

Abstract
Background
Psoriatic arthritis is an inflammatory disease associated with joint damage, impaired function, pain, and reduced quality of life. Methotrexate is a disease‐modifying anti‐rheumatic drug (DMARD) commonly prescribed to alleviate symptoms, attenuate disease activity, and prevent progression of disease.
Objectives
To assess the benefits and harms of methotrexate for psoriatic arthritis in adults.
Search methods
We searched CENTRAL, MEDLINE, Embase, the WHO International Clinical Trials Registry Platform, and www.clinicaltrials.gov for relevant records. We searched all databases from inception to 29 January 2018. We handsearched included articles for additional records and contacted study authors for additional unpublished data. We applied no language restrictions.
Selection criteria
We included all randomised controlled trials (RCTs) and quasi‐RCTs that compared methotrexate versus placebo, or versus another DMARD, for adults with psoriatic arthritis. We reported on the following major outcomes: disease response (measured by psoriatic arthritis response criteria (PsARC)), function (measured by the Health Assessment Questionnaire for Rheumatoid Arthritis (HAQ)), health‐related quality of life, disease activity (measured by disease activity score (28 joints) with erythrocyte sedimentation rate (DAS28‐ESR)), radiographic progression, serious adverse events, and withdrawals due to adverse events.
Data collection and analysis
Two review authors independently reviewed search results, assessed risk of bias, extracted trial data, and assessed the quality of evidence using the GRADE approach. We undertook meta‐analysis only when this was meaningful.
Main results
We included in this review eight RCTs conducted in an outpatient setting, in Italy, the United Kingdom, the United States of America, China, Russia, and Bangladesh. Five studies compared methotrexate versus placebo, and four studies compared methotrexate versus other DMARDs. The average age of participants varied across studies (26 to 52 years), as did the average duration of psoriatic arthritis (one to nine years). Doses of methotrexate varied from 7.5 mg to 25 mg orally per week, but most studies administered approximately 15 mg or less orally per week. Risk of bias was generally unclear or high across most domains for all studies. We considered only one study to have low risk of selection and detection bias. The main study informing results of the primary comparison (methotrexate vs placebo up to six months) was at low risk of bias for all domains except attrition bias and reporting bias.
We restricted reporting of results to the comparison of methotrexate versus placebo for up to six months. Low‐quality evidence (downgraded due to bias and imprecision) from a single study (221 participants; methotrexate dose 15 mg orally or less per week) informed results for disease response, function, and disease activity. Disease response, measured by the proportion who responded to treatment according to PsARC (response indicates improvement), was 41/109 in the methotrexate group and 24/112 in the placebo group (risk ratio (RR) 1.76, 95% confidence interval (CI) 1.14 to 2.70). This equates to an absolute difference of 16% more responders with methotrexate (4% more to 28% more), and a number needed to treat for an additional beneficial outcome (NNTB) of 6 (95% CI 5 to 25). Mean function, measured by the HAQ (scale 0 to 3; 0 meaning no functional impairment; minimum clinically important difference 0.22), was 1.0 points with placebo and 0.3 points better (95% 0.51 better to 0.09 better) with methotrexate; absolute improvement was 10% (3% better to 17% better), and relative improvement 30% (9% better to 51% better). Mean disease activity as measured by the DAS28‐ESR (scale of 0 to 10; lower score means lower disease activity; minimum clinically important difference unknown) was 3.8 points in the methotrexate group and 4.06 points in the placebo group; mean difference was ‐0.26 points (95% CI ‐0.65 to 0.13); absolute improvement was 3% (7% better to 1% worse), and relative improvement 6% (16% better to 3% worse).
Low‐quality evidence (downgraded due to risk of bias and imprecision) from three studies (n = 293) informed our results for serious adverse events and withdrawals due to adverse events. Due to low event rates, we are uncertain if methotrexate results show increased risk of serious adverse events or withdrawals due to adverse events compared to placebo. Results show 1/141 serious adverse events in the methotrexate group and 4/152 in the placebo group: RR 0.26 (95% CI 0.03 to 2.26); absolute difference was 2% fewer events with methotrexate (5% fewer to 1% more). In all, 9/141 withdrawals in the methotrexate group were due to adverse events and 7/152 in the placebo group: RR 1.32 (95% CI 0.51 to 3.42); absolute difference was 1% more withdrawals (4% fewer to 6% more).
One study measured health‐related quality of life but did not report these results. No study measured radiographic progression.
Authors' conclusions
Low‐quality evidence suggests that low‐dose (15 mg or less) oral methotrexate might be slightly more effective than placebo when taken for six months; however we are uncertain if it is more harmful. Effects of methotrexate on health‐related quality of life, radiographic progression, enthesitis, dactylitis, and fatigue; its benefits beyond six months; and effects of higher‐dose methotrexate have not been measured or reported in a randomised placebo‐controlled trial.
Plain language summary
Methotrexate for psoriatic arthritis
Background
Psoriatic arthritis is an inflammatory condition that causes painful, swollen, and stiff joints, along with painful tendons and swollen fingers and toes. It is associated with psoriasis ‐ a disease of the skin or nails. If severe, rheumatologists prescribe methotrexate, a disease‐modifying anti‐rheumatic drug (DMARD), to improve symptoms and prevent worsening. Other DMARDs might include leflunomide, ciclosporin A, sulfasalazine, and gold (although gold treatment is rarely used).
Review question
We aimed to assess the benefits and harms of methotrexate compared with placebo (a fake drug) or similar drugs for adults with psoriatic arthritis. Methotrexate compared with placebo was the primary comparison. Major outcomes were disease response (number of patients with a positive response to treatment), function, health‐related quality of life, disease activity, radiographic progression (bone damage over time as seen on X‐rays), serious adverse events (side effects requiring hospital admission, necessitating intensive therapy, causing permanent disability or death), and withdrawals due to adverse events (side effects that cause people to stop taking the treatment).
Search date
We searched for evidence up to 29 January 2018.
Study characteristics
We included eight studies published between 1964 and 2014. All studies involved people from rheumatology clinics. Studies were conducted in Italy, United Kingdom, United States of America, China, Russia, and Bangladesh. Five studies compared methotrexate against placebo (345 people), and four studies compared methotrexate against another DMARD (leflunomide (61 people), ciclosporin A (35 people), gold (30 people), and sulfasalazine (24 people)). The average age of people included in these studies varied from 26 to 52 years. The average duration of psoriatic arthritis ranged from one to nine years. The dose of methotrexate consisted of 7.5 mg to 25 mg orally, but for most studies, 15 mg was given orally per week. In most western countries, a dose of 15 mg to 20 mg orally per week is normally used in routine practice.
Key results
After six months of treatment, comparison with placebo (a fake drug) showed that methotrexate resulted in the following (note that one study measured but did not report quality of life, and no studies measured radiographic progression).
Proportion who responded to treatment as measured by the Psoriatic Arthritis Response Criteria
16% more people, or 16 more people out of 100, improved with treatment (4% more to 28% more)
37 out of 100 people taking methotrexate improved
21 out of 100 people taking placebo improved
Function (lower scores mean better function)
Function was improved by 10% (ranging from 3% better to 17% better), or by 0.30 points (ranging from 0.09 better to 0.51 better) on a 0 to 3 scale (this is expected to be meaningful to patients)
People taking methotrexate rated their function as 0.7 point
People taking placebo rated their function as 1.0 point
Disease activity (lower scores mean less active disease)
Disease activity improved by 3% (7% better to 1% worse), or by 0.26 points (0.65 better to 0.13 worse) on a 0 to 10 scale
People taking methotrexate had a disease activity score of 3.8 points
People taking placebo had a disease activity score of 4.06 points
Serious adverse events (more events mean more harm)
2% fewer people, or two fewer people out of 100 (5% fewer to 1% more), reported a serious adverse event with methotrexate
One person out of 100 people taking methotrexate had a serious adverse event
Three out of 100 people taking placebo had a serious adverse event
Withdrawals due to adverse events (more events means more harm)
1% more people, or one more person out of 100 (4% fewer to 6% more), withdrew from treatment with methotrexate
Six out of 100 people taking methotrexate withdrew
Five out of 100 people taking placebo withdrew
Quality of the evidence
Low‐quality evidence suggests that methotrexate might lead to slightly greater benefit than placebo for some outcomes (e.g. improving function) but may be no better than placebo for other outcomes (e.g. reducing disease activity). We assessed the quality of the evidence as low due to flawed trial design and imprecision (some results are meaningful to patients and some are not). We are uncertain whether methotrexate causes more harm than placebo due to the small number of reported events.
Summary of findings
Summary of findings for the main comparison. Methotrexate compared to placebo for psoriatic arthritis (up to six months).
| Methotrexate compared to placebo for psoriatic arthritis (up to six months) | ||||||
| Patient or population: psoriatic arthritis Setting: rheumatology clinics (outpatient setting) Intervention: methotrexate (oral ≤ 15 mg per week) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with methotrexate | |||||
| Disease response assessed with PsARC (response event indicates improvement) Follow‐up: mean 6 months | 214 per 1000 | 377 per 1000 (244 to 579) | RR 1.76 (1.14 to 2.70) | 221 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | Absolute difference ‐ 16% more responded to treatment with methotrexate (4% more to 28% more); relative change ‐ 76% more responded to treatment with methotrexate (14% more to 170% more) NNTB 6 (5 to 25) When using imputed values, study authors calculated OR 1.77 (95% CI 0.97 to 3.23)c |
| Function assessed with HAQ Scale from 0 to 3 (0 shows no functional impairment) Follow‐up: mean 6 months | Mean HAQ score was 1.0 | Mean difference in HAQ score was 0.3 lower (0.51 lower to 0.09 lower) | ‐ | 221 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | Absolute change ‐ 10% better with methotrexate (3% better to 17% better); relative change ‐ 30% with methotrexate (95% CI 9% to 51% improvement)d |
| Health‐related quality of life ‐ not reported | ‐ | ‐ | ‐ | ‐ | ‐ | Measured in one study but reported as abstract only; data for extraction could not be obtained (personal communication) |
| Disease activity assessed with DAS28‐ESR Scale from: 0 to 10 (0 shows no disease activity) Follow‐up: mean 6 months | Mean DAS28‐ESR was 4.06 | Mean difference in DAS28‐ESR was 0.26 lower (0.65 lower to 0.13 higher) | ‐ | 221 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | Absolute improvement ‐ 3% better with methotrexate (7% better to 1% worse); relative improvement ‐ 6% better with methotrexate (16% better to 3% worse)d |
| Radiographic progression ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured in any study |
| Serious adverse events (SAEs) assessed by number of events Follow‐up: mean 6 months | 26 per 1000 | 7 per 1000 (1 to 59) | RR 0.26 (0.03 to 2.26) | 293 (3 RCTs) | ⊕⊕⊝⊝ LOWa,b | Absolute difference ‐ 2% fewer events with methotrexate (5% fewer to 1% more); relative difference ‐ 74% fewer (97% fewer to 116% more) |
| Withdrawals due to adverse events (WAEs) assessed by number of events Follow‐up: mean 6 months | 46 per 1000 | 61 per 1000 (23 to 158) | RR 1.32 (0.51 to 3.42) | 293 (3 RCTs) | ⊕⊕⊝⊝ LOWa,b | Absolute difference ‐ 1% more events with methotrexate (4% fewer to 6% more); relative difference ‐ 32% more events with methotrexate (49% fewer to 242% more) |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DAS28‐ESR: disease activity score (28 joints) with erythrocyte sedimentation rate; HAQ: Health Assessment Questionnaire for Rheumatoid Arthritis; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; OR: odds ratio; PsARC: Psoriatic Arthritis Response Criteria; RCT: randomised controlled trial; RR: risk ratio; SAEs: serious adverse events; WAEs: withdrawals due to adverse events. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded due to risk of bias: judged as unclear or high risk in at least one study.
bDowngraded due to imprecision: low numbers of events with confidence intervals including potentially clinically meaningless benefits.
cStudy authors did not report summary data from the ITT population. We assumed that missing participants had no response, and we calculated the ITT analysis using the number randomised.
dRelative changes calculated as absolute change (mean difference) divided by mean at baseline in the placebo group (values were 1.0 on 0 to 3 HAQ; 4.06 on 0 to 10 DAS28‐ESR).
Summary of findings 2. Methotrexate compared to other DMARDs for psoriatic arthritis (up to six months).
| Methotrexate compared to other DMARDs for psoriatic arthritis (up to six months) | ||||||
| Patient or population: psoriatic arthritis Setting: rheumatology clinics (outpatient setting) Intervention: methotrexate (oral 7.5 mg to 25 mg per week) Comparison: other DMARDs | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with other DMARDs | Risk with methotrexate (any dose) | |||||
| Disease response (leflunomide) assessed with ACR50 Follow‐up: mean 6 months | 813 per 1000 | 853 per 1000 (626 to 1000) | RR 1.05 (0.77 to 1.45) | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | Absolute difference ‐ 4% more responders with methotrexate (22% fewer to 31% more); relative change ‐ 5% more responders (23% fewer to 45% more). NNTB not calculated |
| Function (leflunomide) assessed with HAQ Scale from 0 to 3 Follow‐up: mean 6 months | Mean HAQ score for leflunomide was 0.17 | Mean difference in HAQ score for leflunomide was 0.13 lower (0.23 lower to 0.03 lower) | ‐ | 31 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | Absolute improvement ‐ 4% better with methotrexate (1% better to 8% better); relative improvement ‐ 76% better with methotrexate (18% to 135% improvement)c |
| Health‐related quality of life ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured by any study |
| Disease activity ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured by any study |
| Radiographic progression ‐ not measured | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured by any study |
| Serious adverse events (leflunomide) assessed by number of events Follow‐up: mean 6 months | 0 per 1000 | 0 per 1000 (0 to 0) | Not estimable | 61 (2 RCTs) | ⊕⊝⊝⊝ VERY LOWa,b | Absolute risk difference and risk ratio could not be calculated due to zero events in both arms. Direction and magnitude of the true effect remain uncertain |
| Withdrawals due to adverse events (leflunomide) assessed by number of events Follow‐up: mean 6 months | Of the 2 included studies, Asaduzzaman 2014 had zero events in both groups ‐ absolute and relative risks could not be calculated. In Zhang 2009, the ratio of events to total number of participants per group was 1/13 for methotrexate and 2/18 for leflunomide. RR 0.69 (95% CI 0.07 to 6.85). Comments apply to Zhang 2009 only | ‐ | 61 (2 RCTs) | ⊕⊝⊝⊝ VERY LOWa,b | For Zhang 2009: absolute risk difference ‐ 3% lower than leflunomide (95% CI 24% lower to 17% higher); risk ratio ‐ 31% lower than leflunomide (95% CI 93% lower to 585% higher). NNTH not calculated | |
| Serious adverse events (ciclosporin A) assessed by number of events Follow‐up: mean 6 months | 0 per 1000 | 0 per 1000 (0 to 0) | Not estimable | 35 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | Absolute risk difference and risk ratio could not be calculated due to zero events in both arms. Direction and magnitude of the true effect remain uncertain |
| Withdrawals due to adverse events (ciclosporin A) assessed by number of events Follow‐up: mean 6 months | 176 per 1000 | 222 per 1000 (58 to 851) | RR 1.26 (0.33 to 4.82) | 35 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | Absolute risk difference ‐ 5% higher than ciclosporin A (95% CI 22% lower to 31% higher); risk ratio ‐ 26% higher than ciclosporin A (95% CI 67% lower to 382% higher). NNTH not calculated |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). ACR50: American College of Rheumatology response criteria for 50% improvement; CI: confidence interval; DMARDs: disease‐modifying anti‐rheumatic drugs; HAQ: Health Assessment Questionnaire for Rheumatoid Arthritis; NNTB: number needed to treat for an additional beneficial outcome; NNTH: number needed to treat for an additional harmful outcome; OR: odds ratio; RCT: randomised controlled trial; RR: risk ratio; SAEs: serious adverse events; WAEs: withdrawals due to adverse events. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded due to risk of bias: judged as unclear or high in at least one domain and at least one study.
bDowngraded twice due to imprecision: low numbers of events with confidence intervals including potentially clinically meaningless benefits.
cRelative changes calculated as absolute change (mean difference) divided by mean at baseline in the placebo group (values were 0.17 on 0 to 3 HAQ).
Background
Description of the condition
Psoriatic arthritis (PsA) is an inflammatory joint disease affecting approximately 30% of people with psoriasis (Gladman 2005; Mease 2013; Truong 2015). Estimates of the prevalence of PsA in the general population vary between 0.01% and 0.19%, depending on geographical location (Stolwijk 2016). White people are affected more often than Middle‐Eastern and East‐Asian people (Stolwijk 2016). Men and women are equally affected across the entire age range, and PsA is more common over 40 years of age among people of both sexes (Stolwijk 2016).
Five distinct patterns of PsA have been described: predominant distal interphalangeal joint involvement, arthritis mutilans, symmetrical polyarthritis, asymmetrical oligoarthritis, and spondyloarthritis (Moll 1973). The reported prevalence of each pattern varies, although polyarthritis and oligoarthritis occur most commonly (Gladman 2005). Overlap of spondyloarthritis and peripheral joint disease occurs in 20% to 40% of people (Gladman 2005; Moll 1973). Periarticular structures may also be involved, leading to enthesitis, tenosynovitis, dactylitis, and fingernail dystrophy (Duarte 2012; Moll 1973). Joint erosions are reported in about 60% of people with PsA (Gladman 1987; Torre Alonso 1991), and approximately 20% develop severe joint destruction and deformity (Gladman 1987). The diagnosis can be made clinically and is aided by the ‘classification of psoriatic arthritis’ (CASPAR) criteria (Coates 2012; Taylor 2006). These criteria require the presence of established inflammatory musculoskeletal disease with at least three of the following: a history of psoriasis, dactylitis, psoriatic nail dystrophy, radiographic evidence of juxta‐articular new bone formation, or rheumatoid factor negativity (Taylor 2006).
Psoriatic arthritis has a negative impact on quality of life, causing pain, joint stiffness, reduced physical function (Gladman 2005; Husted 1997; Zachariae 2002), and loss of productivity (Walsh 2014). Compared to the general population, people with PsA are at increased risk of cardiovascular disease (incidence rate ratio 1.33, 95% confidence interval (CI) 1.23 to 1.44), as reported in Li 2015, and premature death (standardised mortality ratio 1.36, 95% CI 1.12 to 1.64), as shown by Ali 2007. Inflammation is probably a key contributor to these increased risks (Van Doornum 2002), and limited evidence suggests that anti‐inflammatory therapies reduce cardiovascular disease in PsA (Roubille 2015). Over the last 40 years, the mortality rate in PsA has decreased, possibly as a result of more aggressive treatment of the inflammatory process (Ali 2007).
Description of the intervention
Pharmacological management of PsA includes non‐steroidal anti‐inflammatory drugs (NSAIDs), glucocorticoids, and disease‐modifying anti‐rheumatic drugs (DMARDs). Three major classes of DMARDs are available: conventional synthetic DMARDs (csDMARDs), biological DMARDs (bDMARDs), and targeted synthetic DMARDs (tsDMARDs) (Smolen 2014a).
Methotrexate (MTX) is a csDMARD that can be administered orally, or via subcutaneous or intramuscular injections. It is prescribed weekly, at doses ranging from 5 mg to 25 mg. At this dose and frequency, common side effects include headache, nausea, vomiting, abdominal pain, and mouth ulcers (Alarcón 2000). Rare but serious adverse effects include myelosuppression, hepatotoxicity, infection, and pulmonary fibrosis (Alarcón 2000). Daily supplementation with folic or folinic acid can alleviate hepatotoxic and gastrointestinal adverse effects (Shea 2013).
How the intervention might work
Methotrexate is used to treat many conditions and may have different effects, depending on the disease and the dose. At high doses used for malignancy, MTX antagonises the folic acid metabolic pathway (Cronstein 2000). This disrupts production of nucleotide bases, triggering several cellular processes that culminate in apoptosis (Cronstein 2000). At low doses used for inflammatory diseases such as PsA, this pathway does not adequately explain its effects (Cronstein 2000). Several alternative mechanisms appear to be important, including accumulation of extracellular adenosine, alteration of the cytokine repertoire of inflammatory cells, and modulation of humoral and cellular immunity (Cronstein 2000; Cutolo 2002). The biochemical mechanisms that explain these effects remain incompletely understood.
Methotrexate is an effective therapy for cutaneous psoriasis (Schmitt 2014). In rheumatoid arthritis, another inflammatory joint condition, MTX provides effective therapy for improving joint disease and quality of life (Lopez‐Olivo 2014), and for reducing risk of cardiovascular disease and death (Micha 2011; Wasko 2013, respectively). Methotrexate has been used historically for PsA on the basis of its benefits in rheumatoid arthritis, and on the assumption that joint pathology is similar to cutaneous pathology in psoriasis. Treating underlying inflammation with DMARDs in PsA aims to achieve improvement in the same clinical outcomes (Smolen 2014b).
Why it is important to do this review
A systematic review by Jones and colleagues showed a paucity of clinical trials for many csDMARDs for treating PsA, including MTX (Jones 2000). Rheumatoid arthritis and PsA have many fundamental differences, such as the distribution of affected joints, their genetic associations, and their pathophysiological mechanisms (Veale 2015). Extrapolating results from clinical trials of MTX in the rheumatoid arthritis population to the PsA population is an inadequate method of proving efficacy. Furthermore, evidence indicates that risks of side effects vary between the two populations (Conway 2014; Conway 2015; Curtis 2009). Despite this, MTX is one of the most commonly used therapies worldwide for treatment of PsA (Helliwell 2008; Kvien 2005; Theander 2014). It is critical that evidence of treatment efficacy and safety be drawn from studies undertaken in a population with the disease of interest. This review is important to further inform clinical decision‐making about the role of methotrexate in treating PsA.
We have conducted this review according to the guidelines recommended by the Cochrane Musculoskeletal Group Editorial Board (Ghogomu 2014).
Objectives
To assess the benefits and harms of methotrexate for psoriatic arthritis in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) and quasi‐RCTs. We included studies reported as full text, those published as abstract only, and unpublished data. We applied no language restrictions.
Types of participants
We included studies of adults aged 18 years or older with a diagnosis of PsA made by a rheumatologist, or by fulfilment of validated classification criteria (e.g. ‘classification of psoriatic arthritis’ (CASPAR) criteria) (Taylor 2006).
We included studies of participants with conditions other than PsA only if they reported outcomes for participants with PsA as a separate subgroup, or if separate data were available from study authors upon request.
Types of interventions
We included trials comparing methotrexate (MTX) at any dose and via any formulation (oral or parenteral) versus placebo, other disease‐modifying anti‐rheumatic drugs (DMARDs) (including bDMARDs), non‐steroidal anti‐inflammatory drugs (NSAIDs), or other analgesics. We allowed co‐intervention with NSAIDs or other analgesics, provided they were used in all treatment arms.
Types of outcome measures
Major and minor outcomes were informed by the Outcome Measures in Rheumatology 8th Conference core domains for PsA (Gladman 2007). The final two minor outcomes were added 'post hoc' to allow meaningful inclusion of studies that preceded the inception of other outcomes.
Major outcomes
Disease response: measured by American College of Rheumatology response criteria for 50% improvement (ACR50) (Felson 1995), Psoriatic Arthritis Response Criteria (PsARC) (Clegg 1996), or European League Against Rheumatism (EULAR) response criteria (Van Gestel 1996)
Function: measured by the Health Assessment Questionnaire for Rheumatoid Arthritis (HAQ) score (Fries 1980), by a modification of the HAQ (Pincus 1983), or by data showing the proportion of participants who achieve a minimally clinically important difference of at least 0.22 (Kosinski 2000)
Health‐related quality of life: measured by Short Form‐36 (SF‐36) (Ware 1992), or by a disease‐specific measure such as the Psoriatic Arthritis Quality Of Life (PSORIQOL) assessment tool (McKenna 2003)
Disease activity: measured by the Disease Activity Score (28 joints) with erythrocyte sedimentation rate (DAS28‐ESR) (Prevoo 1995), by the Clinical Disease Activity Index (CDAI) (Aletaha 2005), or by EULAR response criteria, which include a change in disease activity in addition to activity of the current disease (Van Gestel 1996)
Radiographic progression: measured by the Sharp method (Sharp 1971), by the Van der Heijde modification (Van der Heijde 2000), by the Larsen method (Larsen 1977), or by the Psoriatic Arthritis Ratingen Score (PARS) (Wassenberg 2001)
Serious adverse events (SAEs) resulting in hospitalisation, disability, or death
Withdrawals due to adverse events (WAEs)
Minor outcomes
Disease response: measured by American College of Rheumatology response criteria for 20% improvement (ACR20) (Felson 1995)
Enthesitis: measured by the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) (Heuft‐Dorenbosch 2003), or by the Leeds Enthesitis Index (LEI) (Healy 2008)
Dactylitis: measured by the Leeds Dactylitis Index (LDI) (Helliwell 2005), or by digit count
Pain: measured by the visual analogue scale (VAS) or by the numerical rating scale (NRS)
Fatigue: measured by the VAS or the Functional Assessment of Chronic Illness Therapy (FACIT) fatigue scale (Webster 2003), by the Krupp Fatigue Severity Scale (FSS) (Krupp 1989), or by the Multidimensional Fatigue Inventory (Smets 1995)
Skin disease: measured by the Psoriasis Area and Severity Index (PASI) (Fredriksson 1978), or by the proportion with a reduction in PASI of 25%, 50%, or 75% (PASI 25, 50, 75)
Total adverse events (AEs)
Global assessment of disease activity, as measured by VAS or NRS (Scott 1977)
Joint count: measured by the total number of tender or swollen joints
We have reported outcomes for time points up to and including six months, and longer than six months. In the 'Summary of findings' tables, we have reported outcomes up to and including six months only.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library, Ovid MEDLINE, and Ovid Embase. We searched all databases from their inception to 29 January 2018.
We also searched www.ClinicalTrials.gov and the World Health Organization Clinical Trials Registry Platform (www.who.int/ictrp/en/), on 29 January 2018.
We set out the search strategies for MEDLINE, Embase, and CENTRAL in Appendix 1,Appendix 2, and Appendix 3, respectively, while incorporating a modified search string for RCTs as described by Glanville 2006 for MEDLINE, and by Wong 2006 for Embase.
Searching other resources
We checked the reference lists of all primary studies and review articles for additional references; searched relevant manufacturers' websites for trial information; and contacted authors of included studies to learn about additional studies.
We searched for errata or retractions from included studies published in full text on PubMed (www.ncbi.nlm.nih.gov/pubmed), also on 29 January 2018.
Data collection and analysis
Selection of studies
Two review authors (TW, TT) independently screened titles and abstracts of all studies identified as a result of the search and coded them as 'retrieve' (eligible or potentially eligible/unclear) or 'do not retrieve'. Retrieved studies underwent full‐text review. We identified and recorded reasons for excluding ineligible studies. We resolved disagreements through discussion, or by consultation with a third review author (AM). We identified and excluded duplicates and collated multiple reports of the same study, so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram and Characteristics of excluded studies tables.
Data extraction and management
We used a data collection form that was piloted on at least one study in the review to document study characteristics and outcome data. Two review authors (TW, SW) extracted study characteristics from the included studies, anda third review author (AM) spot‐checked study characteristics for accuracy against the trial report. We extracted the following study characteristics.
Methods: study design, total duration of study, details of any 'run‐in' period, number of study centres and locations, study setting, withdrawals, and dates of study.
Participants: N, mean age, age range, sex, disease duration, severity of condition, diagnostic criteria, important baseline data, inclusion criteria, and exclusion criteria.
Interventions: drugs used as intervention, concomitant medications, and excluded medications.
Comparisons: drug(s) used in non‐intervention arm(s).
Outcomes: major and minor outcomes specified and collected, and time points reported.
Characteristics of the design of the trial as outlined below in Assessment of risk of bias in included studies.
Notes: funding for the trial and notable declarations of interest of trial authors.
Two review authors (TW, SW) independently extracted outcome data from the included studies. We extracted the number of events and the number of participants in each treatment group for dichotomous outcomes, and means and standard deviations and the number of participants in each treatment group for continuous outcomes. We have noted in the Characteristics of included studies table if outcome data were not reported in a usable way and when data were transformed or estimated from a graph. We resolved disagreements by reaching consensus or by involving a third person (TT). One review author (TW) transferred data into Review Manager 5 (RevMan 2014). We double‐checked that data were entered correctly by comparing data presented in the systematic review against the study reports.
For numerical data presented only in figures/graphs, we contacted the authors of the report and requested the original data. If necessary, we planned to use plot digitiser software to extract data from graphs or figures. We planned to extract these data in duplicate.
When reports included multiple measures of a single outcome, our order of preference was as follows.
For function, we chose the HAQ score, followed by the modified HAQ or the proportion of participants who achieved a minimally clinically important difference of at least 0.22.
For disease activity, we chose the DAS28, followed by CDAI or EULAR response criteria.
For serious adverse events and withdrawals due to adverse events, we reported the sum total of each.
For pain, we preferred a VAS, followed by an NRS.
For skin disease, we preferred absolute PASI score, followed by PASI 25, PASI 50, and PASI 75.
If both final values and changes from baseline values were reported for the same outcome, we used final values.
If both unadjusted and adjusted values for the same outcome were reported, we used unadjusted values.
If data were analysed based on an intention‐to‐treat (ITT) sample and another sample (e.g. per‐protocol, as‐treated), we extracted ITT data.
Time points extracted were up to and including six months, and more than six months.
Assessment of risk of bias in included studies
Two review authors (TW, AM) independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We resolved disagreements by discussion or by consultation with another review author (SW). We assessed risk of bias according to the following domains.
Random sequence generation.
Allocation concealment.
Blinding of participants and personnel.
Blinding of outcome assessment: this was considered separately for subjective self‐reported outcomes (such as pain and function) and objective outcomes (such as radiographic progression and adverse events).
Incomplete outcome data.
Selective outcome reporting.
Other potential bias, including differences in baseline characteristics, co‐intervention use, and compliance with study therapy.
We graded each potential source of bias as having high, low, or unclear risk, and we provided a quote from each study report together with a justification for our judgement in the 'Risk of bias' table. We summarised 'Risk of bias' judgements across different studies for each of the domains listed. We considered blinding separately for different key outcomes when necessary (e.g. for unblinded outcome assessment, risk of bias for all‐cause mortality may be different than for a participant‐reported pain scale). When we judged risk of bias to be no different between outcomes, we reported this as a single domain applicable to all outcomes. We also considered the impact of missing data by examining key outcomes.
When information on risk of bias was related to unpublished data or correspondence with a study author, we have noted this in the 'Risk of bias' table.
When considering treatment effects, we have taken into account the risk of bias for studies that contributed to these outcomes.
We have presented figures generated by the 'Risk of bias' tool to provide summary assessments of risk of bias.
Assesment of bias in conducting the systematic review
We conducted the review according to the published protocol (Wilsdon 2017), and we reported any deviations from it under Differences between protocol and review.
Measures of treatment effect
We analysed dichotomous data as risk ratios (RRs), or as Peto odds ratios (Peto ORs), when the outcome was a rare event (approximately less than 10%), and we used 95% confidence intervals (CIs). We analysed continuous data as mean differences (MDs) or as standardised mean differences (SMDs), also with 95% CIs. We entered data presented as a scale with a consistent direction of effect across studies.
When researchers used different scales to measure the same conceptual outcome (e.g. disability), we calculated the SMD, along with a corresponding 95% CI. We back‐translated the SMD to a typical scale (e.g. 0 to 10 for pain) by multiplying the SMD by a typical among‐person standard deviation (SD) (e.g. SD of the control group at baseline from the most representative trial) (Schünemann 2011a).
In the Effects of interventions section and in the 'Comments' column of the 'Summary of findings' tables, we provided the absolute percentage difference, the relative per cent change from baseline, and the number needed to treat for an additional beneficial outcome (NNTB) or the number needed to treat for an additional harmful outcome (NNTH). We provided the NNTB or the NNTH only when the outcome showed a statistically significant difference.
For dichotomous outcomes, we calculated the NNTB or the NNTH from the control group event rate and the risk ratio, using the Visual Rx NNT calculator (Cates 2008). We produced the NNTB or the NNTH for continuous measures using the Wells calculator, available from the Cochrane Musculoskeletal Editorial Office (musculoskeletal.cochrane.org).
For dichotomous outcomes, we calculated the absolute risk difference using the risk difference statistic in Review Manager 5 (RevMan 2014) and expressed the result as a percentage. For continuous outcomes, we calculated absolute benefit as improvement in the intervention group minus improvement in the control group, in original units, expressed as a percentage.
We calculated the relative per cent change for dichotomous data as 'risk ratio ‐ 1' and expressed this as a percentage. For continuous outcomes, we calculated the relative difference in the change from baseline as absolute benefit divided by baseline mean of the control group, expressed as a percentage.
Unit of analysis issues
When a single trial reported multiple trial arms, we included only relevant arms.
Dealing with missing data
We contacted investigators or study sponsors to verify key study characteristics and to obtain missing numerical outcome data when possible (e.g. when we identified a study as abstract only, when data were not available for all participants). When this was not possible, and when we thought that missing data might introduce serious bias, we explored the impact of including such studies in the overall assessment of results by performing a sensitivity analysis. We clearly described any assumptions and imputations for handling missing data, and we explored the effect of imputation by performing sensitivity analyses.
For dichotomous outcomes (e.g. number of withdrawals due to adverse events), we calculated the event rate using the number of participants randomised in the group as the denominator.
For continuous outcomes (e.g. mean change in pain score), we calculated MD or SMD based on the number of participants analysed at that time point. If study authors did not present the number of participants analysed for each time point, we used the number of randomised participants in each group at baseline.
When possible, we computed missing standard deviations from other statistics such as standard errors, confidence intervals, or P values, according to the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). If we could not calculate standard deviations, we planned to impute them (e.g. from other studies in the meta‐analysis).
Assessment of heterogeneity
We assessed clinical and methodological diversity in terms of participants, interventions, outcomes, and study characteristics of the included studies, to determine whether meta‐analysis was appropriate. We assessed statistical heterogeneity by visually inspecting the forest plot to look for obvious differences in results between studies and by using I² and Chi² statistical tests.
As recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011), we interpreted I² values as follows: 0% to 40% 'might not be important'; 30% to 60% may represent 'moderate' heterogeneity; 50% to 90% may represent 'substantial' heterogeneity; and 75% to 100% represents 'considerable' heterogeneity. We have considered that the importance of I² depends on the magnitude and direction of effects and on the strength of evidence for heterogeneity.
We interpreted the Chi² test such that a P value of 0.10 or less indicates evidence of statistical heterogeneity.
When identified, we reported substantial heterogeneity and investigated possible causes by following the recommendations provided in Section 9.6 of the Cochrane Handbook for Systematic Reviews of Interventions.
Assessment of reporting biases
We created and examined a funnel plot to explore possible small‐study biases. In interpreting funnel plots, we examined different possible reasons for funnel plot asymmetry and related this information to review results. If we were able to pool more than 10 trials, we planned to undertake formal statistical tests to investigate funnel plot asymmetry, and to follow the recommendations provided in Section 10.4 of the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011).
To assess bias in outcome reporting, we checked trial protocols (if available) against published reports. For studies published after 1 July 2005, we screened the Clinical Trial Register at the International Clinical Trials Registry Platform of the World Health Organization (apps.who.int/trialssearch) for the a priori trial protocol. We evaluated whether selective reporting of outcomes was evident.
Data synthesis
We undertook meta‐analyses only when this was meaningful (e.g. when treatments, participants, and the underlying clinical question were similar enough for pooling to make sense). We arranged data according to comparator (i.e. placebo or other DMARD) and duration of follow‐up (i.e. up to six months or beyond six months).
We planned to use a random‐effects model and to perform a sensitivity analysis using a fixed‐effect model.
GRADE and 'Summary of findings' tables
We created 'Summary of findings' (SoF) tables using the following outcomes.
Disease response.
Function.
Health‐related quality of life.
Disease activity.
Radiographic progression.
Serious adverse events.
Withdrawals due to adverse events.
The comparator in the first SoF table is placebo. The second SoF table shows comparisons of other DMARDs (including bDMARDs).
Two review authors (TW, SW) independently assessed the quality of the evidence. We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of a body of evidence as it relates to studies that contributed data to the meta‐analyses for the prespecified outcomes, and we reported the quality of evidence as high, moderate, low, or very low. We used methods and recommendations described in Sections 8.5 and 8.7 and in Chapters 11 and 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a; Schünemann 2011a; Schünemann 2011b). We planned to use GRADEpro GDT software to prepare the SoF tables (GRADEpro GDT 2015). We planned to justify all decisions to downgrade or upgrade the quality of studies by using footnotes and by providing comments to aid the reader's understanding of the review when necessary. We planned to provide the NNTB based on absolute and relative per cent changes in the 'Comments' column of the SoF tables, as described above (see Measures of treatment effect).
Subgroup analysis and investigation of heterogeneity
We planned to carry out the following subgroup analyses.
Oral versus parenteral routes of administration.
Lower dose (15 mg or less) versus higher dose (greater than 15 mg).
Peripheral arthritis (symmetrical polyarthritis, oligoarthritis, distal interphalangeal arthritis, arthritis mutilans) versus axial arthritis (spondyloarthritis).
Methotrexate has variable absorption when administered orally at doses above 15 mg (Hamilton 1997). Parenteral administration is predictable and linear throughout the dose range (Hamilton 1997; Schiff 2014), hence effectiveness may differ between these subgroups.
Variation in treatment response by pattern of arthritis might highlight an area for further investigation.
We planned to use the following outcomes in subgroup analyses for all comparisons.
Measure of disease activity/response (ACR, DAS28, PsARC).
Assessment of function (HAQ).
Reported serious adverse events and withdrawals due to adverse events.
We planned to use the formal test for subgroup interactions provided in Review Manager 5 (RevMan 2014), and to use caution in interpreting subgroup analyses, as advised in Section 9.6 of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2011). We planned to compare the magnitude of effects between subgroups by assessing overlap of the confidence intervals of summary estimates. Non‐overlap of confidence intervals indicates statistical significance.
We were unable to extract these data from the included studies, and so we could not complete these subgroup analyses.
Sensitivity analysis
When we had identified sufficient studies, we planned to perform sensitivity analyses to assess the impact of any bias attributable to inadequate or unclear treatment allocation, including studies with quasi‐randomised designs (selection bias), blinding of participant/assessor (detection bias), and loss to follow‐up (attrition bias) compared to studies without these limitations (low risk vs high risk or unclear risk). We planned to apply this approach to the major outcomes and to both comparisons. We found insufficient studies and were unable to complete these sensitivity analyses.
We explored the effect that imputed values had on outcomes for all studies when this information was available. Data to perform this sensitivity analysis were available for only one study (Kingsley 2012), for which we examined disease response (PsARC, ACR20), function, disease activity, pain, skin disease, patient and physician global assessments of disease activity, and swollen and tender joint counts.
Results
Description of studies
Results of the search
We identified 6668 records through database searches and two additional records from other sources. After removing duplicates, we screened 4245 records by abstract and title and excluded 4197 records. We retrieved the full‐text publications for 48 records, and we included eight records in this review (Figure 1). We did not identify any studies available as unpublished data only.
1.

Study flow diagram.
Included studies
Details of individual included trials can be viewed in the Characteristics of included studies table. We had two studies ‐ Burdeinyi 1992, published in Russian, and Zhang 2009, published in Mandarin ‐ translated to English. We identified four records as relating to a single RCT and collated them as a single study record at the start of the full‐text review phase (Kingsley 2012).
Design
We included eight RCTs (Asaduzzaman 2014; Black 1964; Burdeinyi 1992; Kingsley 2012; Scarpa 2008; Spadaro 1995; Willkens 1984; Zhang 2009). One study used a cross‐over design (Black 1964), and the other seven studies described a parallel design.
Sample size
The largest trial included 221 participants (Kingsley 2012), and the smallest 21 participants (Black 1964). Only one trial was registered with a clinical trial registry (Kingsley 2012).
Setting
All studies were conducted in an outpatient setting in Italy, the United Kingdom, the United States of America, China, Russia, or Bangladesh.
Participants
All studies included adults with psoriatic arthritis recruited from rheumatology clinics. One study enrolled participants from 16 years of age (Spadaro 1995); however, screening review authors (TW, TT) agreed to include it, as no participants were younger than 30 years of age. No studies used the CASPAR criteria for psoriatic arthritis (PsA) classification. Although no study explicitly stated that the diagnosis of PsA was made by a rheumatologist, all studies either recruited participants from rheumatology clinics, or ensured that a rheumatologist was involved in the study.
Interventions
Five studies compared methotrexate versus placebo (Black 1964; Burdeinyi 1992; Kingsley 2012; Scarpa 2008; Willkens 1984). Four studies compared methotrexate versus other DMARDs (Asaduzzaman 2014; Burdeinyi 1992; Spadaro 1995; Zhang 2009), and two of these were multi‐arm trials. One study compared methotrexate versus NSAIDs (we considered this as a placebo arm because NSAIDs were permitted in all intervention groups), parenteral gold, and sulfasalazine (Burdeinyi 1992). One study compared methotrexate monotherapy versus leflunomide monotherapy and both therapies combined (Zhang 2009). Of the remaining two trials with a DMARD comparator, one compared methotrexate versus leflunomide (Asaduzzaman 2014), and another compared methotrexate versus ciclosporin (Spadaro 1995).
Doses of oral methotrexate used across studies varied from 7.5 mg to 25 mg weekly. Although this dose range is considered standard, we considered doses greater than 15 mg as 'higher‐dose'. It was not possible to determine how many participants received methotrexate doses greater than 15 mg. Two studies used parenteral methotrexate, at doses of 10 mg weekly in Scarpa 2008 and 1 mg/kg to 3 mg/kg every 10 days in Black 1964. Doses of other DMARDs included leflunomide 20 mg daily in Asaduzzaman 2014 and Zhang 2009, and elemental gold equivalent 34 mg and sulfasalazine 1 g twice a day in Burdeinyi 1992.
Use of concomitant analgesia was permitted in all trials, and these agents varied from NSAIDs to oral, parenteral, or intra‐articular corticosteroids.
Outcomes
Seven studies reported outcomes for time points up to six months (Asaduzzaman 2014; Black 1964; Kingsley 2012; Scarpa 2008; Spadaro 1995; Willkens 1984; Zhang 2009), and two studies reported outcomes for time points beyond six months (Burdeinyi 1992; Spadaro 1995). All studies did not report all outcomes. Three studies did not report outcomes in an extractable way (Black 1964; Burdeinyi 1992; Willkens 1984), and study authors either could not be contacted or were unable to provide additional information upon request. Kingsley 2012 provided additional information for ITT and complete case cohorts via written correspondence. Spadaro 1995 was unable to provide additional information. The authors of three studies did not respond to requests for additional information (Asaduzzaman 2014; Scarpa 2008; Zhang 2009).
Our original protocol specified that we would extract outcomes for quality of life, radiographic progression, enthesitis, dactylitis, and fatigue (Wilsdon 2017). No studies reported these outcomes; however, we observed that studies consistently reported tender and swollen joint counts and patient and physician global assessments of disease activity. The most recent Outcome Measures in Rheumatoid Arthritis Clinical Trials (OMERACT) recommendations acknowledge these outcomes as having clinical significance (Orbai 2017). Further, these findings directly assisted in answering the primary question of this review, and so we extracted data for these outcomes and included them.
Excluded studies
We excluded 40 studies after reviewing the full‐text records. We excluded studies because they used the wrong study design (eight studies; Abu‐Shakra 1995; Calguneri 2004; Combe 2013; Combe 2016; Feldges 1974; Goupille 1995; Mazzanti 1994; Merola 2016), the wrong intervention (13 studies; Atzeni 2011; Collins 2015; Conti 2008; Gottlieb 2016a; Gottlieb 2016b; Kavanaugh 2012; Khraishi 2016; McInnes 2015; Mease 2015; Min 2016; Schett 2011; Schett 2012; Szentpetery 2014), the wrong comparator (11 studies; Baranauskaite 2012; Coates 2013; Coates 2014; Coates 2015a; Coates 2015b; Coates 2016a; Coates 2017; Ischenko 2010; O'Brien 1962; Raffayova 2009a; Raffayova 2009b), or the wrong patient population (six studies; Fraser 2005; Glinatsi 2015; Kavanaugh 2006a; Kavanaugh 2006b; Mease 2016; Saurat 2010), or because we were unable to extract data specific to PsA participants (two studies; Bird 1977; Hall 1978). The excluded studies can be matched to their respective reasons in the Characteristics of excluded studies table.
Ongoing studies
One study is currently in progress and is due for completion of data collection in 2018 (NCT02376790). The corresponding entry at ClinicalTrials.gov describes a multi‐armed RCT comparing etanercept monotherapy, methotrexate monotherapy, and the combination of both therapies for psoriatic arthritis. The monotherapy arms might be suitable for inclusion in future versions of this review.
Risk of bias in included studies
The risk of bias for included studies can be viewed as a summary table (Figure 2) or graph (Figure 3).
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
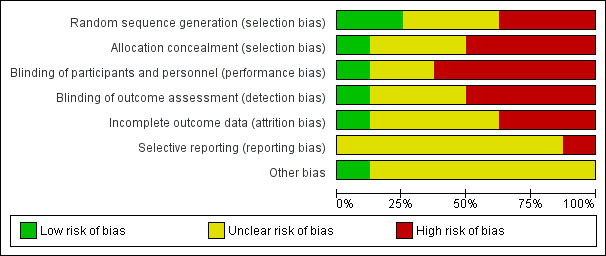
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Although all studies were described as randomised, only two studies adequately described their randomisation process (Asaduzzaman 2014; Kingsley 2012; low risk of bias). Only a single study adequately described allocation concealment methods (Kingsley 2012; low risk of bias). We judged all other studies to be at unclear or high risk of bias because they poorly described their method, or because we believed their method was likely to introduce bias.
Blinding
We judged only one study to be at low risk of bias for blinding, as the description of blinding methods was adequate (Kingsley 2012). Two studies were of an open‐label design (Asaduzzaman 2014; Spadaro 1995; high risk of performance and detection bias). One study was of a double‐blind design but did not describe blinding methods in any detail (Black 1964; uncertain risk of performance and detection bias). Willkens 1984 used a placebo tablet but did not provide further detail regarding blinding methods (uncertain risk of bias). Burdeinyi 1992 did not use placebo tablets and did not describe blinding methods in any detail; we judged this study to be at high risk of performance bias and uncertain risk of detection bias. The remaining studies did not report any blinding methods in any detail (Scarpa 2008; Zhang 2009; high risk of bias).
Incomplete outcome data
Only one study described a method of handling missing data (via multiple imputation) that may have influenced our interpretation of study results in favour of methotrexate (Kingsley 2012; unclear risk of attrition bias). One study had a low attrition rate that we judged unlikely to alter our interpretation of the results, which we deemed as having low risk of bias (Asaduzzaman 2014). The remaining studies did not describe handling of missing data, although attrition was low in three studies (Black 1964; Scarpa 2008; Willkens 1984; unclear risk of attrition bias), and was high in three studies (Burdeinyi 1992; Spadaro 1995; Zhang 2009; high risk of attrition bias).
Selective reporting
Only one trial was registered in a clinical trial registry and showed selective reporting of complete case analysis data but not of ITT analysis data (Kingsley 2012). Also these investigators collected but did not report quality of life data; we judged this study to be at high risk of reporting bias (Kingsley 2012). No other included study had a published trial protocol nor clinical trial registration. Due to our inability to substantiate that reporting bias had not occurred, we judged the remaining seven studies to be at unclear risk of reporting bias (Asaduzzaman 2014; Black 1964; Burdeinyi 1992; Scarpa 2008; Spadaro 1995; Willkens 1984; Zhang 2009).
Other potential sources of bias
We considered the similarity of baseline characteristics, use of co‐interventions, and compliance as a combined other potential source of bias. We judged only one study to be at low risk of bias (Kingsley 2012), and we determined that all other trials were at unclear risk of bias due to differences between treatment groups in each of these domains.
Effects of interventions
Results for the major outcomes can be reviewed in Table 1 and Table 2. The remaining results can be viewed in Data and analyses. We planned subgroup analyses for oral versus parenteral methotrexate, lower‐dose versus higher‐dose methotrexate, and peripheral versus axial disease. The included studies did not provide data to enable performance of these analyses. We have grouped results for methotrexate versus other disease‐modifying anti‐rheumatic drugs (DMARDs) according to the DMARD comparator.
Methotrexate versus placebo (up to six months)
Four studies with a placebo comparator reported outcomes up to six months (Black 1964; Kingsley 2012; Scarpa 2008; Willkens 1984). Not all studies reported all outcomes. One study used a cross‐over design, and we planned to extract data from the first phase of the study for comparison of methotrexate versus placebo (Black 1964). Outcomes reported were not extractable for use in this review, and study authors could not be reached. Another study reported adverse events but reported other outcomes of interest as median change from baseline without any measure of dispersion (Willkens 1984). Study authors were unable to provide additional information. Only adverse event data were extractable. Scarpa 2008 reported several outcomes of interest but presented data as medians with an interquartile range. We assumed the data were skewed, and study authors did not respond to our requests for clarification. We did not estimate means and SD from these values. For this reason, and because the risk of bias was unclear or high across most domains, we did not pool data from Scarpa 2008 with those from Kingsley 2012 for most outcomes.
Major outcomes (comparison 1)
Disease response (psoriatic arthritis response criteria (PsARC))
Only one study (Kingsley 2012: 221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) reported data for this outcome. Study authors responded to requests for unpublished data on disease response for the intention‐to‐treat (ITT) cohort (n = 221), which included imputed values for missing data. To communicate uncertainty in the result, they provided a mean and a standard error. They did not provide absolute numbers of Psoriatic Arthritis Response Criteria (PsARC) responders from the ITT cohort but provided them for the complete case cohort. PsARC response is a dichotomous outcome (i.e. responder, non‐responder); therefore we used complete case PsARC responders at six months and assumed that all other participants from the ITT cohort were non‐responders. For methotrexate, 41 of 109 achieved PsARC response, and for placebo, 24 of 112 achieved PsARC response. We calculated a risk ratio (RR) for achieving PsARC response with methotrexate of 1.76 (95% confidence interval (CI) 1.14 to 2.70; Analysis 1.1), an absolute risk difference of 0.16 (95% CI 0.04 to 0.28), and a number needed to treat for an additional beneficial outcome (NNTB) of 6 (95% CI 4 to 25).
1.1. Analysis.
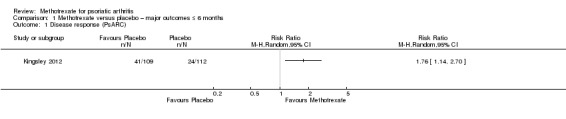
Comparison 1 Methotrexate versus placebo – major outcomes ≤ 6 months, Outcome 1 Disease response (PsARC).
We performed a sensitivity analysis using data from the complete case cohort only. Study authors provided unpublished data on the number of PsARC responders in the complete case cohort (N = 128) at six months. For methotrexate, 41 of 67 achieved PsARC response, and for placebo, 24 of 61 achieved PsARC response. We calculated a risk ratio for achieving a PsARC response with methotrexate of 1.56 (95% CI 1.08 to 2.24; Analysis 9.1), an absolute risk difference of 0.22 (95% 0.05 to 0.39), and an NNTB of 5 (95% CI 3 to 20).
9.1. Analysis.
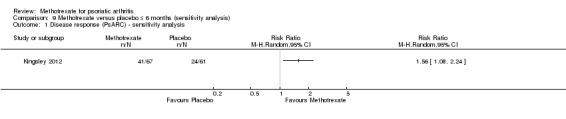
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 1 Disease response (PsARC) ‐ sensitivity analysis.
In the published manuscript for Kingsley 2012, study authors used the imputed values and calculated an increased odds ratio (OR) of PsARC response for methotrexate (OR 1.77, 95% CI 0.97 to 3.23). This was a statistically non‐significant result (P = 0.06).
We have included results from our ITT analysis in Table 1. We judged evidence quality to be low (downgraded due to risk of bias and imprecision).
Function
Only one study (Kingsley 2012: 221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) reported data for this outcome. We extracted data for the ITT cohort (n = 221), including imputed values for missing data. We estimated the standard deviation (SD) from the 95% CI in the published manuscript. At six months, mean function (Health Assessment Questionnaire for Rheumatoid Arthritis (HAQ), scale 0 to 3; higher scores indicate greater disability) in the placebo group was 1 point. We calculated a mean difference (MD) in HAQ scores of ‐0.30 (95% CI ‐0.51 to ‐0.09; Analysis 1.2). The negative sign indicates a lower HAQ score for methotrexate. This corresponds to an absolute improvement of 10% with methotrexate (95% CI 3% to 17% improvement) and a relative improvement of 30% with methotrexate (95% CI 9% to 51% improvement).
1.2. Analysis.
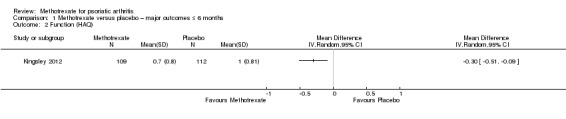
Comparison 1 Methotrexate versus placebo – major outcomes ≤ 6 months, Outcome 2 Function (HAQ).
We performed a sensitivity analysis using data for the complete case cohort (N = 128). We estimated the SD from the 95% CI reported in the supplement provided with the published manuscript. We calculated MD in HAQ scores of ‐0.20 (95% CI ‐0.42 to 0.02; Analysis 9.2). The negative sign indicates a lower HAQ score for methotrexate. This corresponds to an absolute improvement of 7% with methotrexate (95% CI 14% improvement to 1% worse) and a relative improvement of 22% with methotrexate (95% CI 47% improvement to 2% worse).
9.2. Analysis.
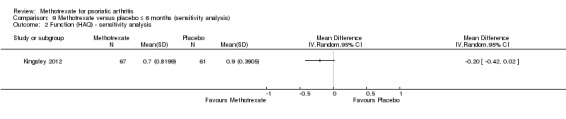
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 2 Function (HAQ) ‐ sensitivity analysis.
We included results from the ITT analysis in Table 1. We judged the quality of the evidence to be low (downgraded due to risk of bias and imprecision).
Health‐related quality of life
Studies reported no data for this outcome.
Disease activity
Only one study (Kingsley 2012: 221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) reported data for this outcome. Study authors confirmed that they used disease activity score (28 joints) with erythrocyte sedimentation rate (DAS28‐ESR; scale 0 to 10; higher scores indicate greater disease activity) as their outcome measure, and they provided unpublished data for the ITT cohort (n = 221) upon request. These included their imputed values for missing data. We estimated SD from the standard error (SE) provided by study authors. At six months, mean disease activity (DAS28‐ESR) in the placebo group was 4.1 points. We calculated an MD of ‐0.26 (95% CI ‐0.65 to 0.13; Analysis 1.3). The negative sign indicates a lower mean DAS28‐ESR score for methotrexate. This corresponds to an absolute improvement of 3% with methotrexate (95% CI 7% improvement to 1% worse) and a relative improvement of 6% with methotrexate (95% CI 16% improvement to 3% worse).
1.3. Analysis.

Comparison 1 Methotrexate versus placebo – major outcomes ≤ 6 months, Outcome 3 Disease activity (DAS28‐ESR).
We performed a sensitivity analysis using unpublished data for the complete case cohort (N = 128), which study authors provided upon request. We calculated an MD of ‐0.24 (95% CI ‐0.63 to 0.15; Analysis 9.3), with the negative sign indicating a lower mean DAS28‐ESR score for methotrexate. This corresponds to an absolute improvement of 2% with methotrexate (95% CI 10% improvement to 2% worse) and a relative improvement of 6% with methotrexate (95% CI 24% improvement to 4% worse).
9.3. Analysis.
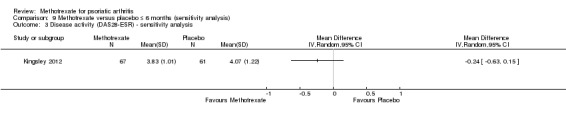
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 3 Disease activity (DAS28‐ESR) ‐ sensitivity analysis.
We included results from the ITT analysis in Table 1. We judged the quality of the evidence to be low (downgraded due to risk of bias and imprecision).
Radiographic progression
Studies reported no data for this outcome.
Serious adverse events
Three studies reported serious adverse events (SAEs) (Kingsley 2012; Scarpa 2008; Willkens 1984). Two studies reported zero SAEs at three months (Scarpa 2008; Willkens 1984). For Kingsley 2012, study authors provided unpublished data for the ITT cohort (N = 221) at six months upon request.
We analysed the data using the Mantel‐Haenszel method and a random‐effects model (Analysis 1.4). For methotrexate, 1 of 141 had an SAE, and for placebo, 4 of 152 had an SAE. We calculated an RR for experiencing an SAE with methotrexate of 0.26 (95% CI 0.03 to 2.26) and an absolute risk difference of ‐0.02% (95% CI ‐0.05 to 0.01). We did not calculate the number needed to treat for an additional harmful outcome (NNTH) for this statistically non‐significant event. We found that heterogeneity was low (Chi² = 0.40; df = 2; P = 0.82; I² = 0), and we included the results of this analysis in Table 1.
1.4. Analysis.
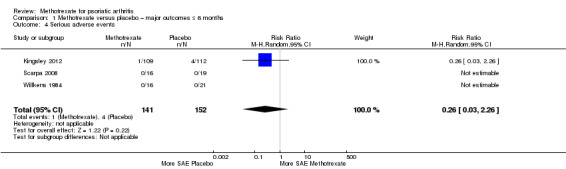
Comparison 1 Methotrexate versus placebo – major outcomes ≤ 6 months, Outcome 4 Serious adverse events.
We performed a sensitivity analysis using a fixed‐effect model and found no variation in the results.
Although Black 1964 reported no extractable adverse event data, the published manuscript describes the death of one participant following three escalating doses of intravenous methotrexate at 10‐day intervals. It is unclear in what phase this occurred (methotrexate before placebo cross‐over, or placebo before methotrexate cross‐over). Study authors could not be contacted.
We judged the quality of evidence to be low (downgraded due to risk of bias and imprecision).
Withdrawals due to adverse events
Three studies reported withdrawals due to adverse events (WAEs) (Kingsley 2012; Scarpa 2008; Willkens 1984). Two studies reported zero WAEs at three months (Scarpa 2008; Willkens 1984).
For methotrexate, results show nine WAEs among 141 participants, and for placebo, seven WAEs among 152 participants. We analysed data using the Mantel‐Haenszel method and a random‐effects model. We calculated an RR for WAEs with methotrexate of 1.32 (95% CI 0.51 to 3.42; Analysis 1.5) and an absolute risk difference of 0.01 (95% CI ‐0.04 to 0.06). We did not calculate an NNTH for this non‐significant result. We found that heterogeneity was low (Chi² = 0.18; df = 2; P = 0.91; I² = 0), and we included the results of this analysis in Table 1.
1.5. Analysis.
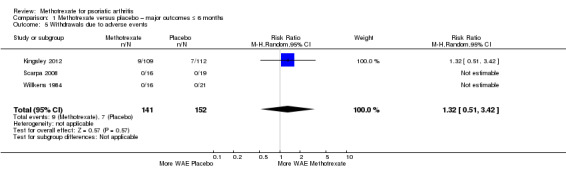
Comparison 1 Methotrexate versus placebo – major outcomes ≤ 6 months, Outcome 5 Withdrawals due to adverse events.
We performed a sensitivity analysis using a fixed‐effect model. We calculated an RR for WAEs with methotrexate of 1.32 (95% CI 0.51 to 3.42; Analysis 1.5) and an absolute risk difference of 0.02 (95% CI ‐0.04 to 0.07). We did not calculate an NNTH for this non‐significant result. We found that heterogeneity was low (Chi² = 0.18; df = 2; P = 0.91; I² = 0).
We judged the quality of the evidence to be low (downgraded due to risk of bias and imprecision).
Minor outcomes (comparison 2)
Disease response (American College of Rheumatology response criteria for 20% improvement (ACR20))
Only one study (Kingsley 2012: 221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) reported this outcome. Study authors responded to requests for unpublished data on disease response for the ITT cohort (n = 221), which included imputed values for missing data. To communicate uncertainty in the result, they provided a mean and an SE. They did not provide absolute numbers of American College of Rheumatology response criteria for 20% improvement (ACR20) responders from the ITT cohort but provided them for the complete case cohort. ACR20 response is a dichotomous outcome (i.e. responder, non‐responder); therefore, we used complete case ACR20 responders at six months and assumed that all other participants from the ITT cohort were non‐responders. For methotrexate, 23 of 109 participants achieved ACR20 response, and for placebo, 13 of 112 achieved ACR20 response. We calculated an RR for achieving ACR20 response with methotrexate of 1.82 (95% CI 0.97 to 3.40; Analysis 2.1) and an absolute risk difference of 0.09 (95% CI 0.00 to 0.19). We did not calculate an NNTB for this result.
2.1. Analysis.

Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 1 Disease response (ACR20).
We performed a sensitivity analysis using data from the complete case cohort. Study authors provided unpublished data on the number of ACR20 responders in the complete case cohort (N = 128) at six months. For methotrexate, 23 of 67 achieved ACR20 response, and for placebo, 13 of 61 achieved ACR20 response. We calculated an RR for achieving an ACR20 response with methotrexate of 1.61 (95% CI 0.90 to 2.89; Analysis 9.4) and an absolute risk difference of 0.13 (95% ‐0.02 to 0.28). We did not calculate an NNTB for this statistically non‐significant result.
9.4. Analysis.
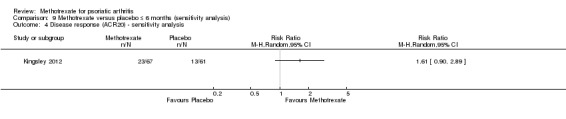
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 4 Disease response (ACR20) ‐ sensitivity analysis.
The published manuscript reported an OR for achieving an ACR20 response with methotrexate of 2.0 (95% CI 0.65 to 6.22). This was a statistically non‐significant result (P = 0.23).
We judged the quality of the evidence to be moderate (downgraded due to imprecision).
Enthesitis
Studies reported no data for this outcome.
Dactylitis
Studies reported no data for this outcome.
Pain
One study (Kingsley 2012: 221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) assessed pain using a visual analogue scale (VAS) (scale 0 mm to 100 mm; higher scores indicate more pain) and reported values for the ITT cohort (n = 221). These included imputed values for missing data. We estimated the SD from the 95% CI provided in the published manuscript. At six months, mean pain score in the placebo group was 38.3 mm. We calculated an MD of ‐9.5 mm (95% CI ‐16.78 mm to ‐2.22 mm; Analysis 2.2). The negative sign indicates a lower pain score with methotrexate. This corresponds to an absolute improvement of 10% with methotrexate (95% CI 2% to 17% improvement) and a relative improvement of 25% with methotrexate (95% CI 6% to 44% improvement).
2.2. Analysis.
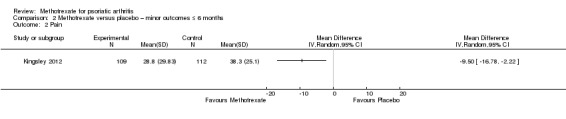
Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 2 Pain.
We performed a sensitivity analysis using data for the complete case cohort (N = 128). We estimated the SD from the 95% CI reported in the supplement provided with the published manuscript. We calculated an MD of ‐9.40 mm (95% CI ‐17.87 mm to ‐0.93 mm; Analysis 9.5). The negative sign indicates a lower pain score with methotrexate. This corresponds to an absolute improvement of 9% with methotrexate (95% CI 1% to 18% improvement) and a relative improvement of 25% with methotrexate (95% CI 3% to 48% improvement).
9.5. Analysis.
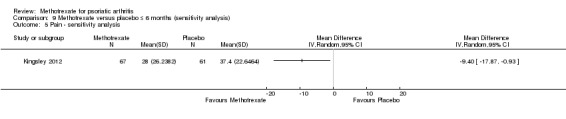
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 5 Pain ‐ sensitivity analysis.
One study (Scarpa 2008: 35 randomised participants; intramuscular methotrexate 10 mg per week (standard low‐dose oral methotrexate 15 mg per week)) assessed pain using a VAS (scale 0 mm to 100 mm; higher scores indicate more pain) and reported values for the ITT cohort (n = 35) at three months. Values reported in the manuscript were median and interquartile range (IQR). We assumed these values had a skewed distribution, and study authors did not respond to our requests for confirmation. We did not estimate the mean nor the SD. The median (IQR) was 50 mm (44) for methotrexate and 32 mm (60) for placebo (NSAIDs). We did not analyse the data further.
A meta‐analysis was not possible. We judged the quality of the evidence to be low (downgraded due to risk of bias and imprecision).
Fatigue
Study authors reported no data for this outcome.
Skin disease
Only one study (Kingsley 2012: 221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) reported this outcome. Study authors provided unpublished data for the ITT cohort (n = 221) upon request. These included imputed values for missing data. We estimated the SD from the SE provided by study authors. At six months, mean skin disease (Psoriasis Area and Severity Index (PASI), scale 0 to 72; higher scores indicate greater burden of psoriasis) in the placebo group was 3.1 points. We calculated an MD of ‐0.92 (95% CI ‐1.90 to 0.06; Analysis 2.3). The negative sign indicates a lower PASI score with methotrexate, which corresponds to an absolute improvement of 1% with methotrexate (95% CI 0% to 3% improvement) and a relative improvement of 29% with methotrexate (95% CI 60% improvement to 2% worse).
2.3. Analysis.
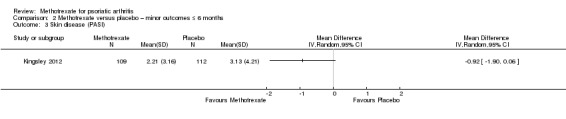
Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 3 Skin disease (PASI).
We performed a sensitivity analysis using unpublished data for the complete case cohort (N = 128) provided by study authors upon request. We calculated an MD of ‐1.38 (95% CI ‐2.69 to ‐0.07; Analysis 9.6). The negative sign indicates a lower PASI score with methotrexate, which corresponds to an absolute improvement of 2% with methotrexate (95% CI 0% to 4% improvement) and a relative improvement of 41% with methotrexate (95% CI 2% to 81% improvement).
9.6. Analysis.
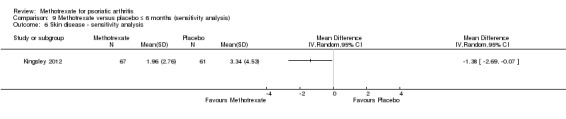
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 6 Skin disease ‐ sensitivity analysis.
We judged the quality of the evidence to be low (downgraded due to indirectness and imprecision).
Total adverse events
Three studies reported total adverse events (AEs) (Kingsley 2012; Scarpa 2008; Willkens 1984).
We analysed the data using the Mantel‐Haenszel method and a random‐effects model. We calculated the RR for experiencing an AE from methotrexate of 2.13 (95% CI 1.27 to 3.59; Analysis 2.4) with low heterogeneity (Chi² = 1.05; df = 1; P = 0.30; I² = 5%). The absolute risk difference was 0.21 (95% CI ‐0.17 to 0.60) (the negative sign indicates more AEs with placebo). We did not calculate the NNTH for this statistically non‐significant event, and we noted considerable heterogeneity (Chi² = 51.55; df = 2; P < 0.00001; I² = 96%).
2.4. Analysis.
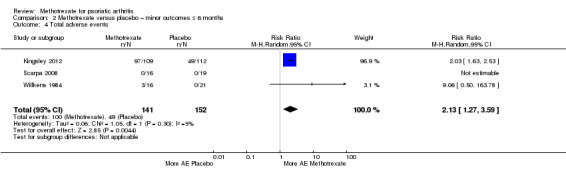
Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 4 Total adverse events.
We performed a sensitivity analysis using a fixed‐effect model. We calculated the RR for experiencing an AE from methotrexate of 2.10 (95% CI 1.68 to 2.62) with low heterogeneity (Chi² = 1.05; df = 1; P = 0.30; I² = 5%). The absolute risk difference was 0.37 (95% CI 0.28 to 0.45), and the NNTH was 3 (95% CI 2 to 3) with considerable heterogeneity (Chi² = 51.55; df = 2; P < 0.00001; I² = 96%).
We judged the quality of the evidence to be low (downgraded due to inconsistency and imprecision).
Patient global assessment of disease activity
One study (Kingsley 2012: 221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) performed patient global assessment of disease activity on a VAS (scale 0 mm to 100 mm; higher scores indicate greater patient‐perceived global disease activity) and reported values for the ITT cohort (n = 221). These included imputed values for missing data. We estimated the SD from the 95% CI reported in the supplement provided with the published manuscript. At six months, mean patient global assessment score in the placebo group was 42.2 mm. We calculated an MD of ‐10.4 mm (95% CI ‐18.67 to ‐2.13; Analysis 2.5). The negative sign indicates a lower assessment score with methotrexate, which shows an absolute improvement of 10% with methotrexate (95% CI 2% to 19% improvement) and a relative improvement of 25% with methotrexate (95% CI 5% to 44% improvement).
2.5. Analysis.

Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 5 Patient global assessment of disease activity.
We performed a sensitivity analysis using data for the complete case cohort (N = 128). We estimated the SD using the 95% CI reported in the supplement provided with the published manuscript. We calculated an MD of ‐9.20 mm (95% CI ‐17.8 to ‐0.59; Analysis 9.7). The negative sign indicates a lower patient global assessment score with methotrexate. This corresponds to an absolute improvement of 9% with methotrexate (95% CI 1% to 18% improvement) and a relative improvement of 22% with methotrexate (95% CI 1% to 44% improvement).
9.7. Analysis.
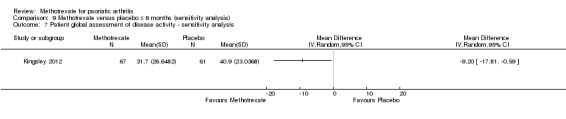
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 7 Patient global assessment of disease activity ‐ sensitivity analysis.
Scarpa 2008 reported patient global assessment of disease activity on a Likert scale from 0 to 5. Researchers reported values for the ITT cohort (n = 35) at three months as median (IQR). We assumed these values had a skewed distribution. Study authors did not respond to our requests for confirmation. We did not estimate mean nor SD. The median (IQR) was 3 (2) for methotrexate and 2 (3) for placebo (NSAIDs). We did not analyse the data further.
We did not perform a meta‐analysis. We judged the quality of the evidence to be very low (downgraded due to risk of bias, inconsistency, and imprecision).
Physician global assessment of disease activity
Kingsley 2012 (221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) performed physician global assessment of disease activity on a VAS (scale 0 to 100 mm; higher scores indicate greater physician‐perceived global disease activity) and reported values for the ITT cohort (n = 221). These included imputed values for missing data. We estimated the SD using the 95% CI from the published manuscript. At six months, the mean physician global assessment score in the placebo group was 33 mm. We calculated an MD of ‐9.70 mm (95% CI ‐15.31 to ‐4.09; Analysis 2.6). The negative sign indicates a lower physician global assessment score for methotrexate, which corresponds to an absolute improvement of 10% with methotrexate (95% CI 4% to 15% improvement) and a relative improvement of 29% with methotrexate (95% CI 12% to 46% improvement).
2.6. Analysis.
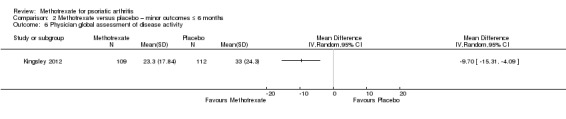
Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 6 Physician global assessment of disease activity.
We performed a sensitivity analysis for Kingsley 2012, using data for the complete case analysis (N = 128). We estimated the SD from the 95% CI reported in the supplement provided with the published manuscript. We calculated an MD of ‐13.8 mm (95% CI ‐20.08 to ‐7.52; Analysis 9.8). The negative sign indicates a lower physician global assessment score for methotrexate, which corresponds to an absolute improvement of 14% with methotrexate (95% CI 8% to 20% improvement) and a relative improvement of 39% with methotrexate (95% CI 21% to 56% improvement).
9.8. Analysis.
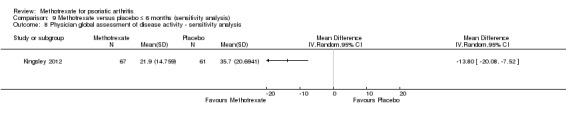
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 8 Physician global assessment of disease activity ‐ sensitivity analysis.
Scarpa 2008 performed physician global assessment of disease activity on a Likert scale of 0 to 5. They included the ITT cohort (n = 35), with values reported at three months as median (IQR). We assumed these values had a skewed distribution, and study authors did not respond to our requests for confirmation. We did not estimate mean nor SD. The median (IQR) was 3 (2) for methotrexate and 2 (3) for placebo (NSAIDs). We did not analyse the data further.
We did not perform a meta‐analysis. We judged evidence quality to be very low (downgraded due to risk of bias, inconsistency, and imprecision).
Swollen joint count
Kingsley 2012 (221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) assessed the number of swollen joints from a 66‐joint examination (scale 0 to 66; higher scores indicate more swollen joints) for the ITT cohort (n = 221). These included imputed values for missing data. We estimated the SD from the 95% CI in the published manuscript. At six months, the mean swollen joint count in the placebo group was 5.2 joints. We calculated an MD of ‐0.60 joints (95% CI ‐2.62 to 1.42; Analysis 2.7). The negative sign indicates fewer swollen joints with methotrexate, which corresponds to an absolute improvement of 1% with methotrexate (95% CI 4% improvement to 2% worse) and a relative improvement of 11% with methotrexate (95% CI 50% improvement to 27% worse).
2.7. Analysis.

Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 7 Swollen joint count.
We performed a sensitivity analysis for Kingsley 2012, using data for the complete case cohort (N = 128). We estimated the SD from the 95% CI reported in the supplement provided with the published manuscript. We calculated an MD of ‐1.4 joints (95% CI ‐4.11 to 1.31; Analysis 9.9). The negative sign indicates fewer swollen joints with methotrexate, which corresponds to an absolute improvement of 2% with methotrexate (95% CI 6% improvement to 2% worse) and a relative improvement of 25% with methotrexate (95% CI 72% improvement to 23% worse).
9.9. Analysis.
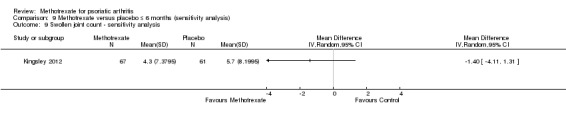
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 9 Swollen joint count ‐ sensitivity analysis.
Scarpa 2008 assessed the number of swollen joints but did not specify how many joints they examined. They reported data for the ITT cohort at three months (n = 35) as median (IQR). We assumed that these values had a skewed distribution. Study authors did not respond to our requests for confirmation. We did not estimate mean nor SD. The median (IQR) was 0 joints (1) for methotrexate and 1 joint (2) for placebo (NSAIDs). We did not analyse the data further.
We did not perform a meta‐analysis, and we judged evidence quality to be very low (downgraded due to risk of bias, inconsistency, and imprecision).
Tender joint count
Kingsley 2012 (221 randomised participants; oral methotrexate 15 mg per week (standard low‐dose)) assessed the number of tender joints from a 68‐joint examination (scale 0 to 68; higher scores indicate more tender joints) for the ITT cohort (n = 221). These included imputed values for missing data. We estimated the SD from the 95% CI provided in the published manuscript. At six months, mean tender joint count in the placebo group was 10 joints. We calculated an MD of ‐2.3 joints (95% ‐5.5 to 0.9; Analysis 2.8). The negative sign indicates fewer tender joints with methotrexate, which corresponds to an absolute improvement of 3% with methotrexate (95% CI 8% improvement to 1% worse) and a relative improvement of 23% with methotrexate (95% CI 55% improvement to 9% worse).
2.8. Analysis.
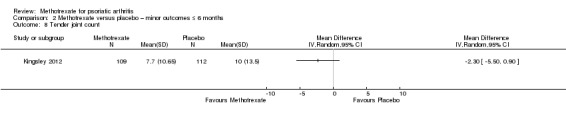
Comparison 2 Methotrexate versus placebo – minor outcomes ≤ 6 months, Outcome 8 Tender joint count.
We performed a sensitivity analysis for Kingsley 2012, using data for the complete case cohort (N = 128). We estimated the SD from the 95% CI reported in the supplement provided with the published manuscript. We calculated an MD of ‐2.80 joints (95% CI ‐6.55 to 0.95; Analysis 9.10). The negative sign indicates fewer tender joints with methotrexate, which corresponds to an absolute improvement of 4% with methotrexate (95% CI 10% improvement to 1% worse) and a relative improvement of 26% with methotrexate (95% CI 62% improvement to 9% worse).
9.10. Analysis.
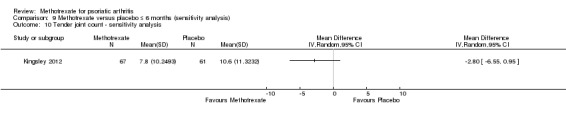
Comparison 9 Methotrexate versus placebo ≤ 6 months (sensitivity analysis), Outcome 10 Tender joint count ‐ sensitivity analysis.
Scarpa 2008 assessed the number of tender joints but did not specify how many joints they examined. They reported data for the ITT cohort (n = 35) at three months as median (IQR). We assumed that these values had a skewed distribution. Study authors did not respond to our requests for confirmation. We did not estimate mean nor SD. The median (IQR) was 1 joint (1) for methotrexate and 2 joints (3) for placebo (NSAIDs). We did not analyse the data further.
We did not perform a meta‐analysis, and we judged evidence quality to be very low (downgraded due to risk of bias, inconsistency, and imprecision).
Methotrexate versus placebo (longer than six months)
Only one study with a placebo comparator reported outcomes beyond six months (Burdeinyi 1992). We extracted data only for WAEs and total AEs. Researchers either did not report the other outcomes in any way, or they reported the data in a format that was not extractable. We note that many of the specified outcome measures were not created until after this study was published. Study authors could not be contacted.
Major outcomes (comparison 3)
Disease response (American College of Rheumatology response criteria for 50% improvement (ACR50), PsARC)
Study authors reported no data for this outcome.
Function
Study authors reported no data for this outcome.
Health‐related quality of life
Study authors reported no data for this outcome.
Disease activity
Study authors reported no data for this outcome.
Radiographic progression
Study authors reported no data for this outcome.
Serious adverse events
Study authors reported no data for this outcome.
Withdrawals due to adverse events
Burdeinyi 1992 (72 randomised participants; oral methotrexate 10 mg per week (standard dose 15 mg per week)) reported WAEs at 12 months in the placebo (NSAIDs) arm. For methotrexate, they reported 12 WAEs among 31 participants, and for placebo, 0 WAEs among 41. We calculated the RR for WAEs due to methotrexate of 32.81 (95% CI 2.02 to 533.71; Analysis 3.1), an absolute risk difference of 0.39 (95% CI 0.21 to 0.56), and an NNTH of 3 (95% CI 3 to 5).
3.1. Analysis.

Comparison 3 Methotrexate versus placebo ‐ major outcomes > 6 months, Outcome 1 Withdrawals due to adverse events.
We judged evidence quality to be very low (downgraded due to risk of bias, indirectness, and imprecision).
Minor outcomes (comparison 4)
Disease response (ACR20)
Study authors reported no data for this outcome.
Enthesitis
Study authors reported no data for this outcome.
Dactylitis
Study authors reported no data for this outcome.
Pain
Study authors reported no data for this outcome.
Fatigue
Study authors reported no data for this outcome.
Skin disease
Study authors reported no data for this outcome.
Total adverse events
Burdeinyi 1992 (72 randomised participants; oral methotrexate 10 mg per week (standard dose 15 mg per week)) reported total AEs at 12 months in the placebo (NSAIDs) arm. For methotrexate, 17 of 31 participants experienced AEs, and for placebo, 15 of 41 experienced AEs. We calculated the RR for experiencing an AE from methotrexate of 1.50 (95% 0.90 to 2.51; Analysis 4.1) and an absolute risk difference of 0.18 (95% CI ‐0.05 to 0.41). We did not calculate an NNTH for this statistically non‐significant result.
4.1. Analysis.
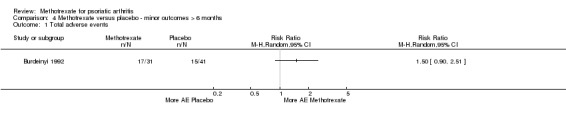
Comparison 4 Methotrexate versus placebo ‐ minor outcomes > 6 months, Outcome 1 Total adverse events.
We judged evidence quality to be very low (downgraded due to risk of bias, indirectness, and imprecision).
Patient global assessment of disease activity
Study authors reported no data for this outcome.
Physician global assessment of disease activity
Study authors reported no data for this outcome.
Swollen joint count
Study authors reported no data for this outcome.
Tender joint count
Study authors reported no data for this outcome.
Methotrexate versus other DMARDs (up to six months)
Three studies with another DMARD comparator reported outcomes up to six months (Asaduzzaman 2014; Spadaro 1995; Zhang 2009). Not all studies reported all outcomes. We judged the overall risk of bias for both Spadaro 1995 and Zhang 2009 to be too high to permit meaningful meta‐analysis.
Major outcomes (comparison 5)
Disease response (ACR50, PsARC)
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported the number of ACR50 responders at six months. For methotrexate, results show that 12 of 14 participants were responders, and for placebo, 13 of 16 were responders. We calculated the RR for achieving an ACR50 response with methotrexate at 1.05 (95% CI 0.77 to 1.45; Analysis 5.1) and an absolute risk difference of 0.04 (95% CI ‐0.22 to 0.31). We did not calculate an NNTB for this statistically non‐significant result.
5.1. Analysis.
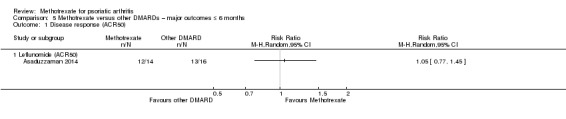
Comparison 5 Methotrexate versus other DMARDs – major outcomes ≤ 6 months, Outcome 1 Disease response (ACR50).
Asaduzzaman 2014 also reported the number of PsARC responders at six months. For methotrexate, results show that 14 of 14 participants were responders, and for placebo, 16 of 16 were responders. We calculated an RR of 0.00 (95% CI 0.88 to 0.13; Analysis 10.1) and an absolute risk difference of 0.00 (95% CI ‐0.12 to 0.12). We did not calculate an NNTB for this statistically non‐significant result.
10.1. Analysis.
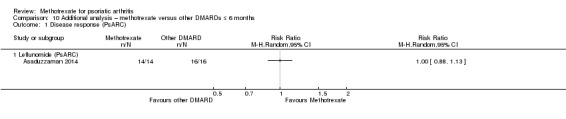
Comparison 10 Additional analysis – methotrexate versus other DMARDs ≤ 6 months, Outcome 1 Disease response (PsARC).
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported data for the number of PsARC responders; however, values reported in the written text of the published manuscript were different from those presented in the graph. We were uncertain which values were correct. Study authors did not respond to our request for clarification, and we did not extract the data.
We included ACR50 results from Asaduzzaman 2014 in Table 2. We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Study authors reported no data for this outcome.
Function
Leflunomide
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) was the only study that used a leflunomide comparator to report this outcome. Study authors reported HAQ scores (scale 0 to 3; higher scores indicate greater disability) for the ITT cohort (n = 31). At six months, mean function in the leflunomide group was 0.17 points. We calculated a mean difference of ‐0.13 (95% CI ‐0.23 to ‐0.03; Analysis 5.2). The negative sign indicates a lower HAQ score with methotrexate. This corresponds to an absolute improvement of 4% with methotrexate (95% CI 1% to 8% improvement) and a relative improvement of 76% (95% CI 18% to 135% improvement). We included the results of this analysis in Table 2.
5.2. Analysis.
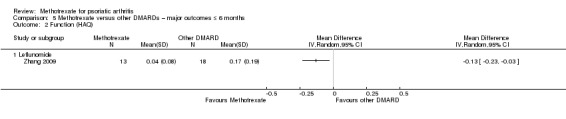
Comparison 5 Methotrexate versus other DMARDs – major outcomes ≤ 6 months, Outcome 2 Function (HAQ).
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Study authors reported no data for this outcome.
Health‐related quality of life
Leflunomide
Study authors reported no data for this outcome.
Ciclosporin A
Study authors reported no data for this outcome.
Disease activity
Leflunomide
Study authors reported no data for this outcome.
Ciclosporin A
Study authors reported no data for this outcome.
Radiographic progression
Leflunomide
Study authors reported no data for this outcome.
Ciclosporin A
Study authors reported no data for this outcome.
Serious adverse events
Leflunomide
Both Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) and Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported zero SAEs for both methotrexate and leflunomide groups. We did not perform a meta‐analysis because it was not meaningful in this circumstance.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week), oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported zero SAEs for both methotrexate and ciclosporin A groups. We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Withdrawals due to adverse events
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported zero WAEs in both methotrexate and leflunomide groups. We were unable to calculate the absolute risk difference or the risk ratio.
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported WAEs at six months. For methotrexate, results show one WAE among 13 participants, and for leflunomide, two WAEs among 18. We calculated the RR for WAE for methotrexate of 0.69 (95% CI 0.07 to 6.85; Analysis 5.4) and an absolute risk difference of ‐0.03 (95% CI ‐0.24 to 0.17). We did not calculate an NNTH for this statistically non‐significant result. We included the results of this analysis in Table 2.
5.4. Analysis.
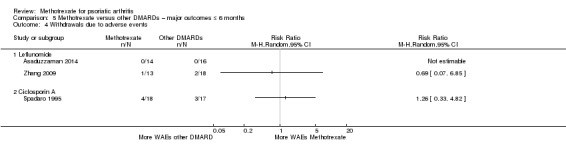
Comparison 5 Methotrexate versus other DMARDs – major outcomes ≤ 6 months, Outcome 4 Withdrawals due to adverse events.
We did not perform a meta‐analysis, and we judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported WAEs at six months. For methotrexate, results show four WAEs among 18 participants, and for ciclosporin A, three WAEs among 17. We calculated an RR for WAEs from methotrexate of 1.26 (95% CI 0.33 to 4.82; Analysis 5.4) and an absolute risk difference of 0.05 (95% CI ‐0.22 to 0.31). We did not calculate an NNTH for this statistically non‐significant result, and we included the results of this analysis in Table 2.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Minor outcomes (comparison 6)
Disease response (ACR20)
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported the number of participants with a disease response. For methotrexate, the number of ACR20 responders was 14 of 14, and for leflunomide, results show 16 of 16 responders. We calculated an RR for achieving an ACR20 response with methotrexate of 1.00 (95% CI 0.88 to 1.13; Analysis 6.1) and an absolute risk difference of 0% (95% CI ‐0.12 to 0.12). We did not calculate an NNTB for this statistically non‐significant result.
6.1. Analysis.
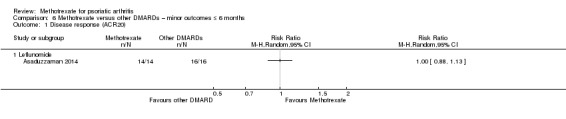
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 1 Disease response (ACR20).
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported data at six months; however, values reported in the written text were different from those presented in the graph. We were uncertain which values were correct. Study authors did not respond to our request for clarification, and we did not extract the data.
A meta‐analysis was not possible. We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Study authors reported no data for this outcome.
Enthesitis
Leflunomide
Study authors reported no data for this outcome.
Ciclosporin A
Study authors reported no data for this outcome.
Dactylitis
Leflunomide
Study authors reported no data for this outcome.
Ciclosporin A
Study authors reported no data for this outcome.
Pain
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported pain outcomes from a VAS (scale 0 to 10 cm; higher scores indicate more pain) for the ITT cohort (n = 30). At six months, mean pain in the leflunomide group was rated as 1 cm. We calculated an MD of ‐0.07 cm (95% CI ‐0.74 to 0.60; Analysis 6.2). The negative sign indicates a lower pain score with methotrexate, which corresponds to an absolute improvement of 1% with methotrexate (95% CI 7% improvement to 6% worse) and a relative improvement of 7% with methotrexate (95% CI 74% improvement to 60% worse).
6.2. Analysis.
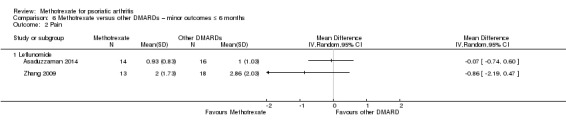
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 2 Pain.
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported pain outcomes from a VAS but did not specify the length. Study authors did not respond to our request for clarification, and we made no assumptions about the scale. At six months, mean pain in the leflunomide group was 1 unit. We calculated an MD of ‐0.86 units (95% CI ‐2.19 to 0.47; Analysis 6.2). The negative sign indicates a lower pain score with methotrexate, which corresponds to a relative improvement of 30% with methotrexate (95% CI 77% improvement to 16% worse).
We did not perform a meta‐analysis, and we judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Study authors reported no data for this outcome.
Fatigue
Leflunomide
Study authors reported no data for this outcome.
Ciclosporin A
Study authors reported no data for this outcome.
Skin disease
Leflunomide
Only Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported this outcome for the leflunomide subgroup, using PASI as their measure (scale 0 to 72; higher scores indicate greater burden of psoriasis) for the ITT cohort (n = 30). At six months, mean PASI score in the leflunomide group was 2.69 points. We calculated an MD of ‐0.01 (95% CI ‐1.18 to 1.16; Analysis 6.3). The negative value indicates a lower PASI score with methotrexate, which corresponds to an absolute change of 0% (95% CI 2% improvement to 2% worse) and a relative change of 0% (95% CI 44% improvement to 43% worse).
6.3. Analysis.
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 3 Skin disease.
We judged evidence quality to be very low (downgraded due to risk of bias, indirectness, and imprecision).
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported this outcome using PASI as their measure (scale 0 to 72; higher scores indicate greater burden of psoriasis) for the per‐protocol cohort (n = 28). At six months, mean PASI score in the ciclosporin A group was 4.2 points. We calculated an MD of ‐1.10 (95% CI ‐1.73 to ‐0.47; Analysis 6.3). The negative sign indicates a lower PASI score with methotrexate, which corresponds to an absolute improvement of 1.5% with methotrexate (95% CI 1% to 2% improvement) and a relative improvement of 26% with methotrexate (95% CI 11% to 41% improvement).
We judged evidence quality to be very low (downgraded due to risk of bias, indirectness, and imprecision).
Total adverse events
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported total AEs at six months. For methotrexate, results show 27 AEs among 14 participants, and for leflunomide, 25 AEs among 16. We were unable to calculate an absolute risk difference or risk ratio.
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported total AEs at six months. For methotrexate, results show five AEs among 13 participants, and for leflunomide, seven AEs among 18. We calculated the RR for experiencing an AE from methotrexate of 0.99 (95% CI 0.40 to 2.43; Analysis 6.4) and an absolute risk difference of 0.00 (95% CI ‐0.35 to 0.34). We did not calculate an NNTH for this statistically non‐significant result.
6.4. Analysis.
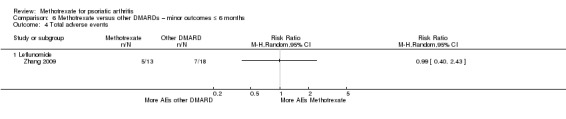
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 4 Total adverse events.
A meta‐analysis was not possible. We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Study authors reported no data for this outcome.
Patient global assessment of disease activity
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported patient global assessment of disease activity on a Likert scale (scale 1 to 5; higher scores indicate greater patient‐perceived global disease activity) for the ITT cohort (n = 30) at six months. The mean for both groups was 2, but the SD for both groups was 0. Study authors did not respond to our request for clarification. We were not able to calculate the MD nor the 95% CI.
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) did not indicate the measurement instrument used for patient global assessment of disease activity. Study authors did not respond to requests for clarification, and we made no assumptions about scale length, but we assumed that higher scores indicated greater patient‐perceived global disease activity. At six months, mean patient global assessment in the leflunomide group was 2.39 points. We calculated an MD of ‐0.96 (95% CI ‐1.64 to ‐0.28; Analysis 6.5). The negative sign indicates a lower disease activity score with methotrexate, which corresponds to a relative improvement of 40% with methotrexate (95% CI 12% to 69% improvement).
6.5. Analysis.
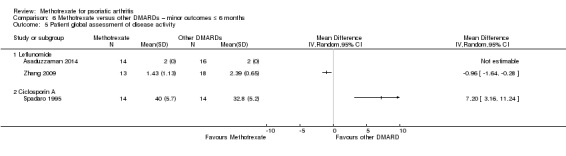
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 5 Patient global assessment of disease activity.
A meta‐analysis was not possible. We judged evidence quality to be very low (downgraded due to risk of bias, inconsistency, and imprecision).
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported patient global assessment on a VAS (scale 0 to 100 mm; higher scores indicate greater patient‐perceived global disease activity) for the per‐protocol cohort (n = 28). At six months, the mean patient global assessment score for the ciclosporin A group was 32.8 mm. We calculated an MD of 7.20 mm (95% CI 3.16 to 11.24; Analysis 6.5), indicating a higher score for methotrexate, which corresponds to an absolute worsening of 7% with methotrexate (95% CI 3% to 11% worse) and a relative worsening of 22% with methotrexate (95% CI 10% to 34% worse).
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Physician global assessment of disease activity
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported physician global assessment of disease activity on a Likert scale (scale 1 to 5; higher scores indicate greater physician‐perceived global disease activity) for the ITT cohort (n = 30) at six months. The mean for both groups was 2, but the SD for both groups was 0. Study authors did not respond to our request for clarification, and we did not impute the missing data. We were not able to calculate the MD nor the 95% CI.
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported physician global assessment for disease activity but did not describe the measurement instrument used. Study authors did not respond to our request for clarification, and we made no assumptions about the scale. They reported data for the ITT cohort (n = 31). At six months, the mean physician global assessment score in the leflunomide group was 2.39 points. We calculated a mean difference of ‐0.30 (95% CI ‐0.96 to 0.36; Analysis 6.6). The negative sign indicates a lower assessment score with methotrexate, which corresponds to a relative improvement of 13% with methotrexate (95% CI 40% improvement to 15% worse).
6.6. Analysis.
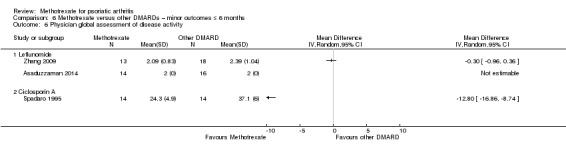
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 6 Physician global assessment of disease activity.
A meta‐analysis was not possible. We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported physician global assessment of disease activity from a VAS (scale 0 to 100 mm; higher scores indicate greater physician‐perceived global disease activity) for the per‐protocol cohort (n = 28). At six months, the mean physician global assessment score in the ciclosporin A group was 37.1 mm. We calculated a mean difference of ‐12.80 (95% CI ‐16.86 to ‐8.74; Analysis 6.6). The negative sign indicates a lower assessment score with methotrexate, which corresponds to an absolute improvement of 13% with methotrexate (95% CI 9% to 17% improvement) and a relative improvement of 35% with methotrexate (95% CI 24% to 45% improvement).
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Swollen joint count
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported the number of swollen joints from a 66‐joint examination (scale 0 to 66; higher scores indicate more swollen joints) for the ITT cohort (n = 30). At six months, the mean swollen joint count in the leflunomide group was 0.5 joints. We calculated a mean difference of 0.14 (95% CI ‐0.36 to 0.64; Analysis 6.7). The negative sign indicates fewer swollen joints with methotrexate, which corresponds to an absolute worsening of 0.2% with methotrexate (95% CI 0.5% improvement to 1% worse) and a relative worsening of 28% with methotrexate (95% CI 72% improvement to 128% worse).
6.7. Analysis.
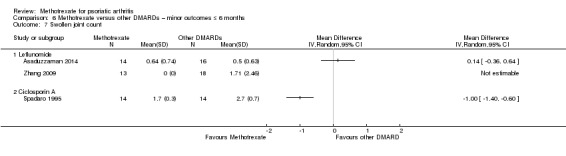
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 7 Swollen joint count.
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported the number of swollen joints from a 74‐joint examination (scale 0 to 74; higher scores indicate more swollen joints) for the ITT cohort (n = 31). Study authors reported an SD of 0 for the methotrexate group. They did not respond to our request for clarification, and we were not able to calculate an MD.
A meta‐analysis was not possible. We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) did not indicate the number of joints from their examination but reported outcomes for the per‐protocol cohort (n = 28) at six months. Study authors were not able to provide clarification. We assumed a 66‐joint count (scale 0 to 66; higher scores indicate more swollen joints) because this is the number of joints examined by Asaduzzaman 2014 and Kingsley 2012. At six months, the mean swollen joint count in the ciclosporin A group was 2.7 joints. We calculated an MD of ‐1.00 joints (95% CI ‐1.40 to ‐0.60; Analysis 6.7). The negative sign indicates fewer swollen joints with methotrexate, which corresponds to an absolute improvement of 1.5% with methotrexate (95% CI 1% to 2% improvement) and a relative improvement of 37% with methotrexate (95% CI 22% to 52% improvement).
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Tender joint count
Leflunomide
Asaduzzaman 2014 (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported the number of tender joints from a 68‐joint examination (scale 0 to 68; higher scores indicate more tender joints) for the ITT cohort (n = 30). At six months, the mean tender joint count for the leflunomide group was 1 joint. We calculated an MD of 0.33 (95% CI ‐0.36 to 1.02; Analysis 6.8). The negative sign indicates fewer tender joints with methotrexate, which corresponds to an absolute worsening of 0.5% with methotrexate (95% CI 0.5% improvement to 2% worse) and a relative worsening of 33% with methotrexate (95% CI 36% improvement to 102% worse).
6.8. Analysis.
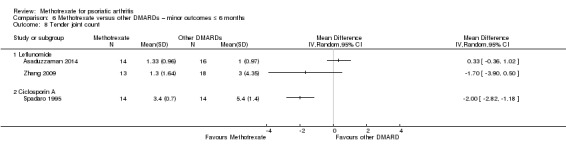
Comparison 6 Methotrexate versus other DMARDs – minor outcomes ≤ 6 months, Outcome 8 Tender joint count.
Zhang 2009 (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week); oral leflunomide 20 mg daily (standard dose)) reported the number of swollen joints from a 76‐joint examination (scale 0 to 76; higher scores indicate more tender joints) for the ITT cohort (n = 31). At six months, the mean tender joint count in the leflunomide group was three joints. We calculated an MD of ‐1.70 (95% CI ‐3.90 to 0.50; Analysis 6.8). The negative sign indicates fewer tender joints with methotrexate, which corresponds to an absolute improvement of 2% with methotrexate (95% CI 5% improvement to 1% worse) and a relative improvement of 57% with methotrexate (95% CI 130% improvement to 17% worse).
We did not perform a meta‐analysis, and we judged evidence quality to be very low (downgraded due to risk of bias, inconsistency, and imprecision).
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) did not indicate the number of joints from their examination but reported outcomes for the per‐protocol cohort (n = 28). Study authors were not able to provide clarification. We assumed a 68‐joint count because this is the number of joints examined by Asaduzzaman 2014 and Kingsley 2012. At six months, mean tender joint count in the ciclosporin A group was 5.4 joints. We calculated an MD of ‐2.00 joints (95% CI ‐2.82 to ‐ 1.18; Analysis 6.8). The negative sign indicates fewer tender joints with methotrexate, which corresponds to an absolute improvement of 3% with methotrexate (95% CI 2% to 4% improvement) and a relative improvement of 37% with methotrexate (95% CI 22% to 52% improvement).
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Methotrexate versus other DMARDs (longer than six months)
We identified two studies for this category (Burdeinyi 1992; Spadaro 1995). Studies did not report all outcomes. In the case of Burdeinyi 1992, study authors actually collected data for many of our specified outcomes but did not report them in an extractable way. Study authors could not be contacted or were unable to provide additional information.
Major outcomes (comparison 7)
Disease response (ACR50, PsARC)
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Function
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Health‐related quality of life
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Disease activity
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Radiographic progression
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Serious adverse events
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported zero SAEs for both methotrexate and ciclosporin groups for the ITT cohort (n = 35) at 12 months. We were not able to calculate the absolute risk difference and the risk ratio.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Withdrawals due to adverse events
Although data for WAEs could be extracted from both studies, we judged risk of bias for most domains to be uncertain or high. We believe it is less informative for results to be pooled rather than reported as individual DMARD comparators. We did not pool the data to perform a meta‐analysis.
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported WAEs for the ITT cohort (n = 35) at 12 months. For methotrexate, five WAEs occurred in 18 participants, and for placebo, results show five WAEs among 17. We calculated the RR for WAE from methotrexate of 0.94 (95% CI 0.33 to 2.69; Analysis 7.2) and an absolute risk difference of ‐0.02 (95% CI ‐0.32 to 0.28). We did not calculate an NNTH for this statistically non‐significant result.
7.2. Analysis.
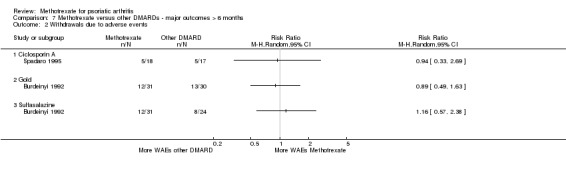
Comparison 7 Methotrexate versus other DMARDs ‐ major outcomes > 6 months, Outcome 2 Withdrawals due to adverse events.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Gold
Burdeinyi 1992 (61 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); intramuscular gold 34 mg weekly (unclear of standard dose)) reported WAEs for the ITT cohort (n = 61) at 12 months. For methotrexate, they showed 12 WAEs among 31 participants, and for gold, 13 WAEs among 30. We calculated the RR for WAEs from methotrexate of 0.89 (95% CI 0.49 to 1.63; Analysis 7.2) and an absolute risk difference of ‐0.05 (95% CI ‐0.29 to 0.20). We did not calculate an NNTH for this statistically non‐significant result.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Sulfasalazine
Burdeinyi 1992 (55 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral sulfasalazine 1 g twice daily (standard dose)) reported WAEs for the ITT cohort (n = 55) at 12 months. For methotrexate, they show 12 WAEs among 31 participants, and for sulfasalazine, 8 WAEs among 24. We calculated the RR for WAEs from methotrexate of 1.16 (95% CI 0.57 to 2.38; Analysis 7.2) and an absolute risk difference of 0.05 (95% CI ‐0.20 to 0.31).
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Minor outcomes (comparison 8)
Disease response (ACR20)
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Enthesitis
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Dactylitis
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Pain
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Fatigue
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Skin disease
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported effects on skin disease as measured by the PASI (scale 0 to 72; higher scores indicate greater burden of psoriasis) for the per‐protocol cohort (n = 23). At 12 months, the mean PASI score in the ciclosporin A group was 3.5 points. We calculated an MD of ‐0.60 (95% CI ‐1.43 to 0.23; Analysis 8.1). The negative sign indicates a lower PASI score with methotrexate, which corresponds to an absolute improvement of 1% with methotrexate (95% CI 2% improvement to 0.5% worse) and a relative improvement of 17% with methotrexate (95% CI 41% improvement to 7% worse).
8.1. Analysis.
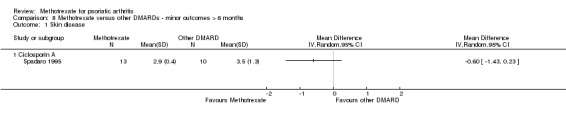
Comparison 8 Methotrexate versus other DMARDs ‐ minor outcomes > 6 months, Outcome 1 Skin disease.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Total adverse events
Although we were able to extract data for total AEs from both studies, we judged risk of bias for most domains to be uncertain or high. We believe it is less informative for results to be pooled rather than reported as individual DMARD comparators. We did not pool the data to perform a meta‐analysis.
Ciclosporin A
Study authors reported no data for this outcome.
Gold
Burdeinyi 1992 (61 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); intramuscular gold 34 mg weekly (unclear of standard dose)) reported total AEs for the ITT cohort (n = 61) at 12 months. For methotrexate, results show 17 AEs among 31 participants, and for gold, 16 AEs among 30. We calculated the RR for experiencing an AE with methotrexate of 1.03 (95% CI 0.65 to 1.63; Analysis 8.2) and an absolute risk difference of 0.02 (95% CI ‐0.24 to 0.27). We did not calculate an NNTH for this statistically non‐significant result.
8.2. Analysis.
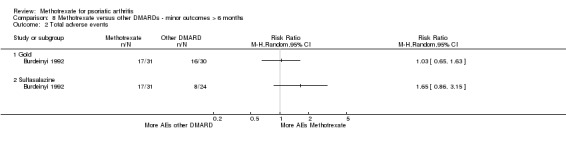
Comparison 8 Methotrexate versus other DMARDs ‐ minor outcomes > 6 months, Outcome 2 Total adverse events.
We judged evidence quality to be very low (downgraded due to risk of bias, indirectness, and imprecision).
Sulfasalazine
Burdeinyi 1992 (55 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week); oral sulfasalazine 1 g twice daily (standard dose)) reported total AEs for the ITT cohort (n = 55) at 12 months. For methotrexate, results show 17 AEs among 31 participants, and for sulfasalazine, 8 AEs among 24. We calculated the RR for experiencing an AE with methotrexate of 1.65 (95% CI 0.86 to 3.15; Analysis 8.2) and an absolute risk difference of 0.22 (95% CI ‐0.04 to 0.47).
We judged evidence quality to be very low (downgraded due to risk of bias, indirectness, and imprecision).
Patient global assessment of disease activity
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported patient global assessment of disease activity on a VAS (scale 0 to 100 mm; higher scores indicate greater patient‐perceived global disease activity) for the per‐protocol cohort (n = 23). At 12 months, the mean patient global assessment score in the ciclosporin A group was 27 mm. We calculated an MD of 3.00 mm (95% CI ‐0.79 to 6.79; Analysis 8.3). The negative sign indicates a lower assessment score with methotrexate, which corresponds to an absolute worsening of 3% with methotrexate (95% CI 1% improvement to 7% worse) and a relative worsening of 11% with methotrexate (95% CI 3% improvement to 25% worse).
8.3. Analysis.
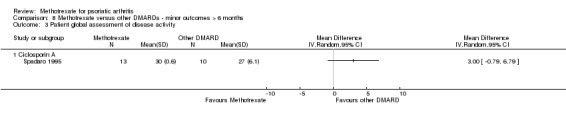
Comparison 8 Methotrexate versus other DMARDs ‐ minor outcomes > 6 months, Outcome 3 Patient global assessment of disease activity.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Physician global assessment of disease activity
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported physician global assessment of disease activity on a VAS (scale 0 to 100 mm; higher scores indicate greater physician‐perceived global disease activity) for the per‐protocol cohort (n = 23). At 12 months, the mean physician global assessment score was 41 mm. We calculated an MD of ‐14.90 mm (95% CI ‐20.23 to ‐9.57; Analysis 8.4). The negative sign indicates a lower assessment score with methotrexate, which corresponds to an absolute improvement of 15% with methotrexate (95% CI 10% to 20% improvement) and a relative improvement of 36% with methotrexate (95% CI 23% lower to 49% lower).
8.4. Analysis.
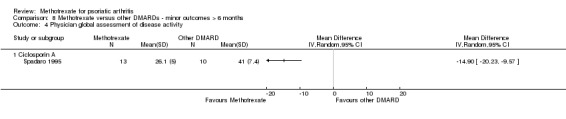
Comparison 8 Methotrexate versus other DMARDs ‐ minor outcomes > 6 months, Outcome 4 Physician global assessment of disease activity.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Swollen joint count
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported the number of swollen joints but did not specify the number of joints examined. Study authors were not able to provide clarification. We assumed a 66‐joint count (scale 0 to 66; higher scores indicate more swollen joints) because this was the number of joints examined by Asaduzzaman 2014 and Kingsley 2012. They reported data for the per‐protocol cohort (n = 23). At 12 months, the mean swollen joint count in the ciclosporin A group was 2.5 joints. We calculated an MD of ‐1.70 joints (95% CI ‐2.21 to ‐1.19; Analysis 8.5). The negative sign indicates fewer swollen joints with methotrexate, which corresponds to an absolute improvement of 2.5% with methotrexate (95% CI 2% to 3% improvement) and a relative improvement of 68% (95% CI 48% to 88% improvement).
8.5. Analysis.

Comparison 8 Methotrexate versus other DMARDs ‐ minor outcomes > 6 months, Outcome 5 Swollen joint count.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Tender joint count
Ciclosporin A
Spadaro 1995 (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week); oral ciclosporin 3 to 5 mg/kg daily (standard dose)) reported the number of tender joints but did not specify the number of joints examined. Study authors were not able to provide clarification. We assumed a 68‐joint count (scale 0 to 68; higher scores indicate more tender joints) because this was the number of joints examined by Asaduzzaman 2014 and Kingsley 2012. They reported data for the per‐protocol cohort (n = 23). At 12 months, the mean tender joint count in the ciclosporin A group was 5.9 joints. We calculated an MD of ‐3.90 joints (95% CI ‐5.05 to ‐2.75; Analysis 8.6). The negative sign indicates fewer tender joints with methotrexate, which corresponds to an absolute improvement of 6% with methotrexate (95% CI 4% to 7% improvement) and a relative improvement of 66% with methotrexate (95% CI 47% to 86% improvement).
8.6. Analysis.
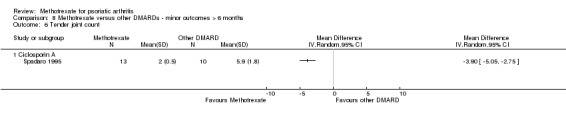
Comparison 8 Methotrexate versus other DMARDs ‐ minor outcomes > 6 months, Outcome 6 Tender joint count.
We judged evidence quality to be very low (downgraded due to risk of bias and imprecision).
Gold
Study authors reported no data for this outcome.
Sulfasalazine
Study authors reported no data for this outcome.
Subgroup analyses
We were not able to complete our planned subgroup analyses because we could not extract the required data.
Discussion
Summary of main results
We identified eight randomised controlled trials (RCTs) that assessed the benefits and safety of methotrexate for psoriatic arthritis. Five studies compared methotrexate to placebo (Black 1964; Burdeinyi 1992; Kingsley 2012; Scarpa 2008; Willkens 1984), whereas four compared methotrexate to another disease‐modifying anti‐rheumatic drug (DMARD) (Asaduzzaman 2014; Burdeinyi 1992; Spadaro 1995; Zhang 2009). One of these studies was a multi‐armed trial comparing placebo, methotrexate, gold, and sulfasalazine. Six studies reported outcomes up to six months, and two studies reported outcomes up to 12 months. We summarised results for major outcomes for the main comparisons in Table 1 and Table 2.
Methotrexate versus placebo (up to six months)
Summary
Compared with placebo, low‐quality evidence based on one trial (221 randomised participants; oral methotrexate 15 mg per week (standard low dose); Kingsley 2012) suggests that methotrexate treatment might provide clinically meaningful benefit with respect to disease response (Psoriatic Arthritis Response Criteria (PsARC)), function, pain, and patient and physician global assessments of disease activity. However, trial results demonstrate no clinically important benefit with respect to disease response (American College of Rheumatology response criteria for 20% improvement (ACR20)), disease activity score (28 joints) with erythrocyte sedimentation rate (DAS28‐ESR), tender and swollen joint counts, or skin disease.
The same trial measured health‐related quality of life but did not report the results (Kingsley 2012). No study measured radiographic progression, enthesitis, dactylitis, or fatigue.
It is difficult to explain why some outcomes confer benefit and others do not. We expected to see a consistent effect if true benefit was derived. One possibility is that Kingsley 2012 was underpowered to identify statistically significant differences. This hypothesis is supported by the fact that both function and disease response (PsARC) were sensitive to missing data (analyses using imputed data vs analyses without imputation).
Another possibility is that methotrexate must be taken at a higher dose for a longer duration to demonstrate efficacy. Kingsley 2012 reached a dose of 15 mg in 78% of participants, with only 11% taking higher doses. Although use of higher doses of methotrexate may be more efficacious than use of lower doses, as suggested by Coates 2015b and Coates 2017b, a placebo‐controlled trial of methotrexate at these doses has not been conducted. Further limitations of Kingsley 2012 are discussed in the published manuscript of this trial and in a review by Pincus 2015.
Although the Pincus systematic review did not analyse whether methotrexate augments the effects of other DMARDs, several studies suggest no additional effect on disease response when DMARDs are combined with biologic DMARDs (Antoni 2005; Kavanaugh 2009; Kavanaugh 2017; McInnes 2015; Mease 2005).
Compared to placebo, low‐quality evidence based on four trials (293 randomised participants) suggests that methotrexate may be more harmful in terms of causing any adverse events, but we are uncertain if these events are more serious or lead to drug cessation (Black 1964; Kingsley 2012; Scarpa 2008; Willkens 1984).
Major outcomes
Kingsley 2012 measured health‐related quality of life but did not report the results. No studies measured radiographic progression.
Low‐quality evidence from one trial (221 randomised participants; oral methotrexate 15 mg per week (standard low dose)) suggests that at six months, mean function (Health Assessment Questionnaire for Rheumatoid Arthritis (HAQ); scale 0 to 3; higher scores indicate greater disability) was rated at 1 point with placebo and 0.30 points lower (0.09 lower to 0.51 lower) with methotrexate, for an absolute improvement of 10% (3% to 17% improvement) and a relative improvement of 30% (9% improvement to 51% improvement) (Kingsley 2012). This improvement is likely to be clinically meaningful (minimum clinically important absolute difference is 0.22 points), but the 95% confidence interval (CI) includes clinically unimportant results. This imprecision limits our certainty in the results. Methotrexate might provide clinically meaningful benefit for function (Table 1).
Low‐quality evidence from the same trial (221 randomised participants; oral methotrexate 15 mg per week (standard low dose)) suggests that at six months, mean disease activity (DAS28‐ESR; scale 0 to 10; higher scores indicate more disease activity) was 4.1 points for placebo and 0.26 points lower (0.65 lower to 0.13 higher) for methotrexate, for an absolute improvement of 3% (7% better to 1% worse) and relative improvement of 6% (16% better to 3% worse) (Kingsley 2012). The 95% CI included potential worsening of disease activity. Imprecision for this outcome limits our certainty in the results. The minimum clinically important difference for DAS28‐ESR has not been defined, but for comparison in rheumatoid arthritis, it is considered to be 1.2 points. Our results suggest that methotrexate may provide no clinically important benefit for disease activity (Table 1).
Although disease response on ACR20 was a minor outcome, this is best interpreted through direct comparison with disease response on PsARC. Results for disease response (PsARC and ACR20; measured as responder/non‐responder, response‐benefit) show inconsistency. One trial (221 randomised participants; oral methotrexate 15 mg per week (standard low dose)) reported that more participants taking methotrexate achieved a PsARC response than those taking placebo (41/109 (or 38 per 100) with methotrexate, 24/112 (or 21 per 100) with placebo; risk ratio (RR) 1.76, 95% CI 1.14 to 2.70; absolute risk difference 0.16, 95% CI 0.04 to 0.28; number needed to treat for an additional beneficial outcome (NNTB) 6, 95% CI 4 to 25) (Kingsley 2012). Our analysis included unpublished data provided by the Kingsley 2012 authors (personal communication), wherein the number of responders was counted for participants who completed the study with complete data collection. We assumed that remaining participants from the intention‐to‐treat (ITT) cohort were non‐responders. Similar results were obtained in a sensitivity analysis based on complete case cohort data with no assumptions made (41/67 (or 61 per 100) for methotrexate, 24/61 (or 39 per 100) for placebo; RR 1.56, 95% CI 1.08 to 2.24; absolute risk difference 0.16, 95% CI 0.04 to 0.28; NNTB 5, 95% CI 3 to 20). The primary analysis in the published manuscript, which used a multiple imputation method for missing data, did not demonstrate a difference between methotrexate and placebo for this outcome (odds ratio (OR) 1.77, 95% CI 0.7 to 3.23). Overall, methotrexate might provide clinically meaningful benefit for disease response (PsARC), but this estimate is uncertain and is likely to be affected by further high‐quality research (Table 1).
In contrast to the PsARC response, all analyses for ACR20 response were consistent with each other. At six months, low‐quality evidence from the same trial suggested there was no difference in ACR20 response between methotrexate (23/109 (or 21 per 100) for methotrexate) and placebo (13/112 (or 12 per 100) for placebo; RR 1.82, 95% CI 0.97 to 3.40; absolute risk difference 0.13, 95% CI 0.00 to 0.19) (Kingsley 2012). Again, this analysis used unpublished data provided by the authors of Kingsley 2012 (personal communication), wherein the number of responders was counted for participants who completed the study and had a complete data collection. We assumed that remaining participants from the ITT cohort were non‐responders. This lack of benefit for disease response (ACR20) is observed when one performs a sensitivity analysis by using only data for the complete case cohort (23/67 (or 34 per 100) for methotrexate, 13/61 (or 21 per 100) for placebo; RR 1.61, 95% CI 0.90 to 2.89; absolute risk difference 0.13, 95% CI ‐0.02 to 0.28; negative sign indicates more responders with placebo), and again, in the published manuscript, which used imputed values (OR 2.0, 95% CI 0.65 to 6.22). Methotrexate might provide no clinically important benefit for disease response (ACR20).
We downgraded evidence for disease response (PsARC and ACR20), function, and disease activity due to risk of bias and imprecision. We judged there to be potential attrition bias and reporting bias for the main study informing the results for this comparison (Kingsley 2012). It is likely that additional data from high‐quality studies will alter the certainty of study results.
With respect to safety, four studies ‐ Black 1964,Kingsley 2012,Scarpa 2008, and Willkens 1984 (data from Black 1964 not pooled due to inability to attribute adverse events (AEs) to a treatment group) ‐ provided low‐quality evidence suggesting that methotrexate may not lead to more withdrawals due to AEs (9/141 (or 64 per 1000) for methotrexate, 7/152 (or 46 per 1000) for placebo; RR 1.32, 95% CI 0.51 to 3.42; absolute risk difference 0.01, 95% CI ‐0.04 to 0.06; the negative sign indicates more withdrawals due to adverse events (WAEs) with placebo)) or serious adverse events (SAEs) (1/141 (or 7 per 1000) for methotrexate, 4/112 (or 36 per 1000) for placebo; RR 0.26, 95% CI 0.03 to 2.26; absolute risk difference ‐0.02, 95% CI ‐0.05 to 0.01; the negative sign indicates more SAEs with placebo) (Table 1).
Although we were unable to formally extract data from Black 1964, in which participants received higher‐dose intravenous (IV) methotrexate and placebo in a cross‐over design, it is notable that one participant died from complications of bone marrow failure after receiving IV methotrexate. We were unable to identify which group this participant had been assigned to before cross‐over, and so we could not include this result in our analysis. Overall, methotrexate might be more harmful than placebo when taken for up to six months (Table 1).
We downgraded the evidence for withdrawals and SAEs due to risk of bias and imprecision. We judged there to be potential attrition bias and reporting bias for the main study informing this comparison (Kingsley 2012). The other studies were at unclear or high risk of bias for all domains (selection bias, performance bias, detection bias, attrition bias, reporting bias, other bias) (Scarpa 2008; Willkens 1984). The small number of events led to imprecision. It is likely that additional data from high‐quality studies will alter the certainty of study results.
Minor outcomes
No studies measured enthesitis, dactylitis, or fatigue.
A single study (221 randomised participants; oral methotrexate 15 mg per week (standard low dose)) provided low‐quality evidence suggesting benefit of methotrexate in improving pain and patient global assessment and physician global assessment of disease activity at six months (Kingsley 2012). Mean pain was rated at 38.3 mm on a visual analogue scale (VAS) (scale 0 to 100 mm; higher scores indicate more pain) and was rated 9.5 mm lower (2.2 mm lower to 16.8 mm lower) with methotrexate, for an absolute pain reduction of 10% (2% better to 17% better) and relative pain reduction of 25% (6% better to 44% better). Mean patient global assessment was 42.2 mm on a VAS (scale 0 to 100 mm; higher scores indicate greater perceived disease activity) with placebo and was 10.4 mm lower (2.1 mm lower to 18.7 mm lower) with methotrexate, for an absolute score that was 10% lower (2% lower to 19% lower) and a relative score that was 25% lower (5% lower to 44% lower). Mean physician global assessment was 33 mm on a VAS (scale 0 to 100 mm; higher scores indicate greater perceived disease activity) with placebo and was 9.7 mm lower (4.1 mm lower to 15.3 mm lower) with methotrexate, for an absolute score of 10% lower (4% lower to 15% lower) and relative improvement of 29% (12% lower to 46% lower). Although these results are unlikely to be clinically meaningful (minimal clinically important absolute difference is 15%), the 95% CI includes a clinically meaningful result. Imprecision for these outcomes limits our certainty in the results. Methotrexate might provide clinically meaningful benefits for pain and for patient and physician global assessments.
At six months, the same study provided low‐quality evidence suggesting that methotrexate may be no better than placebo for tender joint count, swollen joint count, and skin disease (Kingsley 2012). The mean tender joint count was 10 for placebo (68 joint count; 0 to 68) and was 2.3 joints lower (5.5 lower to 0.9 higher) for methotrexate, for an absolute improvement of 3% (8% better to 1% worse) and relative improvement of 23% (55% better to 9% worse). The mean swollen joint count was 5.2 for placebo (66 joint count; 0 to 66) and was 0.6 joints lower (2.6 lower to 1.4 higher) for methotrexate, for an absolute improvement of 1% (4% better to 2% worse) and relative improvement of 11% (50% better to 27% worse). Mean skin disease was 3.1 points on the PASI (0 to 72 points; lower scores indicate less psoriasis) for placebo and was 0.9 points lower (1.9 lower to 0.06 higher) for methotrexate, for an absolute improvement of 1% (0% to 3% better) and relative improvement of 29% (60% improvement to 2% worse). Kingsley 2012 was not designed to assess benefits for skin disease specifically as a primary outcome. Outcome assessors may not have been as skilled in assessing skin disease with PASI, and the patient population may not have had severe skin disease from baseline. We believe skin disease could be considered in a separate review dedicated to this topic.
With respect to adverse events, we pooled data from three studies for meta‐analysis (293 randomised participants; Kingsley 2012; Scarpa 2008; Willkens 1984). We found that methotrexate was associated with more adverse events (100/149 (or 67 per 100)) when compared to placebo (49/152 (or 33 per 100)) (RR 2.13, 95% CI 1.27 to 3.59; absolute risk difference 0.21, 95% CI ‐0.17 to 0.60). For meta‐analysis, we used a random‐effects model; we did not find an absolute risk difference, primarily because each study contributed equally to the analysis and two studies (72 randomised participants) reported a total of only three adverse events for either group. We performed a sensitivity analysis using a fixed‐effect model and found different results (RR 2.10, 95% CI 1.68 to 2.62; absolute risk difference 0.37, 95% CI 0.28 to 0.45; NNTH 3, 95% CI 2 to 3). It is likely that methotrexate is poorly tolerated compared with placebo; however, imprecision for this outcome reduces our certainty in the result and is difficult to extrapolate directly to clinical practice, which has a long history of methotrexate use for this indication.
We downgraded the evidence for adverse event outcomes due to risk of bias and imprecision. We judged there to be potential attrition bias and reporting bias for the main study informing this comparison (Kingsley 2012). The other studies were at unclear or high risk of bias for all domains (selection bias, performance bias, detection bias, attrition bias, reporting bias, and other bias) (Scarpa 2008; Willkens 1984). The small number of events led to imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Methotrexate versus placebo (beyond six months)
Summary
No study measured disease response (PsARC or ACR20), function, health‐related quality of life, disease activity, radiographic progression, enthesitis, dactylitis, pain, fatigue, skin disease, patient or physician global assessments of disease activity, swollen or tender joint counts, or serious adverse events. Very low‐quality evidence from one study (72 randomised participants; oral methotrexate 10 mg per week (standard dose 15 mg per week) suggests that methotrexate might lead to more withdrawals than placebo beyond six months, but we are uncertain if it causes more adverse events in total (Burdeinyi 1992).
It is important to note that this study was published before many of our outcomes of interest were formally developed and validated (Burdeinyi 1992). We downgraded the evidence due to potential risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias), indirectness, and imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Methotrexate versus other DMARDs (up to six months)
Summary
Results from three studies provided very low‐quality evidence suggesting that methotrexate may be as effective as leflunomide 20 mg orally daily, and may be more effective than ciclosporin A at 3 mg to 5 mg/kg orally daily, and that participants found both drugs to be similarly tolerable (Asaduzzaman 2014; Spadaro 1995; Zhang 2009). The two studies that compared methotrexate to leflunomide show marked differences across several risk of bias domains (Asaduzzaman 2014; Zhang 2009). Because of these differences, we believe it is inappropriate to pool these results, and so we analysed them separately.
Major outcomes
Leflunomide
No study measured health‐related quality of life, disease activity, radiographic progression, or SAEs.
At six months, very low‐quality evidence from one trial (30 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week)) suggests there is no difference in achieving an ACR50 response (12/14 (or 86 per 100) for methotrexate, 13/16 (or 82 per 100) for placebo; RR 1.05, 95% CI 0.77 to 1.45; absolute risk difference 0.04, 95% CI ‐0.22 to 0.31; negative sign means lower response with methotrexate) (Table 2) (Asaduzzaman 2014). Every participant in both groups in the same study achieved an ACR20 response (Asaduzzaman 2014). Given the low‐quality evidence on methotrexate versus placebo at this same time point for disease response (ACR20), we are uncertain if this result is accurate. We believe this result should not be used to inform clinical practice.
At six months in one trial (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week (standard low dose is 15 mg per week)); mean function (HAQ score) was 0.17 points with leflunomide (scale 0 to 3; 0 = no functional impairment) and was 0.13 points lower (0.23 lower to 0.03 lower) with methotrexate, for an absolute improvement of 4% (1% better to 8% better) and relative improvement of 76% (18% better to 135% better) (Zhang 2009). This reduction is not likely to be clinically meaningful (minimal clinically important absolute difference is 0.22 points), but the 95% CI includes a meaningful result, and this imprecision limits our certainty in the results. We are uncertain whether methotrexate is better than leflunomide in improving function (Table 2).
One trial (30 randomised participants; oral methotrexate 10 mg per week) reported zero withdrawals due to adverse events, and so we are uncertain of the true direction or magnitude of the effect (Asaduzzaman 2014). Very low‐quality evidence from the other trial (31 randomised participants; oral methotrexate 7.5 mg to 25 mg per week) suggests that withdrawals may not be increased with methotrexate (1/13 (or 8 per 100) with methotrexate, 2/18 (or 11 per 100) with placebo; RR 0.69, 95% CI 0.07 to 6.85; absolute risk difference ‐0.03, 95% CI ‐0.24 to 0.17; negative sign indicates less harm with methotrexate) (Table 2) (Zhang 2009).
We downgraded the evidence for all outcomes due to potential risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias), indirectness, and imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Ciclosporin A
Trials provided no data for disease response (PsARC, ACR50), function, health‐related quality of life, disease activity, or radiographic progression.
Very low‐quality evidence from one trial (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week)) suggests that it is uncertain whether methotrexate is more harmful than ciclosporin A (3 mg to 5 mg/kg orally daily) (Spadaro 1995). This study reported zero SAEs, and we are uncertain of the direction and magnitude of the true effect. Based on very low‐quality evidence from the same study, we are uncertain if methotrexate leads to more withdrawals when compared with ciclosporin A (4/18 (or 22 per 100) with methotrexate, 3/17 (or 18 per 100) with placebo; RR 1.26, 95% CI ‐0.33 to 4.82; absolute risk difference 0.05, 95% CI ‐0.22 to 0.31; negative sign indicates less harm with methotrexate) (Table 2).
It is important to note that this study ‐ Spadaro 1995 ‐ was published before many of the outcomes of interest in our systematic review were formally developed and validated. We downgraded the evidence for all outcomes due to potential risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias), indirectness, and imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Minor outcomes
Leflunomide
Very low‐quality evidence from one study suggests that methotrexate might lead to greater improvement in patient global assessment of disease activity (Zhang 2009), but it is unclear which scale researchers used, and we are not able to clarify this with the study authors. Two studies provided very low‐quality evidence suggesting that there are no between‐group differences for disease response (ACR20), pain, skin disease, physician global assessment of disease activity, swollen and tender joint counts, and total AEs (Asaduzzaman 2014; Zhang 2009). Researchers provided no data for enthesitis, dactylitis, or fatigue.
We downgraded the evidence for all outcomes due to potential risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias), indirectness, and imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Ciclosporin A
No data show measurements for enthesitis, dactylitis, pain, fatigue, or total AEs.
Compared with ciclosporin A, very low‐quality evidence based on one trial (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week)) suggests that methotrexate might provide clinically meaningful benefit with respect to physician global assessment but no clinically meaningful benefit for skin disease, patient global assessment, swollen joint count, or tender joint count (Spadaro 1995).
It is important to note that this study ‐ Spadaro 1995 ‐ was published before many of our outcomes of interest were formally developed and validated. We downgraded the evidence for all outcomes due to potential risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias), indirectness, and imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Methotrexate versus other DMARDs (beyond six months)
Summary
Ciclosporin A
Researchers have provided no data for disease response (ACR50, PsARC, ACR20), function, health‐related quality of life, disease activity, radiographic progression, enthesitis, dactylitis, pain, fatigue, and total adverse events.
Compared with ciclosporin A (oral ciclosporin 3 mg to 5 mg/kg daily), very low‐quality evidence based on one trial (35 randomised participants; oral methotrexate 7.5 mg to 15 mg per week (standard low dose is 15 mg per week)) suggests that beyond six months, methotrexate might provide clinically meaningful benefit with respect to physician global assessment (Spadaro 1995). Spadaro 1995 also indicates there may be no clinically meaningful benefit for swollen joint count, tender joint count, skin disease, or patient global assessment; these study authors reported zero serious adverse events for both groups, and we are uncertain of the true magnitude or direction of effect for this outcome.
We downgraded the evidence for both outcomes due to potential risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias), indirectness, and imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Gold and sulfasalazine
We found inadequate evidence to inform how methotrexate compares to either sulfasalazine or gold therapy beyond six months with respect to disease response (ACR50, PsARC, ACR20), function, health‐related quality of life, disease activity, radiographic progression, enthesitis, dactylitis, pain, fatigue, skin disease, patient and physician global assessments, tender and swollen joint counts, and serious adverse events.
Very low‐quality evidence from one trial (61 randomised participants; oral methotrexate 10 mg per week (standard low dose is 15 mg per week)) limits our certainty about whether methotrexate leads to more withdrawals due to adverse events or total adverse events than gold (intramuscular gold 34 mg per week) (Burdeinyi 1992).
Researchers studied no other DMARDs beyond six months.
It is important to note that this study ‐ Burdeinyi 1992 ‐ was published before many of our outcomes of interest were formally developed and validated. We downgraded the evidence for both adverse event outcomes for both sulfasalazine and gold groups due to potential risk of bias (selection bias, performance bias, detection bias, attrition bias, and reporting bias), indirectness, and imprecision. It is likely that additional data from high‐quality studies will alter the certainty of these results.
Overall completeness and applicability of evidence
Overall we found that researchers have not addressed a large number of outcomes, and this review remains incomplete as a consequence. All study participants had psoriatic arthritis with peripheral disease, although disease severity varied and findings may not apply to all patients encountered in clinical practice. Doses of methotrexate were generally low, and some clinicians may use higher doses internationally. The effect of higher‐dose methotrexate compared to placebo remains unknown. With the exception of the primary comparison (methotrexate vs placebo up to six months), researchers reported very low numbers of adverse events, particularly serious adverse events. This is likely to lead to inaccurate estimations of the safety of methotrexate.
Quality of the evidence
According to the GRADE Working Group, we applied the following grades when assessing evidence: high‐quality evidence indicates high confidence that the true effect lies close to that of the estimate of the effect; moderate‐quality evidence indicates moderate confidence in the effect estimate (i.e. the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different); low‐quality evidence indicates that confidence in the effect estimate is limited (i.e. the true effect may be substantially different from the estimate of the effect); and very low‐quality evidence suggests very little confidence in the effect estimate (i.e. the true effect is likely to be substantially different from the estimate of effect). Using the GRADE approach, we judged the quality of evidence for each outcome and each comparison and provided reasons as follows.
Methotrexate versus placebo (up to six months)
Major outcomes
For outcomes of disease response (PsARC), function, disease activity (DAS28‐ESR), serious adverse events, and withdrawals due to adverse events, results were informed by low‐quality evidence (downgraded due to risk of bias and imprecision). Study authors did not report health‐related quality of life and did not measure radiographic progression.
Minor outcomes
For outcomes of disease response (ACR20), pain, skin disease (PASI), and total adverse events, results were informed by low‐quality evidence (downgraded due to risk of bias and imprecision). Researchers did not measure enthesitis, dactylitis, and fatigue. For outcomes of patient and physician global assessments of disease activity, and tender and swollen joint counts, results were informed by very low‐quality evidence (downgraded due to risk of bias, inconsistency, and imprecision).
Methotrexate versus placebo (beyond six months)
Major outcomes
For outcomes of disease response (PsARC), function, health‐related quality of life, disease activity, radiographic progression, and serious adverse events, study authors did not provide results. Withdrawal due to adverse events was informed by very low‐quality evidence, downgraded due to risk of bias, indirectness, and imprecision.
Minor outcomes
For outcomes of disease response (ACR20), enthesitis, dactylitis, pain, fatigue, skin disease (PASI), patient and physician global assessments of disease activity, and swollen and tender joint counts, study authors did not report results. Total adverse events was informed by very low‐quality evidence, downgraded due to risk of bias, indirectness, and imprecision.
Methotrexate versus other DMARDs (up to six months)
Major outcomes
For outcomes of disease response (PsARC), function, serious adverse events, and withdrawals due to adverse events, results were informed by very low‐quality evidence (downgraded due to risk of bias and imprecision). For outcomes of health‐related quality of life, disease activity, and radiographic progression, trials provided no results.
Minor outcomes
For outcomes of disease response (ACR20), pain, and skin disease (PASI), results were informed by very low‐quality evidence (downgraded due to risk of bias and imprecision). For outcomes of enthesitis, dactylitis, and fatigue, trial authors did not report results. For outcomes of total adverse events, patient and physician global assessments of disease activity, and swollen and tender joint counts, results were informed by very low‐quality evidence (downgraded due to risk of bias, inconsistency, and imprecision).
Methotrexate versus other DMARDs (beyond six months)
Major outcomes
For outcomes of disease response (PsARC), function, health‐related quality of life, disease activity, and radiographic progression, trials did not report results. For outcomes of serious adverse events and withdrawals due to adverse events, results were informed by very low‐quality evidence (downgraded due to risk of bias and imprecision).
Minor outcomes
For outcomes of disease response (ACR20), enthesitis, dactylitis, pain, and fatigue, study authors did not provide results. For outcomes of skin disease (PASI), total adverse events, patient and physician global assessments of disease activity, and swollen and tender joint counts, results were informed by very low‐quality evidence (downgraded due to risk of bias, indirectness, and imprecision).
Potential biases in the review process
For this review, we searched Embase, MEDLINE and the Cochrane Library without language restrictions and identified eight relevant studies. It is always possible to identify additional studies by expanding the search to more databases. We are not aware of any other potential biases in the review process.
Agreements and disagreements with other studies or reviews
The previous Cochrane systematic review on this topic concluded that parenteral high‐dose methotrexate was effective for psoriatic arthritis, and that oral low‐dose methotrexate may be effective but requires further multi‐centre clinical trials to establish its efficacy (Jones 2000). The authors of the present review do not uniformly agree with these conclusions. With respect to high‐dose methotrexate, we were unable to extract data from Black 1964. Despite this, use of high‐dose parenteral methotrexate (up to 150 mg) was shown to be potentially lethal (one patient died from complications of bone marrow failure); therefore, we conclude that this treatment is unlikely to be useful in the management of psoriatic arthritis. With respect to low‐dose methotrexate, several additional studies have been published since Jones 2000 was prepared. Upon inclusion of these studies, we conclude that oral low‐dose methotrexate is not definitively more effective than placebo, although the estimate of effects may be altered by additional large high‐quality trials.
Methotrexate is commonly recommended in international guidelines as first‐line DMARD therapy for patients with peripheral arthritis (Coates 2016b; Gossec 2016). Coates 2017a explains that the persistence of recommending methotrexate in international guidelines is due to its apparent benefit reported by observational trials and the positive expert experience reported. Our systematic review does not provide definitive support for the superiority of methotrexate over placebo.
Authors' conclusions
Implications for practice.
Low‐quality evidence suggests that low‐dose oral methotrexate may be more effective than placebo when taken for six months in terms of disease response (PsARC), function, pain, and patient and physician global assessments of disease activity. The effect size for each of these outcomes is small. Research shows no clinically important differences with respect to disease response (ACR20), disease activity (DAS28‐ESR), tender and swollen joint counts, or skin disease. Methotrexate is generally well tolerated in this population. Neither effects of methotrexate on health‐related quality of life, radiographic progression, enthesitis, dactylitis, and fatigue, and its efficacy beyond six months, nor effects of higher‐dose methotrexate have been adequately studied in a placebo‐controlled trial.
With the exception of leflunomide, head‐to‐head data are inadequate to inform comparison versus other DMARDs, including biologic DMARDs. Data comparing methotrexate versus leflunomide are of very low quality, such that we do not believe they provide clinically meaningful information, and we do believe they should be interpreted and applied with extreme caution. Very low‐quality evidence suggests that methotrexate may be as effective as leflunomide when taken for six months and is generally well tolerated, although very few adverse events have been reported and its comparative safety is uncertain. The comparative efficacy of methotrexate in terms of health‐related quality of life, disease activity, radiographic progression, enthesitis, dactylitis, and fatigue, along with its efficacy beyond six months, has not been studied in a placebo‐controlled trial.
Implications for research.
Studies included in this review were at unclear or high risk of bias for at least one domain, and they provided evidence of low or very low quality. The certainty of these results could be enhanced by additional high‐quality studies of sufficient sample size. Selection of participants using validated classification criteria, such as the CASPAR criteria, would ensure a homogeneous population for future study comparisons. Several outcomes require evaluation for more complete assessment of efficacy (e.g. quality of life, radiographic progression, enthesitis, dactylitis) and could be informed by the latest OMERACT recommendations (Orbai 2017). A longer duration of follow‐up would also be useful to clinicians. Finally, dose‐dependent efficacy remains underexplored. Given the greater bioavailability of methotrexate at doses above 15 mg when administered parenterally rather than orally (Hamilton 1997; Schiff 2014), studies utilising subcutaneous or intramuscular administration of methotrexate above 15 mg weekly might provide a suitable method for exploring dose‐dependent efficacy. Further placebo‐controlled trials of methotrexate for psoriatic arthritis would offer an ideal way to resolve this question, although ethical considerations may mean that this trial design is impractical.
What's new
| Date | Event | Description |
|---|---|---|
| 17 January 2019 | Amended | Contact person updated his contact email |
History
Protocol first published: Issue 7, 2017 Review first published: Issue 1, 2019
| Date | Event | Description |
|---|---|---|
| 22 May 2012 | Amended | CMSG ID A079‐P |
Notes
This protocol is based on a common protocol template recommended by the Cochrane Musculoskeletal Group Editorial Team.
Acknowledgements
Raechel Damarell (Medical Librarian, Flinders University) for her contributions to the development of our search strategies.
Ms. Lee Li, Ms. Helen Hibbard, and Dr. Thomas Goddard for their assistance in translating articles to English.
Cochrane Musculoskeletal for editorial support in writing this review.
Appendices
Appendix 1. MEDLINE search strategy
Methotrexate
(4‐Amino‐10‐methylfolic Acid or 4‐Amino‐4‐deoxy‐10‐methylpteroyl‐L‐glutamic Acid or Methopterin or Amethopterin or Ametopterin or Ametopterina or CL‐14377 or Methotrexat or Methotrexatum or Metotreksaatti or Metotreksatas or Metotrexat or Metotrexato or MTX or NSC‐740 or WR‐19039).mp.
(Abitrexate or Alltrex or Artrait or Atrexel or Bendatrexat or Bertanel or Biometrox or Biotrexate or Brimexate or Caditrex or Dermatrex or Dermotrex or Ebetrex or Ebetrexac or Ebetrexat or Ebetrexate or Emtehexate or Emtexate or Emthexate or Ervemin or Farmitrexat or Fauldexato or Fauldmetro or Folex or Folitrax or Hextrate or Hi‐Trex or Hytas or Ifamet or Imeth or Imutrex or Lantarel or Ledertrexate or Ledertrexato or Leulin or Lexato or Lumexon or Matrex or Maxtrex or Medsatrexate or Meisusheng or Merex or Metex or Methaccord or Methacor or Methobax or Methobion or Methoblastin or Methoblastine or Methocel or Methocip or Methorex or Metoart or Metodik or Metoject or Metojectpen or Metolate or Metorex or Metotab or Metrex or Metrexato or Metrexx or Metrotex or Mexate or Miantrex or Midu or MPL Methoxil or MTX or Neometho or Neotrexat or Neottrexate or Novatrex or O‐trexat or Oncotrex or Onkomet or Onotrex or Otaxem or Otrexup or Pterin or Rasuvo or Reumaflex or Reutrexato or Rheumatrex or Rhodamer or Sactiva or Sanotrexat or Securact or Tecnomet or Tevametho or Texate or Texorate or Tratoben or Tremetex or Trexall or Trexamette or Trexan or Trexeron or Trixate or Trixilem or Unitrexate or Xaken or Xantromid or Zexat).mp.
Antirheumatic Agents/
(antirheumatic* or anti‐rheumatic*).tw,kw.
or/1‐5
Arthritis, Psoriatic/
(psoria* adj5 (arthr* or polyarthr* or poly‐arthr* or oligoarthr* or oligo‐arthr* or rheumat*)).tw,kw.
or/7‐8
Randomized controlled trial.pt.
Controlled clinical trial.pt.
random*.ti,ab.
Placebo.ti,ab.
Drug therapy.fs.
trial.ti,ab.
Groups.ti,ab.
or/10‐16
exp animals/ not humans/
17 not 18
6 and 9 and 19
Appendix 2. Embase search strategy
methotrexate derivative/ or methotrexate/ or methotrexate gamma aspartic acid/ or methotrexate polyglutamate/
(4‐Amino‐10‐methylfolic Acid or 4‐Amino‐4‐deoxy‐10‐methylpteroyl‐L‐glutamic Acid or Methopterin or Amethopterin or Ametopterin or Ametopterina or CL‐14377 or Methotrexat or Methotrexatum or Metotreksaatti or Metotreksatas or Metotrexat or Metotrexato or MTX or NSC‐740 or WR‐19039).mp.
(Abitrexate or Alltrex or Artrait or Atrexel or Bendatrexat or Bertanel or Biometrox or Biotrexate or Brimexate or Caditrex or Dermatrex or Dermotrex or Ebetrex or Ebetrexac or Ebetrexat or Ebetrexate or Emtehexate or Emtexate or Emthexate or Ervemin or Farmitrexat or Fauldexato or Fauldmetro or Folex or Folitrax or Hextrate or Hi‐Trex or Hytas or Ifamet or Imeth or Imutrex or Lantarel or Ledertrexate or Ledertrexato or Leulin or Lexato or Lumexon or Matrex or Maxtrex or Medsatrexate or Meisusheng or Merex or Metex or Methaccord or Methacor or Methobax or Methobion or Methoblastin or Methoblastine or Methocel or Methocip or Methorex or Metoart or Metodik or Metoject or Metojectpen or Metolate or Metorex or Metotab or Metrex or Metrexato or Metrexx or Metrotex or Mexate or Miantrex or Midu or MPL Methoxil or MTX or Neometho or Neotrexat or Neottrexate or Novatrex or O‐trexat or Oncotrex or Onkomet or Onotrex or Otaxem or Otrexup or Pterin or Rasuvo or Reumaflex or Reutrexato or Rheumatrex or Rhodamer or Sactiva or Sanotrexat or Securact or Tecnomet or Tevametho or Texate or Texorate or Tratoben or Tremetex or Trexall or Trexamette or Trexan or Trexeron or Trixate or Trixilem or Unitrexate or Xaken or Xantromid or Zexat).mp.
antirheumatic agent/
(antirheumatic* or anti‐rheumatic*).tw,kw.
or/1‐5
psoriatic arthritis/
(psoria* adj5 (arthr* or polyarthr* or poly‐arthr* or oligoarthr* or oligo‐arthr* or rheumat*)).tw,kw.
or/7‐8
random*.ti,ab. or clinical trial*.mp. or exp health care quality/
6 and 9 and 10
Appendix 3. CENTRAL search strategy
MeSH descriptor: [Methotrexate] this term only
MeSH descriptor: [Antirheumatic Agents] this term only
4‐Amino‐10‐methylfolic Acid or 4‐Amino‐4‐deoxy‐10‐methylpteroyl‐L‐glutamic Acid or Methopterin or Amethopterin or Ametopterin or Ametopterina or CL‐14377 or Methotrexat or Methotrexatum or Metotreksaatti or Metotreksatas or Metotrexat or Metotrexato or MTX or NSC‐740 or WR‐19039
Abitrexate or Alltrex or Artrait or Atrexel or Bendatrexat or Bertanel or Biometrox or Biotrexate or Brimexate or Caditrex or Dermatrex or Dermotrex or Ebetrex or Ebetrexac or Ebetrexat or Ebetrexate or Emtehexate or Emtexate or Emthexate or Ervemin or Farmitrexat or Fauldexato or Fauldmetro or Folex or Folitrax or Hextrate or Hi‐Trex or Hytas or Ifamet or Imeth or Imutrex or Lantarel or Ledertrexate or Ledertrexato or Leulin or Lexato or Lumexon or Matrex or Maxtrex or Medsatrexate or Meisusheng or Merex or Metex or Methaccord or Methacor or Methobax or Methobion or Methoblastin or Methoblastine or Methocel or Methocip or Methorex or Metoart or Metodik or Metoject or Metojectpen or Metolate or Metorex or Metotab or Metrex or Metrexato or Metrexx or Metrotex or Mexate or Miantrex or Midu or MPL Methoxil or MTX or Neometho or Neotrexat or Neottrexate or Novatrex or O‐trexat or Oncotrex or Onkomet or Onotrex or Otaxem or Otrexup or Pterin or Rasuvo or Reumaflex or Reutrexato or Rheumatrex or Rhodamer or Sactiva or Sanotrexat or Securact or Tecnomet or Tevametho or Texate or Texorate or Tratoben or Tremetex or Trexall or Trexamette or Trexan or Trexeron or Trixate or Trixilem or Unitrexate or Xaken or Xantromid or Zexat
antirheumatic* or anti‐rheumatic*
#1 or #2 or #3 or #4 or #5
MeSH descriptor: [Arthritis, Psoriatic] this term only
(psoria* near/5 (arthr* or polyarthr* or poly‐arthr* or oligoarthr* or oligo‐arthr* or rheumat*))
#7 or #8
#6 and #9
Data and analyses
Comparison 1. Methotrexate versus placebo – major outcomes ≤ 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disease response (PsARC) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Function (HAQ) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Disease activity (DAS28‐ESR) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Serious adverse events | 3 | 293 | Risk Ratio (M‐H, Random, 95% CI) | 0.26 [0.03, 2.26] |
| 5 Withdrawals due to adverse events | 3 | 293 | Risk Ratio (M‐H, Random, 95% CI) | 1.32 [0.51, 3.42] |
Comparison 2. Methotrexate versus placebo – minor outcomes ≤ 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disease response (ACR20) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Pain | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Skin disease (PASI) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Total adverse events | 3 | 293 | Risk Ratio (M‐H, Random, 95% CI) | 2.13 [1.27, 3.59] |
| 5 Patient global assessment of disease activity | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6 Physician global assessment of disease activity | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7 Swollen joint count | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8 Tender joint count | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 3. Methotrexate versus placebo ‐ major outcomes > 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Withdrawals due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 4. Methotrexate versus placebo ‐ minor outcomes > 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
Comparison 5. Methotrexate versus other DMARDs – major outcomes ≤ 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disease response (ACR50) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Leflunomide (ACR50) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Function (HAQ) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 Leflunomide | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Leflunomide | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Ciclosporin A | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Withdrawals due to adverse events | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Leflunomide | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Ciclosporin A | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
5.3. Analysis.
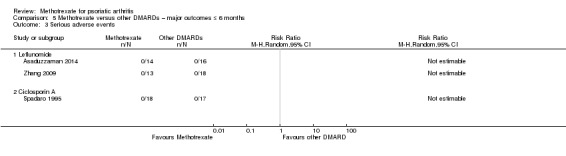
Comparison 5 Methotrexate versus other DMARDs – major outcomes ≤ 6 months, Outcome 3 Serious adverse events.
Comparison 6. Methotrexate versus other DMARDs – minor outcomes ≤ 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disease response (ACR20) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Leflunomide | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Pain | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 Leflunomide | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Skin disease | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Leflunomide | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Leflunomide | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Patient global assessment of disease activity | 3 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5.1 Leflunomide | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Physician global assessment of disease activity | 3 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6.1 Leflunomide | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Swollen joint count | 3 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7.1 Leflunomide | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Tender joint count | 3 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8.1 Leflunomide | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 7. Methotrexate versus other DMARDs ‐ major outcomes > 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Serious adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Ciclosporin A | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawals due to adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Ciclosporin A | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Gold | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Sulfasalazine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
7.1. Analysis.
Comparison 7 Methotrexate versus other DMARDs ‐ major outcomes > 6 months, Outcome 1 Serious adverse events.
Comparison 8. Methotrexate versus other DMARDs ‐ minor outcomes > 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Skin disease | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.1 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Total adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Gold | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Sulfasalazine | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Patient global assessment of disease activity | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Physician global assessment of disease activity | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4.1 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Swollen joint count | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5.1 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Tender joint count | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6.1 Ciclosporin A | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 9. Methotrexate versus placebo ≤ 6 months (sensitivity analysis).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disease response (PsARC) ‐ sensitivity analysis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Function (HAQ) ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Disease activity (DAS28‐ESR) ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Disease response (ACR20) ‐ sensitivity analysis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Pain ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6 Skin disease ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7 Patient global assessment of disease activity ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8 Physician global assessment of disease activity ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 9 Swollen joint count ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 10 Tender joint count ‐ sensitivity analysis | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
Comparison 10. Additional analysis – methotrexate versus other DMARDs ≤ 6 months.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disease response (PsARC) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Leflunomide (PsARC) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Asaduzzaman 2014.
| Methods |
Study design: open‐label, randomised, controlled trial of methotrexate vs leflunomide Duration: 6 months Run‐in period: none Location: Bangladesh Number of study centres: 1 Study setting: outpatient Withdrawals: 2 ‐ method of handling missing data not described Study dates: June 2002 to December 2003 |
|
| Participants |
Randomised: n = 32 Completed: n = 30 Baseline characteristics Mean age (SD):
Sex: 27 males; 3 females Mean disease duration, years (SD):
Mean pain (SD) (VAS 10 cm)
Mean skin disease (SD) (PASI)
Mean patient global assessment (SD) (Likert 1 to 5)
Mean physician global assessment (SD) (Likert 1 to 5)
Mean swollen joint count (SD) (66 joints)
Mean tender joint count (SD) (68 joints)
Severity of condition: active disease from mild to severe Inclusion:
Exclusion:
|
|
| Interventions |
Methotrexate group: 10 mg orally in 2 divided doses (12 hours apart) weekly for 6 months. As per the abstract, total dose was 10 mg weekly Leflunomide group: loading dose of 100 mg orally daily for 3 days, then 20 mg orally daily for 6 months Co‐interventions: both groups were allowed to take ibuprofen orally with a maximum allocated dose of 1400 mg (assumed daily) Excluded interventions: not reported |
|
| Outcomes |
Time points: 6 months Major:
Minor:
|
|
| Notes |
Clinical trials registration: not reported Funding: not reported Declarations of interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was done using a random number table…32 patients of psoriatic arthritis were taken consecutively and grouped into two by card test" |
| Allocation concealment (selection bias) | High risk | Allocation concealment was not attempted |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "This open, randomized clinical trial…" Open‐label design allowed participants to remain aware of their allocated intervention; no attempt was made at blinding study personnel |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors was not attempted |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Out of total 32 patients, one patient from each group was excluded from analysis due to lack of follow‐up" This was not accounted for in the reported analysis, although it was considered unlikely to impact the results |
| Selective reporting (reporting bias) | Unclear risk | This could not be substantiated, as no trial registry record was available for comparison |
| Other bias | Unclear risk | Baseline characteristics and use of co‐interventions displayed variability between groups, and compliance with study therapy was not reported. It is unclear if this may have influenced the results, particularly in light of the small number of participants |
Black 1964.
| Methods |
Study design: randomised, double‐blind, cross‐over trial of methotrexate vs placebo Duration of study: minimum 80 days (20 days pretreatment; 60 days on treatment) Run‐in period: 20 days with trial dose of parenteral methotrexate to test for hypersensitivity Location: United States of America Number of study centres: 1 Study setting: inpatient Withdrawals: 1 ‐ method of handling missing data not described Dates of study: not reported |
|
| Participants |
Randomised: n = 21 Completed: n = 20 Mean age: not reported Sex: 10 males; 11 females Mean disease duration: 8 years (dispersion measure not reported) Severity of condition: not reported Diagnostic criteria: rheumatologist‐diagnosed PsA Inclusion criteria:
Exclusion criteria:
|
|
| Interventions |
Group AB: methotrexate intravenously or intramuscularly at progressively increasing doses from 1 to 3 mg/kg of body weight for the first treatment period, followed by parenteral placebo for the second treatment period Group BA: parenteral placebo first, followed by parenteral methotrexate as above for the second treatment period Concomitant medications: corticosteroid or salicylate therapy at the lowest dose that just allowed an increase in skin or joint activity Excluded medications: not reported |
|
| Outcomes |
Time points: 60 days on one treatment, then cross‐over to the other treatment No extractable data. Study authors could not be contacted |
|
| Notes |
Clinical trials registration: not reported Funding: not reported Declarations of interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "From the first bottle, either methotrexate or placebo according to a randomized schedule…" The schedule is not described in further detail |
| Allocation concealment (selection bias) | Unclear risk | This was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind design, with infusions drawn from a vial No description was provided regarding the extent to which participants or personnel were blinded to treatment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Due to lack of description of the extent of double‐blinding, risk of bias was judged 'uncertain' |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Handling of data from the 2 withdrawn participants (1 withdrawal, 1 death) was not clearly documented. As total study numbers are low, it is unclear if this might impact study outcomes |
| Selective reporting (reporting bias) | Unclear risk | This could not be substantiated, as no trial registry record was available for comparison |
| Other bias | Unclear risk | Compliance with therapy was largely complete (excluding the single withdrawal), and baseline characteristics were not relevant, as this study used a cross‐over design. However, the requirement for co‐intervention with corticosteroids and NSAIDs was variable, and it is unclear if this has had an impact on study outcomes |
Burdeinyi 1992.
| Methods |
Study design: open‐label, randomised controlled trial with 4 arms of therapy (MTX, parenteral gold, sulfasalazine/salazopiridazine, NSAIDs (considered as placebo)) Duration of study: 12 months Run‐in period: none Location: Russia Number of study centres: not reported Study setting: outpatient Withdrawals: 49 ‐ method of handling missing data not described Dates of study: not reported |
|
| Participants |
Randomised: n = 126
Completed: n = 77
Baseline characteristics Mean age (reported only for the 77 completers), dispersion measure not reported:
Sex (reported only for the 77 completers), dispersion measure not reported:
Mean disease duration (reported only for the 77 completers), dispersion measure not reported:
Severity of condition: not reported Diagnostic criteria: rheumatologist‐diagnosed PsA Inclusion criteria: adults with clinically active PsA; no further criteria reported Exclusion criteria: not reported |
|
| Interventions |
Group 1: intramuscular gold injections equivalent to 34 mg elemental gold every week Group 2: sulfasalazine/salazopiridazine 0.5 g daily and increasing to 1 g twice a day Group 3: methotrexate 2.5 mg every 12 hours to a total weekly dose of 10 mg (unclear if this was per oral or parenteral) Group 4: controls (considered as placebo) ‐ NSAIDs (mainly diclofenac or indomethacin ‐ doses not reported) ‐ no placebo tablets Concomitant medications: after enrolment, all participants received intra‐articular corticosteroid injections. NSAIDs (mainly diclofenac or indomethacin) for all participants and intra‐articular corticosteroids at the discretion of the trial clinician. The last intra‐articular injection was no later than 4 weeks before the last examination, and NSAID dose was changed no later than 2 weeks before the last examination Excluded medications: not reported |
|
| Outcomes |
Time points: 12 months Major:
Minor:
N.B. Efficacy of treatment was based on pain scores, severity of morning stiffness, duration of morning stiffness, fatigability, Ritchie Articular Index, swollen joint count, tender joint count, compression force, and ESR. These outcomes were compared to baseline for the individual, and were subsequently combined and reported as the 'treatment effectiveness index'. This was a composite outcome designed by study authors that was not cited as a validated outcome measure. Individual variables were not reported. Study authors could not be contacted to provide individual data for individual outcomes |
|
| Notes |
Clinical trials registration: not reported Funding: not reported Declarations of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study involved 126 patients with clinically active psoriatic arthritis, who were randomised into 4 groups" Specific details regarding the randomisation process were not reported |
| Allocation concealment (selection bias) | Unclear risk | No explanation of allocation concealment procedures was provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Placebo controls were not used, hence it is possible that participants and personnel were aware of the treatment assignment |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No explanation regarding blinding of outcome assessors was provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Losses to follow‐up were recorded with reasons; however, incomplete efficacy data do not appear to be included in the final analysis |
| Selective reporting (reporting bias) | Unclear risk | The primary outcome of Treatment effectiveness index was selectively reported; this is a composite index of several component scores (such as the Ritchie articular index) Components were not reported |
| Other bias | Unclear risk | Differences in baseline characteristics and co‐intervention use were reported Compliance with therapy was not clearly documented The overall impact of this on study results is unclear |
Kingsley 2012.
| Methods |
Study design: double‐blind, randomised, placebo‐controlled trial of methotrexate vs placebo Duration of study: 6 months Run‐in period: none Location: United Kingdom Number of study centres: 22 Study setting: outpatient Withdrawals: 70 ‐ missing data were imputed by multiple imputation using chained equations with 20 cycles Dates of study: January 2003 to July 2008 |
|
| Participants |
Randomised: n = 221
Completed: n = 151
Baseline characteristics Mean age (SD):
Sex:
Mean disease duration (IQR):
Mean function (95% CI) (HAQ)
Health‐related quality of life (SF‐36)
Disease activity (DAS28‐ESR)
Mean pain (95% CI) (VAS 100 mm)
Mean skin disease (95% CI) (PASI)
Mean patient global assessment (95% CI) (VAS 100 mm)
Mean physician global assessment (95% CI) (VAS 100 mm)
Mean swollen joint count (95% CI) (66 joints)
Mean tender joint count (95% CI) (68 joints)
Severity of condition: not reported Diagnostic criteria: rheumatologist‐diagnosed PsA Inclusion criteria:
Exclusion criteria:
|
|
| Interventions |
Methotrexate group: methotrexate tablets 7.5 mg weekly for 4 weeks, 10 mg for 4 weeks, and 15 mg ongoing. Dose could be increased to 20 mg at 4 months and 25 mg at 4 months at the discretion of the clinician Placebo group: matching placebo tablet Concomitant medications:
Excluded medications: oral or intramuscular steroids were not used |
|
| Outcomes |
Time points: 6 months Major:
Minor:
|
|
| Notes |
Clinical trials registration: ISRCTN54376151 Funding: Arthritis Research UK, London South Comprehensive Local Research Network of the National institute for Health Research, Wyeth (UK) (supplied tablets), UKMRC, King's College Declarations of interest: single author (NJM) received honoraria from Abbott and Pfizer |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Allocation sequence was generated by the trial statistician using random number tables" |
| Allocation concealment (selection bias) | Low risk | "Metrologists and trial coordinator were unaware of the allocation sequence. Treatment assignments were in a locked cabinet in the co‐ordinating centre pharmacy for emergency access" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The MTX and placebo were identical in appearance. Each patient received the treatment in the corresponding pre‐packed container" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Metrologists were unaware of the allocation sequence" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | We considered attrition of 35 participants in each group (total attrition 32%) to be high; however missing data were imputed "All missing data were imputed regardless of the reason(s) the data were missing…Assuming unobserved measurements were missing at random, we imputed missing data by multiple imputation using chained equations with 20 cycles, where at the end of the cycle one imputed data set is created and process was repeated to create 20 imputed data sets" |
| Selective reporting (reporting bias) | High risk | The number of events was not reported for ITT analysis (e.g. for PsARC outcome) but was reported for completers. Also, study authors measured but did not report quality of life |
| Other bias | Low risk | Minor differences in baseline characteristics were evident; co‐intervention use and compliance were similar between groups. This trial was judged as having low risk of bias for these outcomes |
Scarpa 2008.
| Methods |
Study design: randomised, controlled trial of methotrexate plus NSAIDs vs NSAIDs alone with later addition of methotrexate Duration of study: 6 months Run‐in period: none Location: Italy Number of study centres: not reported Study setting: outpatient Withdrawals: none Dates of study: not reported |
|
| Participants |
Randomised: n = 35
Completed: n = 35 Baseline characteristics Mean age (SD): 25.6 years (±5.7) Sex: 18 males, 17 females Mean disease duration: not reported Median pain (IQR) (VAS 100 mm)
Median patient global assessment (IQR) (Likert 0 to 5)
Median physician global assessment (IQR) (Likert 0 to 5)
Median swollen joint count (IQR) (assumed 66)
Median tender joint count (IQR) (assumed 68)
Severity of condition: not reported Diagnostic criteria: rheumatologist‐diagnosed PsA Inclusion criteria:
Exclusion criteria: not reported |
|
| Interventions |
Methotrexate up‐front group: methotrexate intramuscular 10 mg weekly with daily NSAID therapy at full dosage NSAID only up‐front group: NSAID therapy at full dosage for 3 months, followed by the addition of methotrexate intramuscular 10 mg weekly for a further 3 months Concomitant medications: NSAIDs at full dose Excluded medications: not reported |
|
| Outcomes |
Time points: data extracted for 3 month outcomes only to allow comparison between methotrexate and NSAIDs (considered placebo) Major:
Minor:
|
|
| Notes |
Clinical trials registration: not reported Funding: not reported Declarations of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | "At enrolment, patients were randomly divided into two groups…" No further detail is provided to describe the randomisation process |
| Allocation concealment (selection bias) | High risk | Allocation concealment was not reported in the article |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants or personnel was not attempted |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors was not attempted |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was no discussion regarding handling of incomplete outcome data |
| Selective reporting (reporting bias) | Unclear risk | This could not be substantiated, as no trial registry record was available for comparison |
| Other bias | Unclear risk | Marked differences in baseline characteristics were evident; use of steroids was not forbidden, and their use was not reported Compliance was not discussed Overall this trial was judged to have unclear risk of bias for outcomes |
Spadaro 1995.
| Methods |
Study design: randomised, controlled trial of low‐dose methotrexate vs ciclosporin A Duration of study: 12 months Run‐in period: none Location: Italy Number of study centres: 1 Study setting: outpatient Withdrawals: 12 ‐ method of handling missing data not reported Dates of study: not reported |
|
| Participants |
Randomised: n = 35
Completed at 6 months: n = 28
Completed at 12 months: n = 23
Baseline characteristics Mean age (range):
Sex:
Mean disease duration (range):
Mean skin disease (SEM) (PASI)
Mean patient global assessment (SEM) (VAS 100 mm)
Mean physician global assessment (SEM) (VAS 100 mm)
Mean swollen joint count (SEM) (assumed 66 joints)
Mean tender joint count (SEM) (assumed 68 joints)
Severity of condition: not reported Diagnostic criteria: rheumatologist‐diagnosed PsA Inclusion criteria:
Exclusion criteria:
|
|
| Interventions |
Methotrexate group: methotrexate tablets in oral doses of 2.5 mg every 12 hours for 3 consecutive doses once a week. Increments of 2.5 mg/week were permitted every month up to a maximum dose of 15 mg/week if the articular response was unsatisfactory Ciclosporin A group: ciclosporin A as an oral tablet at 3 mg/kg/d with increments of 1 mg/kg/d at monthly intervals until maximum permitted dose of 5 mg/kg/d if articular response was unsatisfactory Concomitant medications: NSAIDs at the same dose used from the beginning of the study Excluded medications: not reported |
|
| Outcomes |
Time points: 6 months and 12 months Major:
Minor:
|
|
| Notes |
Clinical trials registration: not reported Funding: not reported Declarations of interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | A description of the randomisation process was not provided |
| Allocation concealment (selection bias) | High risk | A description of allocation concealment was not provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | A description of blinding procedures was not provided. This was an open study, hence it was assumed to be unblinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | A description of blinding procedures was not provided. This was an open study, hence it was assumed to be unblinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A method of handling of incomplete outcome data was not reported, and a large number of participants were withdrawn |
| Selective reporting (reporting bias) | Unclear risk | This could not be substantiated, as no trial registry record was available for comparison |
| Other bias | Unclear risk | Use of co‐intervention with NSAIDs was permitted, although not quantified Minor differences between groups were evident at baseline Compliance was acceptable, although a large number of dropouts were reported Overall this trial was judged as having unclear risk of bias for these outcomes |
Willkens 1984.
| Methods |
Study design: double‐blind, randomised placebo‐controlled trial of methotrexate vs placebo Duration of study: 12 weeks Run‐in period: none Location: United States of America Number of study centres: up to 10 based on author affiliations, although not specifically reported Study setting: outpatient Withdrawals: 4 ‐ method of handling missing data not reported Dates of study: not reported |
|
| Participants |
Randomised: n = 37
Completed: n = 33
Baseline characteristics Mean age, measure of dispersion not reported:
Sex:
Disease duration, measure of dispersion not reported:
Severity of condition:
Diagnostic criteria: rheumatologist‐diagnosed PsA Inclusion criteria:
Exclusion criteria:
|
|
| Interventions |
Methotrexate group: oral methotrexate tablets given as 2.5 mg every 12 hours for 3 consecutive doses each week. This could be increased to 15 mg/week after 6 weeks, with 3 doses of 5 mg taken at 12‐hour consecutive intervals Placebo group: placebo tablet was given every 12 hours for 3 consecutive doses each week Concomitant medications: constant background therapy of either ibuprofen (1600 to 2400 mg/d) or indomethacin (75 to 200 mg/d), which started at least 2 weeks before entry into the trial Analgesic therapy with acetaminophen or propoxyphene was allowed Excluded medications: not reported |
|
| Outcomes |
Time points: 3 months Major:
Minor:
N.B. Efficacy of treatment was based on tender joint count, swollen joint count, grip strength, physician global assessment, patient global assessment, and psoriasis involvement. These outcomes were reported within the trial as median differences between baseline and conclusion for each treatment group. No measures of dispersion were provided. Baseline measures for each outcome were not reported. Study authors were contacted but were unable to provide further details (such as mean (SD), or median (IQR), for each outcome and for each group at baseline and at 3 months) because original data were permanently unavailable |
|
| Notes |
Clinical trials registration: not reported Funding: supported by grants from the National Institute of Arthritis, Metabolism and Digestive Diseases contract no. 6‐2218, the Public Health Service Research #RR‐00064 (from the Division of Research Resources), and an Arthritis Foundation Clinical Research Center to the University of Tennessee Center for Health Sciences Declarations of interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Sequence generation was done via a randomised schedule, but this was not described in any detail |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment procedures were not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Participants received a placebo MTX tablet, although study authors did not describe the appearance of the 2 tablets, nor did they describe blinding procedures |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding procedures were not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Handling of incomplete outcome data was not described |
| Selective reporting (reporting bias) | Unclear risk | Several outcomes were reported in a manner that data could not be extracted |
| Other bias | Unclear risk | Baseline imbalances may favour the treatment group Co‐interventions were the same between groups, although their usage was not reported Compliance was considered acceptable, as a tablet count occurred at each visit Overall this trial was judged to have unclear risk of bias for these results |
Zhang 2009.
| Methods |
Study design: open‐label, quasi‐randomised, controlled, multi‐armed trial of methotrexate compared to leflunomide, or the combination of methotrexate and leflunomide Duration of study: 6 months Run‐in period: none Location: China Number of study centres: 2 Study setting: outpatient Withdrawals: 19 ‐ method of handling missing data not reported Dates of study: not reported |
|
| Participants |
Randomised: n = 65 ‐ (14 participants immediately lost to follow‐up and not accounted for throughout study; timing of dropout unclear in the manuscript)
Completed: n = 46
Baseline characteristics Mean age (SD): 40.1 years (±10.6) Sex:
Mean disease duration (SD) ‐ based on 51 participants (per published report):
Mean function (SD) (HAQ)
Mean pain (SD) (VAS 10 cm)
Mean physician global assessment (SD) (Likert 1 to 5)
Mean patient global assessment (SD) (Likert 1 to 5)
Mean swollen joint count (SD) (74 joints)
Mean tender joint count (SD) (76 joints)
Severity of condition: not reported Diagnostic criteria: rheumatologist‐diagnosed PsA Inclusion criteria:
Exclusion criteria:
|
|
| Interventions |
Methotrexate monotherapy group: methotrexate 7.5 mg/week up to 25 mg/week ‐ route not specified Leflunomide monotherapy group: leflunomide 20 mg daily Combination group ‐ data not extracted Concomitant medications: established NSAIDs or corticosteroids (prednisolone ≤10 mg daily or equivalent) were continued Excluded medications: other DMARDs, bDMARDs, or other psoriasis treatments were ceased 2 weeks before commencement of trial medications |
|
| Outcomes |
Time points: 6 months Major:
Minor:
|
|
| Notes |
Clinical trials registration: not reported Funding: not reported Declarations of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | "Randomization was done with using a random number table…32 patients of psoriatic arthritis were taken consecutively and grouped into two by card test" This was not described in further detail, and the trial was judged to be at high risk of bias |
| Allocation concealment (selection bias) | High risk | Allocation concealment was not attempted |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "This open, randomized clinical trial…" The open‐label design allowed participants and personnel to remain aware of the allocated intervention |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors was not attempted |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Out of total 32 patients, one patient from each group was excluded from analysis due to lack of follow‐up" Incomplete outcome data were inadequately addressed An ITT analysis was not performed |
| Selective reporting (reporting bias) | Unclear risk | This could not be substantiated, as no trial registry record was available for comparison HAQ and some safety assessments were selectively reported without clear prior specification Safety assessment was done in a subjective manner, without reference to a validated and accepted definition |
| Other bias | Unclear risk | Baseline characteristics varied, with the methotrexate group having more active disease The methotrexate group also used more NSAIDs Compliance was not specifically measured or reported Overall these factors present unclear risk of bias |
ACR20: American College of Rheumatology response criteria for 20% improvement.
ACR50: American College of Rheumatology response criteria for 50% improvement.
bDMARD: biological disease‐modifying anti‐rheumatic drug.
CRP: C‐reactive protein.
DAS28‐ESR: disease activity score (28 joints) with erythrocyte sedimentation rate.
DIP: distal interphalangeal joint.
DMARD: disease‐modifying anti‐rheumatic drug.
ELISA: enzyme‐linked immunosorbent drug.
ESR: erythrocyte sedimentation rate.
HAQ: Health Assessment Questionnaire for Rheumatoid Arthritis.
IQR: interquartile ratio.
ITT: intention‐to‐treat.
MTX: methotrexate.
NSAIDs: non‐steroidal anti‐inflammatory drugs.
PASI: Psoriasis Area and Severity Index.
PsA: psoriatic arthritis.
PsARC: Psoriatic Arthritis Response Criteria.
PUVA: psoralen and long‐wave ultraviolet radiation.
SCr: serum creatinine.
SD: standard deviation.
SE: standard error.
SEM: standard error of the mean.
SF‐36: Short Form‐36.
VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abu‐Shakra 1995 | Wrong study design |
| Atzeni 2011 | Wrong intervention |
| Baranauskaite 2012 | Wrong comparator |
| Bird 1977 | Could not extract PsA outcomes |
| Calguneri 2004 | Wrong study design |
| Coates 2013 | Wrong comparator |
| Coates 2014 | Wrong comparator |
| Coates 2015a | Wrong comparator |
| Coates 2015b | Wrong comparator |
| Coates 2016a | Wrong comparator |
| Coates 2017 | Wrong comparator |
| Collins 2015 | Wrong intervention |
| Combe 2013 | Wrong study design |
| Combe 2016 | Wrong study design |
| Conti 2008 | Wrong intervention |
| Feldges 1974 | Wrong study design |
| Fraser 2005 | Wrong patient population |
| Glinatsi 2015 | Wrong patient population |
| Gottlieb 2016a | Wrong intervention |
| Gottlieb 2016b | Wrong intervention |
| Goupille 1995 | Wrong study design |
| Hall 1978 | Could not extract PsA outcomes |
| Ischenko 2010 | Wrong comparator |
| Kavanaugh 2006a | Wrong patient population |
| Kavanaugh 2006b | Wrong patient population |
| Kavanaugh 2012 | Wrong intervention |
| Khraishi 2016 | Wrong intervention |
| Mazzanti 1994 | Wrong study design |
| McInnes 2015 | Wrong intervention |
| Mease 2015 | Wrong intervention |
| Mease 2016 | Wrong patient population |
| Merola 2016 | Wrong study design |
| Min 2016 | Wrong intervention |
| O'Brien 1962 | Wrong comparator |
| Raffayova 2009a | Wrong comparator |
| Raffayova 2009b | Wrong comparator |
| Saurat 2010 | Wrong patient population |
| Schett 2011 | Wrong intervention |
| Schett 2012 | Wrong intervention |
| Szentpetery 2014 | Wrong intervention |
Characteristics of ongoing studies [ordered by study ID]
NCT02376790.
| Trial name or title | A multicenter, double‐blind, randomized controlled study of etanercept and methotrexate in combination or as monotherapy in subjects with psoriatic arthritis |
| Methods | Randomised, double‐blind, controlled study |
| Participants | Adults with psoriatic arthritis per CASPAR criteria |
| Interventions | Etanercept + methotrexate orally in combination vs etanercept monotherapy + placebo tablet orally vs methotrexate 20 mg orally + placebo injection |
| Outcomes | Primary: ACR20 at 24 weeks Secondary: minimal disease activity for arthritis, non‐arthritis activity, and patient‐reported outcomes at 24 weeks |
| Starting date | 3 March 2015 |
| Contact information | Amgen Pty. Ltd. |
| Notes | Etanercept monotherapy vs methotrexate monotherapy arms would be included in this review |
ACR20: American College of Rheumatology response criteria for 20% improvement.
Differences between protocol and review
Several additional minor outcomes were not specified in the original protocol (Wilsdon 2017), specifically patient and physician global assessments of disease activity, and tender and swollen joints counts. Since publication of the protocol, the OMERACT recommended outcomes for PsA trials have been updated (Orbai 2017). These updated recommendations support inclusion of the above‐mentioned outcomes in PsA trials, and given that our objectives were still being directly answered, we considered their addition to be relevant. We have included them as minor outcomes.
The original protocol described that review authors intended to extract the proportion of participants achieving a reduction in PASI of 25% (Wilsdon 2017); however, the included studies rarely reported this. Researchers consistently reported the absolute PASI across studies, and so this became the preferred outcome measure for skin disease.
We explored the effect of including imputed values by performing a sensitivity analysis for outcomes for which this information was available. We did not specify this in the protocol (Wilsdon 2017).
Contributions of authors
TW and TT screened titles and abstracts of all records for relevant studies.
TW and TT screened relevant full‐text records for included studies.
TW and SW extracted data from included studies.
TW and AM completed a risk of bias assessment.
TW entered data into Review Manager 5 and guarantees their accuracy (RevMan 2014).
AM spot‐checked entered data.
TW drafted the final manuscript, with equal input from SW, TT, and AM.
Sources of support
Internal sources
Nil, Other.
External sources
Nil, Other.
Declarations of interest
Tom D Wilsdon: none known.
Tilenka RJ Thynne: none known.
Arduino A Mangoni: none known.
Samuel L Whittle: none known.
Edited (no change to conclusions)
References
References to studies included in this review
Asaduzzaman 2014 {published data only (unpublished sought but not used)}
- Asaduzzaman AT, Sikder A, Mahmud MM, Paul HK, Islam MN. Efficacy and safety of leflunomide in psoriatic arthritis. Journal of the Pakistan Association of Dermatology 2014;24(1):51‐6. [Google Scholar]
Black 1964 {published data only}
- Black RL, O'Brien WM, Scott EJ, Auerback R, Eizen AZ, Bunim JJ. Methotrexate therapy in psoriatic arthritis: double‐blind study on 21 patients. Journal of the American Medical Association 1964;189:743‐7. [PUBMED: 14174051] [PubMed] [Google Scholar]
Burdeinyi 1992 {published data only}
- Burdeinyi AP, Agababova ER, Korotaeva TV, Kharamil'o LF. The comparative efficacy of slow‐acting (basic) preparations in psoriatic arthritis. Terapevticheskii Arkhiv 1992;64(5):54‐9. [PUBMED: 1360714] [PubMed] [Google Scholar]
Kingsley 2012 {published and unpublished data}
- Kingsley GH, Ibrahim F, Scott DI. Quality of life in psoriatic arthritis: comparative results with rheumatoid arthritis using SF36 in trials using methotrexate. Rheumatology (Oxford) 2012;41(Suppl 3):iii89‐iii90. [Google Scholar]
- Kingsley GH, Kowalcyzk A, Taylor H, Ibrahim F, Packham JC, McHugh N, et al. Methotrexate is not disease modifying in psoriatic arthritis: the MIPA trial. Arthritis and Rheumatism 2010;62:664. [Google Scholar]
- Kingsley GH, Kowalczyk A, Taylor H, Ibrahim F, Packham JC, McHugh NJ, et al. A randomized‐controlled trial of methotrexate in psoriatic arthritis. Rheumatology (Oxford) 2012;51(8):1368‐77. [DOI: 10.1093/rheumatology/kes001; PUBMED: 22344657] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley GH, Packham JC, McHugh NJ, Mulherin DM, Kitas GD, Chakravart K, et al. Methotrexate is not disease modifying in psoriatic arthritis: a new treatment paradigm is required. Rheumatology (Oxford) 2010;49(Suppl 1):i1. [Google Scholar]
Scarpa 2008 {published data only (unpublished sought but not used)}
- Scarpa R, Peluso R, Atteno M, Manguso F, Spano A, Iervolino S, et al. The effectiveness of a traditional therapeutical approach in early psoriatic arthritis: results of a pilot randomised 6‐month trial with methotrexate. Clinical Rheumatology 2008;27(7):823‐6. [DOI: 10.1007/s10067-007-0787-7; PUBMED: 18030515] [DOI] [PubMed] [Google Scholar]
Spadaro 1995 {published data only (unpublished sought but not used)}
- Spadaro A, Riccieri V, Sili‐Scavalli A, Sensi F, Taccari E, Zoppini A. Comparison of cyclosporin A and methotrexate in the treatment of psoriatic arthritis: a one‐year prospective study. Clinical and Experimental Rheumatology 1995;13(5):589‐93. [PUBMED: 8575136] [PubMed] [Google Scholar]
Willkens 1984 {published data only (unpublished sought but not used)}
- Willkens RF, Williams HJ, Ward JR, Egger MJ, Reading JC, Clements PJ, et al. Randomized, double‐blind, placebo controlled trial of low‐dose pulse methotrexate in psoriatic arthritis. Arthritis and Rheumatism 1984;27(4):376‐81. [PUBMED: 6712754] [DOI] [PubMed] [Google Scholar]
Zhang 2009 {published data only (unpublished sought but not used)}
- Zhang GL, Huang F, Zhang JL, Li XF. A clinical study of leflunomide and methotrexate therapy in psoriatic arthritis. Zhonghua Nei Ke Za Zhi 2009;48(7):570‐4. [PUBMED: 19957798] [PubMed] [Google Scholar]
References to studies excluded from this review
Abu‐Shakra 1995 {published data only}
- Abu‐Shakra M, Gladman DD, Thorne JC, Long J, Gough G, Farewell VT. Long term methotrexate therapy in psoriatic arthritis: clinical and radiological outcome. Journal of Rheumatology 1995;22:241‐5. [PubMed] [Google Scholar]
Atzeni 2011 {published data only}
- Atzeni F, Boccassini L, Anitvalle M, Salaffi F, Sarzi‐Puttini P. Etanercept plus ciclosporin versus etanercept plus methotrexate for maintaining clinical control over psoriatic arthritis: a randomised pilot study. Annals of Rheumatic Diseases 2011;70(4):712‐4. [DOI] [PubMed] [Google Scholar]
Baranauskaite 2012 {published data only}
- Baranauskaite A, Raffayova H, Kungurov NV, Kubanova A, Venalis A, Helmle L, et al. Infliximab plus methotrexate is superior to methotrexate alone in the treatment of psoriatic arthritis in methotrexate‐naive patients: the RESPOND study. Annals of Rheumatic Diseases 2012;71:541‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bird 1977 {published data only}
- Bird HA, Ring EF, Daniel R, Bacon PA. Comparison of intra‐articular methotrexate with intra‐articular triamcinolone hexacetonide by thermography. Current Medical Research and Opinion 1977;5(2):141‐6. [DOI] [PubMed] [Google Scholar]
Calguneri 2004 {published data only}
- Calguneri M, Cobankara V, Ozturk MA, Ertenli I, Kiraz S, Apras S. Combination therapies in spondyloarthropathies. Kobe Journal of Medical Science 2004;50(2):31‐7. [PubMed] [Google Scholar]
Coates 2013 {published data only}
- Coates LC, Moverley AR, McParland L, Brown S, Collier H, Law J, et al. Results of a randomised controlled trial comparing tight control of early psoriatic arthritis (TICOPA) with standard care: tight control improves outcome. Arthitis and Rheumatism 2013;65:S346. [Google Scholar]
Coates 2014 {published data only}
- Coates L, Moverley A, McParland L, Brown S, Collier H, Brown SR, et al. Effect of tight control of inflammation in early psoriatic arthritis (TICOPA): a multicentre, open‐label, randomised controlled trial. Lancet 2014;383:S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coates 2015a {published data only}
- Coates LC, Helliwell PS. Methotrexate in early psoriatic arthritis ‐ open label data from the TICOPA study. Annals of Rheumatic Diseases 2015;74:861. [Google Scholar]
Coates 2015b {published data only}
- Coates LC, Moverley AR, McParland L, Brown S, Navarro‐Coy N, O'Dwyer JL, et al. Effect of tight control of inflammation in early psoriatic arthritis (TICOPA): a UK multicentre, open‐label, randomised, controlled trial. Lancet 2015;386(10012):2489‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coates 2016a {published data only}
- Coates LC, Helliwell PS. Methotrexate in the tight control in psoriatic arthritis study. Journal of Rheumatology 2016;43(2):356‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coates 2017 {published data only}
- Coates LC, Mahmood F, Emery P, Conaghan PG, Helliwell PS. The dynamics of response as measured by multiple composite outcome tools in the tight control of inflammation in early psoriatic arthritis (TICOPA) trial. Annals of Rheumatic Diseases 2017;76(10):1688‐92. [DOI] [PubMed] [Google Scholar]
Collins 2015 {published data only}
- Collins A, Heijde D, Landewe R, Mease P, McInnes I, Conaghan P, et al. Secukinumab, a monoclonal antibody to interleukin‐17a, provides significant and sustained inhibition of joint structural damage in active psoriatic arthritis regardless of prior TNF inhibitors or concomitant methotrexate: a phase 3 randomized, double‐blind, placebo‐controlled study. Internal Medicine Journal 2015;45(Suppl 2):42. [Google Scholar]
Combe 2013 {published data only}
- Combe B, Kerkmann U, Brock F, Gallo G. Response to etanercept with or without methotrexate combination therapy in patients with psoriatic arthritis. Arthritis and Rheumatism 2013;65:S154. [Google Scholar]
Combe 2016 {published data only}
- Combe B, Behrens F, McHugh N, Brock F, Kerkmann U, Kola B, et al. Comparison of etanercept monotherapy and combination therapy with methotrexate in psoriatic arthritis: results from 2 clinical trials. Journal of Rheumatology 2016;43(6):1063‐7. [DOI] [PubMed] [Google Scholar]
Conti 2008 {published data only}
- Conti F, Ceccarelli F, Priori R, Iagnocco A, Signore A, Velsini G. Intra‐articular infliximab in patients with rheumatoid arthritis and psoriatic arthritis with monoarthritis resistant to local glucocorticoids. Clinical efficacy extended to patients on systemic antitumour necrosis factor alpha. Annals of Rheumatic Diseases 2008;67:1787‐90. [DOI] [PubMed] [Google Scholar]
Feldges 1974 {published data only}
- Feldges DH, Barnes CG. Treatment of psoriatic arthropathy with either azathioprine or methotrexate. Rheumatology and Rehabilitation 1974;13(3):120‐4. [DOI] [PubMed] [Google Scholar]
Fraser 2005 {published data only}
- Fraser AD, Kuijk AW, Westhovens R, Karim Z, Wakefield R, Gerards AH, et al. A randomised, double blind, placebo controlled, multicentre trial of combination therapy with methotrexate plus ciclosporin in patients with active psoriatic arthritis. Annals of Rheumatic Diseases 2005;64(6):859‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Glinatsi 2015 {published data only}
- Glinatsi D, Bird P, Gandjbakhch F, Mease PJ, Boyesen P, Peterfy CH, et al. Validation of the OMERACT psoriatic arthritis magnetic resonance imaging score (PsAMRIS) for the hand and foot in a randomized placebo‐controlled trial. Journal of Rheumatology 2015;42(12):2473‐9. [DOI] [PubMed] [Google Scholar]
Gottlieb 2016a {published data only}
- Gottlieb AB, McInnes I, Mease P, Mpofu S. Secukinumab improves signs and symptoms of psoriatic arthritis: results of phase 3 FUTURE 2 study stratified by concomitant methotrexate use. Journal of the American Academy of Dermatology 2016;1:AB270. [Google Scholar]
Gottlieb 2016b {published data only}
- Gottlieb A, Mease P, McInnes I, Mpofu S. Secukinumab inhibits radiographic progression in patients with psoriatic arthritis: results of phase 3 FUTURE 1 study stratified by concomitant methotrexate use. Journal of the American Academy of Dermatology 2016;1:AB271. [Google Scholar]
Goupille 1995 {published data only}
- Goupille P, Valat JP, Gladman DD. Is methotrexate really effective in patients with psoriatic arthritis?. Journal of Rheumatology 1995;22(12):2369‐70. [PubMed] [Google Scholar]
Hall 1978 {published data only}
- Hall GH, Jones BJ, Head AC, Jones VE. Intra‐articular methotrexate. Clinical and laboratory study in rheumatoid and psoriatic arthritis. Annals of Rheumatic Diseases 1978;37(4):351‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ischenko 2010 {published data only}
- Ischenko A, Kleymenova I, Raymuev K, Solovieva L, Antipova T, Simbirtsev A. Comparison study of recombinant human interleukin‐1 receptor antagonist for treating psoriatic and rheumatoid arthritis. Cytokine 2010;52:22. [DOI: 10.1016/j.cyto.2010.07.095] [DOI] [Google Scholar]
Kavanaugh 2006a {published data only}
- Kavanaugh A, Antoni CE, Gladman DD. The infliximab multinational psoriatic arthritis controlled trial (IMPACT): results of radiographic analyses after 1 year. Annals of Rheumatic Diseases 2006;65(8):1038‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kavanaugh 2006b {published data only}
- Kavanaugh A, Antoni C, Krueger GG, Yan S, Bala M, Dooley LT, et al. Infliximab improves health related quality of life and physical function in patients with psoriatic arthritis. Annals of Rheumatic Diseases 2006;65:471‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kavanaugh 2012 {published data only}
- Kavanaugh A, Gladman D, Beutler A, Han C, Chandran V. Golimumab treatment inhibits progression in joint damage in patients with psoriatic arthritis regardless of baseline disease severity. Journal of Rheumatology 2012;39:1736‐7. [Google Scholar]
Khraishi 2016 {published data only}
- Khraishi M, Gottlieb AB, Hoepken B, Peterson L, Mease PJ. The effect of certolizumab pegol on skin manifestations of psoriatic arthritis over 4 years of treatment. Arthritis and Rheumatology 2016;68(Suppl 10):1724. [Google Scholar]
Mazzanti 1994 {published data only}
- Mazzanti G, Colono L, Sabbata G, Paladini G. Methotrexate and cyclosporine combined therapy in severe psoriatic arthritis. A pilot study. Acta Dermato‐Venerologica (Stockholm) 1994;186:116‐7. [PubMed] [Google Scholar]
McInnes 2015 {published data only}
- McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti‐interleukin 17a monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2015;386(9999):1137‐46. [DOI] [PubMed] [Google Scholar]
Mease 2015 {published data only}
- Mease P, Gottlieb AB, Berman A, Drescher E, Xing J, Banerjee J, et al. A phase IIb, randomized, double‐blind, placebo‐controlled, dose‐ranging, multicenter study to evaluate the efficacy and safety of clazakinumab, an anti‐IL‐6 monoclonal antibody, in adults with active psoriatic arthritis. Arthritis and Rheumatology 2015;66:S423. [Google Scholar]
Mease 2016 {published data only}
- Mease P, Gottlieb AB, Heijde D, FitzGerald O, Johnsen A, Nys M, et al. Abatacept in the treatment of active psoriatic arthritis: 24‐week results from a phase III study. Arthritis and Rheumatology 2016;68(Suppl 10):1366‐9. [Google Scholar]
Merola 2016 {published data only}
- Merola J, Sundaram M, Yang M, Li J, Galebach P, Okun M. Adalimumab is associated with reduced risk of joint‐related signs and symptoms compared with methotrexate in patients with moderate to severe psoriasis. Journal of the American Academy of Dermatology 2016;1:AB235. [Google Scholar]
Min 2016 {published data only}
- Min T. Observation of efficacy of biological combined with different DMARDs against psoriatic arthritis. International Journal of Rheumatic Diseases 2016;19:163. [Google Scholar]
O'Brien 1962 {published data only}
- O'Brien WM, Scott EJ, Black RL, Eizen AZ, Bunim JJ. Clinical trial of amethopterin (methotrexate) in psoriatic and rheumatoid arthritis (preliminary report). Arthritis and Rheumatism 1962;5(3):312. [Google Scholar]
Raffayova 2009a {published data only}
- Raffayova H, Kungurov N, Kubanova A, Baranauskaite A, Venalis A, Helmle L, et al. Infliximab plus methotrexate significantly improves rates of remission for methotrexate naive psoriatic arthritis (PsA) patients compared to methotrexate alone: the RESPOND trial. Arthritis and Rheumatism 2009;60:1780. [Google Scholar]
Raffayova 2009b {published data only}
- Raffayova H, Kunvurov N, Kubanova A, Baranauskaite A, Venalis A, Helmle L, et al. Infliximab plus methotrexate significantly improves synovitis and psoriatic lesions in methotrexate naive psoriatic arthritis (PsA) patients: results of the RESPOND trial. Arthritis and Rheumatism 2009;60:1256. [Google Scholar]
Saurat 2010 {published data only}
- Saurah JH, Unnebrink K, Goldblum O, Bissonnette R. Adalimumab response is consistent across subgroups of patients with moderate to severe psoriasis: subanalysis of the CHAMPION study. Journal of the American Academy of Dermatology 2010;1:AB124. [Google Scholar]
Schett 2011 {published data only}
- Schett GA, Hu A, Stevens R. Oral apremilast is effective with and without concomitant methotrexate therapy in the treatment of subjects with active psoriatic arthritis. Arthritis and Rheumatism 2011;63(Suppl 10):780. [Google Scholar]
Schett 2012 {published data only}
- Schett G, Hu A, Stevens R. Oral apremilast is effective in the treatment of subjects with active psoriatic arthritis with and without concomitant methotrexate therapy. Zeithschrift fur Rheumatologie 2012;71:136. [Google Scholar]
Szentpetery 2014 {published data only}
- Szentpetery A, Heffernan EJ, Haroon M, Gallagher P, Baker A‐M, Cooney M, et al. Abatacept improves synovitis as assessed by magnetic resonance imaging (MRI) in psoriatic arthritis ‐ preliminary analysis from a single centre, placebo‐controlled, crossover study. Arthritis and Rheumatology 2015;66:S607. [Google Scholar]
References to ongoing studies
NCT02376790 {published data only}
- A multicenter, double‐blind, randomized controlled study of etanercept and methotrexate in combination or as monotherapy in subjects with psoriatic arthritis. Ongoing study 3 March 2015.
Additional references
Alarcón 2000
- Alarcón GS. Methotrexate use in rheumatoid arthritis. A clinician's perspective. Immunopharmacology 2000;47(2‐3):259‐71. [DOI: 10.1016/S0162-3109(00)00184-3] [DOI] [PubMed] [Google Scholar]
Aletaha 2005
- Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Research and Therapy 2005;7(4):R796‐806. [DOI: 10.1186/ar1740] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ali 2007
- Ali Y, Tom BD, Schentag CT, Farewell VT, Gladman DD. Improved survival in psoriatic arthritis with calendar time. Arthritis and Rheumatism 2007;56(8):2708‐14. [DOI: 10.1002/art.22800] [DOI] [PubMed] [Google Scholar]
Antoni 2005
- Antoni CE, Kavanaugh A, Kirkham B, Tutuncu Z, Burmester GR, Schneider U, et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis ‐ results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis and Rheumatism 2005;52(4):1227‐36. [DOI: 10.1002/art.20967; PUBMED: 15818699] [DOI] [PubMed] [Google Scholar]
Cates 2008 [Computer program]
- Dr. Christopher Cates EBM website. www.nntonline.net. Visual Rx. Version 3. Dr. Christopher Cates EBM website. www.nntonline.net, 2008.
Clegg 1996
- Clegg DO, Reda DJ, Mejias E, Cannon GW, Weisman MH, Taylor T, et al. Comparison of sulfasalazine and placebo in the treatment of psoriatic arthritis: a Department of Veterans Affairs cooperative study. Arthritis and Rheumatism 1996;39(12):2013‐20. [DOI: 10.1002/art.1780391210] [DOI] [PubMed] [Google Scholar]
Coates 2012
- Coates LC, Conaghan PG, Emery P, Green MJ, Ibrahim G, MacIver H, et al. Sensitivity and specificity of the classification of psoriatic arthritis criteria in early psoriatic arthritis. Arthritis and Rheumatism 2012;64(10):3150‐5. [DOI: 10.1002/art.34536] [DOI] [PubMed] [Google Scholar]
Coates 2016b
- Coates LC, Kavanaugh A, Mease PJ, Soriano ER, Laura Acosta‐Felquer M, Amstrong AW, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis and Rheumatology 2016;68(5):1060‐71. [DOI: 10.1002/art.39573; PUBMED: 26749174] [DOI] [PubMed] [Google Scholar]
Coates 2017a
- Coates LC, Gossec L, Ramiro S, Mease P, Heijde D, Smolen JS, et al. New GRAPPA and EULAR recommendations for the management of psoriatic arthritis. Rheumatology (Oxford) 2017;56(8):1251‐3. [DOI: 10.1093/rheumatology/kew390; PUBMED: 28077693] [DOI] [PubMed] [Google Scholar]
Coates 2017b
- Coates LC, Mahmood F, Emery P, Conaghan PG, Helliwell PS. The dynamics of response as measured by multiple composite outcome tools in the tight control of inflammation in early psoriatic arthritis (TICOPA) trial. Annals of the Rheumatic Diseases 2017;76:1688‐92. [DOI: 10.1136/annrheumdis-2017-211137; PUBMED: 28606970] [DOI] [PubMed] [Google Scholar]
Conway 2014
- Conway R, Low C, Coughlan RJ, O'Donnell MJ, Carey JJ. Methotrexate and lung disease in rheumatoid arthritis: a meta‐analysis of randomized controlled trials. Arthritis and Rheumatology 2014;66(4):803‐12. [DOI: 10.1002/art.38322] [DOI] [PubMed] [Google Scholar]
Conway 2015
- Conway R, Low C, Coughlan RJ, O'Donnell MJ, Carey JJ. Methotrexate use and risk of lung disease in psoriasis, psoriatic arthritis, and inflammatory bowel disease: systematic literature review and meta‐analysis of randomised controlled trials. BMJ 2015;350:h1269. [DOI: 10.1136/bmj.h1269] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cronstein 2000
- Cronstein BN, Chan ES. Treatment of inflammatory disease. In: Cronstein BN, Bertino JR editor(s). Methotrexate. Basel: Birkhauser, 2000:65‐82. [Google Scholar]
Curtis 2009
- Curtis JR, Beukelman T, Onofrei A, Cassell S, Greenberg JD, Kavanaugh A, et al. Elevated liver enzyme tests among patients with rheumatoid arthritis or psoriatic arthritis treated with methotrexate and/or leflunomide. Annals of the Rheumatic Diseases 2009;69(1):43‐7. [DOI: 10.1136/ard.2008.101378] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cutolo 2002
- Cutolo M, Seriolo B, Pizzorni C, Craviotto C, Sulli A. Methotrexate in psoriatic arthritis. Clinical and Experimental Rheumatology 2002;20(6 Suppl 28):S76‐80. [PUBMED: 12463453] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG, editor(s). Chapter 9. Analysing data and undertaking meta‐analyses. In Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). Available from handbook.cochrane.org.
Duarte 2012
- Duarte GV, Faillace C, Freire de Carvalho J. Psoriatic arthritis. Best Practice and Research Clinical Rheumatology 2012;26(1):147‐56. [DOI: 10.1016/j.berh.2012.01.003] [DOI] [PubMed] [Google Scholar]
Felson 1995
- Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis and Rheumatism 1995;38(6):727‐35. [DOI: 10.1002/art.1780380602] [DOI] [PubMed] [Google Scholar]
Fredriksson 1978
- Fredriksson T, Pettersson U. Severe psoriasis ‐ oral therapy with a new retinoid. Dermatologica 1978;157(4):238‐44. [PUBMED: 357213] [DOI] [PubMed] [Google Scholar]
Fries 1980
- Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis and Rheumatism 1980;23(2):137‐45. [PUBMED: 7362664] [DOI] [PubMed] [Google Scholar]
Ghogomu 2014
- Ghogomu EA, Maxwell LJ, Buchbinder R, Rader T, Pardo Pardo J, Johnston RV, et al. Updated method guidelines for Cochrane Musculoskeletal Group systematic reviews and metaanalyses. Journal of Rheumatology 2014;41(2):194‐205. [DOI: 10.3899/jrheum.121306] [DOI] [PubMed] [Google Scholar]
Gladman 1987
- Gladman DD, Shuckett R, Russell ML, Thorne JC, Schachter RK. Psoriatic arthritis (PSA) ‐ an analysis of 220 patients. Quarterly Journal of Medicine 1987;62(238):127‐41. [PUBMED: 3659255] [PubMed] [Google Scholar]
Gladman 2005
- Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Annals of the Rheumatic Diseases 2005;64(Suppl 2):ii14‐7. [DOI: 10.1136/ard.2004.032482] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gladman 2007
- Gladman DD, Mease PJ, Strand V, Healy P, Helliwell PS, Fitzgerald O, et al. Consensus on a core set of domains for psoriatic arthritis. Journal of Rheumatology 2007;34(5):1167‐70. [0315162X‐34‐1167 [pii]] [PubMed] [Google Scholar]
Glanville 2006
- Glanville JM, Lefebvre C, Miles JN, Camosso‐Stefinovic J. How to identify randomized controlled trials in MEDLINE: ten years on. Journal of the Medical Library Association 2006;94(2):130‐6. [PUBMED: 16636704] [PMC free article] [PubMed] [Google Scholar]
Gossec 2016
- Gossec L, Smolen JS, Ramiro S, Wit M, Cutolo M, Dougados M, et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Annals of the Rheumatic Diseases 2016;75(3):499‐510. [PUBMED: 26644232] [DOI] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime, Inc.). Available from www.gradepro.org. GRADEpro Guideline Development Tool. Hamilton (ON): McMaster University (developed by Evidence Prime, Inc.). Available from www.gradepro.org, 2015.
Hamilton 1997
- Hamilton RA, Kremer JM. Why intramuscular methotrexate may be more efficacious than oral dosing in patients with rheumatoid arthritis. British Journal of Rheumatology 1997;36(1):86‐90. [PUBMED: 9117183] [DOI] [PubMed] [Google Scholar]
Healy 2008
- Healy PJ, Helliwell PS. Measuring clinical enthesitis in psoriatic arthritis: assessment of existing measures and development of an instrument specific to psoriatic arthritis. Arthritis Care and Research 2008;59(5):686‐91. [DOI: 10.1002/art.23568] [DOI] [PubMed] [Google Scholar]
Helliwell 2005
- Helliwell PS, Firth J, Ibrahim GH, Melsom RD, Shah I, Turner DeE. Development of an assessment tool for dactylitis in patients with psoriatic arthritis. Journal of Rheumatology 2005;32(9):1745‐50. [PUBMED: 16142872] [PubMed] [Google Scholar]
Helliwell 2008
- Helliwell PS, Taylor WJ, CASPAR Study Group. Treatment of psoriatic arthritis and rheumatoid arthritis with disease modifying drugs ‐ comparison of drugs and adverse reactions. Journal of Rheumatology 2008;35(3):472‐6. [PUBMED: 18203324] [PubMed] [Google Scholar]
Heuft‐Dorenbosch 2003
- Heuft‐Dorenbosch L, Spoorenberg A, Tubergen A, Landewé R, ver Tempel H, Mielants H, et al. Assessment of enthesitis in ankylosing spondylitis. Annals of the Rheumatic Diseases 2003;62(2):127‐32. [DOI: 10.1136/ard.62.2.127] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC, editor(s). Chapter 8. Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, editor(s). Chapter 7. Selecting studies and collecting data. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Husted 1997
- Husted JA, Gladman DD, Farewell VT, Long JA, Cook RJ. Validating the SF‐36 health survey questionnaire in patients with psoriatic arthritis. Journal of Rheumatology 1997;24(3):511‐7. [PUBMED: 9058658] [PubMed] [Google Scholar]
Jones 2000
- Jones G, Crotty M, Brooks P. Interventions for treating psoriatic arthritis. Cochrane Database of Systematic Reviews 2000, Issue 3. [DOI: 10.1002/14651858.CD000212] [DOI] [PubMed] [Google Scholar]
Kavanaugh 2009
- Kavanaugh A, McInnes I, Mease P, Krueger GG, Gladman DD, Gomez‐Reino J, et al. Golimumab, a new human tumour necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: twenty‐four‐week efficacy and safety results of a randomized, placebo‐controlled study. Arthritis and Rheumatism 2009;60(4):976‐86. [DOI: 10.1002/art.24403; PUBMED: 19333944] [DOI] [PubMed] [Google Scholar]
Kavanaugh 2017
- Kavanaugh A, Husni ME, Harrison DD, Kim L, Lo KH, Leu JH, et al. Safety and efficacy of intravenous golimumab in patients with active psoriatic arthritis. Arthritis and Rheumatology 2017;69(11):2151‐61. [DOI: 10.1002/art.40226; PUBMED: 28805045] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kosinski 2000
- Kosinski M, Zhao SZ, Dedhiya S, Osterhaus JT, Ware JE Jr. Determining minimally important changes in generic and disease‐specific health‐related quality of life questionnaires in clinical trials of rheumatoid arthritis. Arthritis and Rheumatism 2000;43(7):1478‐87. [PUBMED: 10902749] [DOI] [PubMed] [Google Scholar]
Krupp 1989
- Krupp LB, LaRocca NG, Muir‐Nash J, Steinberg AD. The fatigue severity scale. Application to patients with multiple sclerosis and systemic lupus erythematosus. Archives of Neurology 1989;46(10):1121‐3. [PUBMED: 2803071] [DOI] [PubMed] [Google Scholar]
Kvien 2005
- Kvien TK, Heiberg MS, Lie E, Kaufmann C, Mikkelsen K, Nordvåg B‐Y, et al. A Norwegian DMARD register: prescriptions of DMARDs and biological agents to patients with inflammatory rheumatic diseases. Clinical and Experimental Rheumatology 2005;23(5 Suppl 39):S188‐94. [PUBMED: 16273806] [PubMed] [Google Scholar]
Larsen 1977
- Larsen A, Dale K, Eek M. Radiographic evaluation of rheumatoid arthritis and related conditions by standard reference films. Acta Radiologica: Diagnosis 1977;18(4):481‐91. [PUBMED: 920239] [DOI] [PubMed] [Google Scholar]
Li 2015
- Li L, Hagberg KW, Peng M, Shah K, Paris M, Jick S. Rates of cardiovascular disease and major adverse cardiovascular events in patients with psoriatic arthritis compared to patients without psoriatic arthritis. Journal of Clinical Rheumatology 2015;21(8):405‐10. [DOI: 10.1097/RHU.0000000000000306] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lopez‐Olivo 2014
- Lopez‐Olivo MA, Siddhanamatha HR, Shea B, Tugwell P, Wells GA, Suarez‐Almazor ME. Methotrexate for treating rheumatoid arthritis. Cochrane Database of Systematic Reviews 2014, Issue 6. [DOI: 10.1002/14651858.CD000957.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
McKenna 2003
- McKenna SP, Cook SA, Whalley D, Doward LC, Richards HL, Griffiths CE, et al. Development of the PSORIQoL, a psoriasis‐specific measure of quality of life designed for use in clinical practice and trials. British Journal of Dermatology 2003;149(2):323‐31. [PUBMED: 12932239] [DOI] [PubMed] [Google Scholar]
Mease 2005
- Mease PJ, Gladman DD, Ritchlin CT, Ruderman EM, Steinfeld SD, Choy EH, et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double‐blind, randomized, placebo‐controlled trial. Arthritis and Rheumatism 2005;52(10):3279‐89. [DOI: 10.1002/art.21306; PUBMED: 16200601] [DOI] [PubMed] [Google Scholar]
Mease 2013
- Mease PJ, Gladman DD, Papp KA, Khraishi MM, Thaçi D, Behrens F, et al. Prevalence of rheumatologist‐diagnosed psoriatic arthritis in patients with psoriasis in European/North American dermatology clinics. Journal of the American Academy of Dermatology 2013;69(5):729‐35. [DOI: 10.1016/j.jaad.2013.07.023] [DOI] [PubMed] [Google Scholar]
Micha 2011
- Micha R, Imamura F, Wyler von Ballmoos M, Solomon DH, Hernán MA, Ridker PM, et al. Systematic review and meta‐analysis of methotrexate use and risk of cardiovascular disease. American Journal of Cardiology 2011;108(9):1362‐70. [DOI: 10.1016/j.amjcard.2011.06.054] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moll 1973
- Moll JM, Wright V. Psoriatic arthritis. Seminars in Arthritis and Rheumatism 1973;3(1):55‐78. [PUBMED: 4581554] [DOI] [PubMed] [Google Scholar]
Orbai 2017
- Orbai A‐M, Wit M, Mease PJ, Duffin KC, Elmamoun M, Tillett W, et al. Updating the Psoriatic Arthritis (PsA) Core Domain Set: a report from the PsA Workshop at OMERACT 2016. Journal of Rheumatology 2017;44:1522‐8. [DOI: 10.3899/jrheum.160904] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pincus 1983
- Pincus T, Summey JA, Soraci SA, Wallston KA, Hummon NP. Assessment of patient satisfaction in activities of daily living using a modified Stanford Health Assessment Questionnaire. Arthritis and Rheumatism 1983;26(11):1346‐53. [PUBMED: 6639693] [DOI] [PubMed] [Google Scholar]
Pincus 2015
- Pincus T, Bergman M, Yazici Y. Limitations of clinical trials in chronic diseases: is the efficacy of methotrexate (MTX) underestimated in polyarticular psoriatic arthritis on the basis of limitations of clinical trials more than on limitations of MTX, as was seen in rheumatoid arthritis?. Clinical and Experimental Rheumatology 2015;33(5 Suppl 93):S82‐93. [PUBMED: 26472658] [PubMed] [Google Scholar]
Prevoo 1995
- Prevoo ML, Van'T Hof MA, Kuper HH, Leeuwen MA, Putte LB, Riel PL. Modified disease activity scores that include twenty‐eight‐joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis and Rheumatism 1995;38(1):44‐8. [DOI: 10.1002/art.1780380107] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Roubille 2015
- Roubille C, Richer V, Starnino T, McCourt C, McFarlane A, Fleming P, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non‐steroidal anti‐inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta‐analysis. Annals of the Rheumatic Diseases 2015;74(3):480‐9. [DOI: 10.1136/annrheumdis-2014-206624] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schiff 2014
- Schiff MH, Jaffe JS, Freundlich B. Head‐to‐head, randomised, crossover study of oral versus subcutaneous methotrexate in patients with rheumatoid arthritis: drug‐exposure limitations of oral methotrexate at doses ≥15 mg may be overcome with subcutaneous administration. Annals of the Rheumatic Diseases 2014;73(8):1549‐51. [DOI: 10.1136/annrheumdis-2014-205228] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schmitt 2014
- Schmitt J, Rosumeck S, Thomaschewski G, Sporbeck B, Haufe E, Nast A. Efficacy and safety of systemic treatments for moderate‐to‐severe psoriasis: meta‐analysis of randomized controlled trials. British Journal of Dermatology 2014;170(2):274‐303. [DOI: 10.1111/bjd.12663] [DOI] [PubMed] [Google Scholar]
Schünemann 2011a
- Schünemann H, Oxman AD, Vist GE, Higgins JBT, Deeks JJ, Glasziou P, et al. editor(s). Chapter 12. Interpreting results and drawing conclusions. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org..
Schünemann 2011b
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH, editor(s). Chapter 11. Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Scott 1977
- Scott PJ, Huskisson EC. Measurement of functional capacity with visual analogue scales. Rheumatology and Rehabilitation 1977;16(4):257‐9. [PUBMED: 414343] [DOI] [PubMed] [Google Scholar]
Sharp 1971
- Sharp JT, Lidsky MD, Collins LC, Moreland J. Methods of scoring the progression of radiologic changes in rheumatoid arthritis. Correlation of radiologic, clinical and laboratory abnormalities. Arthritis and Rheumatism 1971;14(6):706‐20. [PUBMED: 5135791] [DOI] [PubMed] [Google Scholar]
Shea 2013
- Shea B, Swinden MV, Ghogomu E, Ortiz Z, Katchamart W, Rader T, et al. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. Cochrane Database of Systematic Reviews 2013, Issue 5. [DOI: 10.1002/14651858.CD000951.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smets 1995
- Smets EM, Garssen B, Bonke B, Haes JC. The Multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. Journal of Psychosomatic Research 1995;39(3):315‐25. [PUBMED: 7636775] [DOI] [PubMed] [Google Scholar]
Smolen 2014a
- Smolen JS, Heijde D, Machold KP, Aletaha D, Landewé R. Proposal for a new nomenclature of disease‐modifying antirheumatic drugs. Annals of the Rheumatic Diseases 2014;73(1):3‐5. [DOI: 10.1136/annrheumdis-2013-204317] [DOI] [PubMed] [Google Scholar]
Smolen 2014b
- Smolen JS, Braun J, Dougados M, Emery P, Fitzgerald O, Helliwell P, et al. Treating spondyloarthritis, including ankylosing spondylitis and psoriatic arthritis, to target: recommendations of an international task force. Annals of the Rheumatic Diseases 2014;73(1):6‐16. [DOI: 10.1136/annrheumdis-2013-203419] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D, editor(s). Chapter 10. Addressing reporting biases. In Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Stolwijk 2016
Taylor 2006
- Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis and Rheumatism 2006;54(8):2665‐73. [DOI: 10.1002/art.21972] [DOI] [PubMed] [Google Scholar]
Theander 2014
- Theander E, Husmark T, Alenius G‐M, Larsson PT, Teleman A, Geijer M, et al. Early psoriatic arthritis: short symptom duration, male gender and preserved physical functioning at presentation predict favourable outcome at 5‐year follow‐up. Results from the Swedish Early Psoriatic Arthritis Register (SwePsA). Annals of the Rheumatic Diseases 2014;73(2):407‐13. [DOI: 10.1136/annrheumdis-2012-201972] [DOI] [PubMed] [Google Scholar]
Torre Alonso 1991
- Torre Alonso JC, Rodriguez Perez A, Arribas Castrillo JM, Ballina Garcia J, Riestra Noriega JL, Lopez Larrea C. Psoriatic arthritis (PA): a clinical, immunological and radiological study of 180 patients. British Journal of Rheumatology 1991;30(4):245‐50. [DOI: 10.1093/rheumatology/30.4.245] [DOI] [PubMed] [Google Scholar]
Truong 2015
- Truong B, Rich‐Garg N, Ehst B, Deodhar A, Ku J, Vakil‐Gilani K, et al. Demographics, clinical disease characteristics, and quality of life in a large cohort of psoriasis patients with and without psoriatic arthritis. Clinical, Cosmetic and Investigational Dermatology 2015;8:563‐9. [DOI: 10.2147/CCID.S90270] [DOI] [PMC free article] [PubMed] [Google Scholar]
Van der Heijde 2000
- Heijde D. How to read radiographs according to the Sharp/van der Heijde method. Journal of Rheumatology 2000;27(1):261‐3. [PUBMED: 10648051] [PubMed] [Google Scholar]
Van Doornum 2002
- Doornum S, McColl G, Wicks IP. Accelerated atherosclerosis: an extraarticular feature of rheumatoid arthritis?. Arthritis and Rheumatism 2002;46(4):862‐73. [DOI: 10.1002/art.10089] [DOI] [PubMed] [Google Scholar]
Van Gestel 1996
- Gestel AM, Prevoo ML, 't Hof MA, Rijswijk MH, Putte LB, Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism criteria. Arthritis and Rheumatism 1996;39(1):34‐40. [DOI: 10.1002/art.1780390105] [DOI] [PubMed] [Google Scholar]
Veale 2015
- Veale DJ, Fearon U. What makes psoriatic and rheumatoid arthritis so different?. RMD Open 2015;1(1):e000025. [DOI: 10.1136/rmdopen-2014-000025] [DOI] [PMC free article] [PubMed] [Google Scholar]
Walsh 2014
- Walsh JA, McFadden ML, Morgan MD, Sawitzke AD, Duffin KC, Krueger GG, et al. Work productivity loss and fatigue in psoriatic arthritis. Journal of Rheumatology 2014;41(8):1670‐4. [DOI: 10.3899/jrheum.140259] [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item Short‐Form Health Survey (SF‐36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473‐83. [PUBMED: 1593914] [PubMed] [Google Scholar]
Wasko 2013
- Wasko MC, Dasgupta A, Hubert H, Fries JF, Ward MM. Propensity‐adjusted association of methotrexate with overall survival in rheumatoid arthritis. Arthritis and Rheumatism 2013;65(2):334‐42. [DOI: 10.1002/art.37723] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wassenberg 2001
- Wassenberg S, Fischer‐Kahle V, Herborn G, Rau R. A method to score radiographic change in psoriatic arthritis. Zeitschrift für Rheumatologie 2001;60(3):156‐66. [PUBMED: 11475603] [DOI] [PubMed] [Google Scholar]
Webster 2003
- Webster K, Cella D, Yost K. The Functional Assessment of Chronic Illness Therapy (FACIT) Measurement System: properties, applications, and interpretation. Health and Quality of Life Outcomes 2003;1:79. [DOI: 10.1186/1477-7525-1-79] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wilsdon 2017
- Wilsdon TD, Whittle SL, Thynne TR, Mangoni AA. Methotrexate for psoriatic arthritis. Cochrane Database of Systematic Reviews 2017, Issue 7. [DOI: 10.1002/14651858.CD012722] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wong 2006
- Wong SS, Wilczynski NL, Haynes RB. Developing optimal search strategies for detecting clinically sound causation studies in EMBASE. Journal of the Medical Library Association 2006;94(1):41‐7. [D030002662 [pii]] [PMC free article] [PubMed] [Google Scholar]
Zachariae 2002
- Zachariae H, Zachariae R, Blomqvist K, Davidsson S, Molin L, Mørk C, et al. Quality of life and prevalence of arthritis reported by 5,795 members of the Nordic Psoriasis Associations. Data from the Nordic Quality of Life Study. Acta Dermato‐Venereologica 2002;82(2):108‐13. [DOI: 10.1080/00015550252948130] [DOI] [PubMed] [Google Scholar]