

Case Report
A 20‐year‐old Caucasian male presented with right‐sided involuntary movements 2 days after intravenous (IV) heroin use. There was a past history of regular heroin and marijuana use, post‐traumatic stress disorder, depression, and amphetamine‐induced psychosis. There was no history of previous suicide attempts or movement disorders. Prescribed long‐term daily medications were quetiapine (600 mg), sodium valproate (1,500 mg), desvenlafaxine (100 mg), and alprazolam, but compliance was uncertain.
He administered IV heroin with oral quetiapine (1,200 mg) and alprazolam (4 mg). He had no recollection of the next 36 hours. On waking, he was aware of involuntary movements of the right limbs. Examination revealed right hemichorea and impairment of left‐sided fractionated finger movements. MRI brain showed bilateral hyperintense caudate nuclei and globus pallidus (GP) on T2, fluid‐attenuated inversion recovery, and diffusion‐weighted imaging sequences, worse on the left side (Fig. 1A). No changes were noted in the substantia nigra (SN). Benztropine was administered for a presumed acute dystonic reaction and the hemichorea subsequently settled.
Figure 1.
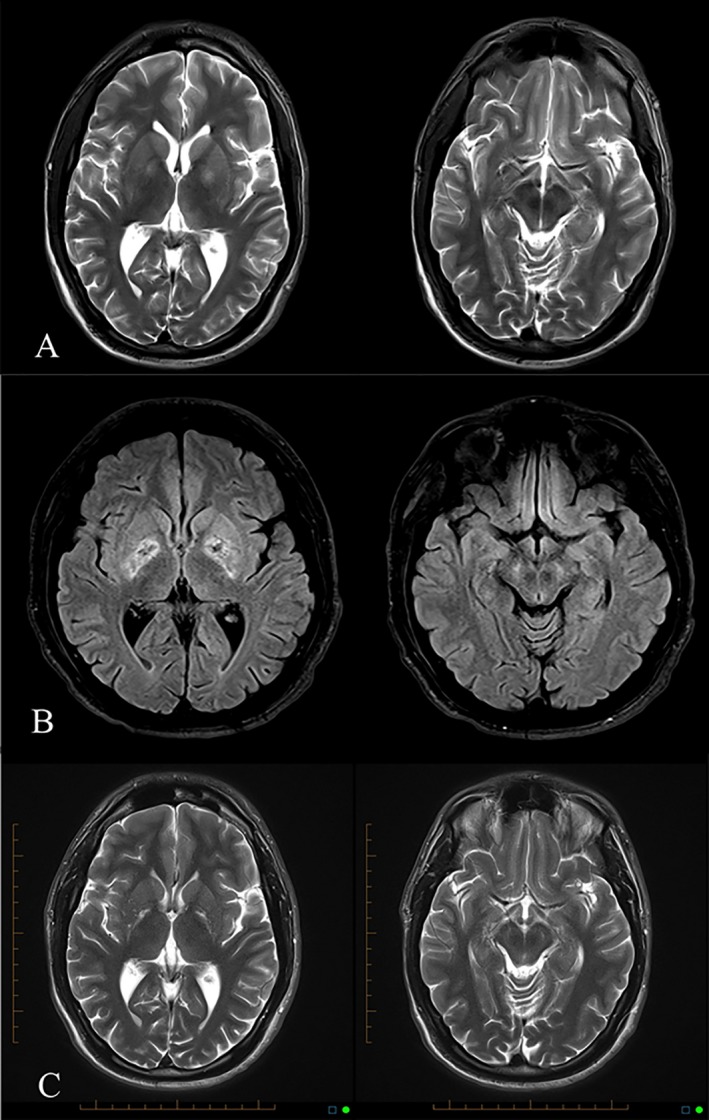
(A) T2‐weighted MRI after overdose showing GP hyperintensity and normal SN. (B) FLAIR MRI 1 month after overdose showing mixed signal in GP and new high signal in SN. (C) T2‐weighted MRI 3 years after overdose showing gliosis of GP and SN.
One month later, he presented with a 1‐week history of jaw spasm, painful posturing of the limbs, and shuffling gait. Examination revealed trismus, dystonic posturing of the hands and feet, and asymmetric (right more than left) cogwheel rigidity and bradykinesia (see Video 1). MRI brain scan showed new SN hyperintensity and mixed high and low signal in both GP consistent with edema and hemorrhagic necrosis (Fig. 1B). Antipsychotic medication was held, benztropine dose was increased, and low‐dose levodopa/carbidopa and pramipexole were commenced (in view of potential pre‐ and postsynaptic dopaminergic deficits). Both dystonia and parkinsonism improved markedly (see Video 1). He was lost to follow‐up 1 year later.
On review nearly 3 years later, he reported deterioration in walking and balance over a period of months. He had ceased benztropine, l‐dopa, and pramipexole months earlier for unclear reasons. On examination, he had continuous stereotypic thumb movements, chorea of the outstretched fingers, broad‐based gait with dystonic posturing of the ankles and toes, and marked trunkal disequilibrium (see Video 1). MRI showed gliosis of GP (symmetrical) and SN (worse on the left; Fig. 1C). Subsequent to introduction of rasagiline and benztropine, there was a marked improvement in his gait, balance, and manual function.
This case demonstrates the movement disorder and neuroimaging manifestations of a severe pallidal and SN insult secondary to heroin overdose. To our knowledge, this is the first published case to describe the evolution of these clinical and radiological features over such a long period of time and, accordingly, may provide insights into the pathogenesis of this disorder.
Toxic or anoxic‐ischemic injury to basal ganglia (BG) neurons are the two mechanisms by which heroin use might have caused these clinical and radiological features.
Heroin users in the 1980s developed subacute parkinsonism as a result of nigrostriatal degeneration after injecting synthetic heroin contaminated by the neurotoxin, MPTP.1 A forensic autopsy series examining the brains of 100 IV heroin users found five of nine brains with pallidal lesions lacked hypoxic or anoxic changes elsewhere,2 suggesting the mechanism of pallidal injury in such patients might be toxic rather than ischemic. Generalized dyskinesia, followed by severe parkinsonism after snorting heroin, was reported without a period of coma or respiratory depression in a patient whose early MRI features were similar to the present case.3 The researchers postulated the injury reflected toxic effects of heroin, or its additives, on metabolically active BG neurons.3
On the other hand, an anoxic, rather than toxic, insult to the BG and SN in the present case is suggested by the 36‐hour period of presumed coma and respiratory depression after drug ingestion. Occult GP ischemia or necrosis is found in up to 10% of heroin addicts at autopsy.2 The subsequent development of SN injury in the present case may have been caused by retrograde, postanoxic degeneration of nigrostriatal axons, as has been described over a similar time course in patients after striatal infarction.4
The clinical manifestations of GP injury are varied and include ballism,5 an akinetic‐rigid syndrome,6 dystonia,6 axial dysfunction/gait freezing without appendicular parkinsonism,7, 8 and a pure behavioral syndrome of inertia with or without obsessive compulsive behaviors.9, 10 Previous studies suggest that age at the time of cerebral anoxia is a determining factor in the subsequent clinical manifestations, with younger patients manifesting a dystonic syndrome and older patients an akinetic‐rigid syndrome.6 Bhatt et al. also reported progression from an akinetic‐rigid syndrome to dystonia in some patients.6
We postulate that the evolution from hemichorea to dystonia in our patient was owing to acute and subacute effects, respectively, of pallidal anoxia. The gradual development of parkinsonism was likely mediated by retrograde postanoxic degeneration of nigrostriatal circuits. Finally, the late development of hand stereotypies may be owing to delayed disinhibition of striatofrontal circuits influencing compulsive motor behaviors.9, 10
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
S.L.S.: 1A, 1B, 1C, 3A
P.D.T.: 3B
T.E.K.: 1A, 1B, 1C, 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: T.E.K. has received honorarium and is a member of the advisory board for Lundbeck Phamaceuticals.
Supporting information
A video accompanying this article is available in the supporting information here.
Video 1. Segment 1: Cranial and limb dystonia‐parkinsonism (corresponding to Fig. 1B). Segment 2: Improvement of dystonia‐parkinsonism at 3 and 6 months post‐overdose, while on treatment with benztropine, l‐dopa, and pramipexole. Segment 3: Limb and gait dystonia with persisting right hemiparkinsonism and thumb stereotypies, after withdrawal of therapy (corresponding to Fig. 1C).
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Deik A, Saunders‐Pullman R, Luciano M. Substances of abuse and movement disorder: complex interactions and comorbidities. Curr Drug Abuse Rev 2012;5:243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson S, Skullerud K. Hypoxic/ischaemic brain damage, especially pallidal lesions, in heroin addicts. Forensic Sci Int 1999;102:51–59. [DOI] [PubMed] [Google Scholar]
- 3. Mätzler W, Nägele T, Gasser T, Krüger R. Acute parkinsonism with corresponding lesions in the basal ganglia after heroin use. Neurology 2007;68:414. [DOI] [PubMed] [Google Scholar]
- 4. Nakane M, Teraoka A, Asato R, Tamura A. Degeneration of the ipsilateral substantia nigra following cerebral infarction in the striatum. Stroke 1992;23:328–332. [DOI] [PubMed] [Google Scholar]
- 5. Vila N, Chamorro A. Ballistic movements due to ischemic infarcts after intravenous heroin overdose: report of two cases. Clin Neurol Neurosurg 1997;99:259–262. [DOI] [PubMed] [Google Scholar]
- 6. Bhatt MH, Obeso JA, Marsden CD. Time course of postanoxic akinetic‐rigid and dystonic syndromes. Neurology 1993;43:314–317. [DOI] [PubMed] [Google Scholar]
- 7. Haaxma R, van Boxtel A, Brouwer W, et al. Motor function in a patient with bilateral lesions of the globus pallidus. Mov Disord 1995;10:761–777. [DOI] [PubMed] [Google Scholar]
- 8. Fève A, Fénelon G, Wallays C, Rémy P, Guillard A. Axial motor disturbances after hypoxic lesions of the globus pallidus. Mov Disord 1993;8:321–326. [DOI] [PubMed] [Google Scholar]
- 9. Laplane D, Levasseur M, Pillon B, et al. Obsessive‐compulsive and other behavioural changes with bilateral basal ganglia lesions. Brain 1989;112:699–725. [DOI] [PubMed] [Google Scholar]
- 10. Strub RL. Frontal lobe syndrome in a patient with bilateral globus pallidus lesions. Arch Neurol 1989;46:1024–1027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Video 1. Segment 1: Cranial and limb dystonia‐parkinsonism (corresponding to Fig. 1B). Segment 2: Improvement of dystonia‐parkinsonism at 3 and 6 months post‐overdose, while on treatment with benztropine, l‐dopa, and pramipexole. Segment 3: Limb and gait dystonia with persisting right hemiparkinsonism and thumb stereotypies, after withdrawal of therapy (corresponding to Fig. 1C).