to FEV1 and sweat changes. European Respiratory Journal 2015;46 Suppl:PA2047. [CFGD Register: BD222j; DOI: 10.1183/13993003.congress2015.PA2047] [DOI] [Google Scholar]
- Edgeworth D, Keating D, Williams E, Clark D, Button BM, Tierney AC, et al. Ivacaftor improves exercise capacity in patients with G551D CF gene mutations. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2015;14 Suppl 1:S27. [Abstract no.: WS14.1; CENTRAL: 1081478; CFGD Register: BD222b; CRS: 5500135000000015] [Google Scholar]
- Keating D, Edgeworth D, Heretier S, Denise C, Tierney A, Kotsimbos T, et al. Sweat chloride response does not reliably correlate with clinical parameters: a placebo controlled crossover trial of ivacaftor in G551D CF patients. Journal of Cystic Fibrosis 2017;16 Suppl 1:S75. [CFGD Register: BD222g] [Google Scholar]
- Tierney AC, Edgeworth D, Williams E, Finlayson F, Keating D, Clark D, et al. Ivacaftor and its effects on body composition in adults with G551D related cystic fibrosis. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2015;14 Suppl 1:S50. [Abstract no.: ePS05.1; CENTRAL: 1081476; CFGD Register: BD222d; CRS: 5500135000000013] [Google Scholar]
- Williams E, Edgeworth D, Fantidis M, Finlayson F, Button BM, Clark D, et al. Patient reported adherence to ivacaftor. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2015;14 Suppl 1:S46. [Abstract no.: ePS03.4; CENTRAL: 1081477; CFGD Register: BD222c; CRS: 5500135000000014] [Google Scholar]
- Wilson J, Keating D, Clark D, Edgeworth D, Allen‐Graham J, Finlayson F, et al. The effect of ivacaftor CFTR gene‐potentiating therapy on cytokine levels in CF patients with the G551D mutation. Journal of Cystic Fibrosis 2017;16 Suppl 1:S83. [CFGD Register: BD222h] [Google Scholar]
- Wilson J, len‐Graham J, Talbot A, Finlayson F, Clark D, Keating D, et al. Treatment with ivacaftor in CF patients with the G551D mutation is associated with improvement in cognition. Journal of Cystic Fibrosis 2018;17(Suppl 3):S57. [CFGD Register: BD222k] [Google Scholar]
- Wilson JW, Keating D, Clark D, Edgeworth D, Allen‐Graham J, Finlayson F, et al. Ivacaftor CFTR gene‐potentiating therapy reduces inflammatory cytokine levels in CF patients with G551D mutation. Pediatric Pulmonology 2017;52 Suppl 47:236‐7. [CFGD Register: BD222i] [Google Scholar]
EudraCT Number: 2014‐000817‐30 {published data only}
- EudraCT Number: 2014‐000817‐30. Comparing the effect of curcumin and genistein to treatment with Ivacaftor in CF patients with a class III mutation. clinicaltrialsregister.eu/ctr‐search/trial/2014‐000817‐30/NL (first received 22 May 2014).
EudraCT Number: 2016‐001440‐18 {published data only}
- EudraCT Number: 2016‐001440‐18. Pharmacokinetic interactions between ivacaftor and cytochrome P450 3A4 inhibitors in cystic fibrosis patients and healthy controls. clinicaltrialsregister.eu/ctr‐search/trial/2016‐001440‐18/NL (first received 16 January 2018).
EudraCT Number: 2016‐001619‐19 {published data only}
- EudraCT Number: 2016‐001619‐19. Genistein as an add‐on treatment for CF?. clinicaltrialsregister.eu/ctr‐search/trial/2016‐001619‐19/NL (first received 27 March 2016).
EUudraCT Number: 2016‐001785‐29 {published data only}
- EudraCT Number: 2016‐001785‐29. Combined effect of CFTR modifiers and intensive antibiotic treatment. clinicaltrialsregister.eu/ctr‐search/trial/2016‐001785‐29/IE (first received 10 May 2016).
FLAMINGO 2017 {published data only}
- Ent KC, Minic P, Verhulst S, Braeckel E, Flume P, Boas S, et al. GLPG2222 in subjects with cystic fibrosis homozygous for F508del: results from a phase II study (FLAMINGO). Journal of Cystic Fibrosis 2018;17(Suppl 3):S42. [CFGD Register: BD254a] [Google Scholar]
- Ent KC, Minic P, Verhulst S, Braeckel E, Flume P, Boas S, et al. Glpg2222 in CF subjects homozygous for f508del: results from a phase ii study (Flamingo). Pediatric Pulmonology 2018;53(S2):250. [CFGD Register: BD254b] [Google Scholar]
Horsley 2018 {published data only}
- Horsley A, Burr L, Kotsimbos T, Ledson M, Schwarz C, Simmonds N, et al. Safety, pharmacokinetics and pharmacodynamics of the CFTR corrector FDL169. Journal of Cystic Fibrosis 2018;17(Suppl 3):S42. [CFGD Register: BD250a] [Google Scholar]
- Horsley AR, Blaas S, Burr L, Carroll M, Downey DG, Drevinek P, et al. Novel CFTR corrector FDL169: safety, pharmacokinetics and pharmacodynamics. Pediatric Pulmonology 2018;53(S2):252. [CFGD Register: BD250b] [Google Scholar]
Hubert 2018 {published data only}
- Hubert D, Dehillotte C, Munck A, David V, Baek J, Mely L, et al. Retrospective observational study of French patients with cystic fibrosis and a Gly551Asp‐CFTR mutation after 1 and 2 years of treatment with ivacaftor in a real‐world setting. Journal of Cystic Fibrosis 2018;17(1):89‐95. [PUBMED: 28711222] [DOI] [PubMed] [Google Scholar]
Kerem 2014 {published data only}
- Ajayi T, Konstan M, Accurso FJ, Boeck K, Kerem E, Rowe S, et al. The use of high resolution computerized tomography of the chest in evaluating the effect of ataluren in nonsense mutation cystic fibrosis (nmCF) lung disease. Journal of Cystic Fibrosis 2013;12 Suppl 1:S64. [Abstract no.: 63; CENTRAL: 921666; CFGD Register: BD167h; CRS: 5500100000011670] [Google Scholar]
- Davies JC, Tiddens HAWM, Malfroot A, Heijerman HGM, Kerem E, Hjelte L, Sun J, et al. Ataluren in nonsense mutation cystic fibrosis patients not receiving tobramycin: significant lung function benefits in the paediatric age range. Journal of Cystic Fibrosis 2016;15 Suppl 1:S21. [Abstract no.: WS13.11; CFGD Register: BD167q] [Google Scholar]
- Boeck K, Heijerman HGM, Davies JC, Sermet‐Gaudelus I, Hjelte L, Kerem E, et al. Ataluren significantly reduces exacerbations in nonsense mutation cystic fibrosis patients not receiving tobramycin. Journal of Cystic Fibrosis 2016;15 Suppl 1:S20. [Abstract no.: WS13.1; CFGD Register: BD167p] [Google Scholar]
- Kerem E, Konstan MW, Boeck K, Accurso FJ, Sermet‐Gaudelus I, Wilschanski M, et al. Ataluren for the treatment of nonsense‐mutation cystic fibrosis: a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet Respiratory Medicine 2014;2(7):539‐47. [DOI: 10.1016/S2213-2600(14)70100-6; PUBMED: 24836205] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerem E, Sermet‐Gaudelus I, Hjelte L, Boeck K. Natural history of patients with cystic fibrosis carrying nonsense mutations: an analysis of placebo‐treated patients from the 009 study. Journal of cystic fibrosis 2016;15 Suppl 1:S118. [Abstract no.: 264; CFGD Register: BD167o] [Google Scholar]
- Kerem E, Wilschanski M, Accurso F, DeBoeck K, Konstan M, Rows S, et al. Natural history of cystic fibrosis in patients with nonsense‐mutation‐mediated disease. Pediatric Pulmonology 2012;47 Suppl 35:312. [Abstract no.: 252; CENTRAL: 921643; CFGD Register: BD167e; CRS: 5500125000000035] [Google Scholar]
- Kerem E, Wilschanski M, Boeck K, Sermet‐Gaudelus I, Constantine S, Elfring GL, et al. Phase 3 study of ataluren (PTC124®) in nonsense mutation cystic fibrosis (nmCF): baseline data. Journal of Cystic Fibrosis 2011;10 Suppl 1:S17. [Abstract no.: 65; CFGD Register: BD167c; MEDLINE: ] [Google Scholar]
- Kerem E, Wilschanski M, Melotti P, Sermet‐Gaudelus I, DeBoeck K, Rowe SM, et al. Phase 3 study of Ataluren (PTC124®) in nonsense mutation cystic fibrosis (NMCF): demographic & other baseline data. Pediatric Pulmonology 2010;45 Suppl 33:314. [Abstract no.: 314; CFGD Register: BD167a; MEDLINE: ] [Google Scholar]
- Kerem E, Wilschanski M, Sermet‐Gaudelus I, Boeck K, Accurso FJ, Konstan MW, et al. The effect of Pseusdomonas aeruginosa infection on pulmonary function outcome in a cohort of patients with nonsense mutation cystic fibrosis. Journal of Cystic Fibrosis 2014;13 Suppl 2:S70. [Abstract no.: 94; CENTRAL: 996543; CFGD Register: BD167k ; CRS: 5500129000000008] [Google Scholar]
- Kerem E, Wilschasnski M, Sermet‐Gaudelaus I, Boeck K, Accurso FJ, Konstan M, et al. Interim results of the phase 3 open‐ label study of ataluren in nonsense mutation cystic fibrosis (nmCF). Journal of Cystic Fibrosis 2013;12 Suppl 1:S15. [Abstract no.: WS7.5; CENTRAL: 921641; CFGD Register: BD167g; CRS: 5500100000011653] [Google Scholar]
- Konstan MW, Accurso FJ, Boeck K, Kerem E, Rowe SM, Sermet‐Gaudelus I, et al. Pretreatment data from phase 3 study of Ataluren document significant disease burden in a subpopulation of patients with nonsense mutation cystic fibrosis. Pediatric Pulmonology 2011;46 Suppl 34:295. [Abstract no.: 232; CFGD Register: BD167d; MEDLINE: ] [Google Scholar]
- Konstan MW, Rowe SM, Accurso FJ, Kerem E, Wilschanski M, Boeck K, et al. Use of different pulmonary exacerbation definitions in the phase 3 clinical trial of ataluren in patients with nonsense mutation cystic fibrosis. Pediatric Pulmonology 2013;48 Suppl 36:298. [Abstract no.: 258; CENTRAL: 921688; CFGD Register: BD167i; CRS: 5500125000000400] [Google Scholar]
- Rowe S, Sermet‐Gaudelus I, Konstan M, Kerem E, Wilschanski M, DeBoeck K, et al. Results of the Phase 3 study of ataluren in nonsense mutation in cystic fibrosis (NMCF). Pediatric Pulmonology 2012;47 Suppl 35:290. [Abstract no.: 193; CENTRAL: 921642; CFGD Register: BD167f; CRS: 5500125000000027] [Google Scholar]
- Rowe SM, Konstan MW, Accurso FJ, Boeck K, Sermet‐Gaudelus I, Kerem E, et al. The use of chronic inhaled antibiotics in the phase 3 clinical trial of ataluren in patients with nonsense‐mutation cystic fibrosis. Pediatric Pulmonology 2013;48 Suppl 36:279. [Abstract no.: 207; CENTRAL: 921748; CFGD Register: BD167j; CRS: 5500125000000409] [Google Scholar]
KONNECTION 2013 {published data only}
- Boeck K, Munck A, Walker S, Faro A, Hiatt P, Chan J, et al. The effect of ivacaftor, a CFTR potentiator, in patients with cystic fibrosis and a non‐G551D‐CFTR gating mutation, the KONNECTION study. Journal of Cystic Fibrosis 2014;13 Suppl 2:S1. [Abstract no.: WS1.1; CENTRAL: 998097; CFGD Register: BD201b; CRS: 5500125000000707] [Google Scholar]
- Boeck K, Paskavitz J, Chen X, Higgins M. Ivacaftor, a CFTR potentiator, in cystic fibrosis patients who have a non‐G551D‐CFTR gating mutation: phase 3, part 1 results. Pediatric Pulmonology 2013;48 Suppl 36:292. [Abstract no.: 241; CENTRAL: 962356; CFGD Register: BD201a; CRS: 5500125000000421] [Google Scholar]
McCarty 2002 {published data only}
- Ahrens RC, Standaert TA, Launspach J, Han SH, Teresi ME, Aitken ML, et al. Use of nasal potential difference and sweat chloride as outcome measures in multicenter clinical trials in subjects with cystic fibrosis. Pediatric Pulmonology 2002;33(2):142‐50. [CFGD Register: BD136d] [DOI] [PubMed] [Google Scholar]
- Aitken ML, Ahrens RC, Karlin DA, Konstan MW, McNamara SC, Regelman WE, et al. Safety of a phase I double‐blind placebo‐controlled dose escalation trial of oral CPX in adult CF patients. Pediatric Pulmonology 1998;26(S17):276. [CFGD Register: BD136b] [Google Scholar]
- McCarty NA, Standaert TA, Teresi M, Tuthill C, Launspach J, Kelley TJ, et al. A phase I randomized, multicenter trial of CPX in adult subjects with mild cystic fibrosis. Pediatric Pulmonology 2002;33(2):90‐8. [CFGD Register: BD136c] [DOI] [PubMed] [Google Scholar]
- McCarty NA, Weatherly MR, Kelley TJ, Konstan MW, Milgram LJH, Teresi M, et al. Multicenter phase I trial of CPX in adults patients with mild CF: results of nasal potential difference measurements. Pediatric Pulmonology 1998;Suppl 17:276. [CFGD Register: BD136a] [Google Scholar]
McGarry 2015 {published data only}
- McGarry ME, Finkbeiner WE, Illek B, Fischer H, Zlock LT, Olshansky S, et al. Ivacaftor response is not predicted by signs of residual CFTR function. Pediatric Pulmonology 2015;50 Suppl 41:292. [Abstract no.: 266; CENTRAL: 1092198; CFGD Register: BD224a; CRS: 5500135000001387] [Google Scholar]
- McGarry ME, Illek B, Ly NP, Zlock L, Olshansky S, Moreno C, et al. In vivo and in vitro ivacaftor response in cystic fibrosis patients with residual CFTR function: n‐of‐1 studies. Pediatric Pulmonology 2017;52(4):472‐9. [CFGD Register: BD224c] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielson DW, Moreno C, McGarry ME, Ly N. Sweat chloride as an outcome measure in n‐of‐one CF drug testing. Pediatric Pulmonology 2015;50 Suppl 41:298. [Abstract no.: 284; CENTRAL: 1092201; CFGD Register: BD224b; CRS: 5500135000001390] [Google Scholar]
NCT01549314 {published data only}
- NCT01549314. Cystic Fibrosis Related Bone Disease: the Role of CFTR. clinicaltrials.gov/ct2/show/NCT01549314 (first received 9 March 2012).
NCT01685801 {published data only}
- NCT01685801. Pilot study testing the effect of ivacaftor on lung function in subjects with cystic fibrosis and residual CFTR function [A pilot study testing the effect of ivacaftor on lung function in subjects with cystic fibrosis, residual CFTR function, and FEV1 ≥40% predicted]. clinicaltrials.gov/ct2/show/NCT01685801 (first received 14 September 2012).
NCT01784419 {published data only}
- NCT01784419. Short term effects of ivacaftor in non‐G551D cystic fibrosis patients. clinicaltrials.gov/ct2/show/NCT01784419 (first received 05 February 2013).
NCT01863238 {published data only}
- NCT01863238. An ocular safety study of ivacaftor‐treated pediatric patients 11 years of age or younger with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT01863238 (first received 27 May 2013).
NCT01946412 {published data only}
- NCT01946412. Roll‐over study of ivacaftor in cystic fibrosis pediatric subjects with a CF transmembrane conductance regulator gene (CFTR) gating mutation. clinicaltrials.gov/ct2/show/NCT01946412 (first received 19 September 2013).
NCT02039986 {published data only}
- NCT02039986. Ivacaftor (Kalydeco) and insulin in cystic fibrosis (CF) [Effects of ivacaftor (Kalydeco) treatment upon insulin and incretin secretion in patients with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT02039986 (first received 20 January 2014).
NCT02141464 {published data only}
- NCT02141464. Energy balance and weight gain with ivacaftor treatment. clinicaltrials.gov/ct2/show/NCT02141464 (first received 19 May 2014).
NCT02310789 {published data only}
- NCT02310789. Does ivacaftor alter wild type CFTR‐open probability in the sweat gland secretory coil?. clinicaltrials.gov/ct2/show/NCT02310789 (first received 8 December 2014).
NCT02311140 {published data only}
- NCT02311140. Effects of Kalydeco on upper airway and paranasal sinus inflammation measured by nasal lavage and on symptoms (KPNSI). clinicaltrials.gov/ct2/show/NCT02311140 (first received 8 December 2014).
NCT02323100 {published data only}
- NCT02323100. Glycerol phenylbutyrate corrector therapy for CF (Cystic Fibrosis). clinicaltrials.gov/show/nct02323100 (first posted 23 December 2014).
NCT02443688 {published data only}
- NCT02443688. A phase 2 study to evaluate the efficacy, safety, and tolerability of CTX‐4430 in adult CF patients. clinicaltrials.gov/ct2/show/NCT02443688 (first received 14 May 2015).
NCT02445053 {published data only}
- NCT02445053. Observational study of outcomes in cystic fibrosis patients with selected gating mutations on a CFTR allele (The VOCAL Study) (VOCAL). clinicaltrials.gov/ct2/show/NCT02445053 (first received 15 May 2015).
NCT02690519 {published data only}
- NCT02690519. Study of GLPG1837 in subjects with cystic fibrosis (S1251N mutation) (SAPHIRA2). clinicaltrials.gov/ct2/show/NCT02690519 (first received 24 February 2016).
NCT02707562 {published data only}
- NCT02707562. Study of GLPG1837 in subjects with cystic fibrosis (G551D Mutation) (SAPHIRA1). clinicaltrials.gov/ct2/show/NCT02707562 (first received 14 March 2016).
NCT02709109 {published data only}
- NCT02709109. A study to evaluate the safety and efficacy of VX‐371 in subjects with cystic fibrosis who are homozygous for the F508del‐CFTR mutation. clinicaltrials.gov/ct2/show/NCT02709109 (first received 15 March 2016).
NCT02718495 {published data only}
- NCT02718495. Study assessing PTI‐428 safety, tolerability, and pharmacokinetics in subjects with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT02718495 (first received 24 March 2016).
NCT02722057 {published data only}
- NCT02722057. A study to confirm the long‐term safety and effectiveness of Kalydeco in patients with cystic fibrosis who have an R117H‐CFTR mutation, including pediatric patients. clinicaltrials.gov/ct2/show/NCT02722057 (first received 29 March 2016).
NCT02724527 {published data only}
- NCT02724527. Study of cavosonstat (N91115) in CF patients who are heterozygous for F508del‐CFTR and a gating mutation and being treated with ivacaftor (SNO‐7). clinicaltrials.gov/ct2/show/NCT02724527 (first received 31 March 2016).
NCT02742519 {published data only}
- NCT02742519. A study to evaluate efficacy and safety of ivacaftor in subjects with cystic fibrosis aged 3 through 5 years who have a specified CFTR gating mutation. clinicaltrials.gov/ct2/show/NCT02742519 (first received 19 April 2015).
NCT02759562 {published data only}
- NCT02759562. Effect of andecaliximab on FEV1 in adults with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT02759562 (first received 03 May 2016).
NCT02934698 {published data only}
- NCT02934698. An efficacy and safety study of ivacaftor in patients with cystic fibrosis and two splicing mutations. clinicaltrials.gov/ct2/show/NCT02934698 (first received 17 October 2016).
NCT03068312 {published data only}
- NCT03068312. A study to evaluate efficacy of ivacaftor in subjects with cystic fibrosis who have a 3849 + 10KB C→T or D1152H CFTR mutation. clinicaltrials.gov/ct2/show/NCT03068312 (first received 01 March 2017).
NCT03256799 {published data only}
- NCT03256799. Evaluation of ivacaftor in patients using ataluren for nonsense mutations. clinicaltrials.gov/ct2/show/NCT03256799 (first received 22 August 2017).
NCT03256968 {published data only}
- NCT03256968. PTC study to evaluate ataluren in combination with ivacaftor. clinicaltrials.gov/ct2/show/NCT03256968 (first received 22 August 2017).
NCT03258424 {published data only}
- NCT03258424. Study assessing PTI‐428 safety, tolerability, and pharmacokinetics in subjects with cystic fibrosis on KALYDECO® as background therapy. clinicaltrials.gov/ct2/show/NCT03258424 (first received 23 August 2017).
NCT03277196 {published data only}
- NCT03277196. A phase 3, 2‐arm, open‐label study to evaluate the safety and pharmacodynamics of long‐term ivacaftor treatment in subjects with cystic fibrosis who are less than 24 months of age at treatment initiation and have an approved ivacaftor‐responsive mutation. clinicaltrials.gov/ct2/show/NCT03277196 (first received 08 September 2017).
NCT03390985 {published data only}
- NCT03390985. Canadian observation trial in CF patients undergoing treatment with ivacaftor (G551D). clinicaltrials.gov/ct2/show/NCT03390985 (first received 5 January 2018).
NCT03474042 {published data only}
- NCT03474042. GLPG2737 on top of Orkambi in subjects with cystic fibrosis (PELICAN). clinicaltrials.gov/ct2/show/NCT03474042 (first received 22 March 2018).
NCT03652090 {published data only}
- NCT03652090. Evaluation of the Primary Human Nasal Epithelial Cell Culture Model in the Context of Personalized Therapy in Cystic Fibrosis. clinicaltrials.gov/ct2/show/NCT03652090 (first received 29 August 2018).
PERSIST 2014 {published data only}
- McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long‐term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp‐CFTR mutation: a phase 3, open‐label extension study (PERSIST). The Lancet. Respiratory medicine 2014;2(11):902‐10. [PUBMED: 25311995] [DOI] [PubMed] [Google Scholar]
Pradal 2002 {published data only}
- Pradal U, Casotti V, Delmarco A, Nicolis E, Livraghi A, Conese M, et al. Effects of gentamicin on ion transport, MRNA and protein CFTR expression in patients with R1162X: A double blind placebo controlled study. Pediatric Pulmonology 2002;34 Suppl 24:263. [CFGD Register: BD145] [Google Scholar]
RIO‐CF 2017 {published data only}
- Derichs N, Taylor‐Cousar J, Tullis E, Davies J, Nazareth D, Downey DG, et al. Safety, tolerability and early signs of efficacy with riociguat for the treatment of adult Phe508del homozygous cystic fibrosis patients: study design and rationale for the Rio‐CF study. Journal of Cystic Fibrosis 2017;16(Supplement 1):S36. [CFGD Register: BD246b] [Google Scholar]
- Taylor‐Cousar JL, Derichs N, Tullis DE, Davies JC, Nazareth D, Downey D, et al. Safety, tolerability and early signs of efficacy with riociguat for the treatment of adult Phe508del homozygous cystic fibrosis patients: safety data from the rio‐CF study. Pediatric Pulmonology 2017;52(Supplement 47):307. [CFGD Register: BD246c] [Google Scholar]
- Taylor‐Cousar JL, Tullis E, Derichs N, Davies JC, Nazareth D, Downey D, et al. Riociguat for the treatment of adult Phe508del homozygous cystic fibrosis: efficacy data from the Phase II Rio‐CF study. Journal of Cystic Fibrosis 2018;17(Suppl 3):S67. [CFGD Register: BD246a] [DOI] [PubMed] [Google Scholar]
Romano 2000 {published data only}
- Romano L, Casciaro R, Vanini P, Zegarra‐Moran O, Negro I, Minuto N, et al. Reduction of sweat ion concentrations following topical application of gentamicin in CF patients carrying nonsense mutations. Proceedings of 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna, Austria. 2001:P11. [CFGD Register: BD144b]
- Romano L, Sacchi R, Zegarra‐Moran O, Vanini P, Guerriero F, Casciaro R, et al. Effects of topical applications of gentamicin on sweat test in CF patients carrying nonsense‐mutations. Pediatric Pulmonology 2000;30 Suppl 20:250. [CFGD Register: BD144a] [Google Scholar]
Rubenstein 1998 {published data only}
- Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4‐phenylbutyrate (Buphenyl) in deltaF508‐homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. American Journal of Respiratory and Critical Care Medicine 1998;157(2):484‐90. [CFGD Register: BD146b; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. A randomized, double blind, placebo‐controlled trial of sodium 4‐phenylbutyrate (Buphenyl) in deltaF508‐homozygous cystic fibrosis patients: Partial restoration of nasal epithelial CFTR function. Pediatric Pulmonology 1997;24 Suppl 14:272. [CFGD Register: BD146a] [DOI] [PubMed] [Google Scholar]
Rubenstein 2006 {published data only}
- Rubenstein RC, Propert KJ, Reenstra WW, Skotleski ML. A pilot trial of the combination of phenylbutyrate and genistein. Pediatric Pulmonology 2006;41 Suppl 29:294. [CFGD Register: BD149] [Google Scholar]
Seliger 2015 {published data only}
- Seliger V, Bai Y, Volkova N, Tian S, Waltz. Prevalance of cataracts in a population of cystic fibrosis patients homozygous for the F508del mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S108. [Abstract no.: 196; CFGD Register: BD213d // BD214c] [Google Scholar]
Sermet‐Gaudelus 2010 {published data only}
- Sermet‐Gaudelus I, Boeck K, Casimir GJ, Vermeulen F, Leal T, Mogenet A, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. American Journal of Respiratory & Critical Care Medicine 2010;182(10):1262‐72. [CFGD Register: BD167b; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
TOPIC 2018 {published data only}
- NCT03085485. The Topic Trial ‐ Study to Determine the Safety and Efficacy of Ivacaftor. clinicaltrials.gov/ct2/show/NCT03085485 (first received 21 March 2017).
Wilschanski 2003 {published data only}
- Wilschanski M, Virgilis D, Strauss‐Liviatan N, Tal A, Bentur L, Blau H, et al. Gentamicin causes functional expression of CFTR in CF patients carrying stop mutations: a double‐blind placebo controlled trial. Pediatric Pulmonology 2000;30 Suppl 20:244. [CFGD Register: BD143a; MEDLINE: ] [Google Scholar]
- Wilschanski M, Yahav J, Blau H, Bentur L, Rivlin J, Aviram M, et al. Restoration of CFTR function by gentamicin in cystic fibrosis patients carrying stop mutations: a double blind placebo controlled trial. Gastroenterology 2003;124(4 Suppl 1):A582. [CFGD Register: BD143c] [Google Scholar]
- Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin‐induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. New England Journal of Medicine 2003;349(15):1433‐41. [CFGD Register: BD143b] [DOI] [PubMed] [Google Scholar]
Wilschanski 2008 {published data only}
- Kerem E, Yaakov Y, Armoni S, Pugatsch T, Shoseyov D, Cohen M, et al. PTC124 induces time‐dependent improvements in chloride conductance and clinical parameters in patients with nonsense‐mutation‐mediated cystic fibrosis. Pediatric Pulmonology 2008;43 Suppl 31:294. [CFGD Register: BD22d; MEDLINE: ] [Google Scholar]
- Sermet‐Gaudelus I, Boeck K, Casimir G, Leal T, Vermeulen F, Mogenet A, et al. Children with nonsense‐mutation‐mediated cystic fibrosis respond to investigational treatment with PTC124. Pediatric Pulmonology 2008;43 Suppl 31:313. [CFGD Register: BD22c; MEDLINE: ] [Google Scholar]
- Sermet‐Gaudelus I, Leal T, Boeck K, Casimir G, Hanssens L, Hage P, et al. PTC124 induces CFTR full‐length production and activity in children with nonsense‐mutation‐mediated CF. Journal of Cystic Fibrosis 2008;7 Suppl 2:S22. [CFGD Register: BD22a; MEDLINE: ] [Google Scholar]
- Wilschanski M, Armoni S, Yaakov Y, Blau H, Shoseyov D, Cohen M, et al. PTC124 treatment over 3 months improves pharmacodynamic and clinical parameters in patients with nonsense ‐mutation‐mediated CF. Journal of Cystic Fibrosis 2008;7 Suppl 2:S22. [CFGD Register: BD22b; MEDLINE: ] [Google Scholar]
Zeitlin 2002 {published data only}
- Zeitlin PL, Diener‐West M, Rubenstein RC, Boyle MP, Lee CK, Brass‐Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4‐phenylbutyrate. Molecular Therapy 2002;6(1):119‐26. [CFGD Register: BD148] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Kazani 2016 {published data only}
- Kazani S, Alcantara J, Debonnett L, Doucet J, Jones I, Kulmatycki K, et al. QBW251 is a safe and efficacious CFTR potentiator for patients with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2016;193(Meeting Abstracts):A7789. [CFGD Register: BD243] [Google Scholar]
- NCT02190604. Safety, Tolerability, Pharmacokinetics, and Preliminary Pharmacodynamics of QBW251 in Healthy Subjects and Cystic Fibrosis Patients.. clinicaltrials.gov/ct2/show/NCT02190604 (first received 15 July 2014).
Uttamsingh 2016 {published data only}
- Uttamsingh V, Pilja L, Brummel CL, Grotbeck B, Cassella JV, Braman G. CTP‐656 multiple dose pharmacokinetic profile continues to support a once‐daily potentiator for cystic fibrosis patients with gating mutations. Pediatric Pulmonology 2016;51 Suppl 45:277. [Abstract no.: 244; CFGD Register: BD241] [Google Scholar]
Additional references
Amaral 2007
- Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends in Pharmacological Sciences 2007;28(7):334‐41. [DOI] [PubMed] [Google Scholar]
Aslam 2017
- Aslam AA, Higgins C, Sinha IP, Southern KW. Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis.. Cochrane Database of Systematic Reviews 2017, Issue 1. [DOI: 10.1002/14651858.CD012040.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bobadilla 2002
- Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations‐‐correlation with incidence data and application to screening. Human Mutation 2002;19(6):575‐606. [DOI] [PubMed] [Google Scholar]
CFMD 2012
- Hospital for Sick Children in Toronto. Cystic Fibrosis Mutation Database. www.genet.sickkids.on.ca/StatisticsPage.html (accessed 01 March 2012).
Deeks 2011
- Deeks J, Higgins J, Altman D. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hamosh 1998
- Hamosh A, FitzSimmons SC, Macek Jr M, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. Journal of Pediatrics 1998;132(2):255‐9. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
KONTINUE 2017
- NCT01707290. Rollover study of ivacaftor in subjects with cystic fibrosis and a non G551D CFTR mutation (KONTINUE) [A phase 3, two‐arm, rollover study to evaluate the safety of long term ivacaftor treatment in subjects 6 years of age and older with cystic fibrosis and a non‐G551D CFTR mutation]. clinicaltrials.gov/ct2/show/NCT01707290 (first received 16 October 2012).
McKone 2004
- McKone EF, Aitken ML. Cystic fibrosis: disease mechanisms and therapeutic targets. Drug Discovery Today: Disease Mechanisms 2004;1(1):137‐43. [Google Scholar]
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire‐Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Riordan 1989
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245(4922):1066. [DOI] [PubMed] [Google Scholar]
Rowntree 2003
- Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Annals of Human Genetics 2003;67(5):471‐85. [DOI] [PubMed] [Google Scholar]
Southern 1997
- Southern KW. Delta F508 in cystic fibrosis: Willing but not able. Archives of Disease in Childhood 1997;76(3):278‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Southern 2007
- Southern KW. Cystic fibrosis and formes frustes of CFTR‐related disease. Respiration: International Review of Thoracic Diseases 2007;74(3):241‐51. [DOI] [PubMed] [Google Scholar]
Southern 2018
- Southern KW, Patel S, Sinha IP, Nevitt SJ. Correctors (specific therapies for class II CFTR mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews 2018, Issue 8. [DOI: 10.1002/14651858.CD010966.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
UK CF Registry Report 2013
- UK CF Trust. Cystic fibrosis: our focus. www.cysticfibrosis.org.uk/media/598466/annual‐data‐report‐2013‐jul14.pdf (accessed 07 Jan 2015).
Van Goor 2009
- Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX‐770.. Proceedings of the National Academy of Sciences of the United States of America 2009;106(44):18825‐30. [DOI: 10.1073/pnas.0904709106] [DOI] [PMC free article] [PubMed] [Google Scholar]
Whiting 2014
- Whiting P, Al M, Burgers L, Westwood M, Ryder S, Hoogendoorn M, Armstrong N, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost‐effectiveness analysis. Health Technology Assessment (Winchester, England) 2014;18(18):1‐106. [DOI: 10.3310/hta18180] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Patel 2012
- Patel S, Sinha IP, Dwan K, Echevarria C, Schechter M, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD009841] [DOI] [PubMed] [Google Scholar]
Patel 2015
- Patel S, Sinha IP, Dwan K, Echevarria C, Schechter M, Southern KW. Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis. Cochrane Database of Systematic Reviews 2015, Issue 3. [DOI: 10.1002/14651858.CD009841.pub2] [DOI] [PubMed] [Google Scholar]