

Abstract
Background
The differential diagnosis of chorea syndromes is complex. It includes inherited forms, the most common of which is autosomal dominant Huntington's disease (HD). In addition, there are disorders mimicking HD, the so‐called HD‐like (HDL) syndromes.
Methods and Results
Here we review main clinical, genetic, and pathophysiological characteristics of HD and the rare HD phenocopies in order to familiarize clinicians with them. Molecular studies have shown that HD phenocopies account for about 1% of suspected HD cases, most commonly due to mutations in C9orf72 (also the main cause of frontotemporal dementia and amyotrophic lateral sclerosis syndromes), TATA box‐binding protein (spinocerebellar ataxia type 17 [SCA17]/HDL4), and JPH3 (HDL2). Systematic screening studies also revealed mutations in PRNP (prion disease), VPS13A (chorea‐acanthocytosis), ATXN8OS‐ATXN8 (SCA8), and FXN (late‐onset Friedreich's Ataxia) in single cases. Further differential diagnoses to consider in patients presenting with a clinical diagnosis consistent with HD, but without the HD expansion, include dentatorubral‐pallidoluysian atrophy and benign hereditary chorea (TITF1), as well as the recently described form of ADCY5‐associated neurodegeneration. Lastly, biallelic mutations in RNF216 and FRRS1L have recently been reported as autosomal recessive phenocopies of HD.
Conclusion
There is a growing list of genes associated with chorea, yet a substantial percentage of patients remain undiagnosed. It is likely that more genes will be discovered in the future and that the clinical spectrum of the described disorders will broaden.
Keywords: chorea, Huntington's disease, HDL disorders, ADCY5, C9orf72
Chorea is a hyperkinetic movement disorder characterized by excessive spontaneous, involuntary movements of abrupt, irregular, unpredictable nature. Severity may range from mild focal involvement (e.g., of the hands) to severe generalized chorea affecting limbs, trunk, head, and face. In some instances, chorea may be restricted to one side of the body (hemichorea), which is important to recognize because it may point to a secondary form due to contralateral structural lesions. Chorea may have numerous causes, including acquired and inherited etiologies.
It has become clear that, among the inherited forms, the most common cause is Huntington's disease (HD), a slowly progressive autosomal dominant neurodegenerative disease characterized by impaired motor function (in particular chorea, dystonia, and parkinsonism), cognitive impairment, and psychiatric symptoms. However, with recent advances in neurogenetics, the list of differential diagnoses for chorea has been expanding. Namely, since the identification in 1993 of the gene underlying HD, huntingtin (HTT), it has emerged that not all patients who present with a clinical phenotype and family history suggestive of HD actually turn out to have the HD mutation. Instead, distinct disorders referred to as HD‐like (HDL) disorders have now been recognized.
Furthermore, recent studies identified C9orf72 and adenylyl cyclase 5 (ADCY5) as new causes of genetic chorea, and these appear to be important differential diagnoses. Furthermore, changes in RNF216 and FRRS1L have been reported in single cases. These recent findings prompted us to this review, the aim of which is to highlight the new developments around HD, the HDLs, and benign hereditary chorea.
In the following, we will summarize the main clinical features, pathophysiological and genetic underpinnings, and other key characteristics.
Huntington's Disease
Huntington's disease is the most common genetically determined neurodegenerative disease, with a prevalence of at least 12.4 per 100,000 individuals.1 The motor phenotype of classic HD is mainly characterized by slowly progressive (usually generalized) chorea. Saccadic eye movements are impaired early. As the disease progresses, dystonia and parkinsonism occur. Patients develop increasing postural instability, dysarthria, and dysphagia.
Early in the course, personality changes or psychiatric symptoms (depression, anxiety, dysphoria) develop, often preceding the motor onset. Cognitive dysfunction is characterized by executive dysfunction (abstract thinking, inhibition of inappropriate behavior) and later by memory dysfunction. The clinical heterogeneity of HD, however, is broad, including parkinsonian akinetic–rigid syndromes and relatively pure dystonic, ataxic, and psychiatric presentations.
The onset of classic HD is in midlife; however, in about 6% to 10% of HD cases the symptoms begin before the age of 20 years, referred to as Westphal variant, which is characterized by an akinetic–rigid syndrome.
The age of onset correlates with the number of trinucleotide repeats in the HD gene, HTT (see below): longer repeats cause earlier onset of disease and vice versa. In successive generations, onset may occur earlier in life (so‐called genetic anticipation, correlating with longer repeat sizes), particularly when the repeat expansion is inherited through the father due to unstable CAG repeat during spermatogenesis. Genetic anticipation also occurs in other repeat disorders, including some of the HDL disorders (e.g, HDL2; see below).
In HD, the pathogenic heterozygous expansion of a CAG repeat is located in exon 1 of the HD gene. Alleles with < 27 CAG repeats are classified as normal, as measured by the number of uninterrupted CAG, whereas alleles with ≥ 36 repeats are detected in patients. Repeats of 36 to 39 CAG are incompletely penetrant and may be found in affected individuals as well as individuals without clinical symptoms. Intermediate alleles (27–35 repeats) rarely but occasionally expand into the reduced or full penetrance range.2 Interestingly, homozygous patients have also rarely been described.3 The rate of new mutations in HD is low. The considerable clinical variability in HD may also be partly attributed to additional genetic and environmental factors. In this regard, a chromosome 15 locus with two independent effects was recently identified, which accelerate or delay onset by 6.1 years and 1.4 years.4 Other genetic factors include a cis‐regulatory variant in the HTT promoter altering NF‐κB binding, recently identified as a bidirectional genetic modifier of the age at onset in HD.5
Current research aims at better understanding the earliest (including presymptomatic) disease stages in order to define therapeutic windows when potential therapy may be beneficial. Large consortia (such as TRACK‐HD and PREDICT‐HD) follow individuals at risk (i.e, genetically confirmed asymptomatic gene mutation carriers) longitudinally to detect subtle changes on neuroimaging (such as volumetric imaging), cognitive function tests, and in other domains (e.g, quantitative motor measures) to establish their usefulness as outcome measures for future HD clinical trials.6
The pathophysiology of HD is not fully understood. The mutant huntingtin protein (mHTT) is large and ubiquitously expressed with damaging effects on neurons. Animal models recapitulate the molecular, cellular, and clinical phenotypes and allow screening for mechanistic treatments.7 Promising targets include RNA interference or antisense oligonucleotides, repressors of zinc‐finger transcription, modulators of mHTT phosphorylation, chaperone upregulators, enhancers of autophagy, and inhibitors of histone deacetylases.8 Furthermore, an anti‐sense oligonucleotides (ASOs) gene‐silencing trial is now underway (https://clinicaltrials.gov/ct2/show/NCT02519036). Restoring neurotrophic support remains another key potential therapeutic approach. Several targets are being pursued, including brain‐derived neurotrophic factor mimesis through tyrosine receptor kinase B agonism and monoclonal antibodies.8
Huntington's Disease‐Like Syndromes
Huntington's disease‐like disorders are a rare cause of chorea (Table 1). Only about 1% of suspected HD cases emerge as phenocopy syndromes.9, 10 Approximately a dozen studies have systematically screened such cohorts11, 12, 13, 14, 15, 16, 17 of patients, with a clinical diagnosis consistent with HD but without the HD expansion for panels of genes causing HD phenocopies; however, a definite genetic diagnosis could only be reached in a small minority of them (2.1% of a total of 1,559 HDL patients screened) (see Fig. 1 and Table 2).
Table 1.
Summary of important genetic causes of chorea syndromes
| Condition | Synonym | Inheritance | Position | Gene | Number of Exons | Triplet Repeat Disorderb | Regions of High Penetrance |
|---|---|---|---|---|---|---|---|
| HD | AD | 4p16 | HTT | 67b | √ | Venezuela and worldwide | |
| HDL1 | AD | 20p13 | PRNP | 2 | √ | ||
| HDL2 | AD | 16q24 | JPH3 | 5b | √ | Black Africa | |
| HDL4 | SCA17 | AD | 6q27 | TBP1 | 8b | √ | |
| Spinocerebellar ataxias, i.e, SCA1, SCA2, SCA3, SCA8, SCA12 | AD | √ | |||||
| DRPLA | NOD, HRS | AD | 12p13 | ATN1 | 10b | √ | Japan |
| C9orf72 repeat expansions | Typically associated with FTD–ALS | AD | 9p21 | C9orf72 | 12c | Finnland, Sweden, Spain | |
| Neuroferritinopathy | AD | 19q13 | FTL1 a | 4 | Cumbrian region of northern England | ||
| Benign hereditary chorea | Thyroid‐lung syndrome | AD | 14q13 | TITF1 | 3 | ||
| Benign hereditary chorea, type 2 | AD | Linked to chr. 8q21 | – | – | |||
| ADCY5‐associated chorea | Familial dyskinesia with facial myokymia | AD | 3q21 | ADCY5 | 21 | ||
| Primary familial brain calcification | Idiopathic basal ganglia calcification, Fahr's disease | AD | 8p11 | SLC20A2 | 11 | ||
| 5q32 | PDGFRB | 23 | |||||
| 22q12 | PDGFB | 7 | |||||
| 1q25 | XPR1 | 25 | |||||
| Chorea‐acanthocytosis | Levine–Critchley syndrome | AR | 9q21 | VPS13A | 73 | ||
| McLeod syndrome | x‐linked | Xp21 | XK | 3 | |||
| HDL3 | AR | 4p15 | – | – | |||
| RNF216‐mediated neurodegeneration | AR | 7p22 | RNF216 (TRIAD3) | 17 | |||
| FRRS1L‐mediated chorea | AR | 9q31 | FRRS1L | 5 | |||
| Wilson disease | Hepatolenticular degeneration | AR | 13q14 | ATP7B | 21 | ||
| Ataxia telangiectsia | ATM syndrome | AR | 11q22 | ATM | 66 | ||
| Aceruloplasminemia | AR | 3q24 | CP | 23 |
Disease‐causing triplet repeat located in exon 1 (HD and HDL2), exon 3 (HDL4/SCA17), exon 5 (DRPLA).
Also associated with hereditary hyperferritinemia cataract syndrome.
Expanded hexanucleotide repeat (GGGGCC) located between the noncoding exons 1a and 1b of C9orf72.
HD, Huntington's disease; AD, autosomal dominant; HDL, Huntington‐like disorder; HTT, huntingtin; SCA 17, spinocerebellar ataxia 17; DRPLA, Dentatorubral‐pallidoluysian atrophy; NOD, Naito‐Oyanagi disease; HRS, Haw River Syndrome; FTD–ALS, frontotemporal dementia–amyotrophic lateral sclerosis; AR, autosomal recessive.
Figure 1.
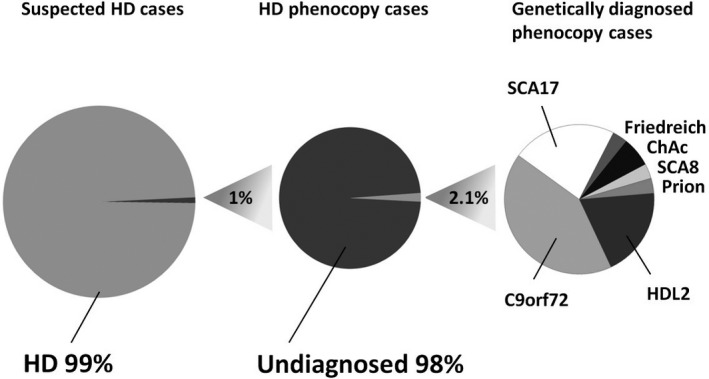
Relative frequencies of Huntington's disease phenocopies (left), successful genetic diagnosis (middle), and individual phenocopy syndromes (right). Adjusted and updated from Wild and Tabrizi.11 Numbers based on genetic screening studies (see Table 2). Friedreich, Friedreich's ataxia; ChAc, chorea‐acanthocytosis.
Table 2.
Summary of genetic screening studies systematically investigating patients with HD phenocopiesa
| Reference | No. of Patients Screened | Genes Screened | No. of Patients (%) in Whom a Definite Diagnosis Could Be Established | Diagnosis Reached | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRNP (Prion/HDL1) | JPH3 (HDL2) | TBP (HDL4/SCA17) | ATN1 (DRPLA) | SCA1, 2 and 3 | C9orf72 | Ferritin | Other | |||||
| 1 | Stevanin et al.11 Brain 2003 | 252 | + | + | + | + | 4 (1.6) |
2 with HDL2, 2 with SCA17 |
||||
| 2 | Keckarevic et al.13 Int J Neurosci 2005 | 48 | + | + | + | None | – | |||||
| 3 | Costa Mdo et al.14 J Hum Gen 2006 | 107 | + | + | + | + | 2 candidate genes: CREBBP and POU3F2 | None | – | |||
| 4 | Wild et al.12 Mov Disord 2008 | 285 | + | + | + | + | + | + | FXN (Friedreich Ataxia) | 8 (2.8) |
5 with HDL4, 1 with Prion disease, 1 with HDL2, 1 with Friedreich ataxia |
|
| 5 | Sulek‐Piatkowska et al.15 Neurol Neurochir Pol 2008 | 224 | + | + | + | 1 (0.44) | 1 with SCA17 | |||||
| 6 | Rodrigues et al.16 Arquivos de neuro‐psiquiatria 2011 | 29 | + | + | + | + | VPS13A (chorea‐acanthocytosis) | 5 (17.2) |
3 with HDL2, 2 with ChAc |
|||
| 7 |
Koutsis et al.17
J Neurol 2012 |
21 | + | + | + | + | SCA8, SCA12 | 1 (4.8) | 1 with SCA8 | |||
| 8 | Moss et al.40 Neurology 2014 | 514 | + | 10 (1.9) | 10 with C9orf72 | |||||||
| 9 | Kostic et al.73 J Neurol 2014 | 39 | + | 1 (2.6) | 1 with C9orf72 | |||||||
| 10 | Koutsis et al.74 Neurobiol Aging 2015 | 40 | + | 2 (5) | 2 with C9orf72 | |||||||
| TOTAL | 1,559a | 33 (2.1) | ||||||||||
It remains unclear in how far the cohort reported by Moss et al., 2014, includes the 285 patients studied by Wild et al., 200812; the studies were performed at the same center.
PRNP, prion protein gene; HD, Huntington's disease; HDL, Huntington's disease‐like disorder; SCA 17, spinocerebellar ataxia 17.
Prion Disease: Huntington's Disease‐Like 1
Huntington's disease‐like 1 (HDL1) is a rare presentation of autosomal dominant familial prion disease, first reported in 2001.18 It is caused by eight (sometimes six) extra repeats of the octapeptide region (Pro‐His‐Gly‐Gly‐Gly‐Trp‐Gly‐Gln) in the prion protein (PrP) gene (PRNP). Mean onset age is in early adulthood between 20 to 45 years. Mean survival time is 1 to 10 years, and rapid progression is suspicious of this cause. Familial prion disease may produce a diverse range of phenotypes, even within the same pedigree. It may resemble HD with prominent personality change, psychiatric symptoms and cognitive decline, chorea, rigidity, and dysarthria. Limb and truncal ataxia and seizures may be present. Prion disease may also present with behavioral or psychiatric symptoms, cognitive impairment, visual disturbance, cerebellar signs, myoclonus, and rigidity and other neurological signs, evolving to mutism and immobility.
The characteristic electroencephalogram (EEG) features seen in sporadic Creutzfeldt‐Jakob disease (CJD) (i.e, generalized bi‐ or triphasic periodic sharp wave complexes) are less frequently seen in the genetic prion variant. Similarly, detection of the 14‐3‐3 protein in the cerebrospinal fluid (CSF) is less consistently present in genetic compared to sporadic CJD, while tau protein in CSF may be prominently elevated in both types.
Neuropathologic examination in HDL1 revealed atrophy and prion deposition in the basal ganglia, frontal and temporal lobes, and cerebellar cortex. In comparison to other prion diseases, spongiosis is not prominent.
Huntington's Disease‐Like 2
Huntington's disease‐like 2 (HDL2)19 caused by mutations in junctophilin 3 (JPH3) accounts for about 0.4% of HD phenocopies in Western countries; however, it is frequent in black South Africans of Sub‐Saharan decent. In a recent large series of 315 unrelated South African individuals referred for diagnostic HD testing, almost one‐third of the black and mixed ancestry individuals had HDL2. That is, more than 60% of the white patients compared to only 36% of the black patients had an expansion in HTT, whereas 15% of the black South African patients but none of the white patients had an expansion in JPH3.20 In fact, all HDL2 cases reported so far have definite or probably African ancestry, and haplotype analysis revealed a common three‐SNP core JPH3 haplotype shared between patients studied. South African patients also shared flanking microsatellites in most cases,20 yielding the hypothesis that the mutation arose in Africa at least 2 thousand years ago.20
The mean age of diagnosis ranged from 31 to 58 years in 41 South African HDL2 patients.20 The condition takes a progressive course and may show remarkable similarities to HD. The classic form of HDL2 presents with similar cognitive, psychiatric, and motor features. Parkinsonism, however, may be more prominent in HDL2 compared to HD. Early‐onset cases with a progressive akinetic–rigid syndrome but clinically insignificant chorea have also been reported (equivalent to the HD Westphal variant).21, 22 In contrast to juvenile‐onset HD in early‐onset HDL2, there is an absence of seizures and mostly normal eye movements. The disease leads to death within 10 to 20 years.19
Huntington's disease‐like 2 is due to highly penetrant CTG‐CAG triplet repeat expansions in the JPH3 gene23 on chromosome 16q24.3. Normal alleles range from six to 28 triplets, whereas pathological repeat expansions range from 40 to 58 triplets.20, 24 The impact of intermediate range stretches remains unclear.19 Similar to HD, there is a negative correlation between age of onset and repeat length.20 In contrast to HD, however, the anticipation phenomenon is more likely when the disease is maternally inherited and the repeat expansions are more unstable. Furthermore, in contrast to HD, normal allele length in the population is not correlated with HDL2 disease frequency.20
Neuropathologically, both HDL2 and HD show marked cortical and striatal neurodegeneration, as well as neuronal protein aggregates staining positive for anti‐ubiquitin antibodies and expanded polyglutamine tracts. However, there may be more brainstem involvement in HD compared to HDL2, which is rather concentrated to cortical involvement (with prominent occipital atrophy). Neuroimaging may reveal generalized brain atrophy, predominantly affecting the caudate heads and putamina. Presence of a putaminal rim hyperintensity has been described.
Little is known about gene function. The encoded protein is involved in stabilization of junctional membrane complexes and regulation of neuronal calcium flux. Abnormal JPH3 protein expression increases cellular vulnerability. An additional toxic effect caused by both a toxic loss of JPH3 expression and a toxic gain of function of JPH3 RNA has been suggested.25, 26
Huntington's Disease‐Like 3
In view of the recessive pattern of inheritance, Huntington's disease‐like 3 (HDL3) will be discussed below.
Spinocerebellar Ataxia Type 17: Huntington's Disease‐Like 4
Triplet repeat expansions in the TATA box‐binding protein (TBP) gene located on chromosome 6q27 cause Huntington's disease‐like 4 (HDL4) as well as spinocerebellar ataxia type 17 (SCA17).
Inheritance is autosomal‐dominant. The normal allele size ranges from 25 to 40 CAG/CAA repeats, whereas 41 to 48 CAG/CAA repeats lead to reduced penetrance and 49 and greater repeats lead to full penetrance.27 However, it has been difficult to determine the exact cutoff of the normal versus pathogenic number of repeats.28 Homozygous intermediate range expansions have also been reported in patients with a HDL phenotype with cerebellar features. Similar to HD, a negative correlation between the size of repeat expansion and the age of onset and intergenerational instability with anticipation have been recognized. The encoded protein, TBP, functions as transcription initiation factor. Polyglutamine‐expanded TBP reduces its own intrinsic DNA‐binding and transcription abilities. Animal models of SCA17 have also been developed and shed further light on the underlying pathophysiology.
The clinical phenotype of HDL4/SCA17 is markedly heterogeneous, and the age at onset ranges from age 3 to 75 years. Cerebellar ataxia is the most common clinical feature (95%), usually presenting with a slowly progressive course, but rapid progression resembling paraneoplastic disorders or prion disease have been reported.29 Extrapyramidal signs (73%), in particular dystonia, chorea, and dementia (76%), frequently occur in SCA17. Furthermore, pyramidal signs, epilepsy, and psychiatric disturbances are not uncommon.30 A true HDL presentation occurs only in a subset of SCA17 cases.9, 10
In keeping with the broad clinical spectrum, there is neuropathologic variation with wide participation across the central nervous system. The cerebellum, cerebral neocortex, basal ganglia (in particular the caudate nucleus), and hippocampus may be involved. Neuronal intranuclear inclusions containing the abnormal protein TBP, ubiquitin, 1C2, and other proteins are widely distributed throughout the brain gray matter.31 Magnetic resonance imaging (MRI) demonstrated atrophy of the cerebellum and the caudate nucleus. Although rim enhancement of the putamen has also been described, it is nonspecific for HDL4/SCA17 because it may also occur in HDL220 and other disorders. Similar to HD, imaging changes may be detected presymptomatically. Thus, MRI volumetry as well as (11)C‐raclopride and (18)F‐fluorodeoxyglucose positron emission tomography reveal neuronal dysfunction and neurodegeneration, even in the presymptomatic stage.32
Notably, other forms of SCAs may also present with chorea and should be kept in mind in patients with an ataxic HDL phenotype, in particular SCA 1, 2, and 3.33, 34
Dentatorubral‐Pallidoluysian Atrophy
Dentatorubral‐pallidoluysian atrophy (DRPLA) (or Naito‐Oyanagi disease) shares many key characteristics of HD and the HDLs. It is a trinucleotide repeat disorder with autosomal dominant inheritance. The repeat expansions in exon 5 of the atrophin 1 (ATN1) gene range from 49 to 88 compared to eight to 25 repeats in healthy individuals. The repeat length correlates inversely with age of onset and directly with disease severity, and marked anticipation occurs with longer stretches, particularly in the context of paternal transmission. Dentatorubral‐pallidoluysian atrophy clusters in Japan, where the prevalence is estimated to be similar to the prevalence of HD. The condition is rare in other countries.35 A recent review identified only 27 non‐Asian families reported in the literature since 1989.36 A founder effect for DRPLA (Haw River syndrome) has been reported in an African American family in North Carolina, the United States.37
The clinical presentation of DRPLA is very heterogeneous and shows an age‐dependent phenotype. Juvenile‐onset cases develop severe progressive myoclonus epilepsy and cognitive decline. Adult‐onset DRPLA features ataxia, choreoathetosis, and dementia as cardinal features that may resemble HD.38 Common MRI findings include cerebellar and brainstem (in particular pontine) atrophy. Adult‐onset DRPLA furthermore displays diffuse hyperintense white matter lesions, a distinguishing feature to HD. Diffuse accumulation of mutant DRPLA protein is found in neuronal nuclei.
The molecular pathophysiology is not fully understood. Studies suggest that the expanded polyglutamine stretches lead to a toxic gain of function and bind to TAFII130—a TATA‐binding protein‐associated factor—and interfere with CREB‐dependent transcriptional activation, which is essential for neuronal survival and plasticity.39
Huntington's Disease‐Like Syndrome Associated with C9orf72 Repeat Expansions
Repeat expansions in C9orf72 have been associated with autosomal dominant frontotemporal dementia and/or amyotrophic lateral sclerosis. However, the recognized clinical phenotype is expanding (Fig. 2). Among others, recently it has been suggested that C9orf72 expansions may be the most common genetic cause of HD phenocopies.40 In a large genetic screening study, 10 of 514 HDL patients were found to carry C9orf72 mutations.40 The mean age at onset was 43 years (range 8–60). Early psychiatric and behavioral problems (including depression, apathy, obsessive behaviour, and psychosis) were common and were the first symptom in six of 10. Cognitive symptoms presented as executive dysfunction.40 Movement disorders were prominent: Three patients exhibited chorea; four exhibited dystonia; four exhibited myoclonus; and three had tremor. Notably, six of the 10 patients had parkinsonian features and four patients had pyramidal features.40 One may critizise that presence of upper motor neuron signs are not typical for classic HD and should have been an exclusion criterion for the genetic study. However, the authors40 elaborate that their cohort screened was composed of patients seen by experienced neurologists in whom the diagnosis of HD was considered and thus reflects clinical reality. They stress40 the clinical diversity seen in HD, and that the definition of HD phenocopy syndromes therefore needs to encompass not only the classical HD triad but also syndromes having a major degree of overlap with HD. We conclude that presence of prominent pyramidal features may be a red flag to consider c9orf72 repeat expansions in an HDL patient.
Figure 2.
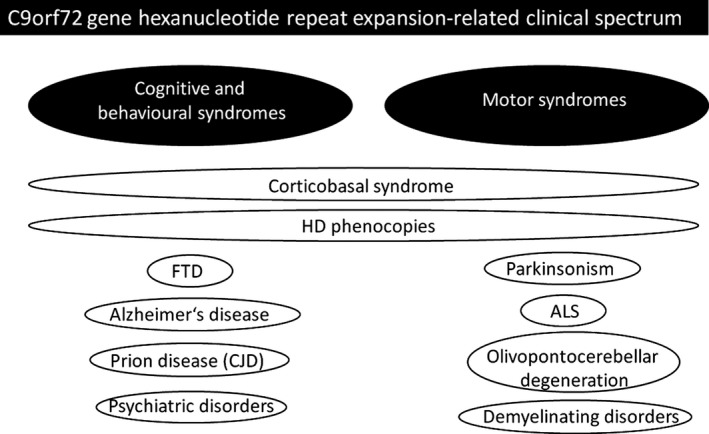
Summary of motor and nonmotor (cognitive and behavioral) syndromes associated with C9orf72 gene hexanucleotide repeat expansion. Figure modified from Souza et al.42 FTD, frontotemporal dementia; ALS, amyotrophic lateral sclerosis; HD, Huntington's disease; CJD, Creutzfeldt‐Jakob disease.
The mutation of C9orf72 is a repeat expansion of the six (hexa‐) letter string of nucleotides GGGGCC. There is a wide global distribution; however, it is mainly found in European‐derived populations (with particular high frequencies in Finland, Sweden, and Spain) but occurs exceptionally rarely in East Asian populations.41 Repeat lengths up to 30 are considered normal. More than 30 repeats—generally 250 to 1,600 repeats—are detected in symptomatic patients.
The protein plays a role as guanine nucleotide exchange factor for small guanosine triphosphatases regulating endosomal trafficking. Additional RNA‐mediated toxicity through accumulation of toxic RNA foci and RNA‐binding proteins with secondary dysregulation of RNA splicing and trafficking has also been hypothesized.42
Neuroferritinopathy
Elevated serum ferritin levels are the red flag for neuroferritinopathy, a progressive autosomal dominant neurodegenerative disease caused by mutations in the ferritin light chain gene (FTL1) located on chromosome 19q13.
Disease onset is usually in midlife, but early onset (in teenage years) and late onset (in the sixth decade) may occur.43 Typically, the disease presents with asymmetric chorea or dystonia. Other clinical features such as bradykinesia/parkinsonism, hyperreflexia, dysarthria, frontal lobe syndrome, and dementia may be variably present. Cognitive deficits and psychiatric features appear to be less prominent compared to HD. Treatment is symptomatic. The literature includes reports on iron chelation therapy in individual cases, resulting in clinical deterioration in one patient and no change in two others.44
Magnetic resonance imaging findings in neuroferritinopathy show a broad variety and may include progressive cystic degeneration of the basal ganglia, particularly cavitation of the globus pallidus and putamen, and thalamic hypointense lesions on T2‐weighted images reflecting iron deposits in addition to cortical atrophy. In rare cases, a pattern resembling the eye‐of‐the‐tiger sign, which is otherwise described in pantothenate kinase‐associated neurodegeneration, has been reported in neuroferritinopathy.45 Ferritin is a ubiquitous iron storage protein, and dysfunction results in formation of iron‐rich intranuclear and intracytoplasmic inclusion bodies, not only within neurons and glia in the brain but also in peripheral nerves, skin, muscles, liver, and even the kidneys. Iron deposition is particularly prominent in the basal ganglia.
So far, only seven different mutations in the FTL1 gene (including a common founder mutation c.460InsA) have been described (in cases from Cumbria/England, France/French‐Canada, Spain/Portugal, Australia, and Japan).46 Six of the mutations affect exon 4 of the gene, mainly affecting the tertiary structure of the ferritin light chain polypeptide. Notably, mutations in the 5′‐nontranslated region of the same gene (also resulting in elevated L‐ferritin production irrespective of iron levels) have been associated with a distinct disorder, hereditary hyperferritinemia cataract syndrome, which is characterized by early onset bilateral cataracts due to intracellular accumulation of ferritin in the lens (in the absence of neurological features). The prevalence of the latter has been estimated to a minimum of one in 200 thousand. In Germany, for example, about 50 to 100 cases of hereditary hyperferritinemia cataract syndrome have been recognized. It remains unclear why deposits are found in the brain (iron and ferritin) in the one disease and in the lens (ferritin) in the other. From the genetic point of view, in neuroferritinopathy mutations are mainly found towards the 3′ region, whereas in hereditary hyperferritinemia cataract syndrome mutations are located in the 5′ nontranslated region (mostly in the iron‐responsive element) of the gene.
Benign Hereditary Chorea: Usually with Infancy‐Onset
Benign hereditary chorea (BHC) is a rare autosomal dominant disease that is characterized by nonprogressive chorea, with early onset in childhood and absence of dementia and caudate atrophy. First clinically described in a large African‐American family from Mississippi, the United States,47 the gene was identified in 2002. Since then, several mutations in the associated small TITF1 (NKX2‐1) gene and also deletions (in some cases also encompassing adjacent genes) have been described. About 30 different mutations have been reported, mostly involving exon 3 (see48 for summary of genetype–phenotype reports of whole gene deletions involving NKX2‐1). The encoded thyroid transcription factor 1 is essential for the organogenesis of the lungs, thyroid, and basal ganglia—and symptoms may involve these organs and systems.
The typical clinical phenotype is infancy‐onset hypotonia and chorea. Other movement disorders, such as myoclonus, dystonia, motor and vocal tics, tremor, and ataxia, may be associated. Recently, recurrent drop attacks with frequent falls in the absence of EEG abnormalities have been reported.49 Chorea may improve or resolve in adulthood; however, it may also persist as mild chorea or convert to disabling myoclonus. Notably, learning difficulties are not infrequent, and involvement of thyroid (67%) and lung (46%) may also occur.50 Importantly, a link to malignancies, most frequently those affecting the lung, is increasingly reported.48 Symptomatic relief may be achieved by tetrabenazine, levodopa, haloperidol, chlorpromazine, or prednisone.
Neuroimaging is usually normal (or shows anomalies of uncertain significance; see48 for review), apart from reduced basal ganglia (striatal) and thalamic uptake in single photon emission computed tomography. Pathological studies also do not reveal significant abnormalities using standard methods. Using immunohistochemical staining, loss of most TITF1‐mediated striatal interneurons was revealed in BHC brains.
Shimohata and colleagues51 proposed genetic heterogeneity of BHC when they reported two Japanese families with autosomal dominant adult‐onset slowly progressive chorea without dementia. Magnetic resonance imaging was normal, and HD, BHC and other HDL syndromes had been excluded by genetic testing. Genetic workup showed linkage to chromosome 8q21.3‐q23.3. Due to the clinical resemblance with BHC, the disease was named benign hereditary chorea type 2.51 Recently, ADCY5 mutations were identified as another cause of BHC (see below).
ADCY5‐Associated Neurological Disease: A New Cause of Childhood‐Onset Chorea and Dystonia
In 2001, Fernandez and colleagues75 reported a 5‐generation family affected by childhood‐ or adolescent‐onset distal choreiform and facial movements that had previously been described in 1978 as BHC. The condition was hence termed familial dyskinesia and facial myokymia (FDFM). However, it was later acknowledged that the facial myokymia were centrally driven dyskinesias (i.e, twitches of periorbital and/or perioral muscles) rather than peripheral myokymia; thus, the denomination of FDFM was misleading. Subsequently, in 2012, using whole exome sequencing, a mutation in the ADCY5 gene (c.2176G>A; p.A726T) was identified in this kindred.52 (see Supp. Video 1) Since then, five additional families and 15 sporadic patients with ADCY5 gene mutations have been reported in the literature. The clinical characteristics are summarized in Table 3 and entail infancy‐ to adolescence‐onset chorea (motor impersistence) that may be combined with dystonia and/or myoclonus mainly involving limbs, neck, and/or face, and sometimes episodic or paroxysmal (however; in contrast to the classic paroxysmal dyskinesia associated with PRRT2, GLUT1, or MR1 gene mutations sudden movement, prolonged physical activity, caffeine, or alcohol are not specific triggers). Worsening of movements during sleep (probably during arousal rather than during drowsiness) may be a characteristic feature. In more severe cases, marked hypotonia and delayed motor milestones may be present. Imaging is usually normal. Milder phenotype and improvement with age has been associated with low‐grade somatic mosaicism. Notably, cardiac involvement was indicated in some cases and should be monitored for in the affected cases. The gene belongs to the adenylate cyclase family involved in cAMP synthesis. Recurrent mutations in codons 418 and 726 suggest functional importance of these residues.53 A functional link to glucose metabolism has been reported.54 Brain pathology has not been reported.
Table 3.
Reported ADCY5 cases
| Reference | No. of Reported Cases | Ethnicity | Genetic Change | Age Onset (years) | Characteristics of Chorea | Triggering/Exacerbating Factors of Chorea | Characteristics of Dystonia | Other Features |
|---|---|---|---|---|---|---|---|---|
|
Fernandez et al.,75 2001 Chen et al.,522012a |
6 | German kindred | c.2176G>A, p.A726T | Childhood–early adulthood (age 19) | (Daily paroxysmal), mostly face and limb, lasting minutes | Stress | Present in 2 (paroxysmal, limbs) | Facial (perioral) dyskinesia in 3, heart disease |
| Chen et al.76 2014a | 2 | European, unrelated | c.1252C>T, p.R418W | Early childhood | Daily paroxysmal generalized, lasting hours to days | Anxiety, stage N2 and N3 sleep | Paroxysmal (generalized) | Myoclonus in 1, pyramidal features in 1, motor regression in 1 |
| Carapito et al.,772015b | 2 | French kindred | c.2088 + 1G>A, haploinsufficiency (de novo) | Infancy | Face and limbs | Not reported | Limbs and neck | Pyramidal features in 1, muscle atrophy of 1 leg |
| Mencacci et al.,78 2015b | 3 | British (familial); Pakistani (sporadic) | c.1252C>T, p.R418W (de novo in the sporadic case) | Infancy–early childhood | Generalized | Awakening or at night, action, stress | Present in 2 (generalized) | Gaze impersistence, motor regression in 1, dysarthria in 2 |
| Chen et al.,53 2015 (includes above‐mentioned Fernandez et al.75 and Chen et al.52) |
18 4 familial 14 sporadic |
European |
p.R418W p.R418Q p.R438P p.L720P p.A726T p.M1029K |
Early childhood | Usually persistent, less commonly episodic | Anxiety, sleep | Limbs, face, trunk | Milder and may improve with age when mosaic |
The patients reported by Fernandez et al., 2001; Chen et al., 2012; and Chen et al., 2014 are included in the most recent article by Chen et al., 2015. Also see Supp. Video 1.
Videos are included in the report by Carapito and Mencacci.
Primary Familial Brain Calcification, Formerly Known as Fahr's Disease
Primary familial brain calcification (PFBC) is genetically heterogeneous and variably characterized by a combination of movement disorders (mostly dystonia, parkinsonism), ataxia, cognitive impairment, and behavioral changes.55, 56 The condition is also often referred to as Fahr's disease, or idiopathic basal ganglia calcification; however, this does not account for the fact that imaging abnormalities often extend beyond the basal ganglia. Inheritance is usually autosomal dominant, and recently four genes (SLC20A2, PDGFB, PDGFRB, XPR1) have been identified as cause of PFBC, accounting for about half of the cases. The encoded proteins are involved in phosphate transportation.
Of note, clinical presentations with prominent or isolated chorea have rarely been reported (albeit the reports date are from prior to gene identification).57 Computed tomography imaging will give the clue and is thus an important step in the workup of patients with a HDL clinical presentation.
Selected Autosomal Recessive Chorea Syndromes
When the family history is incomplete or no information can be retrieved, autosomal recessive chorea syndromes should also be considered in patients with choreic phenotype resembling HD because some of these may produce similar phenotypes. However, in view of word limitations, only an incomplete selection can be discussed in the following.
Huntington's Disease‐Like 3
Huntington's disease‐like 3 is an autosomal recessive HDL neurodegenerative disorder described in a Saudi Arabian family. Considering the early onset and the recessive pattern of inheritance, HDL3 clearly differs from the other HDL syndromes and is thus described in this section. The clinical phenotype was complex, with childhood‐onset mental deterioration, speech disturbance, dystonia, chorea, and other extrapyramidal and pyramidal features. Magnetic resonance imaging showed progressive atrophy of the caudate nuclei bilaterally and the frontal cortex, and a link to HD was suggested by the authors. The causative gene still remains unclear, but the disease locus initially was mapped to chromosome 4p15.3.58 No similar families have been described to date.
Huntington's Disease‐Like Chorea‐Dementia Syndrome Associated with FRRS1L Mutations
Most recently, Salih and colleagues reported a multiplex, consanguineous Saudi Arabian family with four siblings presenting with juvenile onset chorea, dementia, and seizures59 with normal HTT alleles in whom combined homozygosity mapping and exome sequencing approach identified homozygous premature truncation mutations in FRRS1L, an AMPA receptor complex constituent. Further studies suggested protein mislocalization, consistent with the loss of the membrane‐interacting domain. Further details remain to be published.
Chorea‐Acanthocytosis and McLeod Syndrome
Both chorea‐acanthocytosis (ChAc) and McLeod syndrome are core neuroacanthocytosis syndromes characterized by neurodegeneration of the basal ganglia and red cell acanthocytosis.60
Chorea‐acanthocytosis is a rare autosomal recessive neurodegenerative disorder due to mutations in the VPS13A gene on chromosome 9 encoding for chorein. It is estimated that about 1 thousand ChAc cases exist worldwide.61 Chorea‐acanthocytosis causes movement disorders (including chorea, dystonia, parkinsonism, and tics), cognitive impairment, and psychiatric features with great similarities to HD. However, clinical characteristics such as dystonia with prominent orofacial involvement with tongue protrusion, involuntary tongue‐ and lip‐biting, head thrusts, and rubber man‐like appearance may indicate a diagnosis distinct from classic HD.62 Furthermore, seizures (which occur infrequently in late‐onset HD) are seen in half of the patients, and myopathy and axonal neuropathy are common. The disease usually starts in the 20s and progresses slowly over 15 to 30 years.60
Blood tests reveal elevated levels of creatine phosphokinase in most cases. The detection of acanthocytes often remains elusive, although the probability to detect the characteristic deformed erythrocytes can be increased by using a 1:1 dilution with physiological saline and phase contrast microscopy.63 However, many hematology laboratories no longer prepare wet blood films due to health and safety policies,64 and analysis of the protein (chorein) levels is therefore recommended. The function of the protein is not fully understood, but a yet unknown role of chorein in the regulation of secretion and the aggregation of blood platelets has recently been suggested.
Neuroradiologically, findings include progressive striatal atrophy with a maximum in the caudate head.65 Postmortem examinations have shown a neuronal loss and gliosis predominantly affecting the caudate nucleus, putamen, globus pallidus, thalamus, and substantia nigra. In comparison to HD, no significant cortical pathology or specific neuropathologic features as inclusion bodies have been detected.
McLeod syndrome is inherited in an X‐linked matter (thus mainly affecting men) caused by mutations in the XK gene.66 The erythrocyte phenotype is defined as reduced Kell and absent Kx antigen expression on the cell surface (of note, Kell is the third most important erythrocyte antigen system after ABO and rhesus); the clinical phenotype significantly overlaps with ChAc. Peripheral sensorimotor neuropathy and areflexia, as well as the presence of cardiomyopathy, are typical and distinctive features (not seen in ChAc). Imaging features are similar to ChAc, that is, atrophy of the caudate nucleus and putamen.
RNF216‐Mediated Neurodegeneration
Mutations in RNF216 have recently been found in families with hypogonadotropic hypogonadism, ataxia, and dementia.67 They are also associated with the so‐called 4H‐syndrome68 (hypodontia and hypomyelination, alongside ataxia and hypogonadotropic hypogonadism). Furthermore, there is one recent report of recessive HDL families harboring biallelic gene RNF216 mutations.69 The clinical phenotype consisted of chorea, behavioral problems, and severe dementia as the core features in all patients. Brain imaging consistently showed white matter lesions and cerebellar atrophy. Low gonadotropin serum levels could be demonstrated in the index family and may be the clue toward this diagnosis pointing away from typical HD.69 The gene encodes a ubiquitin E3 ligase.
Conclusion
There is an increasing number of genetically defined choreic syndromes, with more subtypes described just very recently, including C9orf72‐ and ADCY5‐associated chorea, as important differentials to consider. Yet, we will most likely see more genes to be identified in the future given the tremendous revolutionary development in molecular sciences with emerging genetic and genomic technologies. At the same time, it is expected that the recognized clinical and genetic phenotype widens as more patients are reported. Knowledge of the ethnic background in view of the clustering of some disorders (see Table 1), family history, and clinical features is key in diagnostic thinking. However, traditional one‐by‐one genetic testing facilitating the diagnostic workup is increasingly replaced by simultaneous gene testing using next‐generation multiple gene panels and whole exome/genome sequencing. It should be kept in mind that of course there also are other nongenetic causes of chorea, which are beyond the scope of this chapter, including infectious, paraneoplastic, and metabolic causes as well as immune‐mediated70 causes.
Treatment of HD and the genetic HD‐like disorders remains symptomatic. Evidence‐based guidelines have recently been proposed.71 Dopamine‐depleting agents (such as tetrabenazine) and the glutamate antagonist riluzole have received a level B‐rating of evidence. Insufficient data was available to make recommendations for the use of (typical and) atypical neuroleptics (including haloperidol and olanzapine, clozapine, and quetiapine), but γ‐aminobutyric acid–ergic agents (clonazepam, gabapentin, and valproate) may be used as adjunctive therapy. Possible side effects should be discussed with patients and caregivers, including the occurrence of depression/suicidality and parkinsonism with tetrabenazine and elevated liver enzymes with riluzole. Deep brain stimulation has been performed72 in selected cases but will not halt the inevitable deterioration. Therapy should also address accompanying symptoms (e.g, depression), and supportive treatments (e.g, physiotherapy, speech therapy) should be offered.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the First Draft, B. Review and Critique.
S.A.S.: 1A, B, C; 3A
T.B.: 3B
Disclosures
Funding Sources and Conflicts of Interest: S.A.S. was supported by the Else Kröner‐Fresenius Stiftung.
Financial disclosures for previous 12 months: The authors have no disclosures to report.
Supporting information
Video S1: This 20‐year old patient with severe generalized chorea with ballism and dystonia was found to carry mutations in the ADCY5 gene (M1029K). She is a member of the ChDys Family described by Chen et al. 2015.53
Acknowledgments
Due to journal policy, the number of references has been limited to 80. A full list of references is available upon request to the corresponding author.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Rawlins M. Huntington's disease out of the closet? Lancet 2010;376:1372–1373. [DOI] [PubMed] [Google Scholar]
- 2. Samanka AH, Hayden MR. Evidence‐based genetic counseling implications for Huntington disease intermediate allele predictive test results. Clin Genet 2014;85:303–311. [DOI] [PubMed] [Google Scholar]
- 3. Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: the 12‐month longitudinal analysis. Lancet Neurol 2011;10:31–42. [DOI] [PubMed] [Google Scholar]
- 4. Consortium GMoHsDG‐H . Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 2015;162:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Becanovic K, Norremolle A, Neal SJ, et al. A SNP in the HTT promoter alters NF‐kappaB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci 2015;18:807–816. [DOI] [PubMed] [Google Scholar]
- 6. Sampaio C, Borowsky B, Reilmann R. Clinical trials in Huntington's disease: interventions in early clinical development and newer methodological approaches. Mov Disord 2014;29:1419–1428. [DOI] [PubMed] [Google Scholar]
- 7. Ross CA, Tabrizi SJ. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10:83–98. [DOI] [PubMed] [Google Scholar]
- 8. Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington's disease: what's in the pipeline? Mov Disord 2014;29:1434–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schneider SA, Walker RH, Bhatia KP. The Huntington's disease‐like syndromes: what to consider in patients with a negative Huntington's disease gene test. Nat Clin Pract Neurol 2007;3:517–525. [DOI] [PubMed] [Google Scholar]
- 10. Wild EJ, Tabrizi SJ. Huntington's disease phenocopy syndromes. Curr Opin Neurol 2007;20:681–687. [DOI] [PubMed] [Google Scholar]
- 11. Stevanin G, Fujigasaki H, Lebre AS, et al. Huntington's disease‐like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 2003;126:1599–1603. [DOI] [PubMed] [Google Scholar]
- 12. Wild EJ, Mudanohwo EE, Sweeney MG, et al. Huntington's disease phenocopies are clinically and genetically heterogeneous. Mov Disord 2008;23:716–720. [DOI] [PubMed] [Google Scholar]
- 13. Keckarevic M, Savic D, Svetel M, Kostic V, Vukosavic S, Romac S. Yugoslav HD phenocopies analyzed on the presence of mutations in PrP, ferritin, and Jp‐3 genes. Int J Neurosci 2005;115:299–301. [DOI] [PubMed] [Google Scholar]
- 14. Costa Mdo C, Teixeira‐Castro A, Constante M, et al. Exclusion of mutations in the PRNP, JPH3, TBP, ATN1, CREBBP, POU3F2 and FTL genes as a cause of disease in Portuguese patients with a Huntington‐like phenotype. J Hum Genet 2006;51:645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sulek‐Piatkowska A, Krysa W, Zdzienicka E, et al. Searching for mutation in the JPH3, ATN1 and TBP genes in Polish patients suspected of Huntington's disease and without mutation in the IT15 gene. Neurol Neurochir Pol 2008;42:203–209. [PubMed] [Google Scholar]
- 16. Rodrigues GR, Walker RH, Bader B, et al. Clinical and genetic analysis of 29 Brazilian patients with Huntington's disease‐like phenotype. Arq Neuropsiquiatr 2011;69:419–423. [DOI] [PubMed] [Google Scholar]
- 17. Koutsis G, Karadima G, Pandraud A, et al. Genetic screening of Greek patients with Huntington's disease phenocopies identifies an SCA8 expansion. J Neurol 2012;259:1874–1878. [DOI] [PubMed] [Google Scholar]
- 18. Moore RC, Xiang F, Monaghan J, et al. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet 2001;69:1385–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Margolis RL, Rudnicki DD, Holmes SE. Huntington's disease like‐2: review and update. Acta Neurol Taiwan 2005;14:1–8. [PubMed] [Google Scholar]
- 20. Krause A, Mitchell C, Essop F, et al. Junctophilin 3 (JPH3) expansion mutations causing Huntington disease like 2 (HDL2) are common in South African patients with African ancestry and a Huntington disease phenotype. Am J Med Genet B Neuropsychiatr Genet 2015;168:573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneider SA, Marshall KE, Xiao J, LeDoux MS. JPH3 repeat expansions cause a progressive akinetic‐rigid syndrome with severe dementia and putaminal rim in a five‐generation African‐American family. Neurogenetics 2012;13:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greenstein PE, Vonsattel JP, Margolis RL, Joseph JT. Huntington's disease like‐2 neuropathology. Mov Disord 2007;22:1416–1423. [DOI] [PubMed] [Google Scholar]
- 23. Holmes SE, O'Hearn E, Rosenblatt A, et al. A repeat expansion in the gene encoding junctophilin‐3 is associated with Huntington disease‐like 2. Nat Genet 2001;29:377–378. [DOI] [PubMed] [Google Scholar]
- 24. Margolis RL, Holmes SE, Rosenblatt A, et al. Huntington's Disease‐like 2 (HDL2) in North America and Japan. Ann Neurol 2004;56:670–674. [DOI] [PubMed] [Google Scholar]
- 25. Seixas AI, Holmes SE, Takeshima H, et al. Loss of junctophilin‐3 contributes to Huntington disease‐like 2 pathogenesis. Ann Neurol 2012;71:245–257. [DOI] [PubMed] [Google Scholar]
- 26. Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL. Huntington's disease—like 2 is associated with CUG repeat‐containing RNA foci. Ann Neurol 2007;61:272–282. [DOI] [PubMed] [Google Scholar]
- 27. Toyoshima Y, Onodera O, Yamada M, Tsuji S, Takahashi H. Spinocerebellar ataxia type 17 In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle: 1993–2015. http://www.ncbi.nlm.nih.gov/pubmed/20301611 [Google Scholar]
- 28. Shin JH, Park H, Ehm GH, et al. The pathogenic role of low range repeats in SCA17. PLoS One 2015;10:e0135275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mehanna R, Itin I. From normal gait to loss of ambulation in 6 months: a novel presentation of SCA17. Cerebellum 2013;12:568–571. [DOI] [PubMed] [Google Scholar]
- 30. Craig K, Keers SM, Walls TJ, Curtis A, Chinnery PF. Minimum prevalence of spinocerebellar ataxia 17 in the north east of England. J Neurol Sci 2005;239:105–109. [DOI] [PubMed] [Google Scholar]
- 31. Rolfs A, Koeppen AH, Bauer I, et al. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol 2003;54:367–375. [DOI] [PubMed] [Google Scholar]
- 32. Brockmann K, Reimold M, Globas C, et al. PET and MRI reveal early evidence of neurodegeneration in spinocerebellar ataxia type 17. J Nucl Med 2012;53:1074–1080. [DOI] [PubMed] [Google Scholar]
- 33. Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 2004;3:291–304. [DOI] [PubMed] [Google Scholar]
- 34. Pedroso JL, de Freitas ME, Albuquerque MV, Saraiva‐Pereira ML, Jardim LB, Barsottini OG. Should spinocerebellar ataxias be included in the differential diagnosis for Huntington's diseases‐like syndromes? J Neurol Sci 2014;347:356–358. [DOI] [PubMed] [Google Scholar]
- 35. Martino D, Stamelou M, Bhatia KP. The differential diagnosis of Huntington's disease‐like syndromes: ‘red flags’ for the clinician. J Neurol Neurosurg Psychiatry 2013;84:650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wardle M, Morris HR, Robertson NP. Clinical and genetic characteristics of non‐Asian dentatorubral‐pallidoluysian atrophy: a systematic review. Mov Disord 2009;24:1636–1640. [DOI] [PubMed] [Google Scholar]
- 37. Burke JR, Wingfield MS, Lewis KE, et al. The Haw River syndrome: dentatorubropallidoluysian atrophy (DRPLA) in an African‐American family. Nat Genet 1994;7:521–524. [DOI] [PubMed] [Google Scholar]
- 38. Tsuji S. Dentatorubral‐pallidoluysian atrophy (DRPLA): clinical features and molecular genetics. Adv Neurol 1999;79:399–409. [PubMed] [Google Scholar]
- 39. Tsuji S. Dentatorubral‐pallidoluysian atrophy (DRPLA)—discovery of the disease, DRPLA gene and the pathophysiology [Article in Japanese]. Rinsho Shinkeigaku (Clin Neurol) 2000;40:1287–1289. [PubMed] [Google Scholar]
- 40. Hensman Moss DJ, Poulter M, Beck J, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 2014;82:292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van der Zee J, Gijselinck I, Dillen L, et al. A pan‐European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 2013;34:363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Souza PV, Pinto WB, Oliveira AS. C9orf72‐related disorders: expanding the clinical and genetic spectrum of neurodegenerative diseases. Arq Neuropsiquiatr 2015;73:246–256. [DOI] [PubMed] [Google Scholar]
- 43. Chinnery PF. Neuroferritinopathy. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle: 1993–2015. [PubMed] [Google Scholar]
- 44. Chinnery PF, Crompton DE, Birchall D, et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain 2007;130:110–119. [DOI] [PubMed] [Google Scholar]
- 45. Shah SO, Mehta H, Fekete R. Late‐onset neurodegeneration with brain iron accumulation with diffusion tensor magnetic resonance imaging. Case Rep Neurol 2012;4:216–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keogh MJ, Morris CM, Chinnery PF. Neuroferritinopathy. Int Rev Neurobiol 2013;110:91–123. [DOI] [PubMed] [Google Scholar]
- 47. Haerer AF, Currier RD, Jackson JF. Hereditary nonprogressive chorea of early onset. N Engl J Med 1967;276:1220–1224.4225827 [Google Scholar]
- 48. Peall KJ, Kurian MA. Benign hereditary chorea: an update. Tremor Other Hyperkinet Mov (N Y) 2015;5:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosati A, Berti B, Melani F, Cellini E, Procopio E, Guerrini R. Recurrent drop attacks in early childhood as presenting symptom of benign hereditary chorea caused by TITF1 gene mutations. Dev Med Child Neurol 2015;57:777–779. [DOI] [PubMed] [Google Scholar]
- 50. Gras D, Jonard L, Roze E, et al. Benign hereditary chorea: phenotype, prognosis, therapeutic outcome and long term follow‐up in a large series with new mutations in the TITF1/NKX2‐1 gene. J Neurol Neurosurg Psychiatry 2012;83:956–962. [DOI] [PubMed] [Google Scholar]
- 51. Shimohata T, Hara K, Sanpei K, et al. Novel locus for benign hereditary chorea with adult onset maps to chromosome 8q21.3 q23.3. Brain 2007;130:2302–2309. [DOI] [PubMed] [Google Scholar]
- 52. Chen YZ, Matsushita MM, Robertson P, et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenyl cyclase 5. Arch Neurol 2012;69:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen DHM A, Friedman J.R, Korvatska O, Gad A, Bonkowski E.S, et al. ADCY5‐related dyskinesia: broader spectrum and genotype/phenotype correlations. Neurology 2015;85:2026–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hodson DJ, Mitchell RK, Marselli L, et al. ADCY5 couples glucose to insulin secretion in human islets. Diabetes 2014;63:3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tadic V, Westenberger A, Domingo A, Alvarez‐Fischer D, Klein C, Kasten M. Primary familial brain calcification with known gene mutations: a systematic review and challenges of phenotypic characterization. JAMA Neurol 2015;72:460–467. [DOI] [PubMed] [Google Scholar]
- 56. Sobrido MJ, Coppola G, Oliveira J, Hopfer S, Geschwind DH. Primary familial brain calcification In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews. Seattle WA: University of Washington, Seattle; 1993. [Google Scholar]
- 57. Kono S, Manabe Y, Tanaka T, et al. A case of Fahr's disease presenting as chorea successfully treated by the use of quetiapine. Clin Med Case Rep 2009;2:63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kambouris M, Bohlega S, Al‐Tahan A, Meyer BF. Localization of the gene for a novel autosomal recessive neurodegenerative Huntington‐like disorder to 4p15.3. Am J Hum Genet 2000;66:445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salih MM, Fields L, Jepperson T, et al. Mutations in the AMPA receptor complex protein FRRS1L cause an inherited Huntington‐like chorea‐dementia syndrome. Abstract American Academy of Neurology 2015;84:P3.030. [Google Scholar]
- 60. Walker RH, Jung HH, Dobson‐Stone C, et al. Neurologic phenotypes associated with acanthocytosis. Neurology 2007;68:92–98. [DOI] [PubMed] [Google Scholar]
- 61. Jung HH, Danek A, Walker RH. Neuroacanthocytosis syndromes. Orphanet J Rare Dis 2011;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schneider SA, Lang AE, Moro E, Bader B, Danek A, Bhatia KP. Characteristic head drops and axial extension in advanced chorea‐acanthocytosis. Mov Disord 2010;25:1487–1491. [DOI] [PubMed] [Google Scholar]
- 63. Storch A, Kornhass M, Schwarz J. Testing for acanthocytosis A prospective reader‐blinded study in movement disorder patients. J Neurol 2005;252:84–90. [DOI] [PubMed] [Google Scholar]
- 64. Alawneh J, Baker MR, Young GR. Blood films in the investigation of chorea. Pract Neurol 2012;12:268. [DOI] [PubMed] [Google Scholar]
- 65. Henkel K, Walterfang M, Velakoulis D, et al. Volumetric neuroimaging in neuroacanthocytosis In: Walker RH, Saiki S, Danek A, eds. Neuroacanthocytosis Syndromes II. Berlin, Heidelberg, Germany: Springer; 2008:175–185. [Google Scholar]
- 66. Walker RH. Untangling the thorns: advances in the neuroacanthocytosis syndromes. J Mov Disord 2015;8:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Margolin DH, Kousi M, Chan YM, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med 2013;368:1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ganos C, Hersheson J, Adams M, Bhatia KP, Houlden H. The 4H syndrome due to RNF216 mutation. Parkinsonism Relat Disord 2015;21:1122–1123. [DOI] [PubMed] [Google Scholar]
- 69. Santens P, Van Damme T, Steyaert W, et al. RNF216 mutations as a novel cause of autosomal recessive Huntington‐like disorder. Neurology 2015;84:1760–1766. [DOI] [PubMed] [Google Scholar]
- 70. Tofaris GK, Irani SR, Cheeran BJ, Baker IW, Cader ZM, Vincent A. Immunotherapy‐responsive chorea as the presenting feature of LGI1‐antibody encephalitis. Neurology 2012;79:195–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Armstrong MJ, Miyasaki JM. Evidence‐based guideline: pharmacologic treatment of chorea in Huntington disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurology 2012;79:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lopez‐Sendon Moreno JL , Garcia‐Caldentey J, Regidor I, del Alamo M, de Garcia Yebenes J. A 5‐year follow‐up of deep brain stimulation in Huntington's disease. Parkinsonism Relat Disord 2014;20:260–261. [DOI] [PubMed] [Google Scholar]
- 73. Kostic VS, Dobricic V, Stankovic I, Ralic V, Stefanova E. C9orf72 expansion as a possible genetic cause of Huntington disease phenocopy syndrome. J Neurol 2014;261:1917–1921. [DOI] [PubMed] [Google Scholar]
- 74. Koutsis G, Karadima G, Kartanou C, Kladi A, Panas M. C9ORF72 hexanucleotide repeat expansions are a frequent cause of Huntington disease phenocopies in the Greek population. Neurobiol Aging 2015;36(547):e13–e16. [DOI] [PubMed] [Google Scholar]
- 75. Fernandez M, Raskind W, Wolff J, et al. Familial dyskinesia and facial myokymia (FDFM): a novel movement disorder. Ann Neurol 2001;49:486–492. [PubMed] [Google Scholar]
- 76. Chen YZ, Friedman JR, Chen DH, et al. Gain‐of‐function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol 2014;75:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Carapito R, Paul N, Untrau M, et al. A de novo ADCY5 mutation causes early‐onset autosomal dominant chorea and dystonia. Mov Disord 2015;30:423–427. [DOI] [PubMed] [Google Scholar]
- 78. Mencacci NE, Erro R, Wiethoff S, et al. ADCY5 mutations are another cause of benign hereditary chorea. Neurology 2015;85:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: This 20‐year old patient with severe generalized chorea with ballism and dystonia was found to carry mutations in the ADCY5 gene (M1029K). She is a member of the ChDys Family described by Chen et al. 2015.53