

Biallelic mutations in the PARK2 gene, which encodes Parkin, are the most common cause of autosomal recessive, early onset Parkinson disease (EOPD), with frequency inversely correlated with age at onset and complete penetrance. Single heterozygous PARK2 mutations are occasionally detected in patients with PD and in healthy individuals and are considered minor susceptibility factors toward the risk of sporadic later onset PD.1
Case Report
We report on a 41‐year‐old male patient with EOPD (Fig. 1A). Motor signs and symptoms started at age 36 years with slowness and rigidity of the left upper limb. A dopamine (l‐dopa) transporter (DAT) scan revealed bilaterally reduced presynaptic dopaminergic captation with right prevalence. Clinical progression of symptoms was slow, and motor signs greatly improved with dopaminergic medications (ropinirole 8 mg daily and l‐dopa/carbidopa 300/75 mg daily in 3 doses).The latest examination showed mild bradykinesia and rigidity of the upper limbs with left predominance, reduced arm movements during walking, and mild hypomimia (Fig. 2A, C, Video S1). Psychiatric assessment revealed depression and mystic delusions, which started soon after the onset of motor symptoms and required treatment with atypical antipsychotics. The patient denied olfaction disturbances, constipation, or sleep abnormalities such as rapid eye movement sleep behavior disorder.
Figure 1.
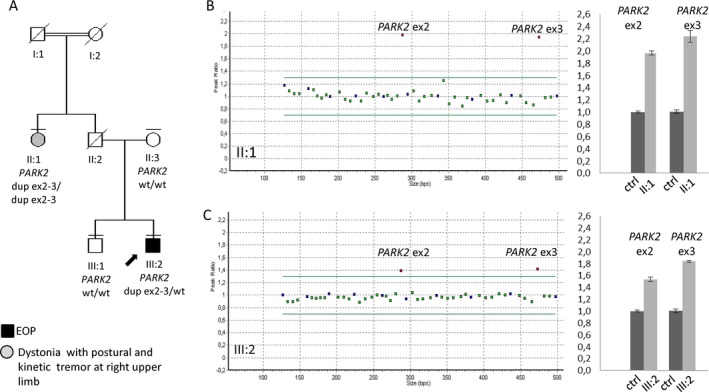
A:) Pedigree of the Italian family. Males and females are represented by squares and circles, respectively. Dead family members are marked with a diagonal bar. Double lines indicate a consanguineous marriage; horizontal bars above symbols denote individuals who underwent genetic testing, the arrow indicates the proband, the black symbol denotes early onset Parkinson disease (EOP), and the gray symbol refers to tremor and dystonia without parkinsonism. Dup indicates duplicate; wt, wild type. B,C: Multiplex ligation‐dependent probe analysis (left) and quantitative real‐time polymerase chain reaction (right) indicate (B) 4 copies of Exons 2 and 3 (ex2 and ex3, respectively) in the aunt (II:1) and (C) three copies of the same exons in the proband (III:2). Quantitative real‐time polymerase chain reaction was performed on genomic DNA by amplification of parkin 2 (PARK2) Exons 2 and 3. Normalization was obtained using telomerase as a housekeeping gene. Ctrl indicates wild‐type control.
Figure 2.
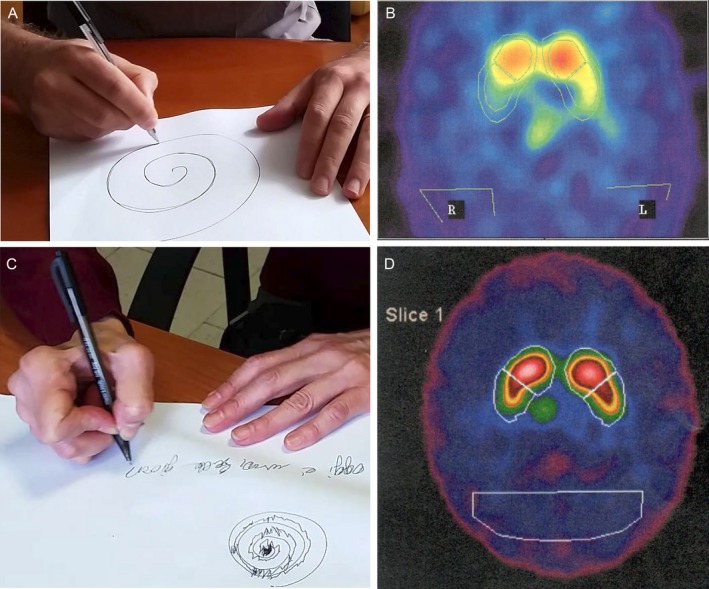
A,B: Spiral test and hand posture during writing were (A) normal in the proband but (B) showed tremor and dystonia in the aunt. C,D: A dopamine transporter scan of (C) the proband reveals reduced bilateral dopaminergic uptake at the basal ganglia, and (D) a scan of his aunt shows reduced dopaminergic uptake on the right side.
Because of the early age at onset and clinical presentation, the patient underwent genetic testing for EOPD genes. Multiplex ligation‐dependent probe analysis (MLPA) (SALSA kit P051; MRC Holland, Amsterdam, the Netherlands) detected a heterozygous duplication of Exons 2 and 3 of the PARK2 gene, which was subsequently confirmed by quantitative real time polymerase chain reaction (qRT‐PCR) on genomic DNA (Fig. 1C). No other pathogenetic variants emerged from Sanger sequencing of all exons or exon‐intron junctions of the genes PARK2, phosphatase and tensin homolog‐induced kinase 1 (PINK1), Daisuke‐Junko‐1 (DJ‐1), guanosine triphosphate cyclohydrolase1; (GCH1), and glucocerebrosidase (GBA).
To properly phase the exon duplications, segregation analysis was performed. However, the patient's mother and brother did not carry any PARK2 exon duplication, suggesting that the proband was heterozygous for a duplication of Exons 2 and 3 in cis on the same allele. To further confirm this observation, we obtained a blood sample from the 71‐year‐old paternal aunt, because the patient's father was deceased. Surprisingly, by MLPA and qRT‐PCR analysis, this lady was identified as homozygous for Exon 2 and 3 duplication, because she carried 4 copies of PARK2 Exons 2 and 3 (Fig. 1B). Clinical examination failed to identify any parkinsonian signs but disclosed a postural‐action tremor and right‐hand dystonic posturing during writing, which started since her 40s. The patient denied olfaction disturbances, constipation, or sleep abnormalities such as rapid eye movement sleep behavior disorder. Qualitative data revealed a mild decrease in DAT in the left caudate and putamen (Fig. 2B, Video S2).
This family is an interesting example of a paradox of Mendelian genetics: on 1 hand, the single heterozygous proband presents with typical EOPD, a phenotype that is usually seen in carriers of biallelic mutations; on the other hand, the homozygous 71‐year‐old aunt is nearly asymptomatic, although PARK2 biallelic mutations are usually considered fully penetrant by the fifth decade of life.
In the proband, molecular testing of other EOPD genes failed to detect additional mutations, and obvious environmental or acquired causes of PD were excluded. However, it must be noted that single heterozygous PARK2 mutations have occasionally been reported in patients with EOPD.2 Even the same heterozygous Exon 2 and 3 duplication was identified in 4 patients with sporadic PD (of whom 1 had EOPD) and 3 asymptomatic relatives.3, 4, 5, 6 We can speculate that, along with the heterozygous PARK2 duplication, other yet unidentified factors may have contributed to the development of EOPD in this patient.
Even more puzzling is the extremely mild phenotype presented by the aunt, who was homozygous for PARK2 Exon 2 and 3 duplication. In the literature, patients with the same homozygous duplication have never been reported, but this rearrangement was observed in compound heterozygosity with the p.(K211N) variant in 3 siblings with EOPD.7 The only observed movement disorders in this lady were action tremor and dystonic posturing. Isolated dystonia is often the presenting symptom in patients who have PARK2 mutations; however, dystonia usually involves the lower limbs and is always followed by the onset of parkinsonian signs.8
Only 2 asymptomatic (or mildly symptomatic) carriers of biallelic PARK2 mutations have been reported to date, and none of their heterozygous family members had PD. The first is the healthy 56‐year‐old sister of 4 patients with typical EOPD: All 5 of these siblings are compound heterozygotes for the p.(T240M) variant and the deletion of Exons 5 and 6.9 The second is the mother of a Greek proband with typical EOPD because of compound heterozygous deletions of PARK2 Exon 2 and Exons 5 through 7. The mother, a 71‐year‐old lady who presented only with slight rigidity, mildly reduced left arm swing, and a 20‐year history of depression, was homozygous for PARK2 Exon 2 deletion. Functional experiments on fibroblasts from this lady revealed a PINK1‐Parkin–independent route of mitophagy activation, suggesting that the reduced penetrance rarely observed in PARK2 mutation carriers could be caused by compensatory mechanisms at the cellular level.10 Neither of these women underwent functional neuroimaging. In our patient, DAT scan examination was clearly abnormal, confirming an underlying dopaminergic denervation, which apparently was not sufficient to reach a threshold for the manifestation of a full parkinsonian phenotype.
The paradox presented here underlines the difficulties that clinicians may encounter in establishing genotype‐phenotype correlates and providing adequate genetic counseling to patients and relatives, and the results confirm that monogenic disorders are more complex than previously thought. The identification of the genetic, epigenetic, and environmental landscape able to modulate the penetrance and expressivity of single gene mutations currently represents a major challenge of genetic research.
Author Roles:
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript preparation: A. Writing the First Draft, B. Review and Critique.
S.P.: 1A, 1C, 2B, 3A, 3B
G.F.: 1B, 1C, 3B
M.G.: 1C, 2B, 3B
M.T.: 1C, 2B, 3B
I.B.: 1C, 3B
A.B.: 1A, 1B, 3B
G.F.: 1A, 1B, 3A, 3B
E.M.V.: 1B, 2C, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for the previous 12 months: Enza Maria Valente reports research support from the Italian Ministry of Health, European Research Council (Ricerca Finalizzata 2013; ERC STG 260888) and Telethon Foundation Italy (GGP13146). The remaining authors report no sources of funding and no conflicts of interest.
Supporting information
Video S1. This video shows the proband displaying mild hypomimia with reduced spontaneous blinking, mild bradykinesia in the left limbs during the finger tapping and foot tapping tasks, no tremor, and normal writing.
Video S2. This video shows the proband's aunt displaying dystonic posturing of the right hand with tremor.
Relevant disclosures and conflicts of interest are listed at the end of this article.
Supporting information may be found in the online version of this article.
Contributor Information
Giovanni Fabbrini, Email: giovanni.fabbrini@uniroma1.it.
Enza Maria Valente, Email: em.valente@hasantalucia.it.
References
- 1. Petrucci S, Consoli F, Valente EM. Parkinson disease genetics: a “continuum” from Mendelian to multifactorial inheritance. Curr Mol Med 2014;14:1079–1088. [DOI] [PubMed] [Google Scholar]
- 2. Grunewald A, Kasten M, Ziegler A, Klein C. Next‐generation phenotyping using the parkin example: time to catch up with genetics. JAMA Neurol 2013;70:1186–1191. [DOI] [PubMed] [Google Scholar]
- 3. Periquet M, Latouche M, Lohmann E, et al. Parkin mutations are frequent in patients with isolated early‐onset parkinsonism. Brain 2003;126:1271–1278. [DOI] [PubMed] [Google Scholar]
- 4. Keyser RJ, Lombard D, Veikondis R, Carr J, Bardien S. Analysis of exons dosage using MLPA in South African Parkinson's disease patients. Neurogenetics 2010;11:305–312. [DOI] [PubMed] [Google Scholar]
- 5. Nuytemans K, Meeus B, Crosiers D, et al. Relative contribution of simple mutations vs copy number variations in five Parkinson disease genes in the Belgian population. Hum Mutat 2009;30:1054–1061. [DOI] [PubMed] [Google Scholar]
- 6. Brooks J, Ding J, Simon‐Sanchez J, Paisan‐Ruiz C, Singleton AB, Scholz SW. Parkin and PINK1 mutations in early‐onset Parkinson's disease: comprehensive screening in publicly available cases and control. J Med Genet 2009;46:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nichols WC, Pankratz N, Uniacke SK, et al. Linkage stratification and mutation analysis at the Parkin locus identifies mutation positive Parkinson's disease families. J Med Genet 2002;39:489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elia AE, Del Sorbo F, Romito LM, Barzaghi C, Garavaglia B, Albanese A. Isolated limb dystonia as presenting feature of Parkin disease. J Neurol Neurosurg Psychiatry 2014;85:827–828. [DOI] [PubMed] [Google Scholar]
- 9. Deng H, Le WD, Hunter CB, Ondo WG, Guo Y, Xie WJ, Jankovic J. Heterogeneous phenotype in a family with compound heterozygous parkin gene mutations. Arch Neurol 2006;63:273–277. [DOI] [PubMed] [Google Scholar]
- 10. Koentjoro B, Park JS, Sue CM. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin‐related Parkinson's disease [serial online]. Sci Rep 2017;7:44373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. This video shows the proband displaying mild hypomimia with reduced spontaneous blinking, mild bradykinesia in the left limbs during the finger tapping and foot tapping tasks, no tremor, and normal writing.
Video S2. This video shows the proband's aunt displaying dystonic posturing of the right hand with tremor.