

Neurodegeneration with brain iron accumulation (NBIA) comprises a group of hereditary heterogeneous disorders (most of them autosomal recessive) characterized by the presence of an extrapyramidal progressive dysfunction and excess iron accumulation in different locations in the brain. Overall, it remains unclear whether increased brain iron content, documented in many neurodegenerative diseases, is a direct cause of neurodegeneration, a secondary event in a pathophysiologic cascade, or just a nonspecific marker of neurodegeneration. NBIA disorders present a wide spectrum of clinical manifestations such as progressive hypo‐ and/or hyperkinetic movement disorders, and a variable degree of pyramidal, cerebellar, peripheral nerve, autonomic, cognitive, and psychiatric involvement, and visual dysfunction. The responsible mutated gene defines each syndrome.1 The second most common types of NBIA after pantothenate kinase‐associated neurodegeneration (PKAN), are PLA2G6‐related disorders. PLA2G6 encodes iPLA2‐VI, a calcium‐independent phospholipase. Traditionally mutations on this gene were thought to cause progressive neurodegeneration with miscellaneous manifestations in three phenotypes: classic infantile neuroaxonal dystrophy (INAD), atypical neuroaxonal dystrophy (atypical NAD), and late‐onset dystonia/parkinsonism starting in adolescence or early adulthood.2 Reports suggest that PLA2G6 mutations cause a phenotypic continuum rather than three discrete phenotypes, further ensuing clinical implications.3 We report two siblings with NBIA2 with a striking disparity in presentation: one as INAD and the second as atypical neuroaxonal dystrophy.
Case Report
The kindred included two siblings from nonconsanguineous parents. Family history was unremarkable except for the presence of Parkinson's disease (PD) in their grandmother.
Case III‐1
Our index case, a 20‐year‐old woman had normal development up to the age of 9 when she begun to experience subtle difficulties in school, social withdrawal, and progressive clumsiness. A few years later she began neurological evaluations that included several MRIs, but up to age 16, brain iron accumulation was not sufficiently detected by MRI to raise the suspicion of PLA2G6 mutation. She had a tonic‐clonic seizure at the age of 19. At age 20, she had normal height and weight, marked ataxia, intention tremor, and slurring speech (Video S1). Examination showed generalized hyperreflexia, gait disturbance, slight dystonic signs such as mild torticollis, and cognitive impairment. EEG was normal and since she was 16, several MRIs showed hypointensity in the substantia nigra and globus pallidus (Fig. 1). There was also a marked cerebellar atrophy in MRI.
Figure 1.
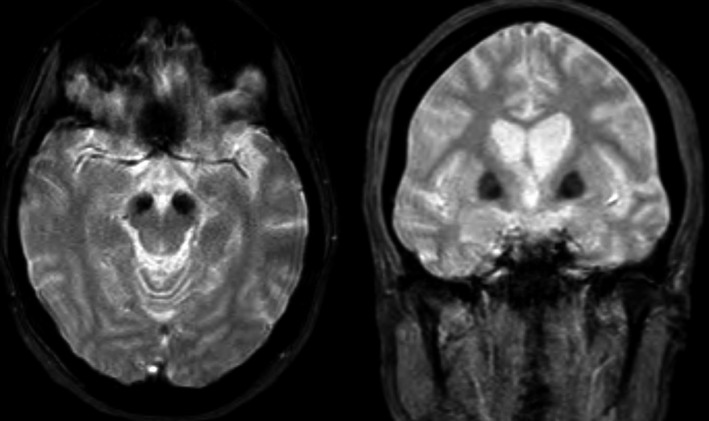
Brain MRI showing iron accumulation. The marked hypointensity in the basal ganglia (especially in the internal globus pallidus) and substantia nigra suggests iron deposition.
Case III‐2
Patient III‐1 had a 32‐year‐old sister. She had been born after a normal pregnancy with Apgar 10. In her first months she had weak cry and frequently adopted opisthotonus posture so she was evaluated throughout her first year with normal EEG, cranial CT, and CSF. A diagnosis of cerebral palsy had been made shortly after birth. She was able to sit at her twelfth month and she never walked independently. At age 3, weight and height were below the third percentile and the fundus was suggestive of pigmentous retinopathy. She slowly deteriorated in motor, cognition, language, and vision. She also developed generalized tonic‐clonic seizures at age 9. At age 33, she was unable to communicate, was confined to a wheelchair, and had spastic paraparesis. Finally she died from a respiratory infection.
In a whole‐exome study in case III‐1, at 50× coverage, NBIA genes were checked for causative mutations in the homozygous or compound heterozygous pattern. We identified two mutations in the PLA2G6 gene, an already‐reported c.C2221T:p.R741W and a novel c.C1435G:p.H479D (Table 1), confirmed by the Sanger method. Both mutations had a frequency of 0 in the 1000 genome database and a SIFT/PolyPhen score of 0.01/0.96. So, despite such a different clinical presentation, the same mutation was confirmed in case III‐2 shortly after. We examined mutation segregation in the parents and the asymptomatic adult brother. Father and brother bore the p.Arg741Trp mutation whereas the mother bore the p.His425 Asp.
Table 1.
PLA2G6 Gene Mutation
| Gene Feature | Gene | Type of Missense Mutation | Annotation | SIFT Score | PolyPhen Score |
|---|---|---|---|---|---|
| Exonic | PLA2G6 | Nonsynonymous SNV ( single nucleotide variants ) | PLA2G6:NM_003560:exon16:c.C2221T:p.R741W | 0.01 | 0.968233 |
| Exonic | PLA2G6 | Synonymous SNV | PLA2G6:NM_003560:exon11:c.C1494T:p.I498I | ||
| Exonic | PLA2G6 | Nonsynonymous SNV | PLA2G6:NM_003560:exon11:c.C1435G:p.H479D | 0.01 | 0.964521 |
Discussion
This kindred exhibits the wide spectrum of manifestations and course of NBIA2 and the difficulties of considering NBIA at early stages, causing delayed diagnosis. Clinical and age at onset discrepancies between siblings has been reported in NBIA2.4 This report confirms this fact, which is very important for prognosis and raises issues of other factors influencing onset and severity. Different expressions of the same genetically determined mutations within the same family have been reported in many genetic disorders. Genetic variations in the promoter regions of the gene or in a different gene may account for this variation. In this regard, exome‐NGS is an unbiased approach that may contribute to the elucidation of different phenotypic expressions, but more samples have to be examined to reach any conclusion. A presenting symptom in one sibling was opisthotonus, a hallmark of NBIA.5 In this case, opisthotonus was early, subsiding as the patient continued to deteriorate. It is also of interest that the patients' grandmother developed PD, because PD has been reported in heterozygote carriers of PLA2G6 mutations.6 She had a resting tremor and gait disturbance with freezing and festination. Her symptoms began at 50 years of age so she remained symptomatic for about the next 35 years until her death. Finally, the kindred underwent multiple biochemical, neurophysiological, and imaging studies that were unable to yield a diagnosis until a whole‐exome picked up their mutation. MRI can be misleading in the early stages of PLA2G6 because the patient could have symptoms before iron deposition appears. Regarding the NCS study, it was normal in our patients even in the advanced stages of the disease. Exome is an unbiased genetic diagnostic tool so it is especially useful in the early stages of the disease when the full clinical symptoms have not developed and the diagnosis of NBIA2 is more challenging. Exome studies are not invasive and have a similar cost and time frame as do other diagnostic tools, so they are now the elective test when a number of genes have to be tested.7 Research performed during the last few years is aimed at discerning the different NBIA genes and their related pathways to move toward therapies targeted toward their pathogenesis. Until then, treatment continues to be symptomatic. Our patient is now receiving treatment with levodopa and she has experienced some mild positive effect, such as the reduction of dystonia and rest tremor. Prevention of secondary complications is important, providing patients with adequate nutrition through swallowing evaluation, dietary assessment, or gastrostomy tube feeding as needed. Patients may also need to be evaluated for treatable causes of pain during episodes of dystonia; they also need routine ophthalmologic assessment and regular assessments of ambulation and speech abilities.8
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript: A. Writing of the first draft, B. Review and Critique.
P.P.‐T.: 1A, 1C, 2A, 2B, 3A
A.E.V.: 1B, 2C, 3B
P.M.U.: 1B, 2C, 3B
M.K.: 1B, 2C, 3B
A.J.‐E.: 1A, 1C, 2B, 2C, 3A, 3B
E.B.: 1B, 2C, 3B, 3C
R.G.‐G.: 1C, 2C, 3B, 3C
A.S.H.: 1B, 1C, 2A, 2B, 3B, 3C
Disclosures
Ethical compliance statement: We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: The authors report no sources of funding or conflicts of interest.
Financial Disclosures for the previous 12 months: Adriano Jiménez‐Escrig received a research grant from Pfizer. The remaining authors report no sources of funding and no conflicts of interest.
Supporting information
Vídeo S1. Eye movement abnormalities with vertical gaze palsy. Facial hypomimia. Increased muscle tone felt during examination by passive movement of the affected segment coexists with resting distal tremor in both upper limbs and hyperreflexia.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Schneider SA, Hardy J, Bhatia KP. Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord 2012;27:42–53. [DOI] [PubMed] [Google Scholar]
- 2. Schneider SA, Dusek P, Hardy J, et al. Genetics and pathophysiology of neurodegeneration with brain iron accumulation (NBIA). Curr Neuropharmacol 2013;11:59–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Erro R, Balint B, Kurian MA, et al. Early ataxia and subsequent parkinsonism: PLA2G6 mutations cause a continuum rather than three discrete phenotypes. Mov Disord 2016; (published online March 2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bower MA, Bushara K, Dempsey MA, et al. Novel mutations in siblings with later‐onset PLA2G6‐associated neurodegeneration (PLAN). Mov Disord 2011;26:1768–1769. [DOI] [PubMed] [Google Scholar]
- 5. Stamelou M, Lai SC, Aggarwal A, et al. Dystonic opisthotonus: a “red flag” for neurodegeneration with brain iron accumulation syndromes? Mov Disord 2013;28:1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gui YX, Xu ZP, Wen LV, et al. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkinsonism Relat Disord 2013;19:21–26. [DOI] [PubMed] [Google Scholar]
- 7. Singleton AB. Rapid genetic diagnosis in single‐gene movement disorders. Mov Disord 2012;27:467–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zorzi G, Zibordi F, Chiapparini L, et al. Therapeutic advances in neurodegeneration with brain iron accumulation. Semin Pediatr Neurol 2012;19:82–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Vídeo S1. Eye movement abnormalities with vertical gaze palsy. Facial hypomimia. Increased muscle tone felt during examination by passive movement of the affected segment coexists with resting distal tremor in both upper limbs and hyperreflexia.