

Abstract
The genetic differentiation of the current horse population was evolutionarily created by natural or artificial selection which shaped the genomes of individual breeds in several unique ways. The availability of high throughput genotyping methods created the opportunity to study this genetic variation on a genome-wide level allowing detection of genome regions divergently selected between separate breeds as well as among different horse types sharing similar phenotypic features. In this study, we used the population differentiation index (FST) that is generally used for measuring locus-specific allele frequencies variation between populations, to detect selection signatures among six horse breeds maintained in Poland. These breeds can be classified into three major categories, including light, draft and primitive horses, selected mainly in terms of type (utility), exterior, performance, size, coat color and appearance. The analysis of the most pronounced selection signals found in this study allowed us to detect several genomic regions and genes connected with processes potentially important for breed phenotypic differentiation and associated with energy homeostasis during physical effort, heart functioning, fertility, disease resistance and motor coordination. Our results also confirmed previously described association of loci on ECA3 (spanning LCORL and NCAPG genes) and ECA11 (spanning LASP1 gene) with the regulation of body size in our draft and primitive (small size) horses. The efficiency of the applied FST-based approach was also confirmed by the identification of a robust selection signal in the blue dun colored Polish Konik horses at the locus of TBX3 gene, which was previously shown to be responsible for dun coat color dilution in other horse breeds. FST-based method showed to be efficient in detection of diversifying selection signatures in the analyzed horse breeds. Especially pronounced signals were observed at the loci responsible for fixed breed-specific features. Several candidate genes under selection were proposed in this study for traits selected in separate breeds and horse types, however, further functional and comparative studies are needed to confirm and explain their effect on the observed genetic diversity of the horse breeds.
Introduction
The present horse population abounds in the variety of phenotypes resulting mainly from selective breeding directed at improvement of particular phenotypic features. Since the domestication, various selection criteria aimed towards the improvement of horse usability in transportation, agriculture or horsemanship have been applied [1–2]. This drove the specialization of particular populations in terms of utility and finally resulted in a creation of formal breeds constituting largely closed populations with high genetic uniformity of individuals within breeds [3]. The current selection in the most of horse breeds is primarily directed at the improvement of traits connected with appearance and performance. However, apart from the highly specialized breeds, there are also horse populations which are valued for their primitive nature manifesting in their robust constitution for primitive living conditions [4]. These horse populations were mainly shaped by natural selection, however, selective breeding for breed standard and mating in closed populations makes their genetic characteristics largely similar to that found in other horse breeds.
Both natural and artificial selection lead to changes in allele frequencies between populations, and over time, different variants and haplotype structures are being fixed within separate breeds [5]. Various concepts and statistics have been used to detect such selection signatures from genome-wide SNP data in livestock species. Some of them are based on within breed genome characteristics and other relay on genetic variation among breeds. Most of the available methods are based on: (i) the high frequency of derived alleles and other consequences of hitchhiking within population, (ii) the length and structure of haplotypes, measured by extended haplotype homozygosity (EHH) or EHH-derived statistics, and (iii) the genetic differentiation between populations, measured by FST or related statistics [6]. The genetic differences arising as a result of selection are presumably concentrated at the functional variants loci being beneficial for the selected traits. Thanks to the linkage disequilibrium across the genome, the approximate position of these functional variants can be identified using neutral genome-wide SNP panels and a comparative analysis of allele frequency distribution between different breeds [5]. A commonly used approach to detect diversifying selection signatures is based on the measure of population differentiation due to locus-specific allele frequencies variation between populations, which is quantified using the FST statistic [7] and further averaged at specific chromosome distances to account for stochasticity [8]. FST provides information on the genomic variation at a locus among populations relative to that found within populations [9]. These among-breed differences, which can be considered as diversifying selection signals, have been previously successfully used to map genomic loci spanning the genes with variants responsible for a number of phenotypic features in different species, including coat color, size, muscling, production and reproduction [10–14]. The identification of selection signals across the genome connected with candidate gene approach was also useful to detect loci associated with size, performance, coat color and gait in several horse breeds [2,4,15,16].
To extend the knowledge in this area and provide more data on candidate gene loci responsible for important breed specific features, in this study, we attempt to identify selection signatures by the analysis of genetic diversity of six different horse breeds which can be classified into three major categories: light, draft and small size primitive horses. Among the light horses, apart from well-known Arabian horses we analyzed Małopolski horse which is currently well-balanced riding horse that was originally developed mainly from native Polish population crossed with Thoroughbreds and Arabians. The primitive breeds analyzed in this study included Hucul and Polish Konik. Both these breeds are represented by a small size horses with several characteristics of feral population. Hucul horses originate from Carpathian Mountains and most likely, they are descendants of various types of horses: Tatar, Oriental, Arabian, Turkish, Przewalski’s horses, as well as horses with Norse blood. The Polish Konik originate from a now-extinct Tarpan horse from native Polish population and is characterized by several primitive features, including mouse-dun coat color and a dorsal stripe. The draft horses analyzed in this study included Sokolski and Sztumski breeds. Sokolski horse derives from cross-breeding of local Polish mares of Polish Coldblood type with imported Ardennais and Breton sires. The Sztumski horse is the largest and the heaviest of cold-blooded horses maintained in Poland which was originally created on the basis of local population crossbred mainly with Ardennes and Belgian sires [17]. The direct comparison between these horse types and breeds can provide candidate gene loci presumably responsible for constitution, size and coat color and may also contribute to the recognition of the genetic background and source of variation of horse phenotypic features. Five out of six breeds analyzed in this study are included in the conservation programs following FAO National Rare Livestock Breeds Preservation guidelines and additionally, according to our best knowledge, these native horse breeds were not studied before in terms of diversifying selection and thus this study provides new data on horse breeds genetic diversity and variation.
Methods
Animals, samples and genotyping
The study material comprised blood samples obtained from 571 horses (both males and females) randomly sampled from different herds and belonging to six different breeds. The breeds were selected to represent three major horse types: light horses–Arabians (n = 124; AR) and Malopolski horse (n = 56; MLP); primitive horses–Hucul (n = 116; HC) and Polish Konik (n = 99; KN) breeds; and draft horses–Sokolski (n = 107; SOK) and Sztumski (n = 69; SZTUM). The blood was collected from jugular vein by a veterinary doctor in an amount of 10 ml to a vial containing EDTA K3. Samples from Arabian horses were obtained from three different studs: SK Janów (Lubelskie province), SK Michałów (Świętokrzyskie Province) and SK Białka (Lubelskie province) which were our project partners. In the case of Małopolski horses–the material was sampled in all above studs and in farms of individual breeders with their personal consent. The biological material of Hucul horses was obtained in the Gładyszów Stud (Małopolskie province) and ZDIZ PIB Odrzechowa (Podkarpackie province) with the consent of their Chairmans. In the case of Polish Konik, the material was sampled in: Popielno Research Station, IRiŻZ PAN (Warmian-Masurian province) and Kalitnik—PTOP Research Station (Podlasie province) with the consent of the Presidents. The biological material from draft horses was collected in herds participating in the programs for the conservation of genetic resources of Sztumski and Sokolski horses. The herds were located in the Podlasie (Sokólski) and Pomorskie (Sztumski) provinces. Each farmer participating in the program signed a cooperation agreement with the NRIAP, where they undertake to provide data and biological material for research purposes. Breeders participating in the conservation program are obliged to keep animals in accordance with the criteria of animal welfare, which is a subject to the control of district veterinarians. The genomic DNA was purified from the blood using a Sherlock AX kit (A&A Biotechnology) and after a quality control was genotyped with the use of the Neogen Equine Community BeadChip assay (Illumina) according to the standard Infinium Ultra protocol. All animal procedures were approved by the Local Animal Care Ethics Committee No. II in Kraków–permission number 1293/2016 in accordance with EU regulations.
Data filtering
All sampled animals were genotyped with the use of Neogen Equine Community array (Illumina) including probes for 65,157 SNPs with an average inter-marker distance of 36.3 kb. Only genotypes with call rate >0.97 were used for the analysis. The initial SNPs set was filtered to remove markers located on the sex chromosomes (EquCab2.0 genome build). The initially filtered SNP panel included 61,268 markers, which was further reduced by applying population-wide filters. The filters included MAF threshold of 5% and <20% of missing genotypes in the whole studied population. Additionally, SNPs with critical p-value for HWE <1.0E-06 in each breed separately were excluded. This resulted in the final SNP panel of 52,023 markers, scattered across the genome with an average inter-marker distance of 43.0 kb.
Identification of diversifying selection signatures
The genome regions with differentially fixed variants or strongly differing in alleles frequency between separate breeds were identified using pairwise Wright’s FST [9], the conventional measure of population genetic differentiation. The FST values obtained for pairwise comparisons at each SNP were breed-standardized according to a methodology proposed by Akey et al. 8]. The standardized FST values were calculated (di) as:
where is expected value and denotes the standard deviation of FST between breeds i and j calculated from all analyzed SNPs. To account for random locus-by-locus variation, a 10-SNP sliding window was applied on the obtained di values. Candidate regions affected by diversifying selection were defined as the 99.9th percentile of the empirical distributions of window-averaged di values. The overlapping regions under selection were merged and (while searching for potentially linked genes) regions were extended on both ends by 25kb. Additional comparisons using di values were made between major horse types (light, primitive and draft) to detect potential selection differences between them. The linkage disequilibrium (LD) and haplotype block structure at the most diversified regions among the studied breeds were analyzed using HaploView 4.2 [18] software examining pairwise LD on the distance up to 500kb and detecting blocks based on a method proposed by Gabriel et al. [19]. Additionally, a detailed analysis was performed for previously detected candidate genes loci for horse body size (LCORL/NCAPG) [2,20] and the dun coat color phenotype (TBX3) [21].
Populations differentiation visualization was performed using principal component analysis on SNP genotypes and cladogram created based on weighted FST distances using neighbor-joining method [22].
The functional annotation of genes detected within the strongest selection signals was performed using the KOBAS 3.0 web server [23] and Panther Classification System [24]. A gene list enrichment analysis was done according to all known horse genes (genome) applying a correction for multiple testing.
Results
SNP panel statistics and general genetic differentiation of the studied breeds
The data filtering allowed obtaining a common set of 52,023 SNPs polymorphic across the whole population with a mean inter-marker distance of 43.0 kb (±45.1). The number of polymorphic SNPs (MAF>0.01) per breed ranged from 47,495 to 50,775 in HC and MLP breeds, respectively. The average MAF across all SNPs was the lowest in KN (0.214) and the highest in MLP (0.262) breed. The averaged observed heterozygosity per breed ranged from 0.296 in KN to 0.353 in MLP (Table 1). Mean and weighted overall pairwise FST distances were the lowest between both draft horse breeds (0.012 and 0.014) and the highest level of genetic differentiation was found between the Arabians and the primitive or draft horses (Table 2). The breeds differentiation was additionally visualized using the principal component analysis based on SNP genotypes and a cladogram of mean pairwise FST distances created using the neighbor joining method (NJ) (Fig 1). The PCA analysis showed clear separation of genetic profiles of both light horses from remaining breeds and a clearly distinct genetic profile of Hucul horses, while the NJ method showed the visible similarity of genetic profiles within major horse types with an exception of primitive horses, which were clearly separated (Fig 1).
Table 1. SNP panel polymorphism characteristics.
| Breed | No of SNPs | Monomorphic | Low frequency variants (0<MAF<0.01) | Polymorphic SNPs (MAF>0.01) | Mean MAF | Observed heterozygosity | Expected heterozygosity | Mean inter-marker distance (kb) |
|---|---|---|---|---|---|---|---|---|
| AR | 52,023 | 2,636 | 1,193 | 48,194 | 0.232 | 0.316 | 0.311 | 44.18 |
| HC | 3,322 | 1,206 | 47,495 | 0.216 | 0.302 | 0.293 | ||
| KN | 3,252 | 821 | 47,950 | 0.214 | 0.296 | 0.292 | ||
| MLP | 673 | 575 | 50,775 | 0.262 | 0.353 | 0.348 | ||
| SOK | 966 | 1,495 | 49,562 | 0.215 | 0.296 | 0.295 | ||
| SZTUM | 1,435 | 1,226 | 49,362 | 0.214 | 0.297 | 0.294 |
Table 2. Mean (above diagonal) and weighted (below diagonal) pairwise FST distances for the studied horse breeds.
| Mean FST | |||||||
| Breed | AR | HC | KP | MLP | SOK | SZTUM | |
| Weighted FST | AR | x | 0.121 | 0.122 | 0.080 | 0.122 | 0.122 |
| HC | 0.148 | x | 0.093 | 0.117 | 0.083 | 0.084 | |
| KP | 0.150 | 0.112 | x | 0.113 | 0.077 | 0.077 | |
| MLP | 0.085 | 0.128 | 0.127 | x | 0.111 | 0.106 | |
| SOK | 0.151 | 0.083 | 0.094 | 0.126 | x | 0.012 | |
| SZTUM | 0.150 | 0.099 | 0.093 | 0.123 | 0.014 | x | |
Fig 1. Genetic differentiation of the studied horse breeds visualized using principal components analysis (A) and FST-based cladogram (B).

Principal components analysis was performed based on SNP genotypes. The cladogram was created based on weighted FST distances as presented in Table 2. Neighbor Joining method was used to order the breeds according to their relative genetic distances.
Breed-specific selection signatures
Signatures of diversifying selection among the studied horse breeds were detected based on the breed-normalized pairwise FST distances (di) (S1 File). After smoothing of the obtained di values by moving average, top 0.1% of the observations were considered as the most pronounced selection signals associated with breed specific features. The merging of overlapping regions allowed for detection from 10 (MLP, SOK) to 15 (KN) genomic loci with strong selection signals per breed with a size ranging from 163.9 kb to 4.4 Mb (Table 3). The highest number of the strongest selection signals across all breeds was detected on ECA1 and ECA11 and only single regions were detected on ECA12, 14, 16, 19, 21 and 24. Several genomic regions with strong section signals overlapped between different breeds and these regions were located on chromosomes 1, 2, 3, 7, 8, 11, 15 and 22 (Table 3, Fig 2). The most commonly observed selection signal common for different breeds (HC, KN, SOK, SZTUM) was detected on ECA11 between 22.9 and 23.7 Mb of the chromosome sequence.
Table 3. Genome regions spanning the top 99.9 percentile of di values representing the strongest diversifying selection signals between the studied horse breeds.
| Breed | CHR | start | Stop | Size |
|---|---|---|---|---|
| AR | 2 | 1,624,948 | 1,973,143 | 348,195 |
| 2 | 101,234,639 | 101,658,575 | 423,936 | |
| 3 | 32,433,254 | 32,870,018 | 436,764 | |
| 4 | 16,091,738 | 16,486,662 | 394,924 | |
| 4 | 40,341,969 | 40,854,832 | 512,863 | |
| 4 | 48,962,364 | 49,486,277 | 523,913 | |
| 6 | 4,845,026 | 5,072,913 | 227,887 | |
| 8 | 21,048,394 | 21,430,992 | 382,598 | |
| 8 | 59,193,971 | 59,722,834 | 528,863 | |
| 11 | 34,293,691 | 34,694,290 | 400,599 | |
| 15 | 54,233,062 | 54,834,053 | 600,991 | |
| 15 | 71,304,187 | 71,920,250 | 616,063 | |
| 18 | 49,493,092 | 50,255,137 | 762,045 | |
| 19 | 50,182,286 | 51,212,610 | 1,030,324 | |
| HC | 1 | 152,621,838 | 153,113,818 | 491,980 |
| 1 | 174,446,252 | 174,904,138 | 457,886 | |
| 2 | 100,516,619 | 100,788,428 | 271,809 | |
| 6 | 1,212,183 | 2,028,427 | 816,244 | |
| 7 | 40,085,801 | 40,454,393 | 368,592 | |
| 7 | 49,901,427 | 51,046,996 | 1,145,569 | |
| 8 | 17,883,095 | 18,489,045 | 605,950 | |
| 9 | 18,212,591 | 18,700,974 | 488,383 | |
| 11 | 23,158,971 | 23,712,507 | 553,536 | |
| 14 | 973,820 | 2,251,732 | 1,277,912 | |
| 17 | 67,810,418 | 68,213,301 | 402,883 | |
| 24 | 2,078,405 | 2,409,598 | 331,193 | |
| 27 | 23,060,597 | 23,534,561 | 473,964 | |
| KN | 1 | 9,825,107 | 10,332,546 | 507,439 |
| 1 | 75,808,309 | 76,154,827 | 346,518 | |
| 2 | 93,508,775 | 93,977,695 | 468,920 | |
| 8 | 17,883,095 | 18,611,435 | 728,340 | |
| 10 | 35,789,240 | 36,007,871 | 218,631 | |
| 10 | 52,072,347 | 52,549,569 | 477,222 | |
| 10 | 58,667,764 | 59,059,229 | 391,465 | |
| 10 | 69,514,973 | 69,856,620 | 341,647 | |
| 11 | 23,158,971 | 23,712,785 | 553,814 | |
| 22 | 24,711,960 | 25,203,806 | 491,846 | |
| 22 | 26,260,281 | 26,769,595 | 509,314 | |
| 22 | 30,647,730 | 31,272,127 | 624,397 | |
| 23 | 14,554,518 | 14,978,574 | 424,056 | |
| 23 | 18,764,766 | 19,132,094 | 367,328 | |
| 27 | 1,149,556 | 1,896,914 | 747,358 | |
| MLP | 1 | 67,781,374 | 68,262,049 | 480,675 |
| 1 | 121,619,834 | 121,783,765 | 163,931 | |
| 4 | 29,496,419 | 30,029,942 | 533,523 | |
| 7 | 41,537,197 | 42,121,193 | 583,996 | |
| 7 | 44,416,041 | 48,776,083 | 4,360,042 | |
| 8 | 20,947,045 | 21,430,992 | 483,947 | |
| 8 | 21,695,072 | 22,112,785 | 417,713 | |
| 16 | 34,084,269 | 34,987,389 | 903,120 | |
| 17 | 74,057,493 | 74,788,851 | 731,358 | |
| 18 | 20,843,949 | 21,361,957 | 518,008 | |
| SOK | 1 | 75,808,309 | 76,229,305 | 420,996 |
| 3 | 104,821,176 | 106,074,186 | 1,253,010 | |
| 5 | 52,447,911 | 52,699,832 | 251,921 | |
| 6 | 6,293,623 | 6,508,480 | 214,857 | |
| 7 | 39,645,983 | 40,575,820 | 929,837 | |
| 9 | 75,386,842 | 75,620,809 | 233,967 | |
| 11 | 22,985,500 | 23,712,785 | 727,285 | |
| 11 | 28,738,223 | 29,312,157 | 573,934 | |
| 11 | 29,629,969 | 29,933,173 | 303,204 | |
| 15 | 54,468,973 | 54,773,734 | 304,761 | |
| SZTUM | 2 | 75,001,575 | 75,861,363 | 859,788 |
| 2 | 101,372,126 | 101,658,575 | 286,449 | |
| 3 | 105,046,433 | 105,830,625 | 784,192 | |
| 5 | 9,797,727 | 10,581,432 | 783,705 | |
| 7 | 40,036,320 | 40,454,665 | 418,345 | |
| 11 | 22,985,500 | 23,732,591 | 747,091 | |
| 11 | 29,561,696 | 30,016,859 | 455,163 | |
| 12 | 20,064 | 276,525 | 256,461 | |
| 17 | 63,828,104 | 64,275,079 | 446,975 | |
| 21 | 18,007,980 | 18,348,602 | 340,622 | |
| 22 | 25,951,536 | 26,850,432 | 898,896 |
Fig 2. Selection signatures among the studied horse breeds.
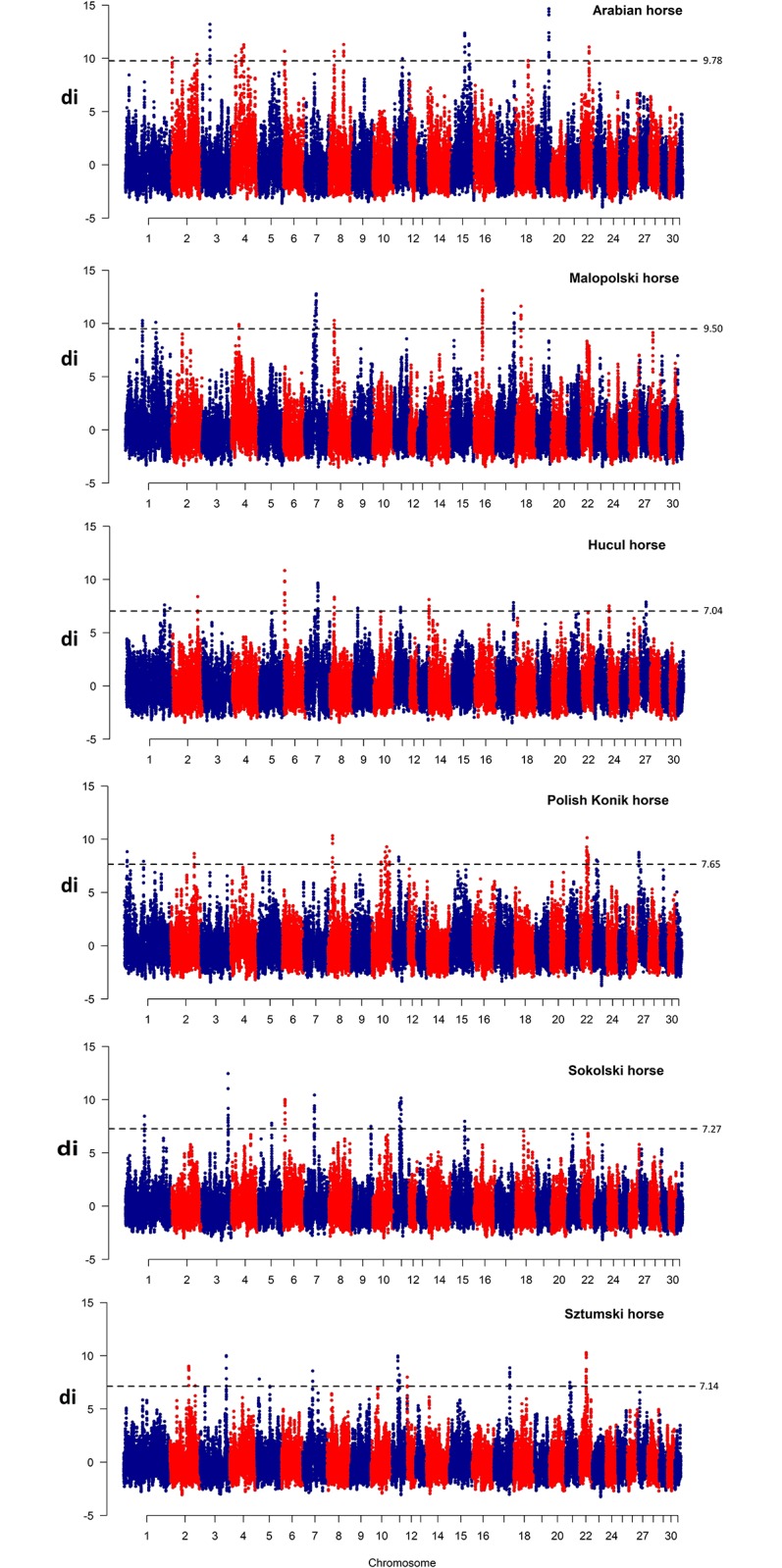
Di values for comparison between each breed and remaining breeds were plotted against centered genomic positions of the analyzed di windows. The blue dashed line indicates 99.9% of the highest di values.
To analyze gene content of the genome regions spanning the strongest detected selection signals (top 0.1% of di values), each region was expanded by 25 kb on up- and downstream directions to account for potentially linked genes. This allowed for detection from 65 (SOK) to 169 (MLP) ENSEMBL genes per breed (S2 File). The analysis of genes common for different breeds showed that as many as 18 genes were common for HC, KP, SOK and SZTUM breeds and no common genes were observed between MLP and both the primitive and draft horses (S3 File) [25]. The functional classification of well-annotated genes using Panther software according to GO terms showed that all the genes detected across breeds (506 unique genes) were mainly involved in cellular processes (32% of genes) like e.g.: cell communication and cell cycle or metabolic processes (22%), including: primary metabolic processes (89 genes), nitrogen compound metabolic processes (50 genes) or phosphate-cogitating compound associated processes (28 genes). All the genes were connected with a large number of Panther pathways, for which the highest number of genes was connected with inflammation mediated by chemokine and cytokine (8 genes), TRH receptor signaling, TGF-beta signaling, CCRK signaling map or GRH receptor signaling pathway (5 genes in each).
When functional classification was performed for individual breeds, visible differences in the enriched GO categories were detected (S4 File). The pathway analysis performed on our dataset for genes found in the Arabian horses showed the presence of some genes connected inter alia with: ATP synthesis coupled electron transport (COX4I1), bile secretion (ABCG8, ABCG5, ADCY1), fat digestion, oocyte meiosis (ADCY1, ANAPC7), ovarian steroidogenesis (ADCY1) and insulin signaling (CBLB) or secretion (ADCY1) pathways. Genes detected in the Malopolski horse were associated with largely similar biological pathways as those found in the Arabian horses and included inter alia: progesterone-mediated oocyte maturation (PRKACA, ANAPC5, ANAPC7), oocyte meiosis (PRKACA, ANAPC5, ANAPC7), fatty acid metabolism (TECR), but also covered other processes associated with immune system functions, like e.g.: leukocyte transendothelial migration (MYL2), antigen processing and presentation (PRKACA) and inflammatory mediator regulation of TRP channels (PRKACA, PRKCD). Within selection signatures characteristic for the primitive Hucul breed, we found several genes involved inter alia in processes like: olfactory transduction (LOC100066263, LOC100066541, LOC100055475, LOC100066487, LOC100060476, LOC100060509, LOC100066238), cardiac muscle contraction (ACTC1) and alanine, aspartate and glutamate metabolism (GFPT2, CPS1). Among 100 genes detected in the strongest selection signatures found in the Polish Konik horse we detected genes connected e.g. with: inflammatory mediator regulation of TRP channels or T cell receptor signaling pathway (PLCG1), chemokine signaling pathway (GNG4) and glycerolipid metabolism (LPIN3). The genes detected in the draft Sokolski horses were connected inter alia with: cardiac muscle contraction (ATP1A1, CACNB1), bile secretion (ATP1A1, ABCG8), fat digestion/absorption (ABCG8) and insulin secretion (ATP1A1), so with pathways largely similar to those found in the light horses. Genes associated with diversifying selection signatures in the Sztumski horse were connected for example with: prolactin signaling pathway (CWC25), cytokine-cytokine receptor interaction (LASP1) or general metabolism (PCGF2) (S5 File).
Signatures of diversifying selection between major horse types
To identify genome regions differentially fixed between different major horse types, di values were calculated between pairs of breeds assigned to three categories: light (AR, MLP), primitive (ponies) horses (HC, KN) and draft horses (SOK, SZTUM). The obtained di values were then smoothed by sliding window of 10 SNPs and 0.1% of the highest observations was analyzed for gene content (S6 File).
This comparison revealed 14 genome regions strongly differing in alleles frequency between the light and draft horses, 11 regions differing between the light and primitive horses and 9 differing between the primitive and draft breeds. The highest number of such divergently selected regions was detected on ECA2 (6 regions), 8 and 22 (4 regions on each). The size of individual regions ranged from 124.2 kb to 1.1 Mb. The regions directly overlapping between at least two different comparisons were found on ECA2, 4, 8, and 19 and these overlaps were found for comparisons between the light horses and two remaining horse types (Table 4). The comparison of selection signature plots (Fig 3) revealed similar pattern of allele frequency differences between the light and two other horse types and distinct pattern of frequency differences between the primitive and draft horses.
Table 4. Genome regions spanning the top 99.9 percentile of di values representing the strongest diversifying selection signals between the major studied horse types.
| Chr | Start | End | Size |
|---|---|---|---|
| Light vs. Draft | |||
| 2 | 42,283,588 | 42,693,342 | 409,754 |
| 2 | 75,104,992 | 75,633,717 | 528,725 |
| 2 | 101,239,026 | 101,659,052 | 420,026 |
| 4 | 15,870,441 | 16,546,202 | 675,761 |
| 5 | 52,350,393 | 52,725,393 | 375,000 |
| 7 | 39,875,266 | 40,815,384 | 940,118 |
| 8 | 21,048,394 | 21,430,992 | 382,598 |
| 8 | 21,877,750 | 22,178,476 | 300,726 |
| 9 | 75,379,414 | 75,680,103 | 300,689 |
| 11 | 28,762,301 | 29,038,958 | 276,657 |
| 11 | 29,561,696 | 30,111,682 | 549,986 |
| 18 | 49,493,092 | 50,504,878 | 1,011,786 |
| 19 | 50,507,674 | 51,157,247 | 649,573 |
| 22 | 45,438,601 | 45,752,522 | 313,921 |
| Primitive vs. Light | |||
| 2 | 40,879,307 | 41,345,280 | 465,973 |
| 2 | 42,144,042 | 42,738,837 | 594,795 |
| 2 | 99,966,856 | 100,812,765 | 845,909 |
| 4 | 15,993,651 | 16,486,662 | 493,011 |
| 5 | 55,503,606 | 56,139,444 | 635,838 |
| 5 | 62,395,899 | 62,666,145 | 270,246 |
| 7 | 36,787,932 | 37,213,908 | 425,976 |
| 8 | 20,849,134 | 21,483,539 | 634,405 |
| 19 | 50,182,286 | 51,157,247 | 974,961 |
| 22 | 1,233,709 | 1,498,126 | 264,417 |
| 22 | 22,921,048 | 23,687,775 | 766,727 |
| Primitive vs. Draft | |||
| 1 | 75,333,244 | 75,724,769 | 391,525 |
| 1 | 75,808,309 | 76,294,080 | 485,771 |
| 3 | 104,821,176 | 105,874,361 | 1,053,185 |
| 6 | 6,384,273 | 6,508,480 | 124,207 |
| 8 | 17,879,679 | 18,615,429 | 735,750 |
| 11 | 22,807,586 | 23,892,808 | 1085,222 |
| 14 | 1,049,008 | 1,262,151 | 213,143 |
| 21 | 22,327,128 | 22,649,580 | 322,452 |
| 22 | 26,246,949 | 26,846,925 | 599,976 |
Fig 3. Selection signatures among the major studied horse types.

Di values for comparison between major horse types were plotted against centered genomic positions of the analyzed di windows. The blue dashed line indicates 99.9% of the highest di values.
Within the detected selection signatures for major horse types we detected in total 220 unique genes–from 77 to 87 for separate comparisons (S7 File). The genes with differentially fixed variants between the light and draft horses were associated with several biological pathways, of which the most pronounced were those connected with progesterone-mediated oocyte maturation, oocyte meiosis (ANAPC5, ANAPC7, ADCY1), cardiac muscle contraction (MYL2, ATP1A1), adrenergic signaling in cardiomyocytes (ADCY1, MYL2, ATP1A1), salivary or bile secretion, insulin secretion and thyroid hormone synthesis (ADCY1, ATP1A1). The genes were also connected with biological processes responsible for energy homeostasis, like: ATPase activity or mitochondrial inner membrane function (ATP5F1E) (S8 File).
The regions differentially fixed between the primitive and light horses included 77 genes associated with variety of biological pathways among which the most enriched ones were those connected with immune system functions, e.g.: HTLV-I infection (ETS1, ANAPC7, ADCY1, WNT2B), ubiquitin mediated proteolysis (ANAPC7, CBLB, UBE4B), bacterial invasion of epithelial cells (CBLB, ARPC3) and reproduction, like: oocyte maturation and oocyte meiosis (ANAPC7, ADCY1). The genes were also engaged in biological processes connected with lactate transmembrane transport (SLC16A1), pyridine-containing compound metabolic process (NMNAT1) and wound healing (RHOA) (S8 File).
The genomic regions differing between the primitive and draft horses encompassed 87 different genes among which we detected ones connected with Ras signaling pathway (GNG4, FGF10), sulfur relay system (NFS1), MAPK signaling pathway (CACNB1, FGF10), cardiac muscle contraction (CACNB1) and terpenoid backbone biosynthesis (GGPS1). The genes were also annotated to a wide range of biological processes including inter alia: keratinocyte differentiation, angiogenesis and bone development (MED1) (S8 File).
Special attention was given to genes located within the strongest signals of diversifying selection between different horse types and involved an analysis of the most divergently selected regions per comparison. For comparison between the draft and light horses we analyzed in details a region on ECA7 (39.8–40.8 Mb), for comparison between the primitive and light horses a region on ECA8 (20.8–21.5 Mb) and for comparison between the draft and primitive horses a region on ECA11 (22.8–23.9 Mb) (Fig 3). Additionally, for these regions we established linkage disequilibrium and haplotype block structure to identify haplotypes potentially being under selection.
Within the region on ECA7 (39.8–40.8 Mb), apart from two pseudogenes and one uncharacterized protein coding gene, we found two genes (namely: NTM, coding for Neurotrimin and OPCML, coding for opioid binding protein) involved in central nervous system functioning [26]. The analysis of LD and haplotype structure at the analyzed locus showed the presence of two haplotype blocks with common haplotypes showing frequency above 0.7 in all analyzed breeds (S9 File). The region also neighbored with a large chromosomal area of high LD in AR and MLP horses located upstream of the analyzed selection signature (40.1–52.5 Mb).
The in-depth analysis of the locus on ECA 8 (20.8–21.5 Mb) revealed the presence of 14 genes within the regions, including one uncharacterized protein. The remaining genes included: MYL2, CCDC63, PPP1CC, HVCN1, TCTN1, PPTC7, RAD9B, VPS29, FAM216A, GPN3, ARPC3, ANAPC7, ATP2A2. The analysis of LD at the ECA8 locus showed rather moderate linkage and unambiguous haplotype block structure, however, some overlapping haplotype blocks with frequency higher than 0.5 were found for three out of four compared breeds (S10 File).
Within the large region with the highest differences in alleles frequency between the draft and small size primitive horses on ECA11 (22.8–23.9 Mb), we detected 30 different genes (including two pseudogenes and five uncharacterized proteins). One of them, LASP1 gene, was previously proposed as a candidate gene for size in horses [20]. The analysis of LD structure at the locus showed rather fast linkage disequilibrium decay and haplotype block structure difficult to associate with the selection signal (Fig 4). Nevertheless, overlapping haplotype blocks were detected in the draft horses, spanning haplotypes with frequency exceeding 0.7.
Fig 4. The strongest selection signal at the ECA11 locus spanning the region divergently selected between primitive and draft horses.

The graph presents the genomic position of the ECA11 locus, haplotype blocks found in separate breeds along with the frequency of the most common haplotype. The genomic positions of genes annotated directly (±25kb) at the region are also marked.
Diversifying selection signatures at candidate gene loci for size and gene responsible for coat color dilution
In previous studies, the ECA3 locus encompassing LCORL and NCAPG genes was shown to be differentially selected between several draft and miniature horse breeds [2] and was also presented as explaining a significant portion of genetic variance for this trait in an across-breed study [20]. In our results, one of the four strongest diversifying selection signals detected between the small size primitive horses and draft horses, encompassed a previously described locus on ECA3 (104.8–105.9 Mb), spanning both mentioned genes. The analysis of the haplotypes structure at the locus showed overlapping haplotype blocks for three of the analyzed breeds (KP, HC, SOK), however, they spanned a neighboring pseudogene locus rather than LCORL/NCAPG positions (Fig 5). This observation may be connected with the possible location of functional variants in the upstream region of LCORL (NCAPG is transcribed in opposite direction), possibly influencing its promoter activity.
Fig 5. A selection signal on ECA3 associated with the candidate LCORL/NCAPG locus for body size in the horse.

The graph presents the genomic position of the candidate ECA3 locus, haplotype blocks found in separate breeds along with the frequency of the most common haplotype. The genomic positions of genes annotated directly (±25kb) at the region are also marked.
Another locus which was analyzed in details was a genome region on ECA8 spanning TBX3 gene. Previous studies showed that the dun phenotype (which is characteristic for the studied Polish Konik horse) is related to mutations occurring in TBX3 gene [21,27]. The analysis of diversifying selection signatures characteristic for individual breeds performed in this study showed that the strongest selection signal found in the Polish Konik directly overlapped with this locus (ECA8: 17.9–18.6 Mb). At the locus, we found the low level of LD and poor haplotype structure, however, the detected di signal almost exactly matched the TBX3 genomic position (Fig 6).
Fig 6. A selection signal on ECA8 associated with the TBX3 locus responsible for dun coat color dilution in the Polish Konik horse.

The graph presents the genomic position of the candidate ECA8 locus and haplotype blocks found in the Polish Konik horse along with the frequency of the most common haplotype. The genomic positions of genes annotated directly (±25kb) at the region are also marked.
Discussion
In this study we performed a genome-wide scan for diversifying selection signatures in horse breeds representing performance, exterior and size selected animals. As a comparative population we included primitive, extensively selected horses with well-developed primitive characteristics like: good fertility, disease resistance and adaptation to harsh environmental conditions [28]. Many genome regions divergently selected between separate breeds were identified in this study as well as selection signatures characteristic for specific horse types (light, draft and primitive). This allowed for an identification of several candidate genes and associated metabolic pathways which may be potentially responsible for the divergent phenotypes of the studied breeds. To allow comprehensive analysis of the obtained results we focused only on the genome regions with the strongest selection signals, presumably being near fixation in separate breeds and spanning variants responsible for the well-established (fixed) breed-specific traits. To manage a variety of genes found within the selection signals we performed a pathways analysis aimed at the identification of enriched processes. Having in mind that the detected selection signatures are associated with a variety of phenotypic features differentiating the studied breeds (which are conditioned by complex molecular mechanisms), we expected only very few genes connected with separate biological processes in the enrichment analysis. Nevertheless, this analysis allowed us to reduce the complexity of the obtained data and allowed us to search for the pathways and underlying genes potentially being the targets of diversifying selection.
The highest number of the strong diversifying selection signals across all breeds was detected in this study on ECA1 and ECA11. These autosomes were previously shown to contain loci affecting size in horses, which both were detected using population genetics [2,16] and quantitative genomics methods [20]. Several genomic regions with strong selection signals overlapped between different breeds and these regions were located on chromosomes 1, 2, 3, 7, 8, 11, 15 and 22 (Table 3, Fig 2). The most commonly observed selection signal common for different breeds was detected on ECA11 between 22.9–23.7 Mb of the chromosome sequence. This region was previously shown as overlapping with the LASP1 gene locus with a presumed effect on growth and body size traits [2,20].
The analysis of linkage disequilibrium at the most pronounced selection signals found in this study on ECA7, 8 and 11 between major horse types showed rather low level of LD and a poorly conserved haplotype structure in the analyzed breeds, suggesting that the variants selected at the regions are evolutionarily old and their frequency increased during breeds formation rather than under the influence of recent selection. This can be supported by the fact that an high LD expected in regions bearing variants under strong ongoing positive selection [29], actually decayed at the analyzed loci as a result of repeated meiosis and recombinational mechanisms acting over several generations. This is in accordance with the suggestions that different statistical methods, like e.g. extended haplotype homozygosity tests are more suitable to detect selection signatures associated with ongoing selection directed at new functional variants [30]. Nevertheless, selection signals associated with variants which are not fully fixed in the studied populations should be present among signatures that did not reach the applied in this study stringent threshold of 99.9% of the highest di values and more in-depth analysis of our data would allow detecting variants currently segregating in the studied populations [31].
Diversifying selection signatures among major horse types
Within the region on ECA7 (39.8–40.8 Mb), divergently selected between the draft and light horses, apart from two pseudogenes and one uncharacterized protein coding gene we found two genes (namely: NTM, coding for neurotrimin and OPCML, coding for opioid binding protein) that were involved in central nervous system functioning. The study performed in humans suggested that NTM gene (coding for Neurotrimin) locus is associated with the level of IQ and two other genome wide association studies (GWAS) reported an association between NTM variation and cognitive function performances in humans [32, 33]. The diversifying selection signature at this locus between the draft and light horses may potentially contribute to their differing temperaments and/or potentially, the ability to develop different gaits as a function of motor coordination managed by the brain.
The locus on ECA 8 (20.8–21.5 Mb) with clear divergences in alleles frequency between the primitive and light horses encompassed 14 genes, including one uncharacterized protein. Among the genes as potential selection candidate we suggest MYL2 (Myosin Light Chain 2) gene (detected at the peak of the selection signature), which is coding for a contractile protein that plays a significant role in heart development and contraction [34, 35]. The mutation in MYL2 was shown to be responsible for hypertrophic cardiomyopathy (HCM) in humans [36]. The association of the gene with heart contraction and development suggests its potential role in physical effort and advocates selection pressure on its locus in light horses in terms of their performance and endurance which are strongly dependent on heart and lung efficiency [37]. The MYL2 gene was also identified in a genome-wide study on quantitative trait loci affecting show-jumping performance in Hanoverian warmblood horses [38].
Interestingly, one of the genome regions on ECA5 differentiating light and primitive horses encompassed SLC16A1 (Solute Carrier Family 16 Member 1) gene. The MCT1 protein encoded by this gene catalyzes the movement of different monocarboxylates such as lactate and pyruvate across the plasma membrane [39]. In a previous study, Ropka-Molik et al. [40] showed that SLC16A1 gene expression is stimulated during training regimens preparing horses for flat racing. In the studied Arabian horses they detected that the SLC16A1 expression gradually increased in muscle tissue, starting from resting conditions up to top training form [40]. Furthermore, the performed association analysis showed that there was a significant association between g.55589063T>G SNP located in the 5’UTR of SLC16A1 gene and the selected racing results [41]. All these evidences suggest the potential significance of SLC16A1 gene and MCT1 protein for horse performance and that the detected selection signal associated with genetic differences between primitive and light horses may be a result of selection (applied especially in the Arabian and Malopolski horses) towards the improvement of racing abilities.
Within the large region on ECA11 (22.8–23.9 Mb) divergently selected between the primitive (small size) and draft horses (so presumably responsible for size), we detected 30 different genes. Among the genes we detected LASP1 gene, previously suggested as a candidate gene for size in horses [20]. However, some doubts were also associated with this gene as a candidate for size [16] probably due to the insufficient knowledge on this gene functions, hampering clear description of its role in body size regulation [42].
Apart from selection signatures representing differentiation of horse types, a detailed analysis was performed to detect genome regions divergently selected in individual horse breeds. The analysis was aimed at the detection of candidate genes affecting breed-specific characteristics.
Selection signatures found in light horses (Arabian and Malopolski)
Both the Arabian and Malopolski horses are characterized by several common features and are lightweight horses with a breeding purpose mainly directed at aesthetics, racing performance and gait quality. Our study allowed identifying in these horses selection signatures spanning genes involved in regulation of pathways associated with metabolic processes and energy production from lipids (such as: bile secretion, fat digestion and absorption, fatty acid metabolism and regulation of lipolysis in adipocyte) or from carbohydrates (insulin secretion, insulin signaling pathway) (S2 and S5 Files). Several previous studies based on animal models and humans suggest that exercise training improves lipid and cholesterol metabolism [43–45]. According to Meissner et al. [46], physical activity increase bile acid synthesis and as a result, increase fatty acid absorption in exercise-trained animals. Furthermore, it was also shown that exercise increases the insulin-signaling cascade activity, which can be associated with alterations in insulin sensitivity [47]. Similarly, Ropka-Molik et al. [48] pointed out the significant role of mechanisms modulating glucose uptake and lipid metabolism in maintaining body homeostasis during long-term exercise in horses, thus providing strong grounds that elements of this mechanism presumably are targets of selection in light horses. Insulin receptor signaling pathway was also shown to be enriched by genes detected in a selection sweeps found in a recent study employing next generation sequencing data from 52 horses of different breeds [49]. This suggests that insulin signaling is a biochemical cascade important for general horse functional features and their element are under selection in various types and horse breeds.
Moreover, our results obtained for the Arabian horses which dominate in endurance riding and racing [50] pinpointed genes classified as involved in vascular smooth muscle contraction (ADCY1), taurine and hypotaurine metabolism (GAD1) as well as in oxidative phosphorylation (COX4I1), that is processes with clear implications for racing and athletic performance. It is well known that blood flow through the muscle (conditioned inter alia by vascular smooth muscle contraction) can change over time and highly increase during maximal exercise [51–53]. This blood flow adjustment is primarily related to increased oxygen demands of the muscle tissue and it is essential for physical performance during racing. The importance of the role of taurine in exercise endurance has been highlighted by many reports. In mice, the individuals that lacked the taurine transporter (TauT) gene and contained severely reduced muscle taurine content exhibited decreased ability to perform physical exercise in both treadmill and forced swimming tests [54, 55]. Furthermore, Dawson et al. [56] and Miyazaki et al. [57] proved that taurine supplementation prolongs the time to exhaustion during treadmill running, by the release of intramuscular taurine into the blood. Processes of oxidative phosphorylation are even more important for physical endurance. The ATP demand rapidly increases to meet the high rate of ATP consumption during rest-to-work transition and hence efficient processes of ATP synthesis are crucial for muscle kinetics [58]. These pieces of evidence suggest that the detected genes connected with vascular smooth muscle contraction, taurine and hypotaurine metabolism as well as oxidative phosphorylation play a significant role in horse athletic performance and are promising candidates for the endurance traits in the studied Arabian horses.
Selection signatures found in primitive horses (Hucul and Polish Konik)
Hucul and Polish Konik, apart from clear genetic differences (also demonstrated in this study) share several common features resulting from their primitive character and common phylogenic origin. Both these horse breeds are presumed to originate from Eastern European wild horse (Tarpan) and are characterized by several primary features adapting them to living in natural conditions. These features are mainly shaped by natural selection and represent adaptation to survival in harsh environment and are connected with ability for searching food, defense against predators, disease resistance and tolerance of adverse climatic conditions [28].
In Hucul horses, selection signatures overlapped with several genes classified as engaged in olfactory transduction pathway (LOC100066263, LOC100066541, LOC100055475, LOC100066487, LOC100060476, LOC100060509, LOC100066238). It is generally thought that the olfactory system is extremely important for the survival of most mammals and is essential for finding foods, avoiding dangers or identifying mates and offspring [59–63]. It helps to survive in harsh environmental/living conditions to which Hucul horses are well adapted.
Our analysis of selection signatures in the Polish Konik breed allowed the detection of a strong selection signal directly at TBX3 gene locus. This gene was previously reported as being causative for the dun phenotype [21] by affecting expression of KITLG gene. There is also strong evidence demonstrating that two single nucleotide polymorphisms within this gene are responsible for coat color dilution and occurrence of three alleles: dun (D), non-dun1 (d1), and non-dun2 (d2) was presented [21]. The detection of the strong selection signal unambiguously associated with this locus in the Polish Konik horse (sole breed with this coat color phenotype among the studied breeds) confirms the involvement of TBX3 gene in coat color dilution in this breed and the usefulness of the applied population-based approach for detection of functional variants loci in the horse genome.
Selection signatures detected in draft horses
Cold-blooded horses thanks to their muscling, size, strength and stamina are perfect for field work and transport. In the analyzed heavy draft horses, we detected selection signatures encompassing genes involved in pathways essential for maintaining body homeostasis like: aldosterone-regulated sodium reabsorption pathway responsible for sodium homeostasis and blood volume and blood pressure control [64], as well as mineral absorption or endocrine and other factor-regulated calcium reabsorption pathways, playing a central role in the homeostasis of ions [65]. We also identified genes involved in aforementioned pathways associated with metabolic processes and energy production (such as: bile secretion, fat digestion and absorption, protein digestion and absorption, insulin secretion or metabolic pathways) (S2 and S5 Files). All these mechanisms play a crucial role in regulation of biological and cellular functions that are necessary for maintaining body homeostasis during heavy physical effort, which is one of the most important traits selected in draft horses.
In both studied draft horse breeds, we detected a strong selection signal on ECA3, overlapping with previously described LCORL/NCAPG locus affecting size in the horse [2, 20, 66]. These genes were associated with size and growth traits in humans and livestock animals. It was shown that the DCAF16-NCAPG region is a QTL for the average daily gain trait in cattle [67]. Furthermore, the region including NCAPG and LCORL loci was reported to be involved in the determination of size, including both height and mass in cattle [68–72] as well as in pigs [10]. The recent studies confirmed also a significant link between these loci and body height in several horse breeds, including Belgian draft horses [2,20,73–76].
Conclusions
Summarizing, in this study we applied a population differentiation-based approach to detect genomic regions divergently selected between six breeds representing light, draft and primitive horses. The analysis of the most pronounced selection signals allowed us to detect several genes connected with processes potentially important for breeds phenotypic differentiation and associated with energy homeostasis during physical effort, heart functioning, neuron development, fertility, disease resistance and motor coordination. All these processes are potentially important for the traits selected in the analyzed breeds, especially for athletic performance, health and gait quality. Our results also confirmed the previously described association of loci on ECA3 and ECA11 with the regulation of body size in our draft and primitive (small size) horses. The efficiency of the applied statistical approach was also confirmed by the identification of a strong selection signal in the blue dun Polish Konik horse at the locus of TBX3 gene, which was previously shown to be causative for dun coat color dilution.
Supporting information
The corresponding centered genomic position of separate di windows is given.
(XLSX)
(XLSX)
(PNG)
{kind=link}
(DOCX)
(XLSX)
The corresponding centered genomic position of separate di windows is given.
(XLSX)
(XLSX)
(DOCX)
The graph presents the genomic position of the ECA7 locus, haplotype blocks found in separate breeds along with the frequency of the most common haplotype. The genomic positions of genes annotated directly (±25kb) at the region are also marked.
(PNG)
{kind=link}
The graph presents the genomic position of the ECA8 locus, haplotype blocks found in separate breeds along with the frequency of the most common haplotype. The genomic positions of genes annotated directly (±25kb) at the region are also marked.
(PNG)
{kind=link}
Abbreviations
- AR
Arabian horse
- ECA
Horse chromosome (E. caballus)
- HC
Hucul horse
- KN
Polish Konik horse
- LD
Linkage disequilibrium
- MAF
Minor allele frequency
- MLP
Malopolski horse
- NJ
Neighbor joining method
- PCA
Principal components analysis
- SNP
Single nucleotide polymorphism
- SOK
Sokolski horse
- SZTUM
Sztumski horse
Data Availability
The availability of these data are restricted because de-identified genotyping data contain information about population structure and potentially allow to detect haplotypes associated with disease traits, this data can be treated as sensitive in breeding animal populations. Most of the studs and organizations which allowed to sample their horses did not permit to share genotypes publicly. For this reason, the data underlying the results presented in the study are available from Artur Gurgul (artur.gurgul@izoo.krakow.pl) on behalf of the National Research Institute of Animal Production or from BIOSTRATEG project manager – Jędrzej Krupiński (jedrzej.krupinski@izoo.krakow.pl).
Funding Statement
The study was financed from funds of the project: “Directions for use and conservation of livestock genetic resources in sustainable development" co-financed by the National Research and Development Center (Poland) under the Strategic Research and Development Program: "Environment, Agriculture and Forestry" – BIOSTRATEG, the decision number BIOSTRATEG2/297267/14/NCBR/2016. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Lippold S, Matzke NJ, Reissmann M, Hofreiter M. Whole mitochondrial genome sequencing of domestic horses reveals incorporation of extensive wild horse diversity during domestication. BMC Evol Biol. 2011;11:328 10.1186/1471-2148-11-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen JL, Mickelson JR, Rendahl AK, Valberg SJ, Andersson LS, Axelsson J, et al. Genome-Wide Analysis Reveals Selection for Important Traits in Domestic Horse Breeds. PLoS Genet. 2013;9(1):e1003211 10.1371/journal.pgen.1003211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen JL, Mickelson JR, Cothran EG, Andersson LS, Axelsson J, Bailey E, et al. Genetic Diversity in the Modern Horse Illustrated from Genome-Wide SNP Data. PLoS ONE 2013a. 8(1):e54997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Metzger J, Karwath M, Tonda R, Beltran S, Águeda L, Gut M, et al. Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses. BMC Genomics. 2015;16:764 10.1186/s12864-015-1977-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Y, Ding X, Qanbari S, Weigend S, Zhang Q, Simianer H. Properties of different selection signature statistics and a new strategy for combining them. Heredity. 2015;115:426–36. 10.1038/hdy.2015.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics. 2013;193(3):929–41. 10.1534/genetics.112.147231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewontin RC, Krakauer J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics. 1973;74:175–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–14. 10.1101/gr.631202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, et al. Tracking footprints of artificial selection in the dog genome. PNAS. 2010;107:1160–1165. 10.1073/pnas.0909918107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubin CJ, Megens HJ, Barrio AM, Maqbool K, Sayyab S, Schwochow D, et al. Strong signatures of selection in the domestic pig genome. PNAS. 2012;109:19529–19536. 10.1073/pnas.1217149109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkinson S, Lu ZH, Megens HJ, Archibald AL, Haley C, Jackson IJ, et al. Signatures of diversifying selection in European pig breeds. PLoS Genetics. 2013;9: e1003453 10.1371/journal.pgen.1003453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Simoni Gouveia JJ, da Silva MVGB, Paiva SR, de Oliveira SMP. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014;37(2):330–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao F, McParland S, Kearney F, Du L, Berry DP. Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet Sel Evol. 2015;47:49 10.1186/s12711-015-0127-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moon S, Lee JW, Shin D, Shin KY, Kim J, Choi IY, et al. A Genome-wide Scan for Selective Sweeps in Racing Horses. Asian-Australas J Anim Sci. 2015;28(11):1525–31. 10.5713/ajas.14.0696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frischknecht M, Flury C, Leeb T, Rieder S, Neuditschko M. Selection signatures in Shetland ponies. Anim Genet. 2016;47(3):370–2. 10.1111/age.12416 [DOI] [PubMed] [Google Scholar]
- 17.Stachurska A, Nogaj A, Brodacki A, Nogaj J, Batkowska J. Genetic distances between horse breeds in Poland estimated according to blood protein polymorphism. Czech J Anim Sci. 2014;59(6):257–267. [Google Scholar]
- 18.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–5. 10.1093/bioinformatics/bth457 [DOI] [PubMed] [Google Scholar]
- 19.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–9. 10.1126/science.1069424 [DOI] [PubMed] [Google Scholar]
- 20.Makvandi-Nejad S, Hoffman GE, Allen JJ, Chu E, Gu E, Chandler AM, et al. Four Loci explain 83% of size variation in the horse. PLoS ONE. 2012;7:e39929 10.1371/journal.pone.0039929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imsland F, McGowan K, Rubin CJ, Henegar C, Sundström E, Berglund J, et al. Regulatory mutations in TBX3 disrupt asymmetric hair pigmentation that underlies Dun camouflage color in horses. Nat Genet. 2016;48(2):152–8. 10.1038/ng.3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
- 23.Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–22. 10.1093/nar/gkr483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucl. Acids Res. 2017;45:183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heberle H, Meirelles GV, da Silva FR, Telles GP, Minghim R. InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015;16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golynski M, Krumrych W, Lutnicki K. The role of beta-endorphin in horses: a review. Vet. Med. 2011;56(9):423–9. [Google Scholar]
- 27.Stefaniuk-Szmukier M, Ropka-Molik K, Piórkowska K, Szmatoła T, Długosz B, Pisarczyk W, et al. Variation in TBX3 Gene Region in Dun Coat Color Polish Konik Horses. 2017;49:60–2. [Google Scholar]
- 28.Mackowski M, Mucha S, Cholewinski G, Cieslak J. Genetic diversity in Hucul and Polish primitive horse breeds. Arch. Anim. Breed., 2015;58:23–31. [Google Scholar]
- 29.Kim ES, Kirkpatrick BW. Linkage disequilibrium in the North American Holstein population. Anim Genet. 2009;40(3):279–88. 10.1111/j.1365-2052.2008.01831.x [DOI] [PubMed] [Google Scholar]
- 30.Zhong M, Lange K, Papp JC, Fan R. A powerful score test to detect positive selection in genome-wide scans. Eur J Hum Genet. 2010;18(10):1148–59. 10.1038/ejhg.2010.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cagan A, Blass T. Identification of genomic variants putatively targeted by selection during dog domestication. BMC Evol Biol. 2016;16:10 10.1186/s12862-015-0579-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan Y, Wang KS, Aragam N. NTM and NR3C2 polymorphisms influencing intelligence: family-based association studies. PROG NEURO-PSYCHOPH. 2011;35(1):154–60. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Arias-Vasquez A, Sleegers K, Aulchenko YS, Kayser M, et al. A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am J Hum Genet. 2007;81:17–31. 10.1086/518720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.UniProt Knowledgebase. The UniProt Consortium https://www.uniprot.org/uniprot/P10916. Accessed 20 May 2018.
- 35.Sheikh F, Lyon RC, Chen J. Functions of myosin light chain-2 (MYL2) in cardiac muscle and disease. Gene. 2015;569(1):14–20. 10.1016/j.gene.2015.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Claes GR, van Tienen FH, Lindsey P, Krapels IP, Helderman-van den Enden AT, Hoos MB, et al. Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. Eur Heart J. 2016;37(23):1815–22. 10.1093/eurheartj/ehv522 [DOI] [PubMed] [Google Scholar]
- 37.Raub RH, Warren J, Pool-Anderson K. The effect of forced exercise on development of heart, lung and diaphragm muscle in weanling horses. J Equine Vet Sci. 1992;12(2):106–8. [Google Scholar]
- 38.Schröder W, Klostermann A, Stock KF, Distl O. A genome-wide association study for quantitative trait loci of show-jumping in Hanoverian warmblood horses. Anim Genet. 2012;43(4):392–400. 10.1111/j.1365-2052.2011.02265.x [DOI] [PubMed] [Google Scholar]
- 39.GeneCards HUMAN GENE DATABASE. Weizmann Institute of Science. https://www.genecards.org/cgi-bin/carddisp.pl?gene=SLC16A1. Accessed 20 May 2018.
- 40.Ropka-Molik K, Stefaniuk-Szmukier M, Żukowski K, Piórkowska K, Bugno-Poniewierska M. Exercise-induced modification of the skeletal muscle transcriptome in Arabian horses. Physiol Genomics. 2017;49(6):318–26. 10.1152/physiolgenomics.00130.2016 [DOI] [PubMed] [Google Scholar]
- 41.Ropka-Molik K, Stefaniuk-Szmukier M, Szmatoła T, Piórkowska K, Bugno-Poniewierska M. Patent protection application no. P.423426, 2017. “A method for identification of the SLC16A1 gene polymorphisms as a genetic marker”. Patent Office of the Republic of Poland
- 42.Grunewald TG, Butt E. The LIM and SH3 domain protein family: structural proteins or signal transducers or both? Mol Cancer. 2008;7:31 10.1186/1476-4598-7-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Durstine JL, Grandjean PW, Cox CA, Thompson PD. Lipids, lipoproteins, and exercise. J Cardiopulm Rehabil. 2002;22:385–98. [DOI] [PubMed] [Google Scholar]
- 44.Ramachandran S, Penumetcha M, Merchant NK, Santanam N, Rong R, Parthasarathy S. Exercise reduces preexisting atherosclerotic lesions in LDL receptor knock out mice. Atherosclerosis. 2005;178:33–8. 10.1016/j.atherosclerosis.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 45.Halverstadt A, Phares DA, Wilund KR, Goldberg AP, Hagberg JM. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56:444–50. 10.1016/j.metabol.2006.10.019 [DOI] [PubMed] [Google Scholar]
- 46.Meissner M, Lombardo E, Havinga R, Tietge UJ, Kuipers F, Groen AK. Voluntary wheel running increases bile acid as well as cholesterol excretion and decreases atherosclerosis in hypercholesterolemic mice. Atherosclerosis. 2011;218(2):323–9. 10.1016/j.atherosclerosis.2011.06.040 [DOI] [PubMed] [Google Scholar]
- 47.Kim YB, Inoue T, Nakajima R, Shirai-Morishita Y, Tokuyama K, Suzuki M. Effect of long-term exercise on gene expression of insulin signaling pathway intermediates in skeletal muscle. Biochem Biophys Res Commun. 1999;254(3):720–7. 10.1006/bbrc.1998.9940 [DOI] [PubMed] [Google Scholar]
- 48.Ropka-Molik K, Stefaniuk-Szmukier M, Żukowski K, Piórkowska K, Gurgul A, Bugno-Poniewierska M. Transcriptome profiling of Arabian horse blood during training regimens. BMC Genetics. 2017a;18:31 10.1186/s12863-017-0499-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C, Ni P, Ahmad HI, Gemingguli M, Baizilaitibei A, Gulibaheti D, et al. Detecting the Population Structure and Scanning for Signatures of Selection in Horses (Equus caballus) From Whole-Genome Sequencing Data. Evol Bioinform Online. 2018;14:1176934318775106 10.1177/1176934318775106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ricard A, Robert C, Blouin C, Baste F, Torquet G, Morgenthaler C, et al. Endurance Exercise Ability in the Horse: A Trait with Complex Polygenic Determinism. Front Genet. 2017;8:89 10.3389/fgene.2017.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laughlin MH, Korthuis RJ, Duncker DJ, Bache RJ. Control of blood flow to cardiac and skeletal muscle during exercise In: Rowell LB, Shepherd JT, editors. Handbook of Pathophysiology. Regulation and Integration of Multiple Systems. Section 12. Oxford University Press; New York: Oxford: Published for the American Physiological Society; 1996. p. 705–769. [Google Scholar]
- 52.Boushel R, Langberg H, Green S, Skovgaard D, Bulow J, Kjaer M. Blood flow and oxygenation in peritendinous tissue and calf muscle during dynamic exercise in humans. J Physiol. 2000;524(1):305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarelius I, Pohl U. Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiol. 2010;199(4):349–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warskulat U, Flögel U, Jacoby C, Hartwig HG, Thewissen M, Merx MW, et al. Taurine transporter knockout depletes muscle taurine levels and results in severe skeletal muscle impairment but leaves cardiac function uncompromised. FASEB J. 2004;18(3):577–9. 10.1096/fj.03-0496fje [DOI] [PubMed] [Google Scholar]
- 55.Ito T, Kimura Y, Uozumi Y, Takai M, Muraoka S, Matsuda T, et al. Taurine depletion caused by knocking out the taurine transporter gene leads to cardiomyopathy with cardiac atrophy. J Mol Cell Cardiol. 2008;44(5):927–37. 10.1016/j.yjmcc.2008.03.001 [DOI] [PubMed] [Google Scholar]
- 56.Dawson R Jr, Biasetti M, Messina S, Dominy J. The cytoprotective role of taurine in exercise-induced muscle injury. Amino Acids. 2002;22(4):309–24. 10.1007/s007260200017 [DOI] [PubMed] [Google Scholar]
- 57.Miyazaki T, Matsuzaki Y, Ikegami T, Miyakawa S, Doy M, Tanaka N, et al. The harmful effect of exercise on reducing taurine concentration in the tissues of rats treated with CC14 administration. J Gastroenterol. 2004;39(6):557–62. 10.1007/s00535-003-1342-1 [DOI] [PubMed] [Google Scholar]
- 58.Korzeniewski B, Zoladz JA. Training-induced adaptation of oxidative phosphorylation in skeletal muscles. Biochem J. 2003;374(1):37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buck L, Axel R. A novel multigene family may encode odorant receptors: a molecular basis for odor recognition. Cell. 1991;65:175–87. [DOI] [PubMed] [Google Scholar]
- 60.Nei M, Niimura Y, Nozawa M. The evolution of animal chemosensory receptor gene repertoires: roles of chance and necessity. Nat Rev Genet. 2008;9:951–63. 10.1038/nrg2480 [DOI] [PubMed] [Google Scholar]
- 61.Touhara K, Vosshall LB. Sensing odorants and pheromones with chemosensory receptors. Annu Rev Physiol. 2009;71:307–32. 10.1146/annurev.physiol.010908.163209 [DOI] [PubMed] [Google Scholar]
- 62.Niimura Y. Olfactory receptor multigene family in vertebrates: from the viewpoint of evolutionary genomics. Curr. Genomics 2012;13(2): 103–14. 10.2174/138920212799860706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Niimura Y, Matsui A, Touhara K. Extreme expansion of the olfactory receptor gene repertoire in African elephants and evolutionary dynamics of orthologous gene groups in 13 placental mammals. Genome Res. 2014;24(9):1485–96. 10.1101/gr.169532.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bollag WB. Regulation of aldosterone synthesis and secretion. Compr Physiol. 2014;4(3):1017–55. 10.1002/cphy.c130037 [DOI] [PubMed] [Google Scholar]
- 65.Blaine J, Chonchol M, Levi M. Renal Control of Calcium, Phosphate, and Magnesium Homeostasis. Clin J Am Soc Nephrol. 2015;10(7):1257–72. 10.2215/CJN.09750913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Al Abri MA, Posbergh C, Palermo K, Sutter NB, Eberth J, Hoffman GE, et al. Genome-Wide Scans Reveal a Quantitative Trait Locus for Withers Height in Horses Near the ANKRD1 Gene. J Equine Vet Sci. 2018;60:67–73.e1 [Google Scholar]
- 67.Zhang W, Li J, Guo Y, Zhang L, Xu L, Gao X, et al. Multi-strategy genome-wide association studies identify the DCAF16-NCAPG region as a susceptibility locus for average daily gain in cattle. Sci Rep. 2016;6:38073 10.1038/srep38073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eberlein A, Takasuga A, Setoguchi K, Pfuhl R, Flisikowski K, Fries R, et al. Dissection of genetic factors modulating fetal growth in cattle indicates a substantial role of the non-SMC condensin I complex, subunit G (NCAPG) Gene. Genetics. 2009;183:951–64. 10.1534/genetics.109.106476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Setoguchi K, Furuta M, Hirano T, Nagao T, Watanabe T, Sugimoto Y, et al. Cross-breed comparisons identified a critical 591 kb region for bovine carcass weight QTL (CW-2) on chromosome 6 and the Ile-442-Met substitution in NCAPG as a positional candidate. BMC Genetics. 2009;10:43 10.1186/1471-2156-10-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Snelling WM, Allan MF, Keele JW, Kuehn LA, McDaneld T. Genome-wide association study of growth in crossbred beef cattle. J. Anim. Sci. 2010;88(3):837–48. 10.2527/jas.2009-2257 [DOI] [PubMed] [Google Scholar]
- 71.Weikard R, Altmaier E, Suhre K, Weinberger KM, Hammon HM, Albrecht E, et al. Metabolomic profiles indicate distinct physiological pathways affected by two loci with major divergent effect on Bos taurus growth and lipid deposition. Physiol Genomics 2010;42(2):79–88. [DOI] [PubMed] [Google Scholar]
- 72.Setoguchi K, Watanabe T, Weikard R, Albrecht E, Kühn C, Kinoshita A, et al. The SNP c.1326T>G in the non-SMC condensin I complex, subunit G (NCAPG) gene encoding a p.Ile442Met variant is associated with an increase in body frame size at puberty in cattle. Anim Genet. 2011;42:650–5. 10.1111/j.1365-2052.2011.02196.x [DOI] [PubMed] [Google Scholar]
- 73.Signer-Hasler H, Flury C, Haase B, Burger D, Simianer H, Leeb T, et al. A genome-wide association study reveals loci influencing height and other conformation traits in horses. PLoS ONE. 2012;7:e37282 10.1371/journal.pone.0037282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tetens J, Widmann P, Kühn C, Thaller G. A genome-wide association study indicates LCORL/NCAPG as a candidate locus for withers height in German Warmblood horses. Anim Genet. 2013;44:467–71. 10.1111/age.12031 [DOI] [PubMed] [Google Scholar]
- 75.Metzger J, Schrimpf R, Philipp U, Distl O. Expression levels of LCORL are associated with body size in horses. PLoS ONE. 2013;8:e56497 10.1371/journal.pone.0056497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boyko AR, Brooks SA, Behan-Braman A, Castelhano M, Corey E, Oliveira KC, et al. Genomic analysis establishes correlation between growth and laryngeal neuropathy in Thoroughbreds. BMC Genomics. 2014;15:259 10.1186/1471-2164-15-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The corresponding centered genomic position of separate di windows is given.
(XLSX)
(XLSX)
(PNG)
(DOCX)
(XLSX)
The corresponding centered genomic position of separate di windows is given.
(XLSX)
(XLSX)
(DOCX)
The graph presents the genomic position of the ECA7 locus, haplotype blocks found in separate breeds along with the frequency of the most common haplotype. The genomic positions of genes annotated directly (±25kb) at the region are also marked.
(PNG)
The graph presents the genomic position of the ECA8 locus, haplotype blocks found in separate breeds along with the frequency of the most common haplotype. The genomic positions of genes annotated directly (±25kb) at the region are also marked.
(PNG)
Data Availability Statement
The availability of these data are restricted because de-identified genotyping data contain information about population structure and potentially allow to detect haplotypes associated with disease traits, this data can be treated as sensitive in breeding animal populations. Most of the studs and organizations which allowed to sample their horses did not permit to share genotypes publicly. For this reason, the data underlying the results presented in the study are available from Artur Gurgul (artur.gurgul@izoo.krakow.pl) on behalf of the National Research Institute of Animal Production or from BIOSTRATEG project manager – Jędrzej Krupiński (jedrzej.krupinski@izoo.krakow.pl).